
Comparative Genomics, Phylogenetics, Biogeography, and Effects of Climate Change on Toddalia asiatica (L.) Lam. (Rutaceae) from Africa and Asia


 ,
, 
 , and
, and
Abstract
:1. Introduction
2. Results
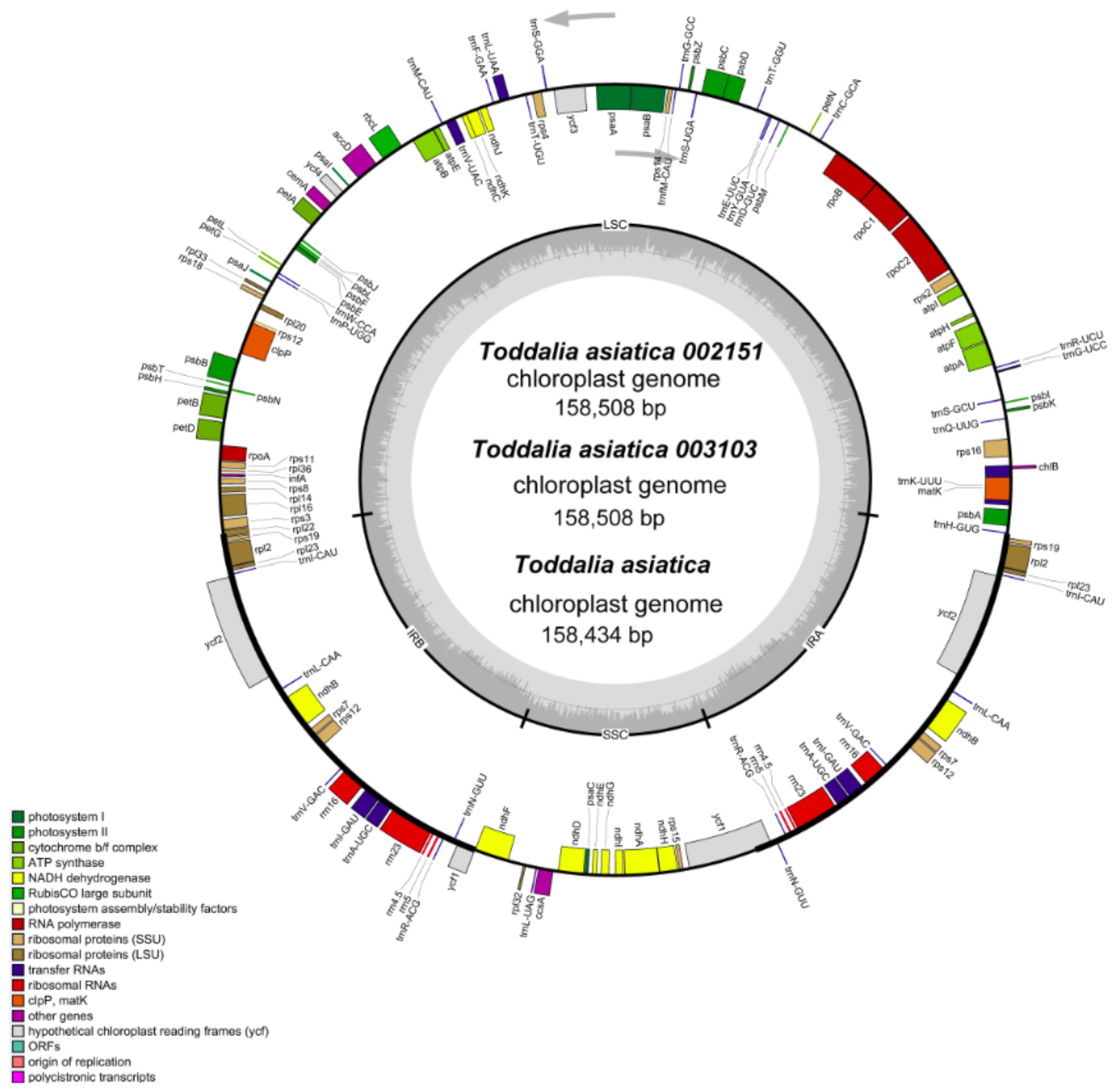
2.1. Complete Chloroplast Genome
2.2. Codon Preference Analysis
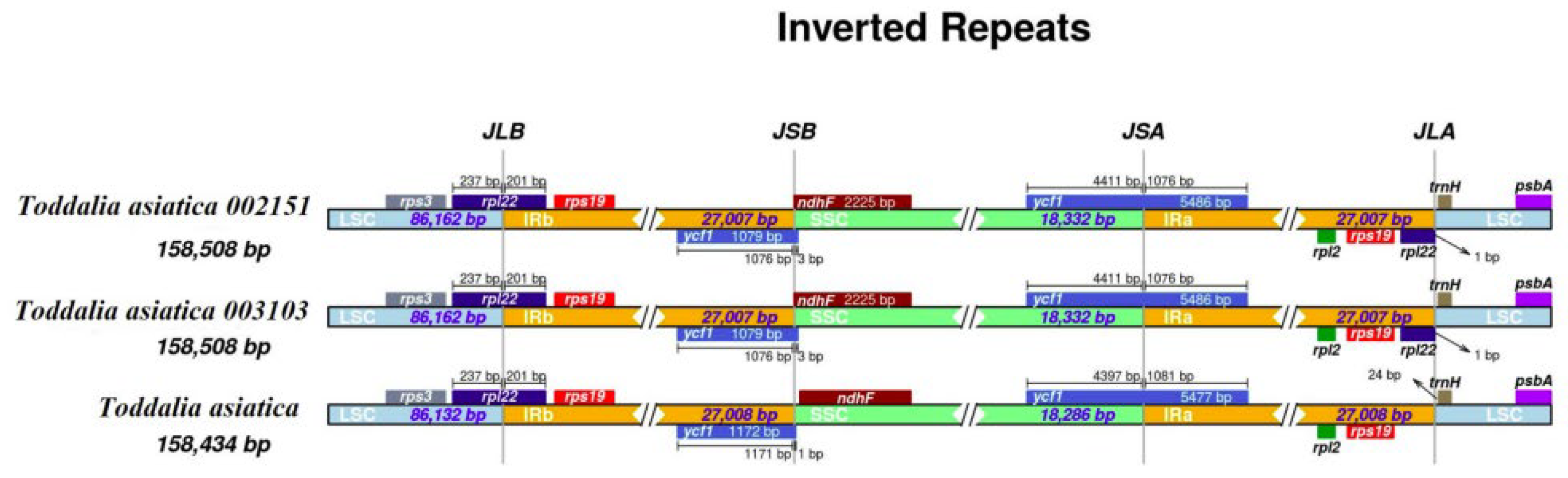
2.3. IR Contraction and Expansion
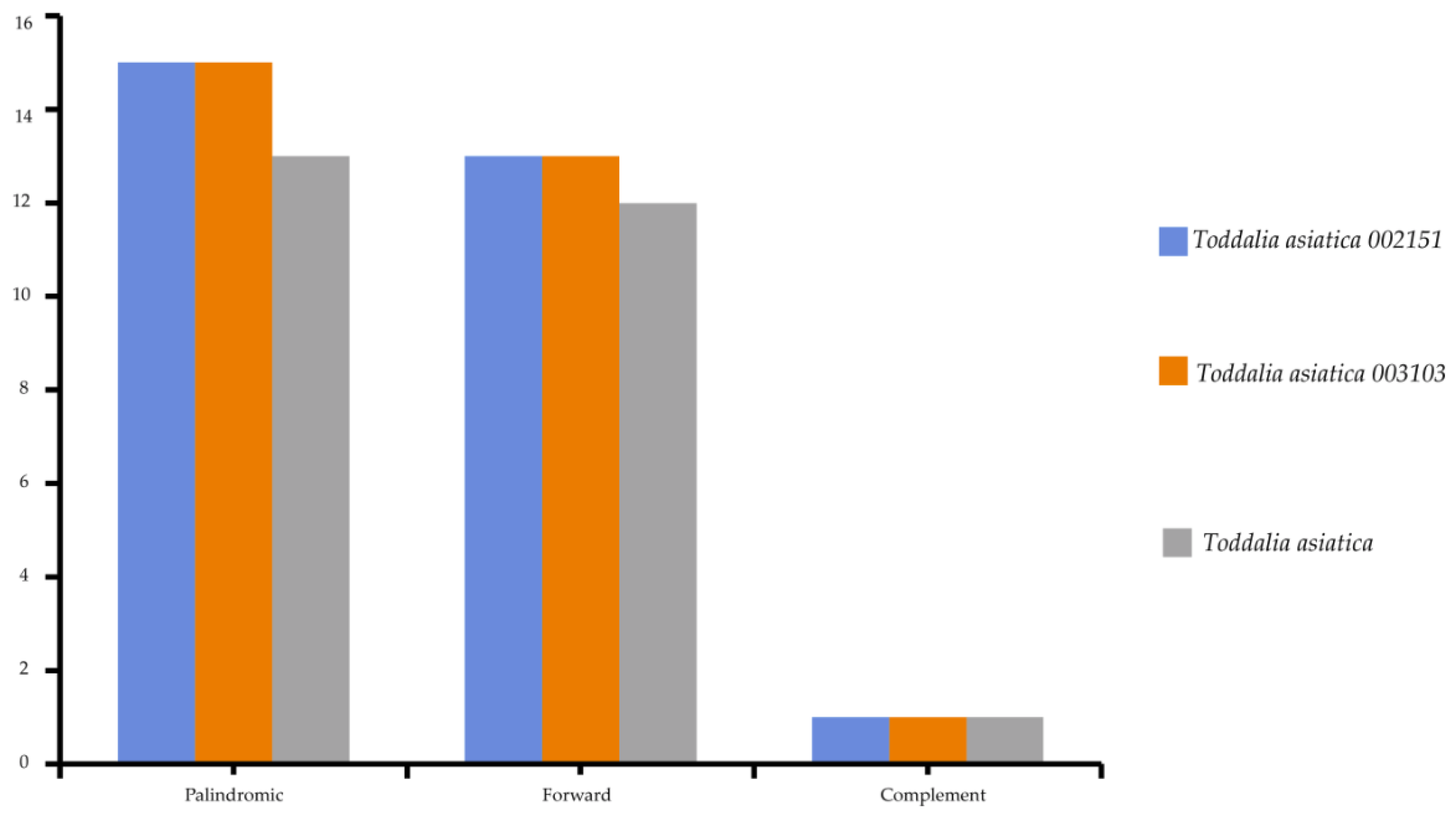
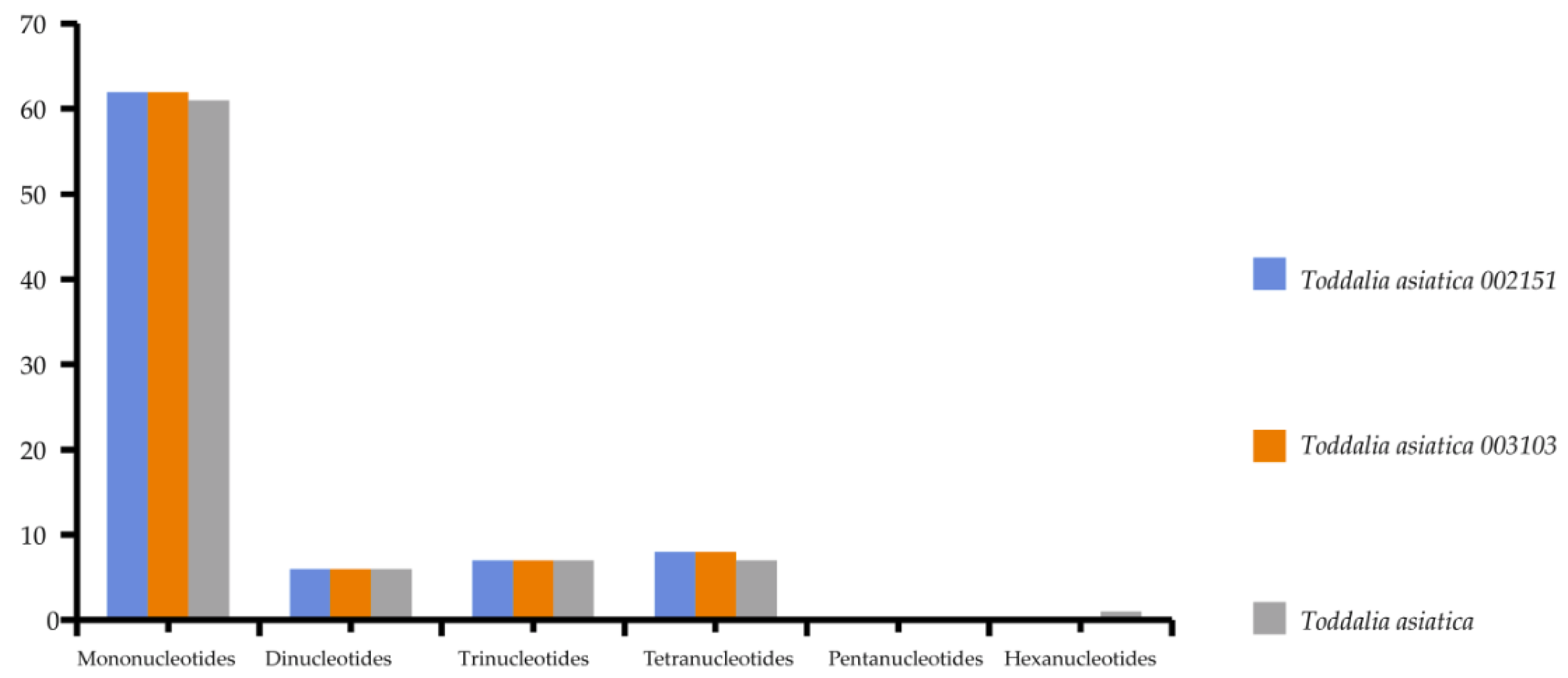
2.4. Repeat Analysis
2.5. Comparative Analysis
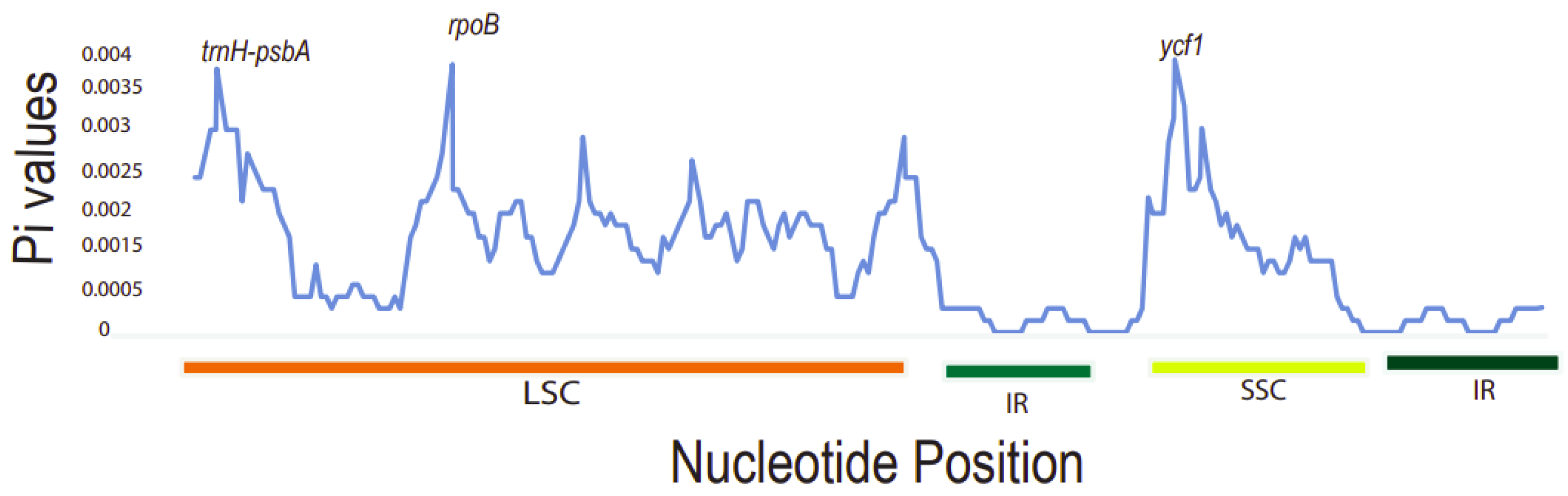
2.6. Sequence Divergence Analysis
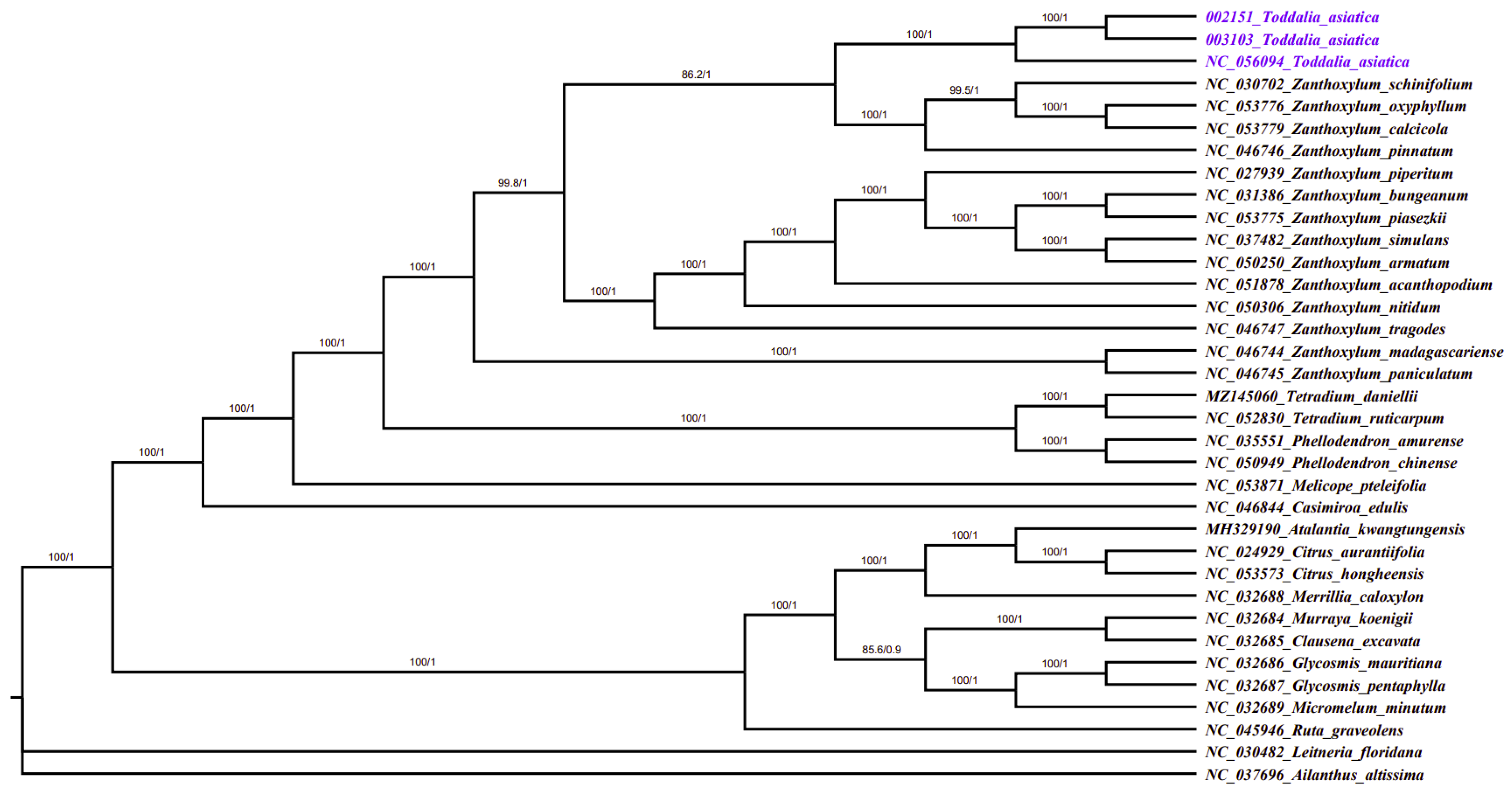
2.7. Phylogenetic Analysis
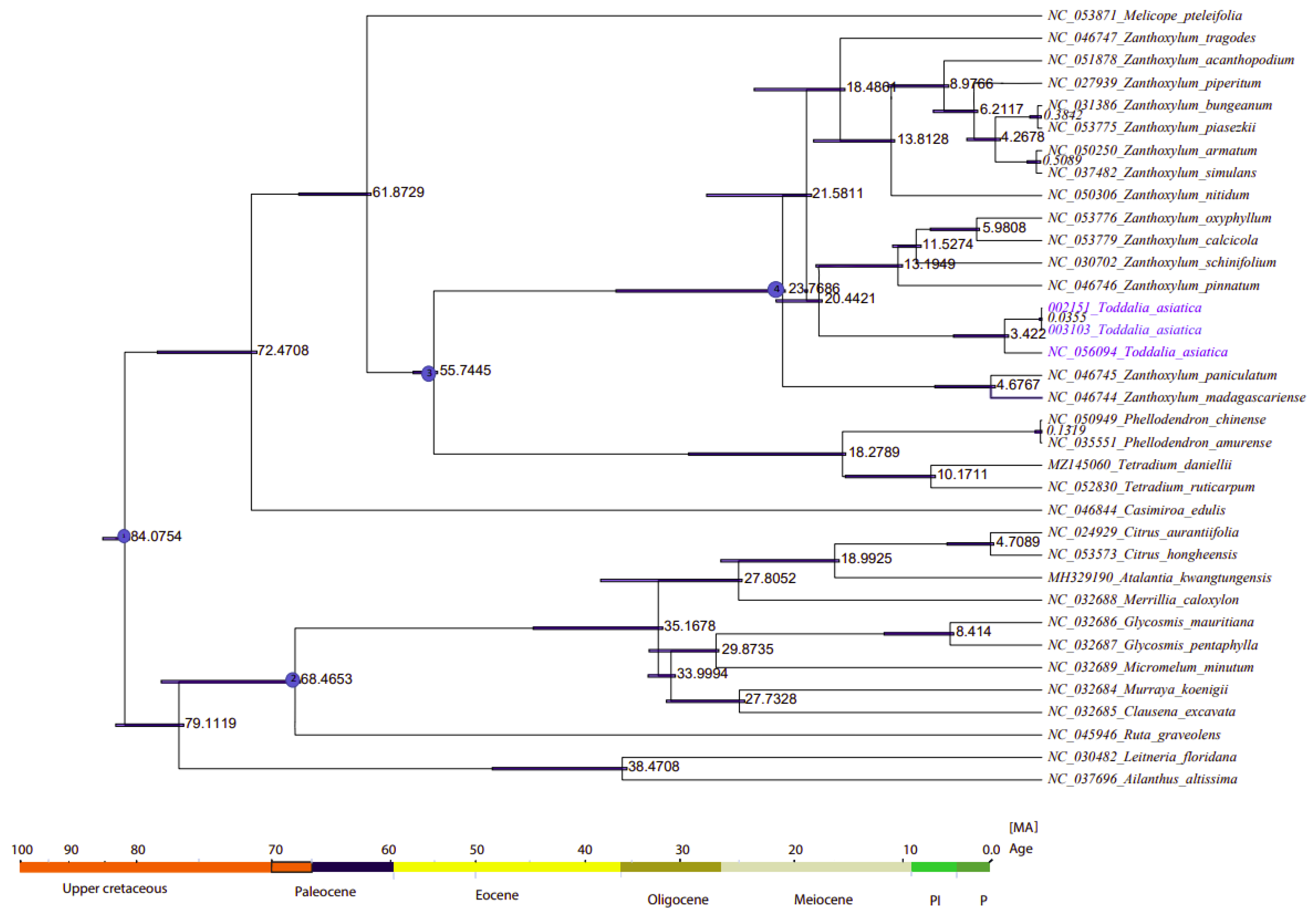
2.8. Divergence Time Estimation
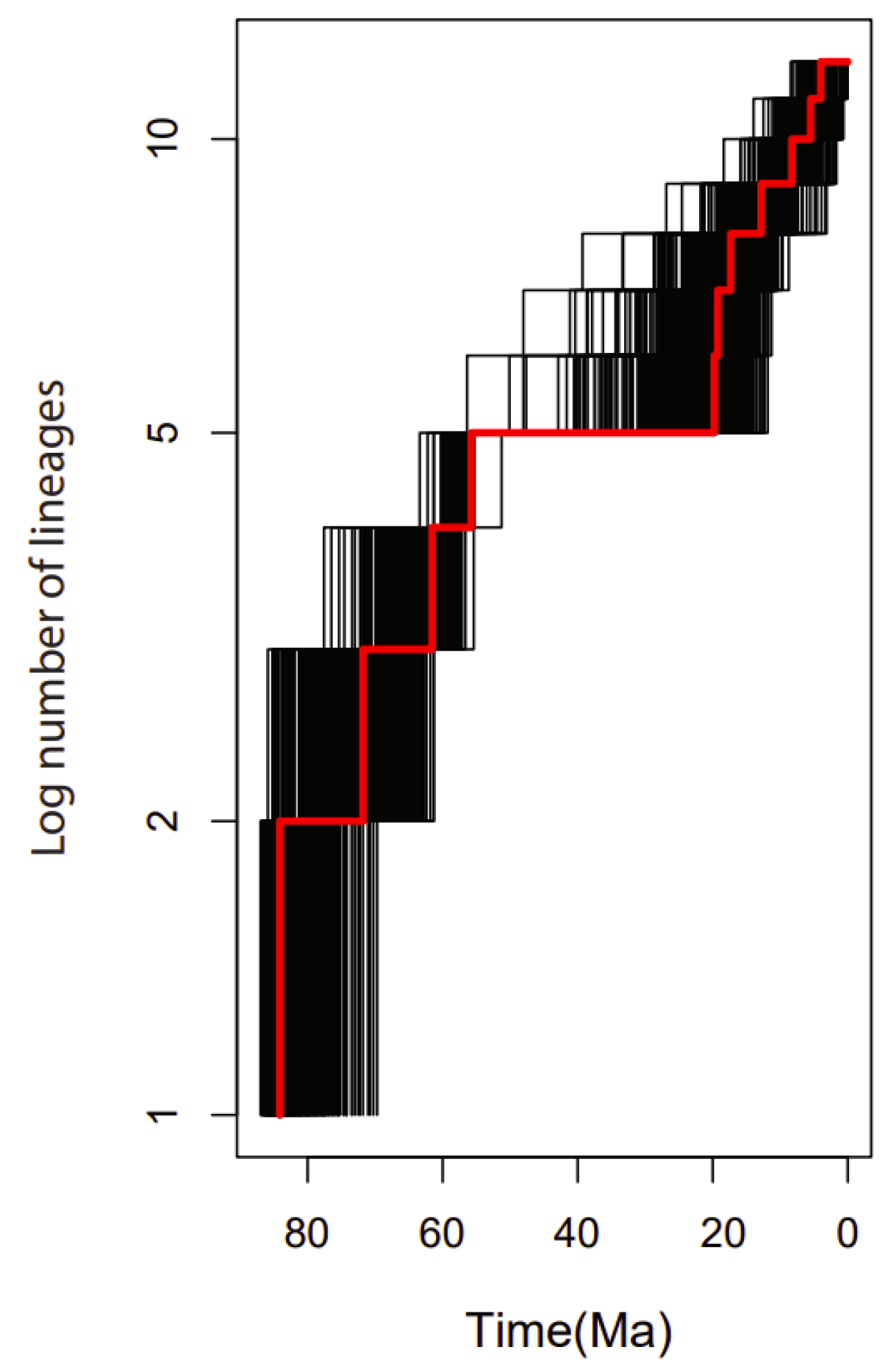
2.9. Diversification Rates Analyses
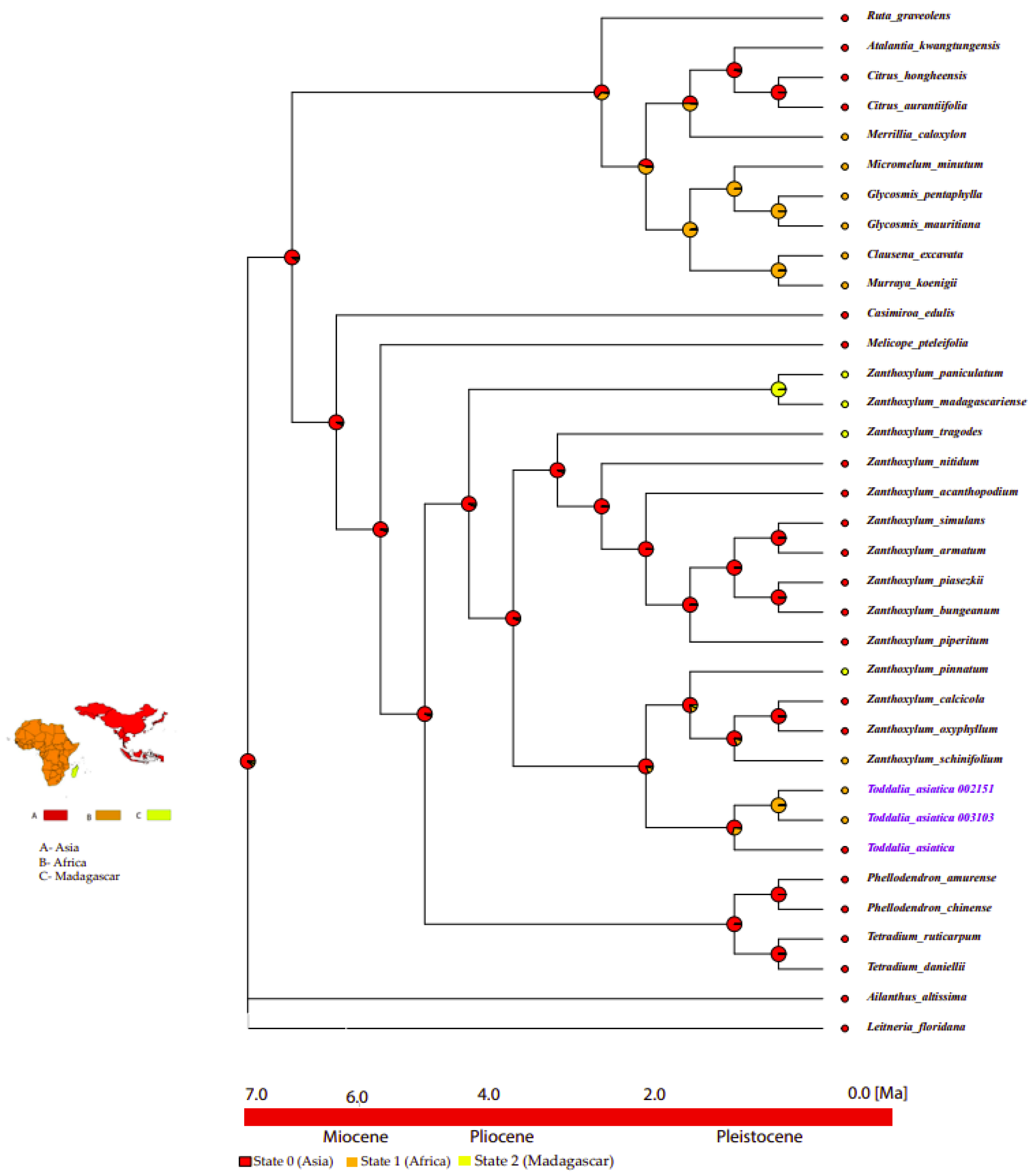
2.10. Ancestral Area Reconstruction
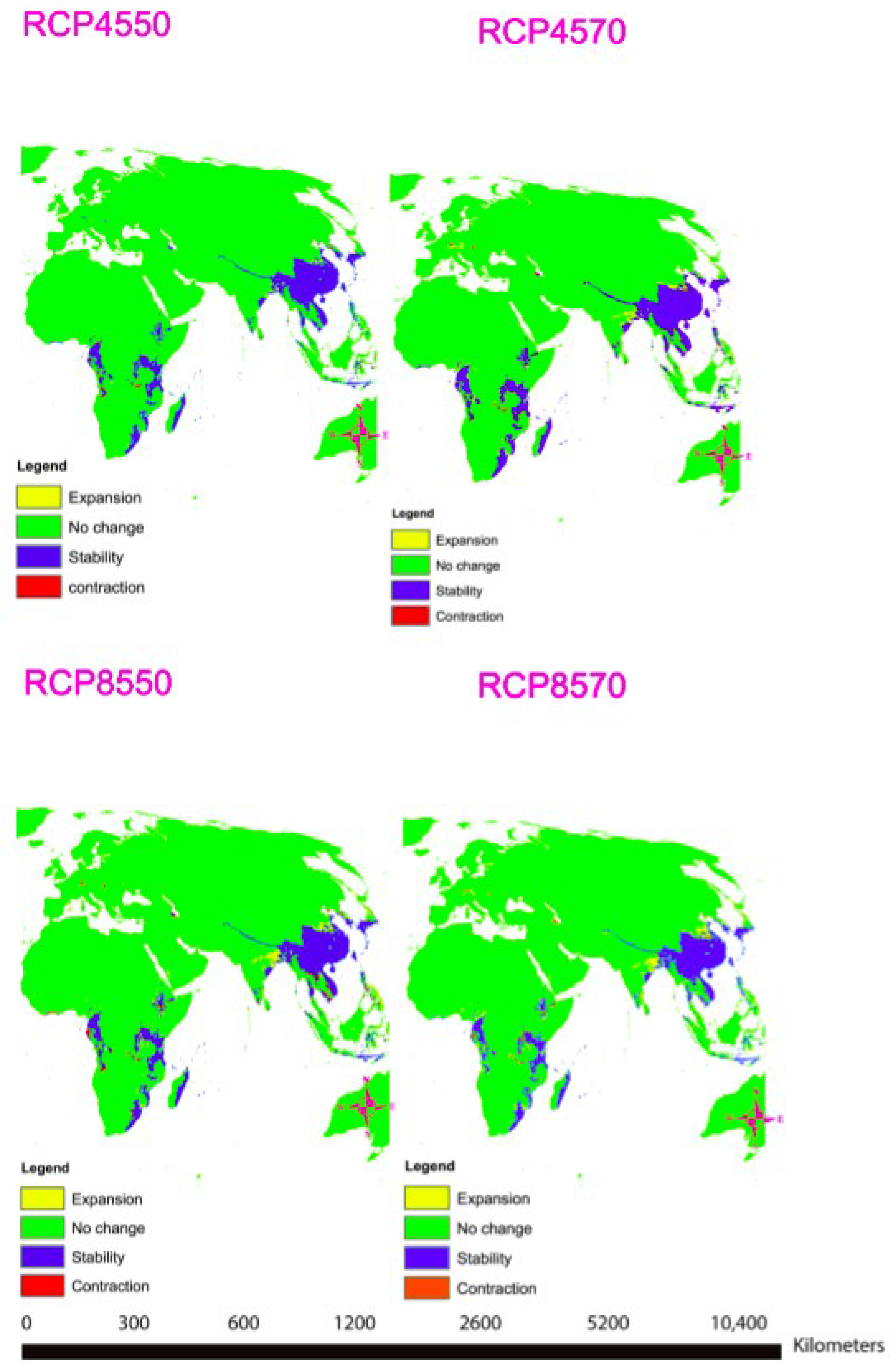
2.11. Climate Variables
3. Materials and Methods
3.1. Plant Materials Collection, DNA Extraction, and Sequencing
3.2. Plastome Annotation and Assembly
3.3. Repeat Analysis
3.4. Comparative Analysis
3.5. Selective Pressure Analysis
3.6. Phylogenetic Analysis
3.7. Divergence Time Estimation
3.8. Diversification Rates Analyses
3.9. Ancestral Area Reconstruction of Toddalia
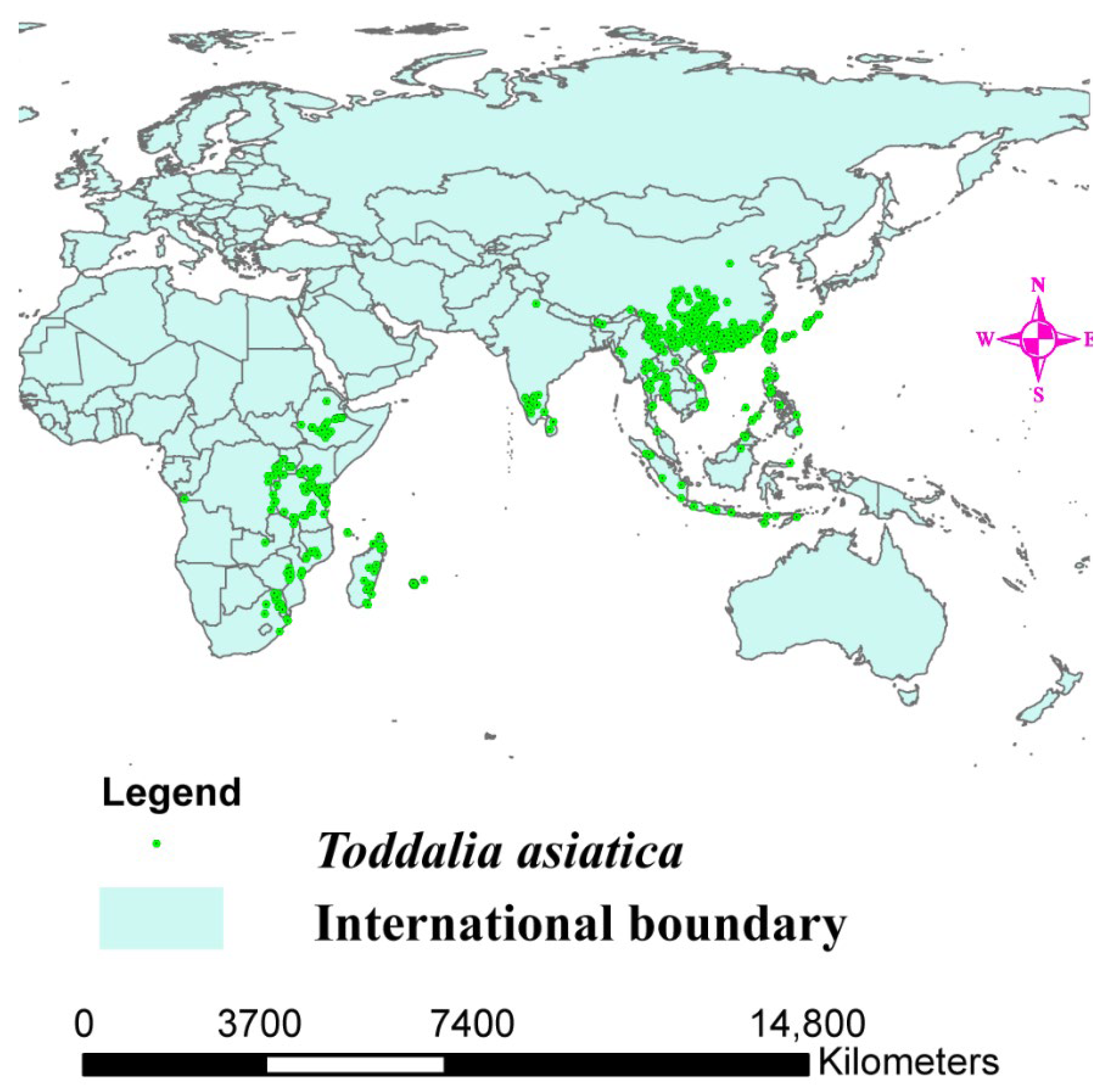
4. Species Distribution Data
4.1. Environmental Factors Consideration
4.2. MaxEnt Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Groppo, M.; Pirani, J.R.; Salatino, M.L.; Blanco, S.R.; Kallunki, J.A. Phylogeny of Rutaceae based on two noncoding regions from cpDNA. Am. J. Bot. 2008, 95, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Kubitzki, K. Flowering Plants. Eudicots: Sapindales, Cucurbitales, Myrtaceae; Springer Science & Business Media: Amtsgericht Bonn, Germany, 2010; Volume 10. [Google Scholar]
- Groppo, M.; Kallunki, J.A.; Pirani, J.R.; Antonelli, A. Chilean Pitavia more closely related to Oceania and Old World Rutaceae than to Neotropical groups: Evidence from two cpDNA non-coding regions, with a new subfamilial classification of the family. PhytoKeys 2012, 9, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W.; Christenhusz, M.; Fay, M.; Byng, J.; Judd, W.S.; Soltis, D.; Mabberley, D.; Sennikov, A.; Soltis, P.S.; Stevens, P.F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Albaayit, S.F.A.; Maharjan, R.; Abdullah, R.; Noor, M.H.M. Anti-Enterococcus Faecalis, Cytotoxicity, Phytotoxicity, and Anticancer Studies on Clausena excavata Burum. f. (Rutaceae) Leaves. BioMed Res. Int. 2021, 2021, 3123476. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Liu, Q.-Y.; Wang, A.; Gao, Y.-W.; Zhao, L.-C.; Guan, W.-B. Comparative analysis and phylogenetic implications of plastomes of five genera in subfamily Amyridoideae (Rutaceae). Forests 2021, 12, 277. [Google Scholar] [CrossRef]
- Appelhans, M.S.; Wen, J.; Wood, K.R.; Allan, G.J.; Zimmer, E.A.; Wagner, W.L. Molecular phylogenetic analysis of Hawaiian Rutaceae (Melicope, Platydesma, and Zanthoxylum) and their different colonization patterns. Bot. J. Linn. Soc. 2014, 174, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Appelhans, M.S.; Reichelt, N.; Groppo, M.; Paetzold, C.; Wen, J. Phylogeny and biogeography of the pantropical genus Zanthoxylum and its closest relatives in the proto-Rutaceae group (Rutaceae). Mol. Phylogenetics Evol. 2018, 126, 31–44. [Google Scholar] [CrossRef]
- Engler, A. Rutaceae. In Die Natürlichen Pflanzenfamilien; Engler, A., Prantl, K., Eds.; Biodiversity Heritage Library: Toulon, France, 1931; Volume 2, pp. 187–359. [Google Scholar]
- Morton, C.M.; Telmer, C. New subfamily classification for the Rutaceae. Ann. Mo. Bot. Gard. 2014, 99, 620–641. [Google Scholar] [CrossRef]
- Waterman, P.G. Alkaloids of the Rutaceae: Their distribution and systematic significance. Biochem. Syst. Ecol. 1975, 3, 149–180. [Google Scholar] [CrossRef]
- Waterman, P.G. The current status of chemical systematics. Phytochemistry 2007, 68, 2896–2903. [Google Scholar] [CrossRef]
- Waterman, P.G.; Grundon, M.F. (Eds.) Phylogenetic implications of the distribution of secondary metabolites within the Rutales. In Chemistry and Chemical Taxonomy of the Rutales; Academic Press: London, UK, 1983; pp. 377–400. [Google Scholar]
- Poon, W.-S.; Shaw, P.-C.; Simmons, M.P.; But, P.P.-H. Congruence of molecular, morphological, and biochemical profiles in Rutaceae: A cladistic analysis of the subfamilies Rutoideae and Toddalioideae. Syst. Bot. 2007, 32, 837–846. [Google Scholar] [CrossRef]
- Appelhans, M.S.; Wen, J.; Wagner, W.L. A molecular phylogeny of Acronychia, Euodia, Melicope and relatives (Rutaceae) reveals polyphyletic genera and key innovations for species richness. Mol. Phylogenetics Evol. 2014, 79, 54–68. [Google Scholar] [CrossRef]
- Huang, C. Rutaceae. Flora Reipublicae Popularis Sinicae; Science Press: Beijing, China, 1997; Volume 43. [Google Scholar]
- Shahzad, A.; Ullah, S.; Dar, A.A.; Sardar, M.F.; Mehmood, T.; Tufail, M.A.; Shakoor, A.; Haris, M. Nexus on climate change: Agriculture and possible solution to cope future climate change stresses. Environ. Sci. Pollut. Res. 2021, 28, 14211–14232. [Google Scholar] [CrossRef]
- Du, Z.; He, Y.; Wang, H.; Wang, C.; Duan, Y. Potential geographical distribution and habitat shift of the genus Ammopiptanthus in China under current and future climate change based on the MaxEnt model. J. Arid. Environ. 2021, 184, 104328. [Google Scholar] [CrossRef]
- Chen, D.-M.; Kang, H.-Z.; Liu, C.-J. An overview on the potential quaternary glacial refugia of plants in China mainland. ZhiwuYanjiu Bull. Bot. Res. 2011, 31, 623–632. [Google Scholar]
- De Blasio, F.V.; Liow, L.H.; Schweder, T.; De Blasio, B.F. A model for global diversity in response to temperature change over geological time scales, with reference to planktic organisms. J. Theor. Biol. 2015, 365, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Reddin, C.J.; Kocsis, Á.T.; Kiessling, W. Marine invertebrate migrations trace climate change over 450 million years. Glob. Ecol. Biogeogr. 2018, 27, 704–713. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabater, B. Evolution and Function of the Chloroplast. Current Investigations and Perspectives. Int. J. Mol. Sci. 2018, 19, 3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talat, F.; Wang, K. Comparative bioinformatics analysis of the chloroplast genomes of a wild diploid Gossypium and two cultivated allotetraploid species. Iran. J. Biotechnol. 2015, 13, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, X.; Liu, Y. Nonoptimal Codon Usage Is Critical for Protein Structure and Function of the Master General Amino Acid Control Regulator CPC-1. mBio 2020, 11, e02605-20. [Google Scholar] [CrossRef] [PubMed]
- Angellotti, M.C.; Bhuiyan, S.B.; Chen, G.; Wan, X.-F. CodonO: Codon usage bias analysis within and across genomes. Nucleic Acids Res. 2007, 35 (Suppl. S2), W132–W136. [Google Scholar] [CrossRef]
- Xu, C.; Dong, J.; Tong, C.; Gong, X.; Wen, Q.; Zhuge, Q. Analysis of synonymous codon usage patterns in seven different Citrus species. Evol. Bioinform. Online 2013, 9, 215–228. [Google Scholar] [CrossRef]
- Xu, C.; Cai, X.; Chen, Q.; Zhou, H.; Cai, Y.; Ben, A. Factors affecting synonymous codon usage bias in chloroplast genome of Oncidium gower ramsey. Evol. Bioinform. Online 2011, 7, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zuo, L.; Lu, D.; Lu, B.; Yang, M.; Wang, J. Comparative analysis of chloroplast genomes of five Robinia species: Genome comparative and evolution analysis. Gene 2019, 689, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, A.; Khan, G.; Lee, I.-J.; Al-Harrasi, A. Expanded inverted repeat region with large scale inversion in the first complete plastid genome sequence of Plantago ovata. Sci. Rep. 2020, 10, 3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khayi, S.; Gaboun, F.; Pirro, S.; Tatusova, T.; El Mousadik, A.; Ghazal, H.; Mentag, R. Complete Chloroplast Genome of Argania spinosa: Structural Organization and Phylogenetic Relationships in Sapotaceae. Plants 2020, 9, 1354. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.-M.; Wicke, S.; Gan, L.; Yang, J.-B.; Jin, J.-J.; Yi, T.-S. The loss of the inverted repeat in the putranjivoid clade of Malpighiales. Front. Plant Sci. 2020, 11, 942. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Lou, W.; Chen, Y.; Jia, X.; Zhai, M.; Guo, Z.; Xuan, J. The chloroplast genome of Carya illinoinensis: Genome structure, adaptive evolution, and phylogenetic analysis. Forests 2020, 11, 207. [Google Scholar] [CrossRef] [Green Version]
- Karthik, L.; Kumar, G.; Keswani, T.; Bhattacharyya, A.; Chandar, S.S.; Bhaskara Rao, K. Protease inhibitors from marine actinobacteria as a potential source for antimalarial compound. PLoS ONE 2014, 9, e90972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, D.-F.; Yu, Y.; Deng, Y.-Q.; Li, J.; Liu, H.-Y.; Zhou, S.-D.; He, X.-J. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Guo, D.; Xing, G.; Yang, C.; Zhang, Y.; Yang, J.; Niu, L.; Zhong, X.; Zhao, Q.; Cui, Y.; et al. Complete Chloroplast Genome Sequence and Comparative and Phylogenetic Analyses of the Cultivated Cyperus esculentus. Diversity 2021, 13, 405. [Google Scholar] [CrossRef]
- Deguilloux, M.-F.; Pemonge, M.-H.; Petit, R.J. Use of chloroplast microsatellites to differentiate oak populations. Ann. For. Sci. 2004, 61, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Piya, S.; Nepal, M.P.; Butler, J.L.; Larson, G.E.; Neupane, A. Genetic diversity and population structure of sickleweed (Falcaria vulgaris; Apiaceae) in the upper Midwest USA. Biol. Invasions 2014, 16, 2115–2125. [Google Scholar] [CrossRef]
- Redwan, R.; Saidin, A.; Kumar, S. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 2015, 15, 196. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhang, X.; Meng, H.; Li, J.; Zhang, D.; Liu, C. Comparative chloroplast genomes of Paris Sect. Marmorata: Insights into repeat regions and evolutionary implications. BMC Genom. 2018, 19, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Munyao, J.N.; Dong, X.; Yang, J.-X.; Mbandi, E.M.; Wanga, V.O.; Oulo, M.A.; Saina, J.K.; Musili, P.M.; Hu, G.-W. Complete chloroplast genomes of Chlorophytum comosum and Chlorophytum gallabatense: Genome structures, comparative and phylogenetic analysis. Plants 2020, 9, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, H.; Teng, W.; Yu, Z.; Liu, X.; Xie, X.; Yue, C.; Li, D.; Liang, M.; Li, Q. Characterization of the complete mitochondrial genome of Alectryonella plicatula (Bivalvia: Ostreidae). Mitochondrial DNA Part B 2021, 6, 1581–1582. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yu, H.; Wang, J.; Lei, W.; Gao, J.; Qiu, X.; Wang, J. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendrakumar, P.; Biswal, A.K.; Balachandran, S.M.; Srinivasarao, K.; Sundaram, R.M. Simple sequence repeats in organellar genomes of rice: Frequency and distribution in genic and intergenic regions. Bioinformatics 2007, 23, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.G.; Awasthi, P.; Bedi, Y.S. Analysis of SSR dynamics in chloroplast genomes of Brassicaceae family. Bioinformation 2010, 5, 16. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, T.; Bai, G.; Zhao, Y. Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 2018, 8, 12379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousseau-Gueutin, M.; Bellot, S.; Martin, G.E.; Boutte, J.; Chelaifa, H.; Lima, O.; Michon-Coudouel, S.; Naquin, D.; Salmon, A.; Ainouche, K. The chloroplast genome of the hexaploid Spartina maritima (Poaceae, Chloridoideae): Comparative analyses and molecular dating. Mol. Phylogenetics Evol. 2015, 93, 5–16. [Google Scholar] [CrossRef]
- Xu, J.-H.; Liu, Q.; Hu, W.; Wang, T.; Xue, Q.; Messing, J. Dynamics of chloroplast genomes in green plants. Genomics 2015, 106, 221–231. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.; Bai, G.; Li, Z.; Kanwal, N.; Zhao, G. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia species. Front. Plant Sci. 2016, 7, 1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschiazzo, E.; Ritland, C.; Bohlmann, J.; Ritland, K. Slow but not low: Genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol. Biol. 2012, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Rono, P.C.; Dong, X.; Yang, J.-X.; Mutie, F.M.; Oulo, M.A.; Malombe, I.; Kirika, P.M.; Hu, G.-W.; Wang, Q.-F. Initial complete chloroplast genomes of Alchemilla (Rosaceae): Comparative analysis and phylogenetic relationships. Front. Genet. 2020, 11, 1390. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.-F.; Zhang, Y.-X.; Zeng, C.-X.; Guo, Z.-H.; Li, D.-Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [Green Version]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kang, Y.; Shin, S.C.; Park, H.; Lee, H. Combined analysis of the chloroplast genome and transcriptome of the Antarctic vascular plant Deschampsia antarctica Desv. PLoS ONE 2014, 9, e92501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelhans, M.S.; Keßler, P.J.; Smets, E.; Razafimandimbison, S.G.; Janssens, S.B. Age and historical biogeography of the pantropically distributed Spathelioideae (Rutaceae, Sapindales). J. Biogeogr. 2012, 39, 1235–1250. [Google Scholar] [CrossRef]
- Gregor, H.-J. Systematics, biostratigraphy and paleoecology of the genus Toddalia Jussieu (Rutaceae) in the European Tertiary. Rev. Palaeobot. Palynol. 1979, 28, 311–363. [Google Scholar] [CrossRef]
- Doyle, J.J. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Köhler, M.; Reginato, M.; Souza-Chies, T.T.; Majure, L.C. Insights into chloroplast genome evolution across Opuntioideae (Cactaceae) reveals robust yet sometimes conflicting phylogenetic topologies. Front. Plant Sci. 2020, 11, 729. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33 (Suppl. S2), W686–W689. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. S2), W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Katoh, K.; Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 2010, 26, 1899–1900. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation Statistical Computing: Vienna, Austria, 2017; Available online: https://www.r-project.org/ (accessed on 26 September 2021).
- Yu, Y.; Harris, A.J.; Blair, C.; He, X. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Mol. Phylogenetics Evol. 2015, 87, 46–49. [Google Scholar] [CrossRef]
- Yi, Y.-j.; Zhou, Y.; Cai, Y.-p.; Yang, W.; Li, Z.-W.; Zhao, X. The influence of climate change on an endangered riparian plant species: The root of riparian Homonoia. Ecol. Indic. 2018, 92, 40–50. [Google Scholar] [CrossRef]
- Booth, T.H. Species distribution modelling tools and databases to assist managing forests under climate change. For. Ecol. Manag. 2018, 430, 196–203. [Google Scholar] [CrossRef]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef] [Green Version]
- Hoveka, L.; Bezeng, B.; Yessoufou, K.; Boatwright, J.; Van der Bank, M. Effects of climate change on the future distributions of the top five freshwater invasive plants in South Africa. S. Afr. J. Bot. 2016, 102, 33–38. [Google Scholar] [CrossRef]
- Vasconcelos, T.S.; Rodríguez, M.Á.; Hawkins, B.A. Species distribution modelling as a macroecological tool: A case study using New World amphibians. Ecography 2012, 35, 539–548. [Google Scholar] [CrossRef]
- Fois, M.; Cuena-Lombraña, A.; Fenu, G.; Bacchetta, G. Using species distribution models at local scale to guide the search of poorly known species: Review, methodological issues and future directions. Ecol. Model. 2018, 385, 124–132. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.G.; Raxworthy, C.J.; Nakamura, M.; Townsend Peterson, A. Predicting species distributions from small numbers of occurrence records: A test case using cryptic geckos in Madagascar. J. Biogeogr. 2007, 34, 102–117. [Google Scholar] [CrossRef]
- Bosso, L.; Rebelo, H.; Garonna, A.P.; Russo, D. Modelling geographic distribution and detecting conservation gaps in Italy for the threatened beetle Rosalia alpina. J. Nat. Conserv. 2013, 21, 72–80. [Google Scholar] [CrossRef]
- Yi, Y.-j.; Cheng, X.; Yang, Z.-F.; Zhang, S.-H. Maxent modeling for predicting the potential distribution of endangered medicinal plant (H. riparia Lour) in Yunnan, China. Ecol. Eng. 2016, 92, 260–269. [Google Scholar] [CrossRef]
- Fielding, A.H.; Bell, J.F. A review of methods for the assessment of prediction errors in conservation presence/absence models. Environ. Conserv. 1997, 24, 38–49. [Google Scholar] [CrossRef]
- Warren, D.L.; Seifert, S.N. Ecological niche modeling in Maxent: The importance of model complexity and the performance of model selection criteria. Ecol. Appl. 2011, 21, 335–342. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Group | Gene Name |
|---|---|
| Ribosomal RNAs | rrn16(2), rrn23(2), rrn5(2), rrn4.5(2) |
| Transfer RNAs | trnA-UGC*(2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnH-GUG, trnS-CGA, trnK-UUU*, trnL-CAA(2), trnL-UAA*, trnL-UAG, trnM-CAU, trnN-GUU(2), trnP-UGG, trnQ-UUG, trnR-ACG(2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(2), trnV-GUA*, trnW-CCA, trnY-GUA |
| Proteins of small ribosomal subunit | rps16*, rps2, rps14, rps15, rps4, rps7(2), rps18, rps12* (2), rps11, rps8, rps3, rps19(2) |
| Proteins of large ribosomal subunit | rpl33, rpl20, rpl36, rpl14, rpl16*, rpl22, rpl2* (2), rpl23(2), rpl32 |
| Subunits of RNA polymerase | rpoC2, rpoC1* |
| Photosystem I | psaB, psaA, psaI, psaJ, psaC |
| Photosystem II | psbA, psbB, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ, psbC |
| Cytochrome b/f complex | petA, petB*, petD*, petG, petL, petN |
| Subunits of ATP synthase Protease | atpA, atpB, atpE, atpF*, atpH, atpI clpP** |
| The large subunit of rubisco | rbcL |
| NADH dehydrogenase | ndhA*, ndhB*(2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Maturase | matK |
| Envelope membrane protein | cemA |
| Acetyl-CoA carboxylase | accD |
| Synthesis gene | ccsA |
| Open reading frames (ORF, ycf) | ycf1, ycf2(2), ycf3**, ycf4, ycf15(2), |
| Features | T. asiatica 002151 | T. asiatica 003103 | T. asiatica |
|---|---|---|---|
| Total cp genome size (bp) | 158,508 | 158,508 | 158,434 |
| Length of LSC (bp) | 86,162 | 86,162 | 86,132 |
| Length of IR (bp) | 27,007 | 27,007 | 27,008 |
| Length of SSC (bp) | 18,332 | 18,332 | 18,286 |
| Total GC content (%) | 38.5 | 38.5 | 38.5 |
| GC content of LSC (%) | 36.8 | 36.8 | 36.8 |
| GC content of IR (%) | 42.9 | 42.9 | 42.9 |
| GC content of SSC (%) | 33.4 | 33.4 | 33.4 |
| Total number of genes | 113 | 113 | 115 |
| Protein encoding genes | 79 | 79 | 81 |
| tRNA genes | 30 | 30 | 30 |
| rRNA genes | 4 | 4 | 4 |
| Code | Environmental Variables | Based on the AUC Metric |
|---|---|---|
| bio2 | mean diurnal range | 3.7% |
| bio3 | Isothermality (bio2/bio7) × 100 | 0.9% |
| bio8 | Mean Temperature of the Wettest Quarter | 7.6% |
| bio9 | Mean Temperature of Driest Quarter | 19.9% |
| bio13 | Precipitation of the Wettest Month | 39.4% |
| bio14 | precipitation of the driest month | 2.3% |
| bio15 | precipitation seasonality | 5.3% |
| bio18 | precipitation of warmest quarter | 2% |
| bio19 | precipitation of the coldest quarter | 1.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mutinda, E.S.; Mkala, E.M.; Dong, X.; Yang, J.-X.; Waswa, E.N.; Nanjala, C.; Odago, W.O.; Hu, G.-W.; Wang, Q.-F. Comparative Genomics, Phylogenetics, Biogeography, and Effects of Climate Change on Toddalia asiatica (L.) Lam. (Rutaceae) from Africa and Asia. Plants 2022, 11, 231. https://doi.org/10.3390/plants11020231
Mutinda ES, Mkala EM, Dong X, Yang J-X, Waswa EN, Nanjala C, Odago WO, Hu G-W, Wang Q-F. Comparative Genomics, Phylogenetics, Biogeography, and Effects of Climate Change on Toddalia asiatica (L.) Lam. (Rutaceae) from Africa and Asia. Plants. 2022; 11(2):231. https://doi.org/10.3390/plants11020231
Chicago/Turabian StyleMutinda, Elizabeth Syowai, Elijah Mbandi Mkala, Xiang Dong, Jia-Xin Yang, Emmanuel Nyongesa Waswa, Consolata Nanjala, Wyclif Ochieng Odago, Guang-Wan Hu, and Qing-Feng Wang. 2022. "Comparative Genomics, Phylogenetics, Biogeography, and Effects of Climate Change on Toddalia asiatica (L.) Lam. (Rutaceae) from Africa and Asia" Plants 11, no. 2: 231. https://doi.org/10.3390/plants11020231