
Characterization of Promising Cytotoxic Metabolites from Tabebuia guayacan Hemsl.: Computational Prediction and In Vitro Testing

 ,
,  , and
, and
Abstract
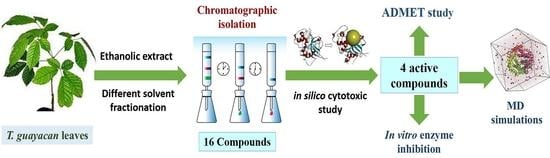
:
1. Introduction
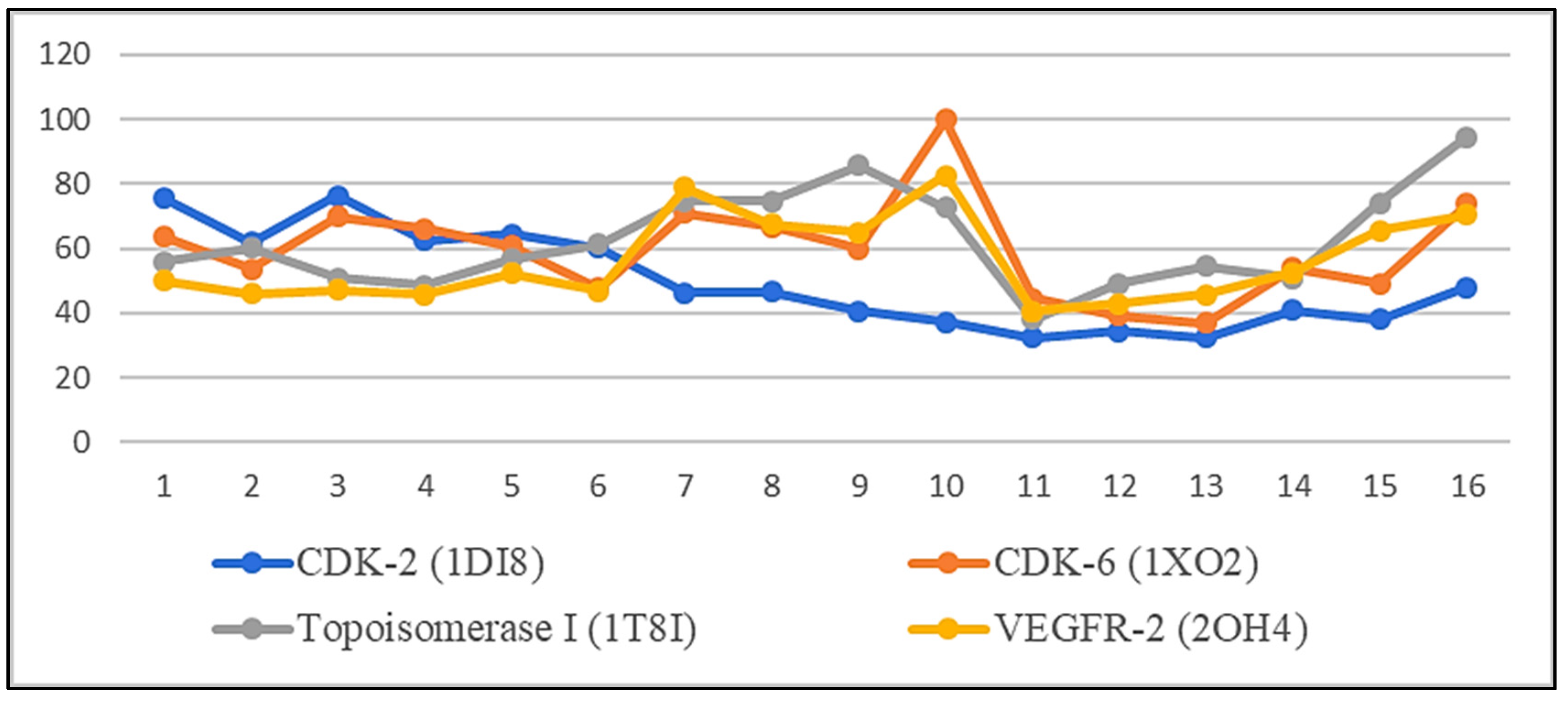
2. Results
2.1. Compounds Identification
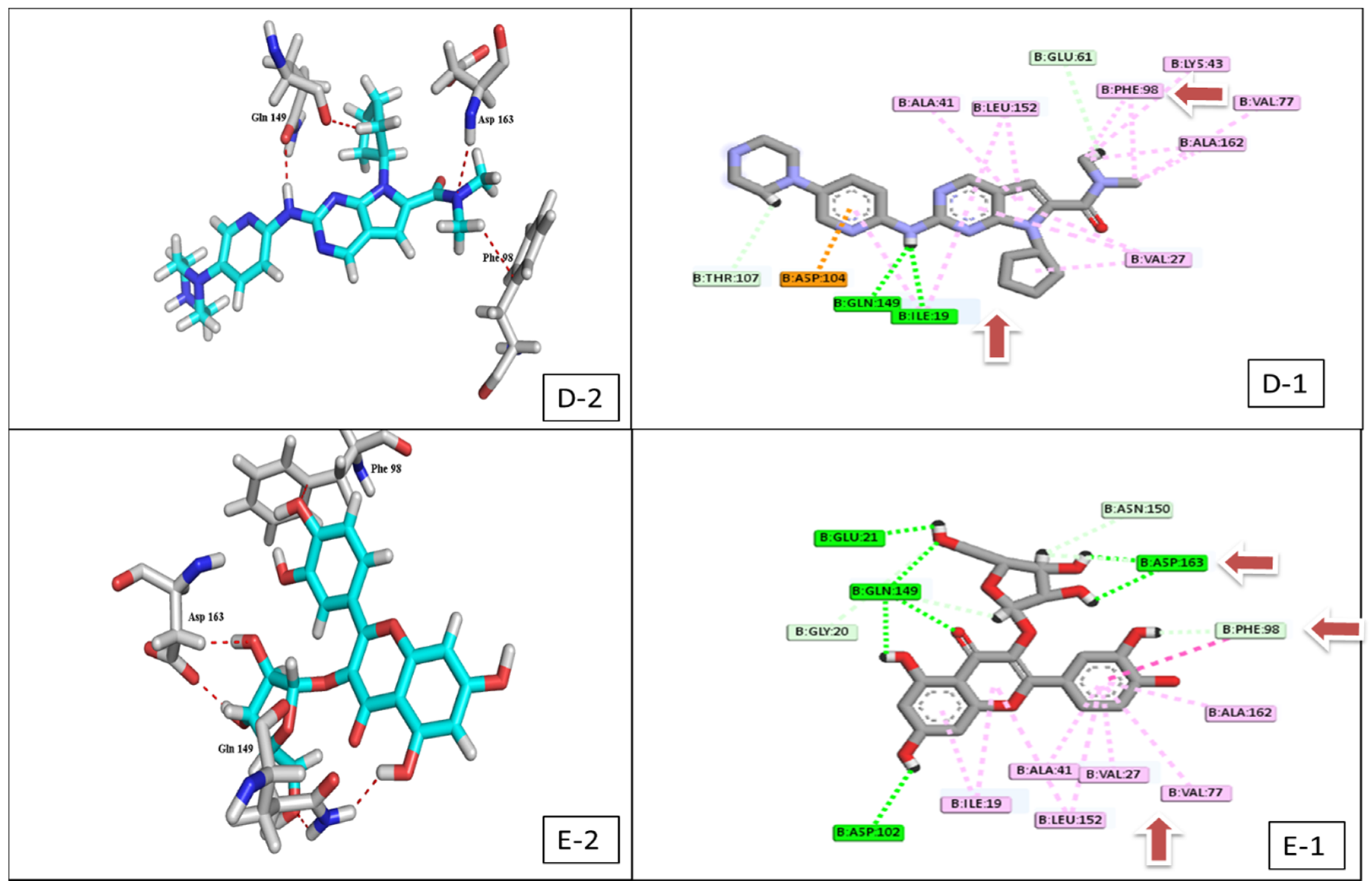
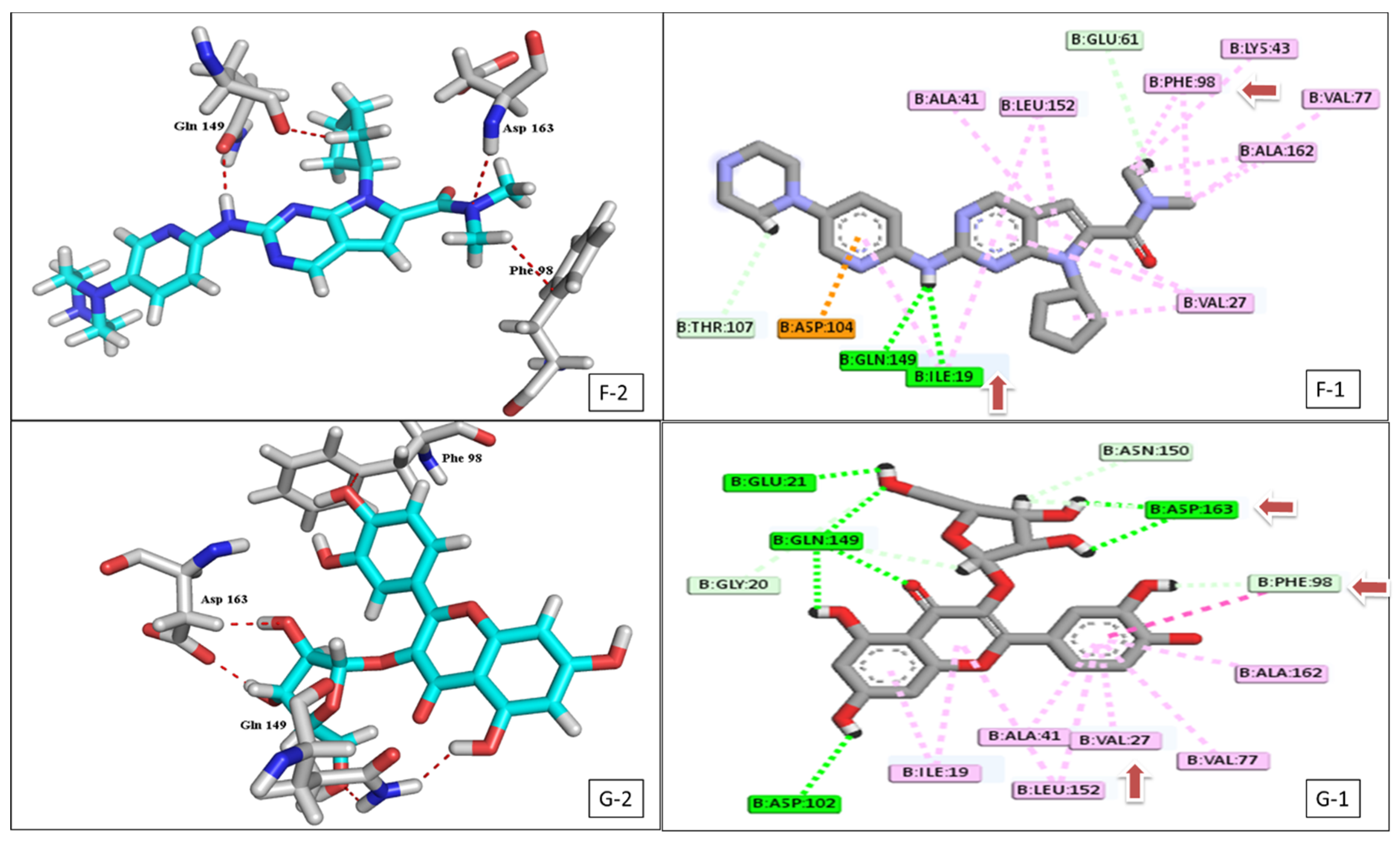
2.2. Docking Study
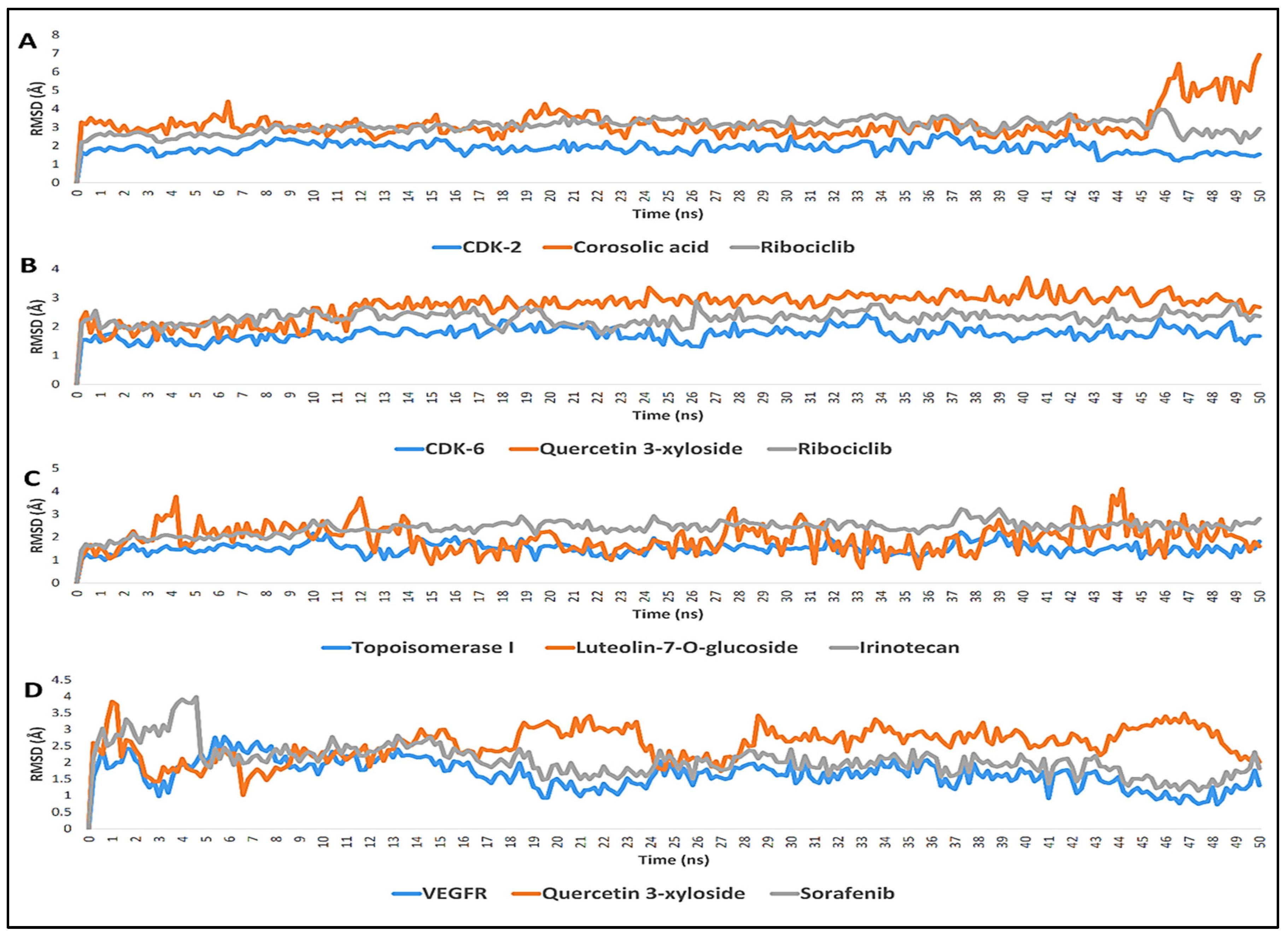
2.3. Molecular Dynamic Simulation
2.4. In Vitro Enzyme Inhibition
2.5. ADMET Properties Evaluation
3. Discussion
4. Materials and Methods
4.1. Collection of Plant Material
4.2. Chemicals
4.3. Chromatographic Materials
4.4. General Experimental Procedures
4.5. Extraction and Isolation Procedure
4.6. Docking Study
4.6.1. Ligand Preparation
4.6.2. Docking Method Validation
4.7. Molecular Dynamic Simulation
4.8. In Vitro Enzyme Inhibition
4.8.1. In Vitro CDK-2 and VEGFR-2 Inhibitory Activity
4.8.2. In Vitro CDK-6 Inhibitory Activity
4.8.3. Statistical Analysis
4.9. Prediction of the Pharmacokinetic Properties and Toxicological Properties Using ADMET
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Shawky, A.M.; Ibrahim, N.A.; Abourehab, M.A.; Abdalla, A.N.; Gouda, A.M. Pharmacophore-based virtual screening, synthesis, biological evaluation, and molecular docking study of novel pyrrolizines bearing urea/thiourea moieties with potential cytotoxicity and CDK inhibitory activities. J. Enzym. Inhib. Med. Chem. 2021, 36, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-dependent kinase 2 inhibitors in cancer therapy: An update. J. Med. Chem. 2018, 62, 4233–4251. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, M.; Khan, P.; Shamsi, A.; Shahbaaz, M.; Hasan, G.M.; Haque, Q.M.R.; Hassan, M.I. Inhibiting CDK6 activity by quercetin is an attractive strategy for cancer therapy. ACS Omega 2020, 5, 27480–27491. [Google Scholar] [CrossRef] [PubMed]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer Res. 2020, 147, 2988–2995. [Google Scholar] [CrossRef] [PubMed]
- Riess, C.; Irmscher, N.; Salewski, I.; Strüder, D.; Classen, C.F.; Große-Thie, C.; Maletzki, C. Cyclin-dependent kinase inhibitors in head and neck cancer and glioblastoma—Backbone or add-on in immune-oncology. Cancer Metastasis Rev. 2021, 40, 153–171. [Google Scholar] [CrossRef]
- Hoff, P.M.; Machado, K.K. Role of angiogenesis in the pathogenesis of cancer. Cancer Treat. Rev. 2012, 38, 825–833. [Google Scholar] [CrossRef]
- Huang, L.; Huang, Z.; Bai, Z.; Xie, R.; Sun, L.; Lin, K. Development and strategies of VEGFR-2/KDR inhibitors. Future Med. Chem. 2012, 4, 1839–1852. [Google Scholar] [CrossRef]
- Moukharskaya, J.; Verschraegen, C. Topoisomerase 1 inhibitors and cancer therapy. Hematol. Oncol. Clin. 2012, 26, 507–525. [Google Scholar] [CrossRef]
- Solowey, E.; Lichtenstein, M.; Sallon, S.; Paavilainen, H.; Solowey, E.; Lorberboum-Galski, H. Evaluating medicinal plants for anticancer activity. Sci. World J. 2014, 2014, 721402. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, E.R.; da Silva Filho, A.A.; Dos Santos, E.R.; Lopes, C.A.C. Antiinflammatory action of lapachol. J. Ethnopharmacol. 1990, 29, 239–241. [Google Scholar] [CrossRef]
- Castellanos, J.R.G.; Prieto, J.M.; Heinrich, M. Red Lapacho (Tabebuia impetiginosa)—A global ethnopharmacological commodity. J. Ethnopharmacol. 2009, 121, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Júnior, J.C.; Conserva, L.M.; Lemos, R.P.L.; de Omena-Neta, G.C.; Cavalcante-Neto, A.; Barreto, E. Isolation of a dihydrobenzofuran lignan, icariside E 4, with an antinociceptive effect from Tabebuia roseo-alba (Ridley) Sandwith (Bignoniaceae) bark. Arch. Pharm. Res. 2015, 38, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Nirmala, M.J.; Samundeeswari, A.; Sankar, P.D. Natural plant resources in anti-cancer therapy—A review. Res. Plant Biol. 2011, 1, 1–14. [Google Scholar]
- El-Hawary, S.S.; Taher, M.A.; AbouZid, S.F.; Amin, E.; Mohammed, R. Genus Tabebuia: A comprehensive review journey from past achievements to future perspectives. Arab. J. Chem. 2021, 14, 103046. [Google Scholar] [CrossRef]
- El-Hawary, S.S.; Mohammed, R.; Tawfike, A.F.; AbouZid, S.F.; Taher, M.A.; Abdelmohsen, U.R.; Amin, E. Metabolic profiling of cytotoxic metabolites from five Tabebuia species supported by molecular correlation analysis. Sci. Rep. 2021, 11, 8405. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Sahu, S.N.; Pattanayak, S.K. Molecular docking and molecular dynamics simulation studies on PLCE1 encoded protein. J. Mol. Struct. 2019, 1198, 126936. [Google Scholar] [CrossRef]
- Wei, M.; Zhao, R.; Cao, Y.; Wei, Y.; Li, M.; Dong, Z.; Yang, C. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo. Eur. J. Med. Chem. 2021, 209, 112903. [Google Scholar] [CrossRef]
- Wiseman, L.R.; Markham, A. Irinotecan. Drugs 1996, 52, 606–623. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, J.; Wang, N.; Kong, X.; Fu, F.; Wang, H.; Yao, J. Design and discovery of quinazoline-and thiourea-containing sorafenib analogs as EGFR and VEGFR-2 dual TK inhibitors. Molecules 2018, 23, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliba, M.O.; Ndukwe, I.G.; Ibrahim, H. Isolation and characterization of Β-sitosterol from methanol extracts of the stem bark of large-leaved rock fig (Ficus abutilifolia Miq). JASEM 2018, 22, 1639–1642. [Google Scholar] [CrossRef] [Green Version]
- Woo, K.W.; Han, J.Y.; Choi, S.U.; Kim, K.H.; Lee, K.R. Triterpenes from Perilla frutescens var. acuta and their cytotoxic activity. Nat. Pro. Sci. 2014, 20, 71–75. [Google Scholar]
- Birhanu, G. Isolation of ursolic acid from the leaves of Ocimum lamiifolium collected from Addis Ababa Area, Ethiopia. Afr. J. Biotechnol. 2020, 19, 65–70. [Google Scholar]
- Xu, S.; Wang, G.; Peng, W.; Xu, Y.; Zhang, Y.; Ge, Y.; Gong, Z. Corosolic acid isolated from Eriobotrya japonica leaves reduces glucose level in human hepatocellular carcinoma cells, zebrafish and rats. Sci. Rep. 2019, 9, 4388. [Google Scholar] [CrossRef]
- Soares, A.D.O.; Tieppo, C.; Soares, L.R.; Corsino, J.; Souza, A.F.; Garcez, F.R.; Garcez, W.S. Iridoides, triterpenos e outros constituintes das cascas do caule e flores de Tabebuia caraiba Bignoniaceae. Quim. Nova 2020, 43, 399–403. [Google Scholar] [CrossRef]
- Aquino, R.; De Simone, F.; Vincieri, F.F.; Pizza, C.; Gaćs-Baitz, E. New polyhydroxylated triterpenes from Uncaria tomentosa. J. Nat. Prod. 1990, 53, 559–564. [Google Scholar] [CrossRef]
- Rai, N.P.; Adhikari, B.B.; Paudel, A.; Masuda, K.; Mckelvey, R.D.; Manandhar, M.D. Phytochemical constituents of the flowers of Sarcococca coriacea of Nepalese origin. J. Nepal Chem. Soc. 2006, 21, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Mou, Y.; Zhao, J.; Wang, J.; Zhou, L.; Wang, M.; Yang, F. Flavonoids from Halostachys caspica and their antimicrobial and antioxidant activities. Molecules 2010, 15, 7933. [Google Scholar] [CrossRef]
- Scotti, L.; Fernandes, M.B.; Muramatsu, E.; Emereciano, V.D.P.; Tavares, J.F.; Silva, M.S.D.; Scotti, M.T. 13C NMR spectral data and molecular descriptors to predict the antioxidant activity of flavonoids. Braz. J. Pharm. Sci. 2011, 47, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Kuruüzüm-Uz, A.; Güvenalp, Z.; Kazaz, C.; Demirezer, L.Ö. Phenolic compounds from the roots of Anchusa azurea var. azurea. Turk. J. Pharm. Sci. 2013, 10, 177–184. [Google Scholar]
- Yan, X.; Murphy, B.T.; Hammond, G.B.; Vinson, J.A.; Neto, C.C. Antioxidant activities and antitumor screening of extracts from cranberry fruit (Vaccinium macrocarpon). J. Agric. Food Chem. 2002, 50, 5844–5849. [Google Scholar] [CrossRef] [PubMed]
- Kalegari, M.; Miguel, M.D.; Dias, J.D.F.G.; Lordello, A.L.L.; Lima, C.P.D.; Miyazaki, C.M.S.; Miguel, O.G. Phytochemical constituents and preliminary toxicity evaluation of leaves from Rourea induta Planch. (Connaraceae). Braz. J. Pharm. Sci. 2011, 47, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Conte, M.M.; Huang, X.C.; Khalil, Z.; Capon, R.J. A search for BACE inhibitors reveals new biosynthetically related pyrrolidones, furanones and pyrroles from a southern Australian marine sponge, Ianthella sp. Org. Biomol. Chem. 2012, 10, 2656–2663. [Google Scholar] [CrossRef] [PubMed]
- Makhmoor, T.; Choudhary, M.I. Radical scavenging potential of compounds isolated from Vitex agnus—Castus. Turk. J. Chem. 2010, 34, 119–126. [Google Scholar]
- Dinh, N.T.; Ngo, X.L.; Thi, H.T.; Thi, H.N.; Thi, H.M.V.; Thi, N.M.N.; Vu, T.K.O. Chemical constituents from ethyl acetate extract of the leaves of Rourea harmandiana Pierre. Vietnam J. Sci. Technol. Eng. 2020, 62, 30–33. [Google Scholar] [CrossRef]
- Ganbaatar, C.; Gruner, M.; Mishig, D.; Duger, R.; Schmidt, A.W.; Knölker, H.J. Flavonoid glycosides from the aerial parts of Polygonatum odoratum (Mill.) Druce growing in Mongolia. Nat. Prod. J. 2015, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.C.; Pai, Y.F.; Tsai, T.H. Isolation of luteolin and luteolin-7-O-glucoside from Dendranthema morifolium Ramat Tzvel and their pharmacokinetics in rats. J. Agric. Food Chem. 2015, 63, 7700–7706. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A.L. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Shiri, F.; Shahraki, S.; Baneshi, S.; Nejati-Yazdinejad, M.; Majd, M.H. Synthesis, characterization, in vitro cytotoxicity, in silico ADMET analysis and interaction studies of 5-dithiocarbamato-1, 3, 4-thiadiazole-2-thiol and its zinc (ii) complex with human serum albumin: Combined spectroscopy and molecular docking investigations. RSC Adv. 2016, 6, 106516–106526. [Google Scholar]
- Lu, H.; Chang, D.J.; Baratte, B.; Meijer, L.; Schulze-Gahmen, U. Crystal structure of a human cyclin-dependent kinase 6 complex with a flavonol inhibitor, fisetin. J. Med. Chem. 2005, 48, 737–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuntawee, W.; Rungrotmongkol, T.; Hannongbua, S. Molecular dynamic behavior and binding affinity of flavonoid analogues to the cyclin dependent kinase 6/cyclin D complex. J. Chem. Inf. Model. 2012, 52, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Imberty, A.; Hardman, K.D.; Carver, J.P.; Perez, S. Molecular modelling of protein-carbohydrate interactions. Docking of monosaccharides in the binding site of concanavalin A. Glycobiology 1991, 1, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Shewchuk, L.; Hassell, A.; Wisely, B.; Rocque, W.; Holmes, W.; Veal, J.; Kuyper, L.F. Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase. J. Med. Chem. 2000, 43, 133–138. [Google Scholar] [CrossRef]
- Belkadi, A.; Kenouche, S.; Melkemi, N.; Daoud, I.; Djebaili, R. K-means clustering analysis, ADME/pharmacokinetic prediction, MEP, and molecular docking studies of potential cytotoxic agents. Struct. Chem. 2021, 32, 2235–2249. [Google Scholar] [CrossRef]
- El-Helby, A.G.A.; Ayyad, R.R.; Sakr, H.; El-Adl, K.; Ali, M.M.; Khedr, F. Design, Synthesis, Molecular Docking, and Anticancer Activity of Phthalazine Derivatives as VEGFR-2 Inhibitors. Arch. Pharm. 2017, 350, 1700240. [Google Scholar] [CrossRef]
- Yadav, D.K.; Khan, F. QSAR, docking and ADMET studies of camptothecin derivatives as inhibitors of DNA topoisomerase-I. J. Chemom. 2013, 27, 21–33. [Google Scholar] [CrossRef]
- Boudjedir, A.; Kraim, K.; Saihi, Y.; Attoui-Yahia, O.; Ferkous, F.; Nacereddine, A.K. A computational molecular docking study of camptothecin similars as inhibitors for topoisomerase 1. J. Struct. Chem. 2021, 32, 689–697. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massagué, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data. Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [Green Version]
- Allam, A.E.; Amen, Y.; Ashour, A.; Assaf, H.K.; Hassan, H.A.; Abdel-Rahman, I.M.; Shimizu, K. In silico study of natural compounds from sesame against COVID-19 by targeting M pro, PL pro and RdRp. RSC Adv. 2021, 11, 22398–22408. [Google Scholar] [CrossRef]
- Galvez, M.; Martın-Cordero, C.; Lopez-Lazaro, M.; Cortes, F.; Ayuso, M.J. Cytotoxic effect of Plantago spp. on cancer cell lines. J. Ethnopharmacol. 2003, 88, 125–130. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Sharma, S.; Mandal, S.; Goswami, A.; Mukhopadhyay, S.; Majumder, H.K. Luteolin, an emerging anti-cancer flavonoid, poisons eukaryotic DNA topoisomerase I. Biochem. J. 2002, 366, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Adegbola, P.I.; Semire, B.; Fadahunsi, O.S.; Adegoke, A.E. Molecular docking and ADMET studies of Allium cepa, Azadirachta indica and Xylopia aethiopica isolates as potential anti-viral drugs for COVID-19. VirusDisease 2021, 32, 85–97. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Z.H.; Qu, X.J. The adverse effects of sorafenib in patients with advanced cancers. Basic Clin. Pharmacol. Toxicol. 2015, 116, 216–221. [Google Scholar] [CrossRef]
- Gunasekaran, T.; Haile, T.; Nigusse, T.; Dhanaraju, M.D. Nanotechnology: An effective tool for enhancing bioavailability and bioactivity of phytomedicine. Asian Pac. J. Trop. Biomed. 2014, 4, S1–S7. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M.A.; AboulMagd, A.M.; Abdel-Rahman, H.M. Quinazoline-Schiff base conjugates: In silico study and ADMET predictions as multi-target inhibitors of coronavirus (SARS-CoV-2) proteins. RSC Adv. 2020, 10, 34033–34045. [Google Scholar] [CrossRef]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of three classes of anticancer agents bound to the human topoisomerase I–DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef]
- Hasegawa, M.; Nishigaki, N.; Washio, Y.; Kano, K.; Harris, P.A.; Sato, H.; Cheung, M. Discovery of novel benzimidazoles as potent inhibitors of TIE-2 and VEGFR-2 tyrosine kinase receptors. J. Med. Chem. 2007, 50, 4453–4470. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. Charmm-Gui: A web-based graphical user interface for Charmm. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Jo, S.; Jiang, W.; Lee, H.S.; Roux, B.t.; Im, W. Charmm-Gui Ligand Binder for absolute binding free energy calculations and its application. ACS Pub. 2013, 53, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Salem, H.S.; Arifuzzaman, M.; Alkahtani, H.M.; Abdalla, A.N.; Issa, I.S.; Alqathama, A.; Rahman, A.F.M. A Series of Isatin-Hydrazones with Cytotoxic Activity and CDK2 Kinase Inhibitory Activity: A Potential Type II ATP Competitive Inhibitor. Molecules 2020, 25, 4400. [Google Scholar] [CrossRef]
- Zeidan, M.A.; Mostafa, A.S.; Gomaa, R.M.; Abou-Zeid, L.A.; El-Mesery, M.; Magda, A.A.; Selim, K.B. Design, synthesis and docking study of novel picolinamide derivatives as anticancer agents and VEGFR-2 inhibitors. Eur. J. Med. Chem. 2019, 168, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Cheng, X.; Zhang, K.; Wang, H.; Liu, B.; Wang, J. Design, synthesis and biological evaluation of pyrimidine derivatives as novel CDK2 inhibitors that induce apoptosis and cell cycle arrest in breast cancer cells. Bioorg. Med. Chem. 2018, 26, 3491–3501. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Macromolecule Target | PDB ID | RMSD of Validation | Initial Scoring Method | Final Scoring Method | Ligand Placement Method | Docking Protocol | Positive Control |
|---|---|---|---|---|---|---|---|
| CDK-2 | 1DI8 | 0.6897 | London dG | GBVI/WSA dG | Triangle Matcher | Rigid receptor | Ribociclib |
| CDK-6 | 1XO2 | 0.3698 | London dG | GBVI/WSA dG | Triangle Matcher | Rigid receptor | Ribociclib |
| Topoisomerase-1 | 1T8I | 0.8515 | London dG | London dG | Template Plugin Feature | Rigid receptor | Irinotecan |
| VEGFR-2 | 2OH4 | 0.4368 | London dG | GBVI/WSA dG | Triangle Matcher | Rigid receptor | Sorafenib |
| No. | Compound Name | CDK-2 (1DI8) | CDK-6 (1XO2) | Topoisomerase-1 (1T8I) | VEGFR-2 (2OH4) |
|---|---|---|---|---|---|
| 1 | β-Sitosterol | −13.27 | −9.61 | −8.17 | −6.30 |
| 2 | Ursolic acid | −10.88 | −8.08 | −8.84 | −5.80 |
| 3 | Corosolic acid | −13.44 | −10.54 | −7.44 | −5.92 |
| 4 | 3-O-p-coumaroyl corosolic acid | −10.99 | −9.98 | −7.10 | −5.75 |
| 5 | 3β-6β-19α-trihydroxy-urs-12-en-28-oic acid | −11.35 | −9.18 | −8.31 | −6.57 |
| 6 | β-Sitosterol-3-O-D-glucopyranoside | −10.61 | −7.20 | −8.99 | −5.90 |
| 7 | Quercetin | −8.13 | −10.70 | −10.96 | −9.94 |
| 8 | Luteolin | −8.22 | −10.05 | −10.93 | −8.51 |
| 9 | Quercetin -3-O-glucoside | −7.14 | −9.03 | −12.56 | −8.19 |
| 10 | Quercetin 3-O-xyloside | −6.54 | −16.23 | −10.66 | −10.39 |
| 11 | 4-Hydroxybenzoic acid | −6.05 | −5.90 | −7.18 | −5.39 |
| 12 | 4-Methoxybenzoic acid | −5.69 | −5.58 | −7.98 | −5.76 |
| 13 | 3,4-Dihydroxybenzoic acid | −5.70 | −6.73 | −5.55 | −5.13 |
| 14 | p-coumaric acid | −7.35 | −8.22 | −7.80 | −6.77 |
| 15 | Rutin | −6.71 | −7.42 | −10.87 | −8.25 |
| 16 | Luteolin-7-O-glucoside | −8.43 | −11.14 | −13.83 | −8.85 |
| Reference Ligands | (−17.58) Ribociclib | (−15.08) Ribociclib | (−14.65) Irinotecan | (−12.58) Sorafenib | |
| CDK-2 IC50 (µg/mL) | CDK-6 IC50 (µg/mL) | VEGFR-2 IC50 (µg/mL) | |
|---|---|---|---|
| Ribociclib | 0.039 ± 0.002 | ||
| Compound 1 | 0.241 ± 0.015 a | ||
| Compound 3 | 0.113 ± 0.007 a,b | ||
| Ribociclib | 0.159 ± 0.008 | ||
| Compound 10 | 0.154 ± 0.007 | ||
| Sorafenib | 0.039 ± 0.002 | ||
| Compound 10 | 0.084 ± 0.003 c |
| Property | Model Name | Predicted Value | Unit | ||||
|---|---|---|---|---|---|---|---|
| Compound 1 | Compound 3 | Compound 10 | Compound 16 | Sorafenib | |||
| Absorption | Water solubility | −6.773 | −3.04 | −2.903 | −3.325 | −4.255 | Numeric (log mol/L) |
| Caco2 permeability | 1.201 | 0.641 | 0.052 | 0.432 | 0.762 | Numeric (log Papp in 10−6 cm/s) | |
| Intestinal absorption (human) | 94.464 | 100 | 51.884 | 46.308 | 85.494 | Numeric (% Absorbed) | |
| Skin Permeability | −2.783 | −2.735 | −2.735 | −2.735 | −2.74 | Numeric (log Kp) | |
| P-glycoprotein substrate | No | No | Yes | Yes | Yes | Categorical (Yes/No) | |
| P-glycoprotein I inhibitor | Yes | No | No | No | Yes | Categorical (Yes/No) | |
| P-glycoprotein II inhibitor | Yes | No | No | No | Yes | Categorical (Yes/No) | |
| Distribution | VDss (human) | 0.193 | −1.282 | 1.508 | −0.106 | −0.009 | Numeric (log L/kg) |
| Fraction unbound (human) | 0 | 0.037 | 0.134 | 0.064 | 0 | Numeric (Fu) | |
| BBB permeability | 0.781 | −0.473 | −1.473 | −1.61 | −1.473 | Numeric (log BB) | |
| CNS permeability | −1.705 | −1.507 | −4.215 | −4.67 | −2.025 | Numeric (log PS) | |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | Categorical (Yes/No) |
| CYP1A2 inhibitor | No | No | No | Yes | No | Categorical (Yes/No) | |
| CYP2C19 inhibitor | No | No | No | No | Yes | Categorical (Yes/No) | |
| CYP2C9 inhibitor | No | No | No | No | Yes | Categorical (Yes/No) | |
| CYP2D6 inhibitor | No | No | No | No | No | Categorical (Yes/No) | |
| CYP3A4 inhibitor | No | No | No | No | Yes | Categorical (Yes/No) | |
| Excretion | Total Clearance | 0.628 | 0.093 | 0.364 | 0.687 | −0.213 | Numeric (log mL/min/kg) |
| Renal OCT2 substrate | No | No | No | No | No | Categorical (Yes/No) | |
| Toxicity | AMES toxicity | No | No | No | No | No | Categorical (Yes/No) |
| Max. tolerated dose (human) | −0.621 | 0.124 | 0.494 | 0.765 | 0.253 | Numeric (log mg/kg/day) | |
| hERG I inhibitor | No | No | No | No | No | Categorical (Yes/No) | |
| hERG II inhibitor | Yes | No | Yes | Yes | Yes | Categorical (Yes/No) | |
| Oral Rat Acute Toxicity (LD50) | 2.552 | 2.513 | 2.585 | 2.54 | 2.14 | Numeric (mol/kg) | |
| Hepatotoxicity | No | No | No | No | Yes | Categorical (Yes/No) | |
| Skin Sensitization | No | No | No | No | No | Categorical (Yes/No) | |
| T.Pyriformis toxicity | 0.43 | 0.285 | 0.285 | 0.285 | 0.307 | Numeric (log ug/L) | |
| Minnow toxicity | −1.802 | 0.276 | 5.071 | 1.266 | −0.515 | Numeric (log mM) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Hawary, S.S.; Mohammed, R.; Taher, M.A.; AbouZid, S.F.; Mansour, M.A.; Almahmoud, S.A.; Huwaimel, B.; Amin, E. Characterization of Promising Cytotoxic Metabolites from Tabebuia guayacan Hemsl.: Computational Prediction and In Vitro Testing. Plants 2022, 11, 888. https://doi.org/10.3390/plants11070888
El-Hawary SS, Mohammed R, Taher MA, AbouZid SF, Mansour MA, Almahmoud SA, Huwaimel B, Amin E. Characterization of Promising Cytotoxic Metabolites from Tabebuia guayacan Hemsl.: Computational Prediction and In Vitro Testing. Plants. 2022; 11(7):888. https://doi.org/10.3390/plants11070888
Chicago/Turabian StyleEl-Hawary, Seham S., Rabab Mohammed, Marwa A. Taher, Sameh Fekry AbouZid, Mostafa A. Mansour, Suliman A. Almahmoud, Bader Huwaimel, and Elham Amin. 2022. "Characterization of Promising Cytotoxic Metabolites from Tabebuia guayacan Hemsl.: Computational Prediction and In Vitro Testing" Plants 11, no. 7: 888. https://doi.org/10.3390/plants11070888