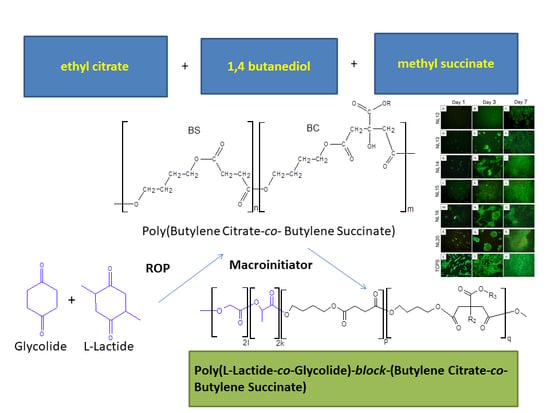
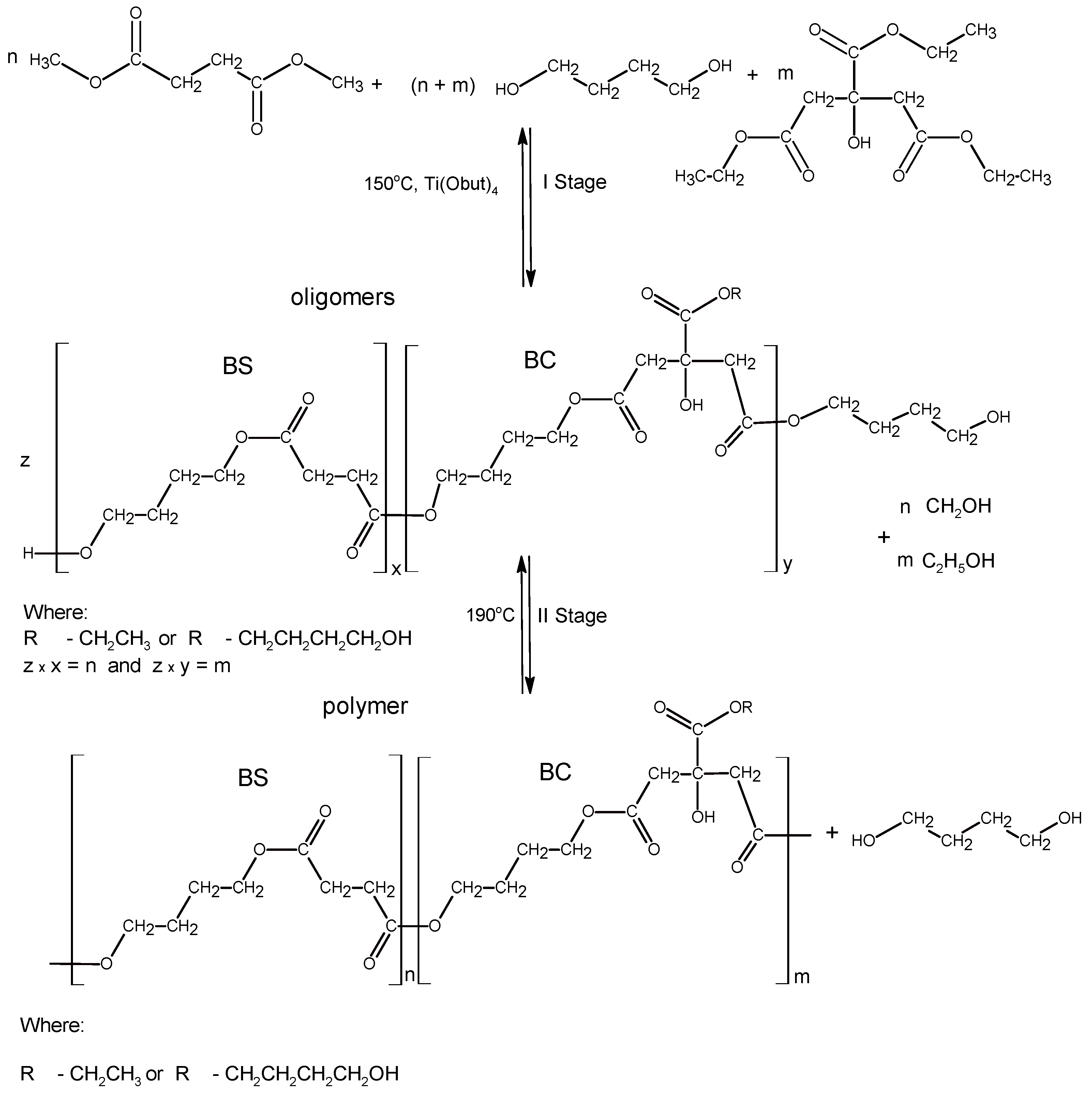
3.1. Synthesis and Characterization of Macro-Initiators—Poly(Butylene Succinate–co–Butylene Citrate)
In the first stage of research, poly(butylene succinate–
co–butylene citrate) and poly(butylene succinate) or poly(butylene citrate) were obtained by a typical polycondensation reaction carried out in bulk, according to
Scheme 1.
The aim of the synthesis was to obtain a series of polyesters, with an average molecular weight of 5–8 × 10
3 g/mol, which in the next stage could be used as macro-initiators in the reaction of
l-lactide polymerization or copolymerization of
l-lactide with glycolide. The polymerization was carried out in accordance with the ROP mechanism. For this reason, the synthesized polyesters had to contain at least two active hydroxyl groups ended the chain. The application of citric acid derivatives as monomers should additionally allow introducing pendant hydroxyl groups into the manufactured copolymer chain. The process was carried out with a molar ratio of 1,4-butanediol to acid esters from 1.07 to 1 in the presence of a Ti(OC
4H
9)
4 catalyst. It turned out that the above reaction conditions guaranteed obtaining non-crosslinked, soluble copolymers without a need to block the pendant hydroxyl groups of citrate ester. By changing the composition of the reaction mixture it is possible to obtain the oligoesters with different composition and molecular weights (
Table 1).
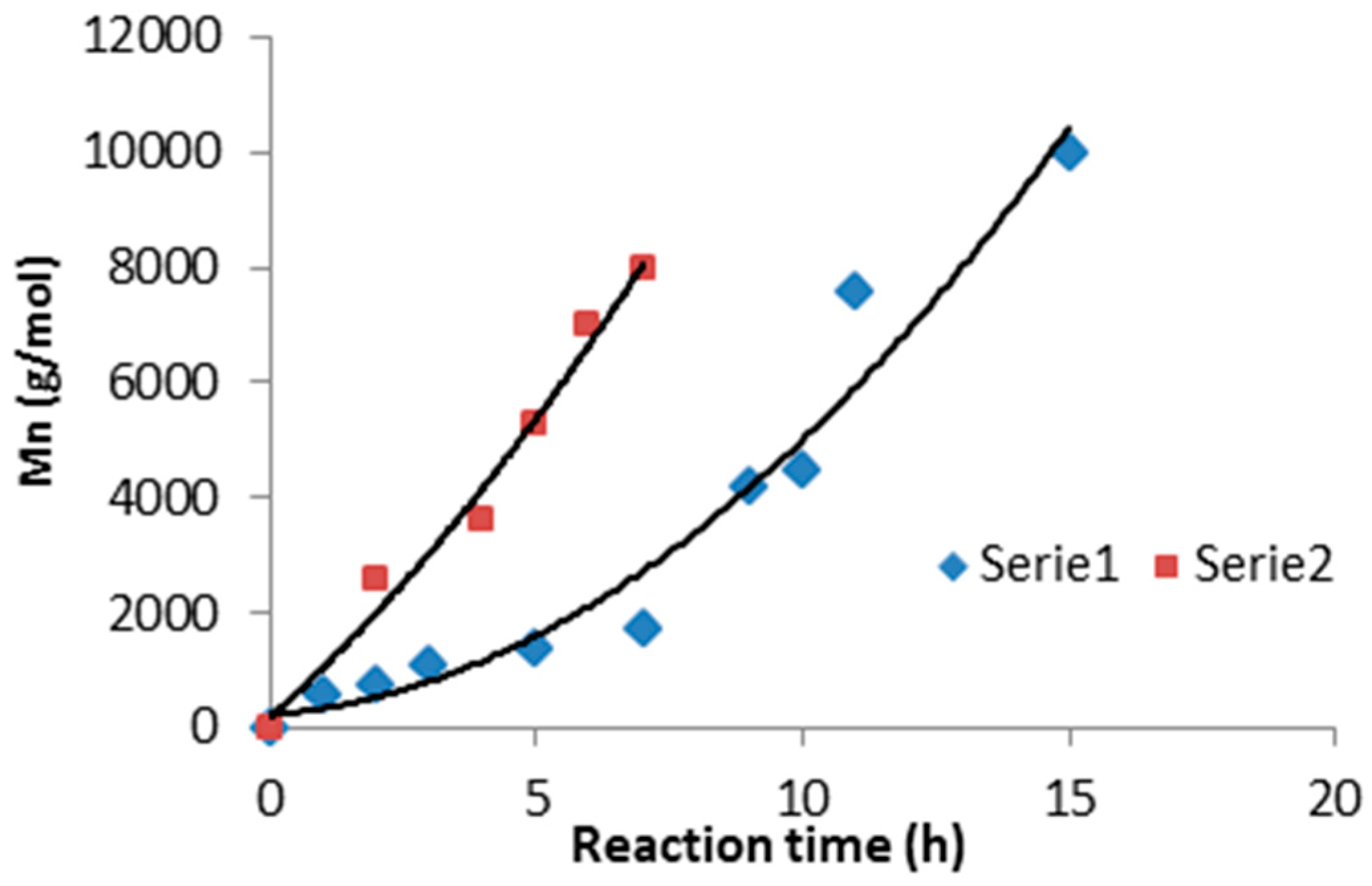
The first phase of transesterification consisted of obtaining butanediol esters at a temperature of 150 °C; this stage lasted about 1 h. Then, after reducing the pressure in the reactor vessel, the temperature was gradually increased up to 180–200 °C. At this temperature, polycondensation was continued until the assumed molar mass was obtained (
Figure 1).
Finally, poly(butylene succinate) with a mass of 7.5–8.0 × 10
3 g/mol was obtained after 6 h of reaction. In the case of obtaining poly(butylene citrate) with similar average mass, the reaction time was much longer and lasted about 13 h. Selected
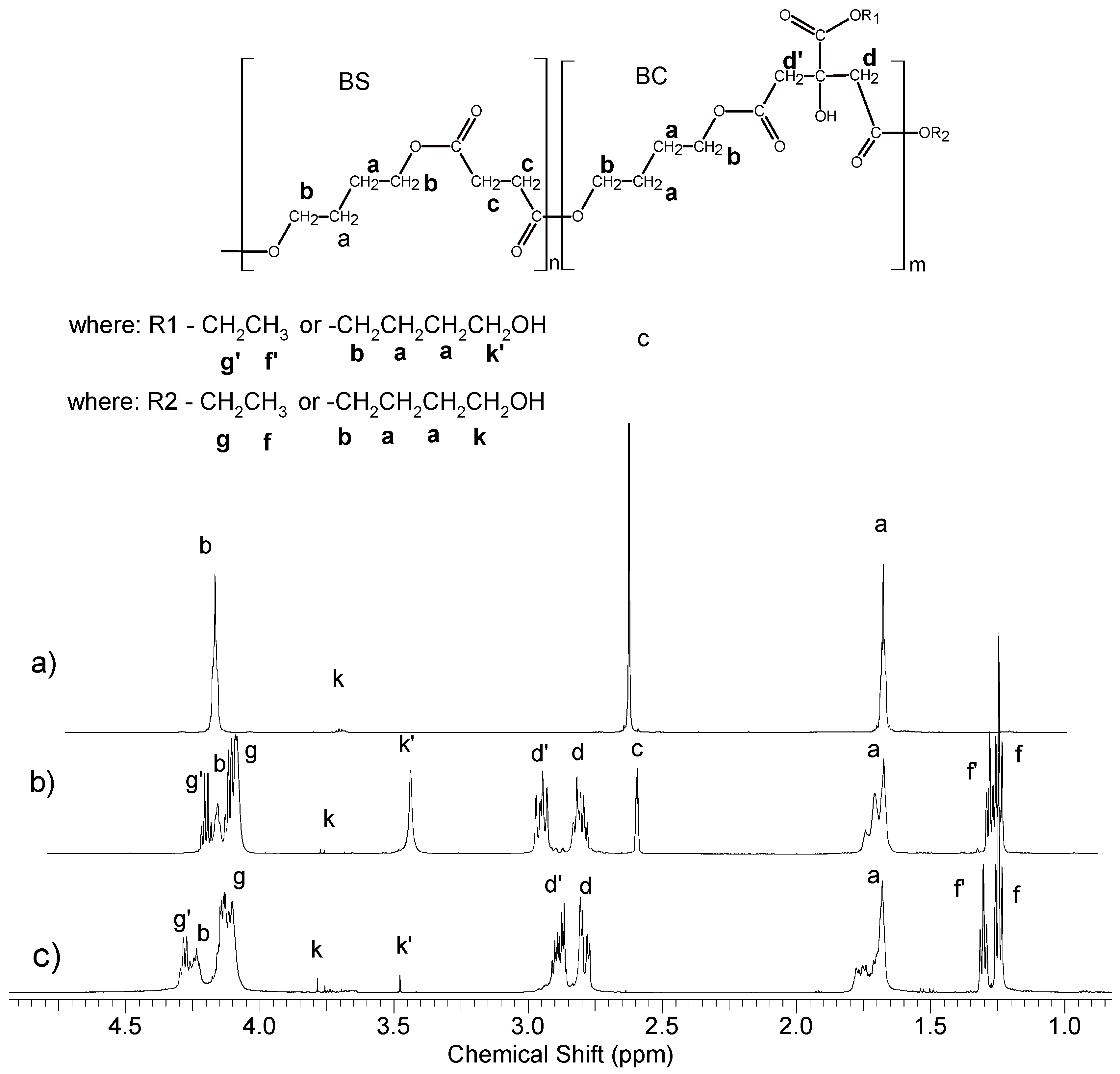
1H NMR spectra of oligomers along with the signal assignments are shown in
Figure 2. Based on the analysis of the signal intensity of the –C
H2-OH end groups appearing in the
1H NMR spectrum at δ = 3.6 ppm (marked as k in
Figure 2), their average molecular weight was determined. The composition of the oligomers did not differ significantly from the initial reaction mixture. Both, on proton and carbon spectra (
Figures S1 and S2 in Supplementary Materials) there was a lack of signals associated with the methyl group of the starting succinate ester. However, it was different in the case of oligomers containing citrate units, and especially in the case of poly(butylene citrate). Based on
1H NMR spectra (
Figure 2), it was found that 1/3 amount of the chain terminations still contains the starting ethyl ester groups (signals f and f’). Since these signals occur at various chemical shifts, they are both esters of the side groups as well as end groups in a quantity ratio of 1:2, respectively. Therefore, the chains growth runs in large part on the ester end groups of the starting ethyl citrate, but also on the side ester groups. The reason for this phenomenon is the non-stoichiometric amount of used 1,4 butanediol (
Table 1). In the case of attempts to increase the amount of this diol the crosslinked insoluble product was obtained.
For polymers containing butylene citrate units, it was necessary to clarify whether the hydroxyl side group of this compound is still present and active after polymerization. For this purpose, additional
1H NMR measurements of the obtained compounds after reaction with trichloroacetyl isocyanate were performed. The total shifts of the methyl signals of butylene citrate units –CH
2C(OH)(COOR)CH
2– (at δ = 2.81 ppm and δ = 2.87 ppm, signal d, d—
Figure 2) in the direction of δ = 3.32 ppm and 3.25 ppm were observed (
Figure S3 in Supplementary Materials). This phenomenon was caused by the reaction of the isocyanate group with active side hydroxyl groups, to form urethane bonds. This means that all side OH groups did not undergo changes during polymerization and remained active. Using the titration analysis, the amounts of OH groups in individual oligomers were estimated (
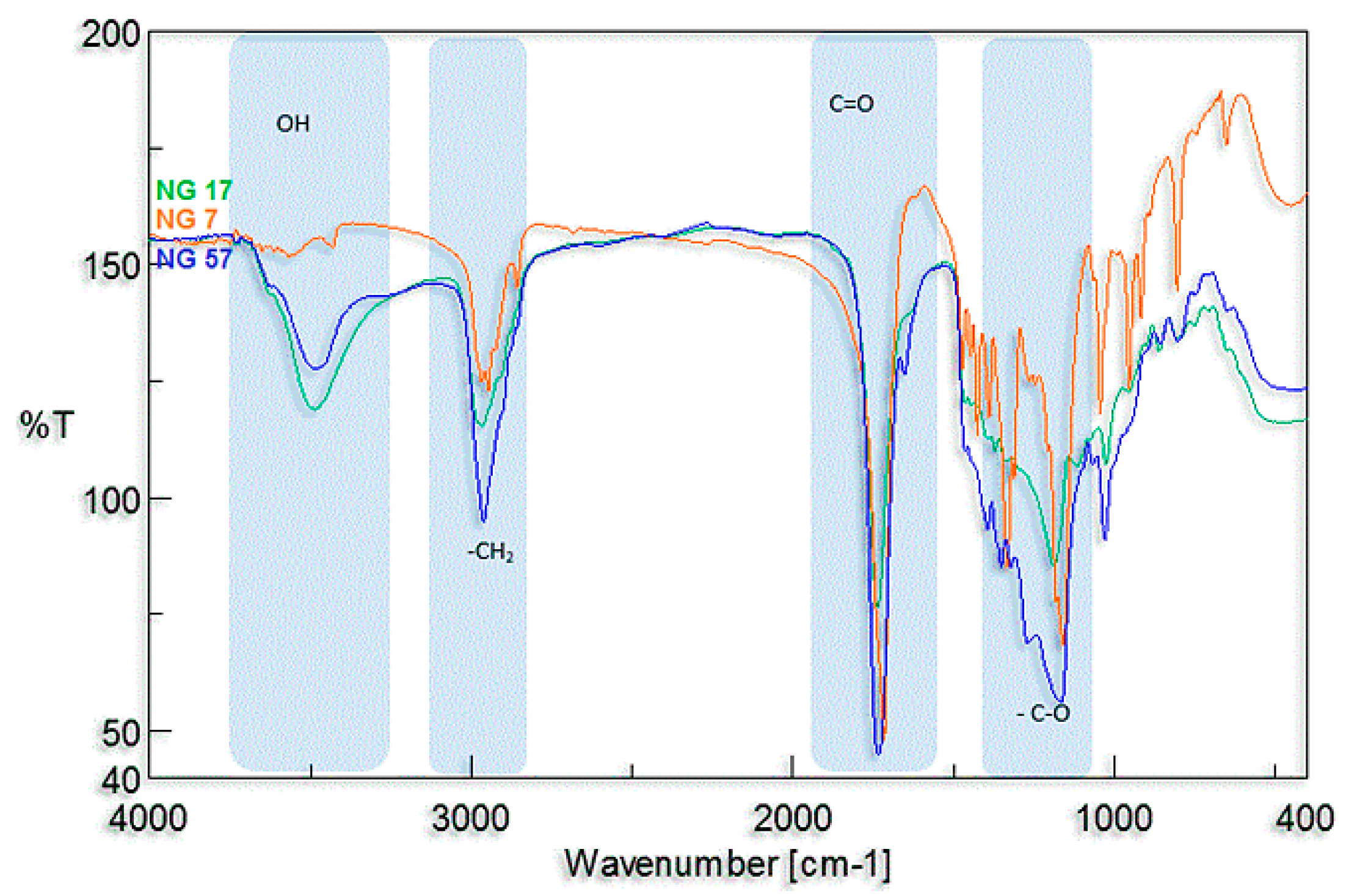
Table 1). The obtained results confirmed the presence of active side hydroxyl groups too. The presence of these groups has also been confirmed with infrared spectroscopy. In the FTIR spectra, a distinct characteristic absorption band was observed in the range of 3300–3000 cm
−1 corresponding to the stretching vibrations of hydroxyl groups (
Figure 3). For poly(butylene succinate–
co–butylene citrate) and poly(butylene citrate) the observed bands were much stronger than for poly(butylene succinate). The observed differences result from the existence of an additional side hydroxyl group in the butylene citrate units in the copolymer chain. The appearance of stronger band depends on the concentration of citrate units, which definitely confirms previous observations that hydroxyl groups of started ethyl citrate do not participate in the conducted polytransesterification.
The influence of the composition and chain structure of the obtained polymers on thermal properties was examined with DSC measurements. Poly(butyl succinate) samples showed the highest degree of crystallinity (fusion enthalpy ΔH about 80 J/g). With the increased content of butylene citrate units in the copolymer chain, the semi-crystallinity of such material gradually decreased (
Table 1). Samples with content of above 50% mol citrate units were completely amorphous. The main reason for this phenomenon is the observed growth of the number of branched structures of chain, with an increase in the share of citrate units.
For poly(butylene succinate) samples, the GPC elugram (
Figure S4 in Supplementary Materials, NG 6) shows relatively low dispersion of molecular weight. For the other polymers containing citrate units in the chain, their molecular mass dispersion significantly increased, however all these polymers were completely soluble.
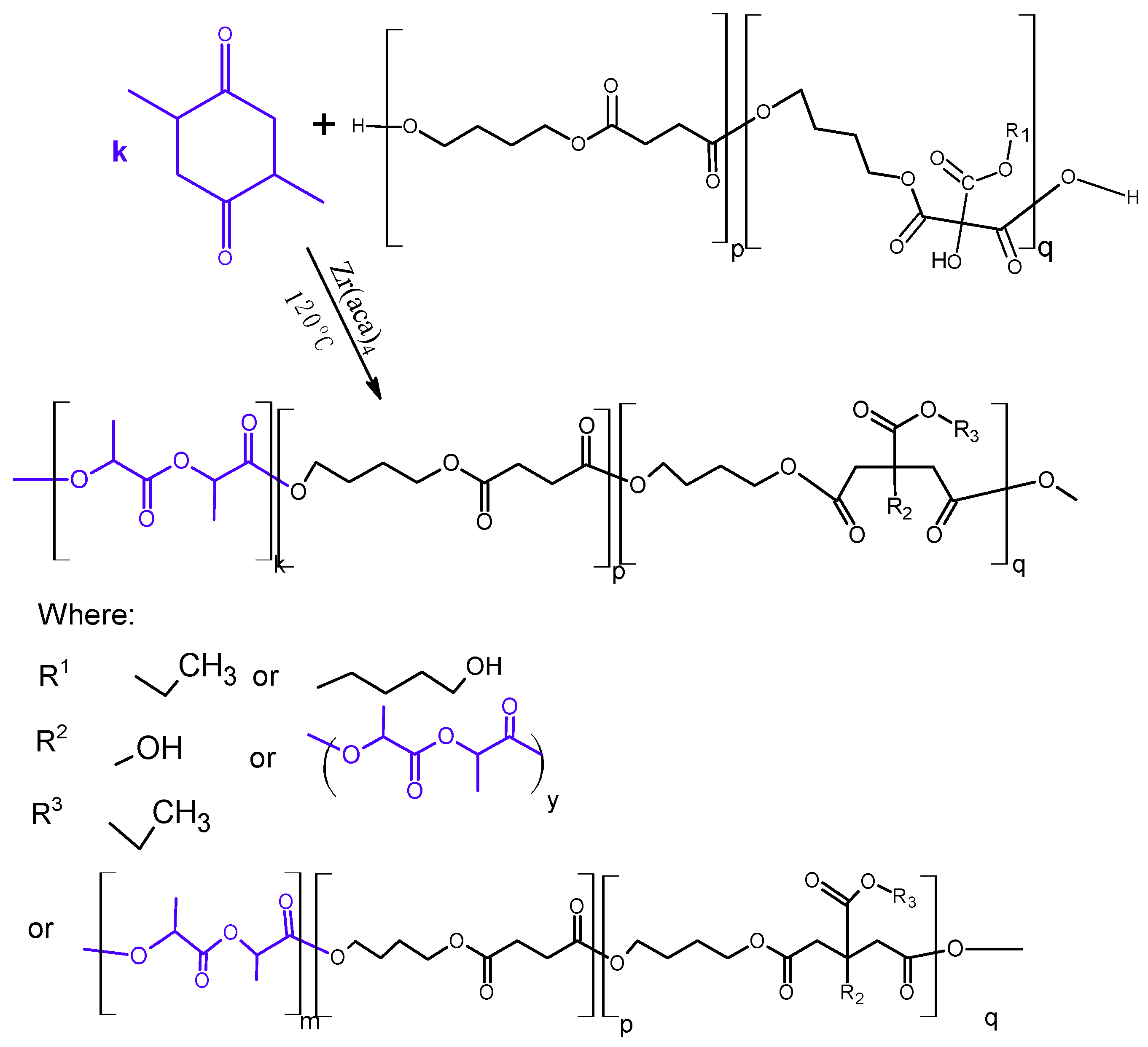
3.2. Synthesis and Characterization of l-Lactide Copolymers with Butylene Succinate and Butylene Citrate
Then, the obtained polyesters (
Table 1) were used as macroinitiators for the
l-lactide polymerization, thus obtaining triblock copolymers (
Scheme 2). Polymerization was carried out in bulk with using zirconium(IV) acetylacetonate as a catalyst, a compound previously tested as an effective, non-toxic catalyst or initiator of the ROP reaction of lactides and lactones [
18,
19]. The choice of this catalyst was also driven by the fact that the compound was active in the ROP polymerization even in the presence of organic acids [
20]. So, the use of such a catalyst guarantees the possibility of conducting polymerization in the presence of compounds containing carboxylate or carboxyl groups, which may be the reaction by-products.
During the polymerization of
l-lactide in the presence of poly(butylene succinate), a high conversion of this monomer was obtained regardless of the composition of the starting reaction mixture (
Table 2). The average molecular weights were close to the predicted. DSC tests (
Figures S5 and S6 in Supplementary Materials) showed a high degree of crystallinity for all synthetized
l-lactide copolymers. The copolymer containing 80%
l-lactidyl units presented crystallinity regions associated with the arrangement of lactidyl blocks. These domains melted at 165 °C (
Table 2, NG7). In the case of an increase amount of succinate units forming chain block, appearance of two melting enthalpies is observed. The one related to the melting of domains formed from butyl succinate is at 108 °C and the second derived from domains of lactidyl blocks melting at 150 °C (
Figure S5b in Supplementary Materials) was observed. In turn in a copolymer with a low content of lactidyl at 108 °C we observed only one enthalpy of melting of areas formed by units of butylene succinate. In this case, the length of the lactidyl blocks was too small to be able to form crystalline areas.
Polymerization of
l-lactide, carried out in the presence of macroinitiators containing both butylene succinate and butylene citrate units in the chain (polyesters NG 17 and NG 49) was significantly slower than previously. About 90% of conversion of
l-lactide was achieved after 140 h (
Figure S7 in Supplementary Materials). When the macroinitiator with low content of citrate units was used, the obtained
l-lactide copolymer possessed a composition similar to the starting reaction mixture, and its molecular weight was slightly lower than the theoretical one (
Table 3, NG20).
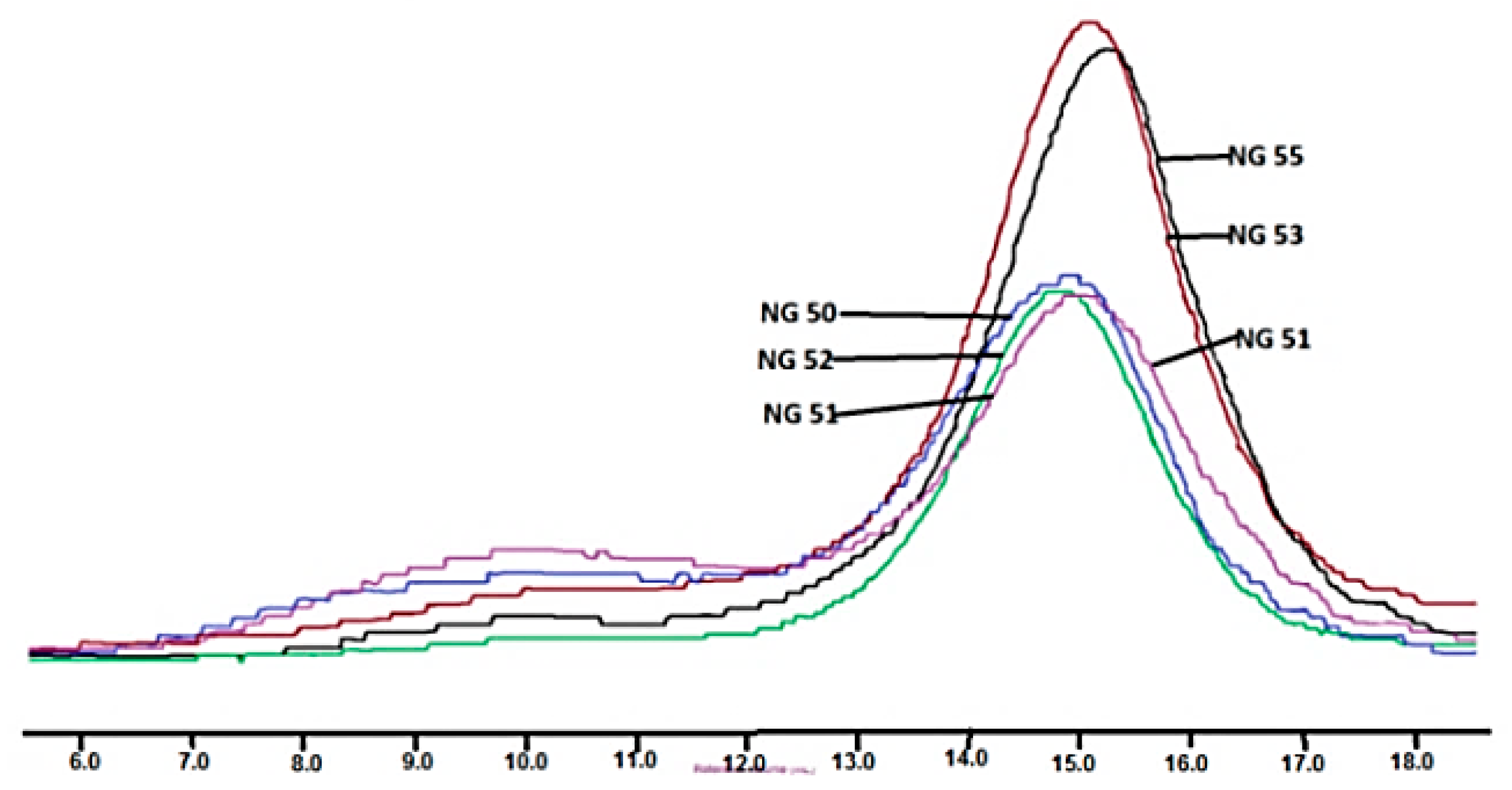
However, when macroinitiators contained a significantly higher amount of citrate units the result was different. In this case, chloroform soluble terpolymers were obtained, but with unexpectedly high inherent viscosity, enormous weight average molecular weight and very high molecular mass dispersion (
Table 3, NG 51, NG53 and NG55). The reason for this phenomenon was the existence of a high molecular mass phase (reaching several hundred thousand g/mol) observed on GPC elugram (
Figure 4).
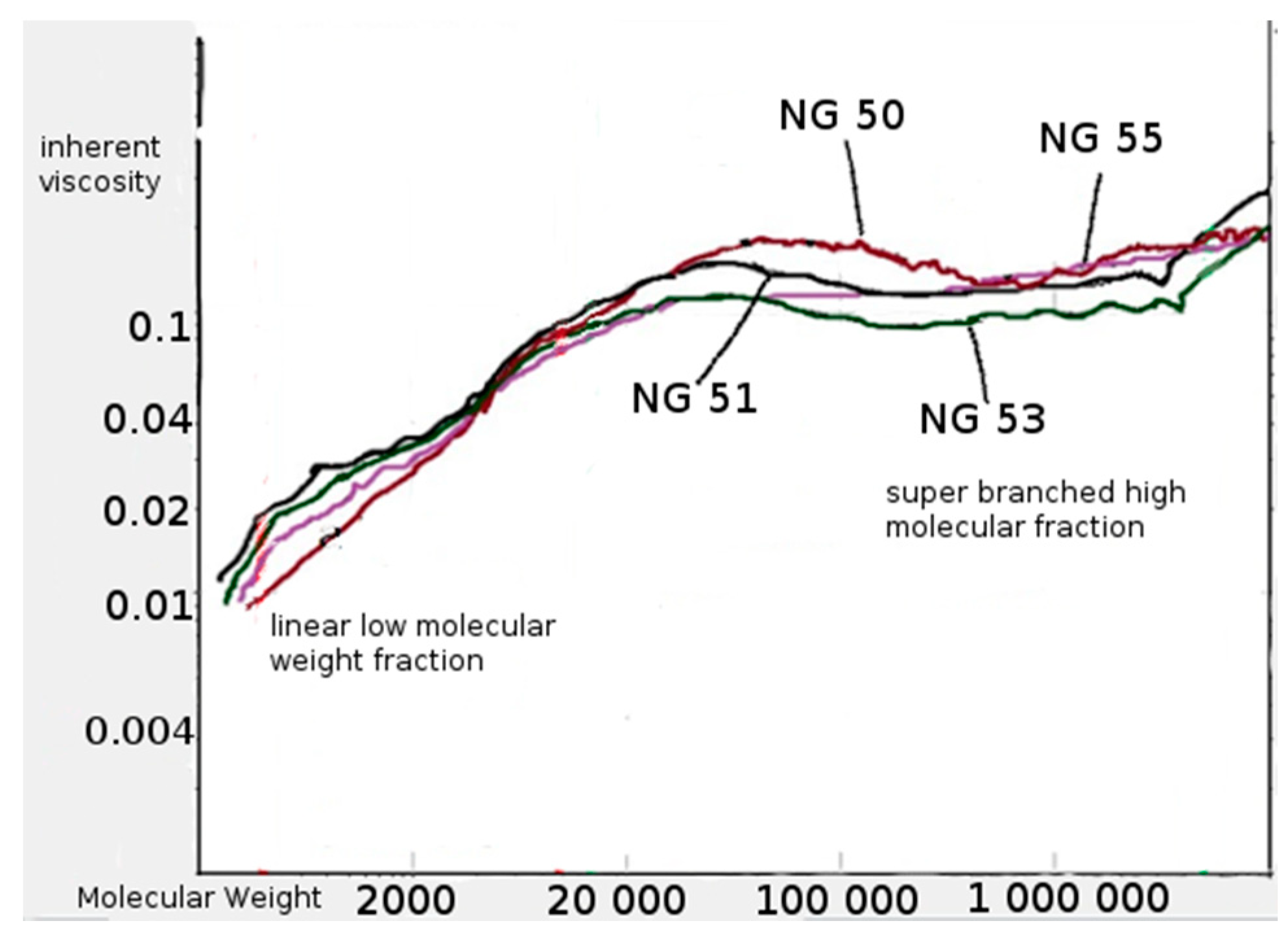
The supplementary GPC tests were carried out using an additional viscosity detector. The polymer fraction with very high
Mw, i.e. between 200 × 10
3 g/mol and 2–3 × 10
6 g/mol was found in the polymerization products. The solution of this fraction, regardless of molecular weight, presented practically the same intrinsic viscosity (
Figure 5).
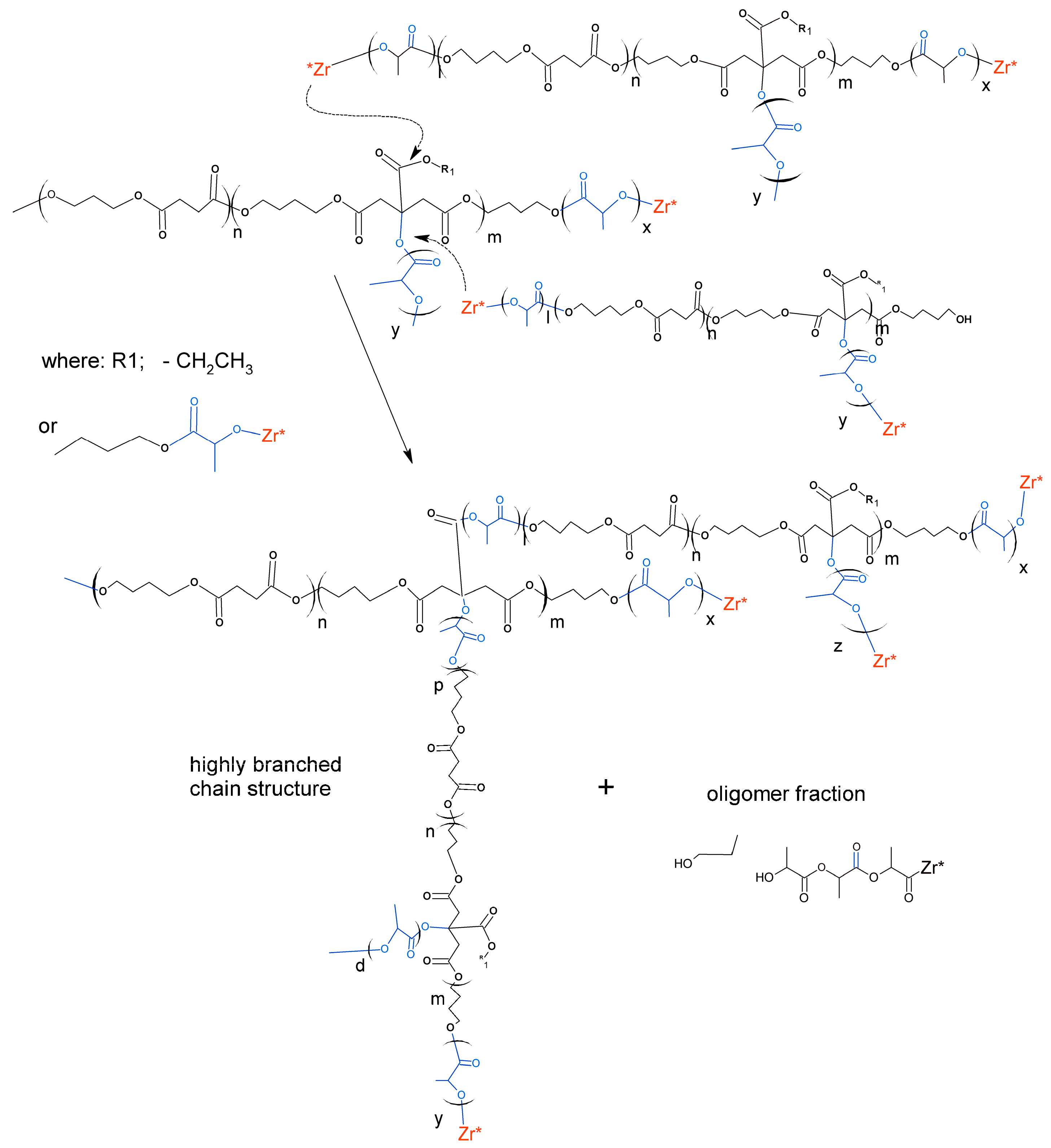
The observed phenomenon indicates a specific, highly branched chain structure of polymers belonging to the discussed high molecular fraction of products [
21]. The low molecular fraction with
Mw from several hundred g/mol to about 150,000 g/mol showed compliance with the Mark–Houwink equation, so it was a fraction of linear chain copolyesters. The products with a very high mass and highly branched chain structure are formed as a result of intermolecular transesterification that occurs in parallel with the main chain growth reaction. This process arrives as a result of attacks of the active ends of the growing chain on the ester groups of the side lactidyl chains formed on the tertiary hydroxyl groups of the citrate units. For the above reason, the intensity of discussed transesterification depends on the content of citrate units in the chain of used macroinitiator. As a result of the transesterification, the molecules merge with each other, resulting in a rapid increase in the mass of associated molecules and the formation of complex branched chain structures. At the same time, short linear oligomers of polylactide and oligomers containing ethyl ester group are created as illustrated in
Scheme 3.
Mw from several hundred g/mol to about 150,000 g/mol showed compliance with the Mark–Houwink equation, so it was a fraction of linear chain copolyesters. The products with a very high mass and highly branched chain structure are formed as a result of intermolecular transesterification that occurs in parallel with the main chain growth reaction. This process arrives as a result of attacks of the active ends of the growing chain on the ester groups of the side lactidyl chains formed on the tertiary hydroxyl groups of the citrate units. For the above reason, the intensity of discussed transesterification depends on the content of citrate units in the chain of used macroinitiator. As a result of the transesterification, the molecules merge with each other, resulting in a rapid increase in the mass of associated molecules and the formation of complex branched chain structures. At the same time, short linear oligomers of polylactide and oligomers containing ethyl ester group are created as illustrated in
Scheme 3.
Previously we described a very similar and analogical phenomenon, which proceeded during the polymerization of cyclic carbonates containing side ester groups [
22]. A mechanism of transesterification in which the attack of active ends is directed to the ester bonds of the main chain is also very probable. However, in the
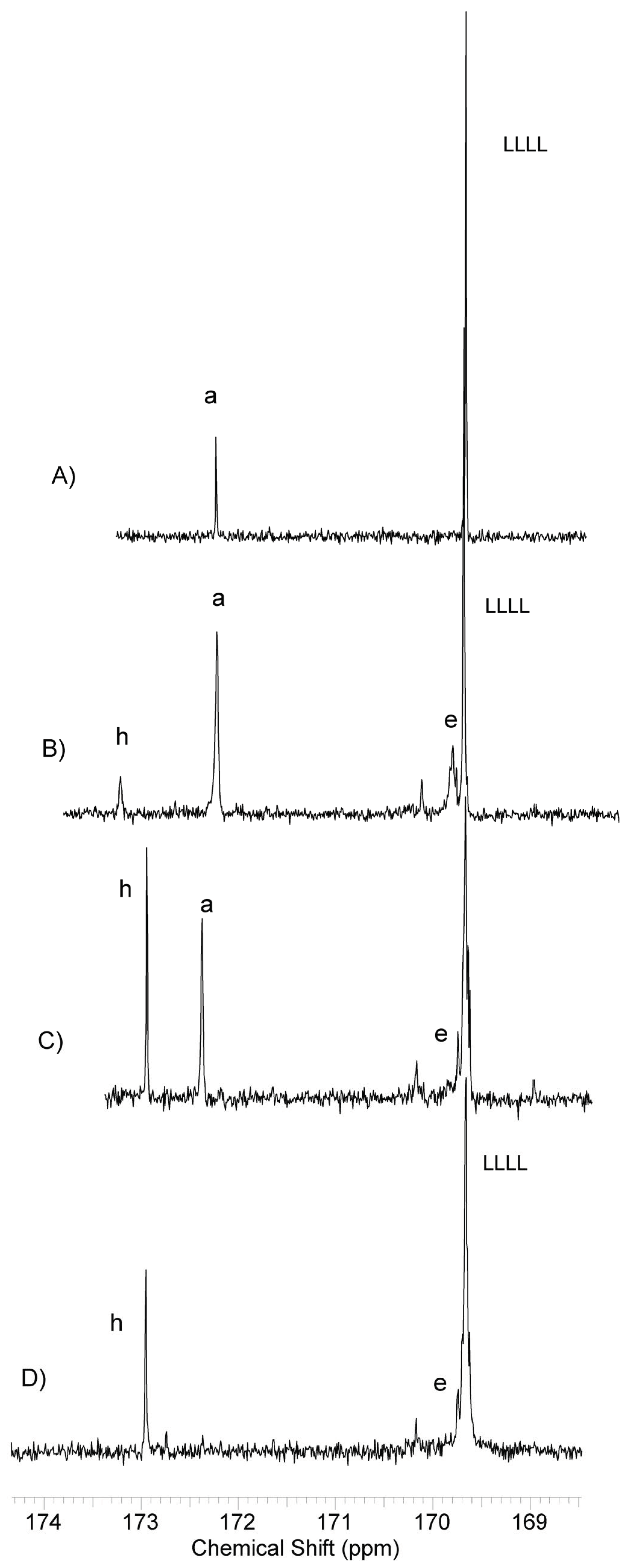
13C NMR spectra, there are no signals that may originate from hypothetically formed on this way lactidyl-citrate or lactidyl-succinate sequences (
Figure 6).
The carbonyl region signals of the citrate and succinate units are not split. Comparing the spectra of samples containing citrate units (
Figure 6D) with the analogue containing only lactidyl block and butyl succinate, it can be seen that the apparent LLLL signal split is rather an effect of overlapping with a signal assigned to carbon of citrate carbonyl. However, an attack of active lactide ends of the growing chain on ester bonds of lactidyl blocks of adjacent molecules is possible. Such transesterification can lead to the formation of multi-block copolymers and shorter active lactidyl chains without visible changes in observed NMR spectra. Therefore, for all obtained copolymers it is impossible to definitely say that they have a three-block structure.
The composition of the final copolymers was determined on the basis of
1H NMR spectra analysis.
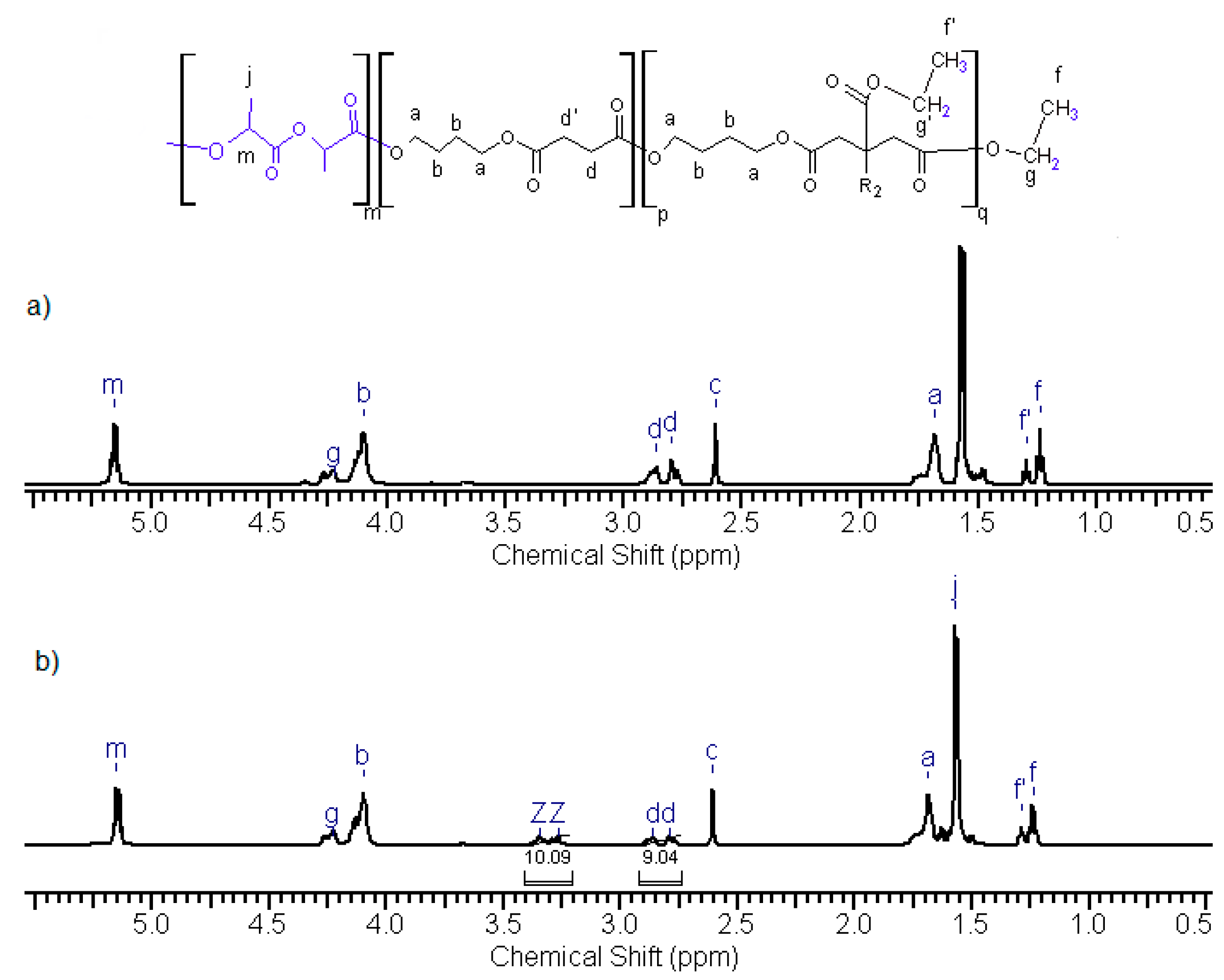
Figure 7 shows as example selected proton NMR spectra of poly (
l-lactide)–
block–poly(butylene succinate–
co–butylene citrate). In this spectrum, signals in the range δ = 5.18 ppm (m, 4H) and 1.6 ppm (j, 6H) are derived from protons of lactidyl units; –C
H– and C
H3 groups. Other signals are related to the presence of citrate and succinate units derived from the macroinitiator—poly (butylene succinate–
co–butylene citrate). These signals are the same as it was previously observed in the spectrum of macroinitiator before the lactide polymerization. In order to check the possible presence of active side hydroxyl groups, the additional
1H NMR analysis was performed after reaction of the samples of copolyesters with trifluoroacetyl isocyanate. A shift of a part of the signals of the methylene protons –
CH2 (OH)(COOR) C
H2– (citrate units) was detected; from the initial δ = 2.81–2.87 ppm (
Figure 7a, denoted as d, d) to δ = 3.25–3.32ppm (
Figure 7b marked as Z, Z). Based on the resulting changes, the content of active side hydroxyl groups in the chain of obtained copolymers was estimated, which in most of the tested samples was about 50%–60% of the number of starting groups. Thus, during the polymerization, only about half of these groups reacted with lactide, forming side polylactide chains. These chains have been involved in observed intermolecular transesterification reactions discussed earlier.
Analysis of DSC curves of the obtained copolyesters shows a strong dependence of their composition on the thermal properties. NG 20 copolymer containing 55% mol of lactidyl groups and 29% succinate units, is characterized by the presence of two enthalpies of melting (
Table 3), one associated with ordered lactidyl domains (at 115 °C) and second derived from succinate blocks (at 40 °C). As the amount of citrate units in the chain increases (at the expense of succinate units,
Table 3, NG 51) only enthalpy of melting of organized lactidyl structures is observed. In contrast, copolymers made of
l-lactide and butylene citrate blocks, i.e., NG 53 and NG 55, are completely amorphous. The glass transition temperature drops strongly as the amount of butyl citrate units in the copolymer chain increases. The observed phenomenon is associated with the increase in the branched chain structures of these copolymers.
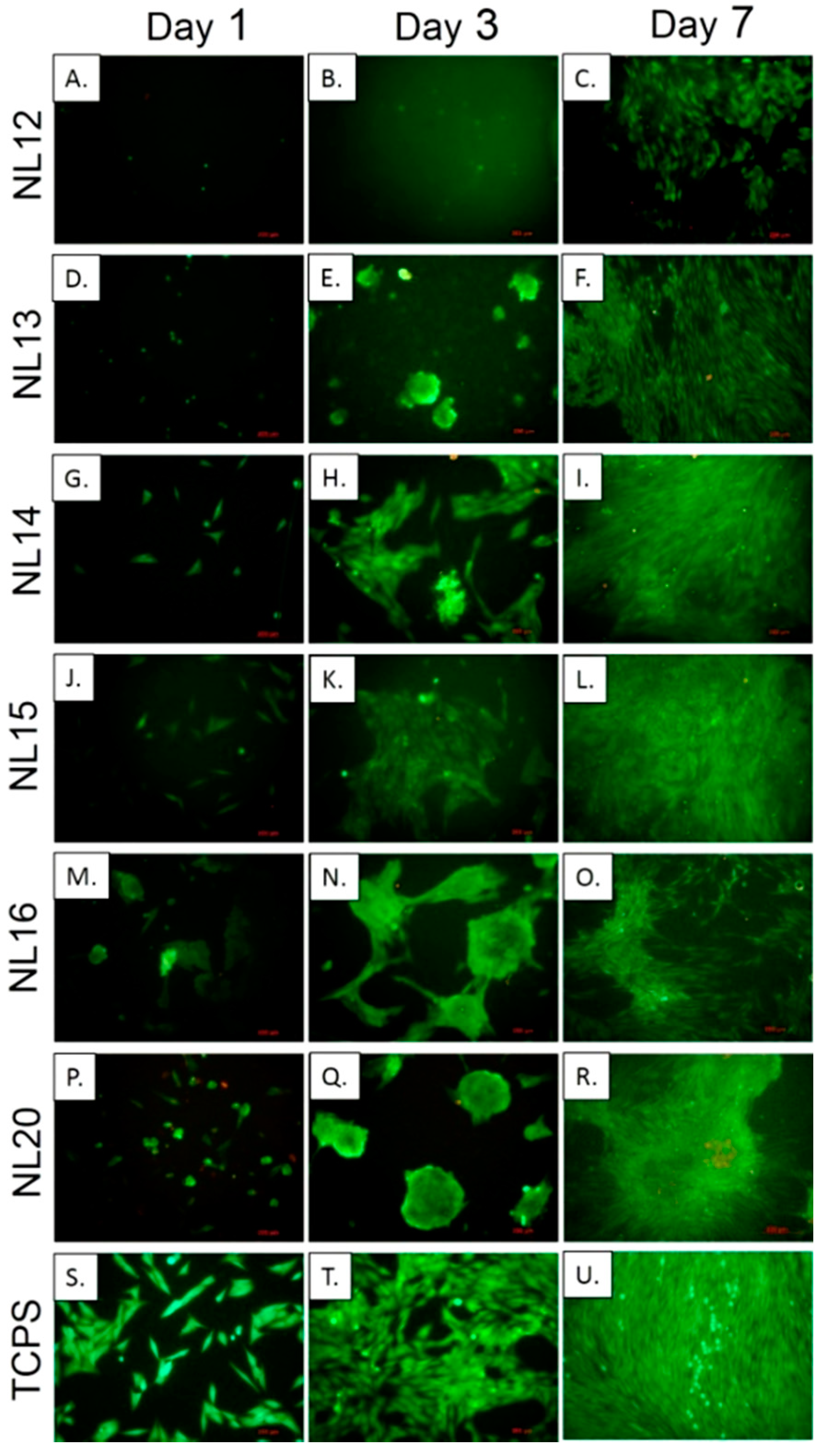
3.3. Synthesis and Characterization of Poly (l-lactide–co–glycolide)–block–poly(butylene succinate–co–butylene citrate)
Due to the relatively high crystallinity of the previously described copolymers containing longer
l-lactide blocks, attempts were made to obtain analogous copolymers but with lower, containing the chain blocks made of
l-lactide and glycolide random sequences. Such terpolymers appear to be better as a potential material for forming scaffolds. The terpolymers were successfully obtained by copolymerization of
l-lactide and glycolide carried out in the presence of poly(butylene succinate–
co–butylene citrate) with an average molecular weight
Mn of about 5000 g/mol, where the molar ratio of BS units to BC was as: 30:70 or 50:50 (
Table 4,
Scheme S1 in Supplementary Materials). Similarly to the previously described polymerization of
l-lactide, after about 6 days a high conversion of monomers was obtained.
Based on the obtained
1H NMR spectra (
Figure 8), the actual copolymer composition and monomer conversion were determined (
Table 4). The proton spectra differed from those described earlier only by the presence of an additional signal of glycolyl methylene protons (
Figure 8, signal n). Quite unexpectedly, even with relatively high contents of citrate units in the copolymer chain, the phenomenon of formation of a highly branched fraction was not observed (
Table 4, NL20, NL 12–13). In the obtained copolymers virtually all OH groups present at tertiary carbon in citrate units of the chain were active in the reaction with isocyanate. After reaction of the resulting product with trichloroacetyl isocyanate the signals of methylene groups of citric acid derivative—dd were completely shifted to signals ZZ. This means that side hydroxyls did not participate in the copolymerization reaction and forming side chains. So, forming high molecular fractions according to transesterification mechanism presented on
Scheme 3 was rather impossible.
Analysis of
13C NMR spectra in the range of carbonyl carbons showed the presence of the same signals as in the case of the previously presented
l-lactide/butyl succinate/butyl citrate copolymers (
Figure 9). Main signals are derived from the carbons of succinate (signal a) and citrate units (signals e and h) as well as lactidyl (LLLL) and additionally glycolide (GGGG or GGLL). Only the carbon signal LLLL was clearly split, as well as the signal associated with the presence of glycolidyl units (GGGG and GGLL signals), analogically as it was the case of poly(
l-lactide–
co–glycolide) or poly(lactide–
co–glycolide)–
block–poly(ethylene glycol) copolymers [
18,
23]. The absence of additional signals that could be derived from shorter chain sequences suggests that the chains of received copolymers have a block chain structure.
In the DSC thermograms of these copolymers presence of two distinct melting enthalpies associated with ordered structured areas were observed; the larger built with the succinate units blocks (at 78–105 °C) and the much smaller created with the lactidyl units blocks (at 113–139 °C). Compared to previously synthesized
l-lactide/butyl succinate/butyl citrate copolymers, there is a significant decrease in the crystallinity of lactidyl domains, which is associated with the presence of the glycolyl-lactidyl sequence in the chain, causing a shortening of the length of lactidyl blocks. The glass transition temperatures are clearly higher, which is mainly due to a practically lack of copolymer fraction with a strong branched structure of the chain (
Table 4).

 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}