
Exploring the Loci Responsible for Awn Development in Rice through Comparative Analysis of All AA Genome Species

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
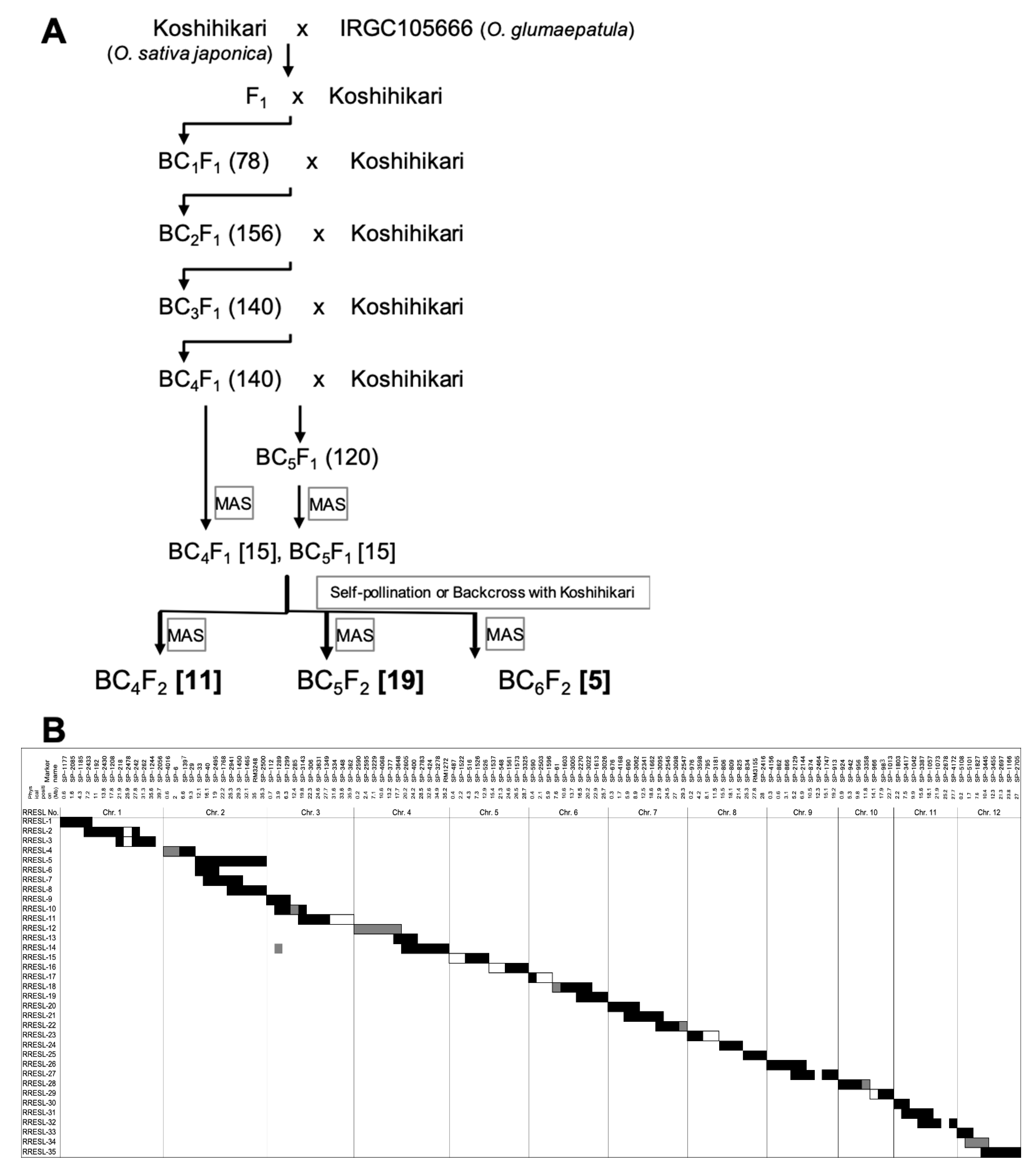
2.1. Development of a CSSL Whose Donor Parent Is O. glumaepatula and Recurrent Parent Is Koshihikari (O. sativa ssp. japonica)
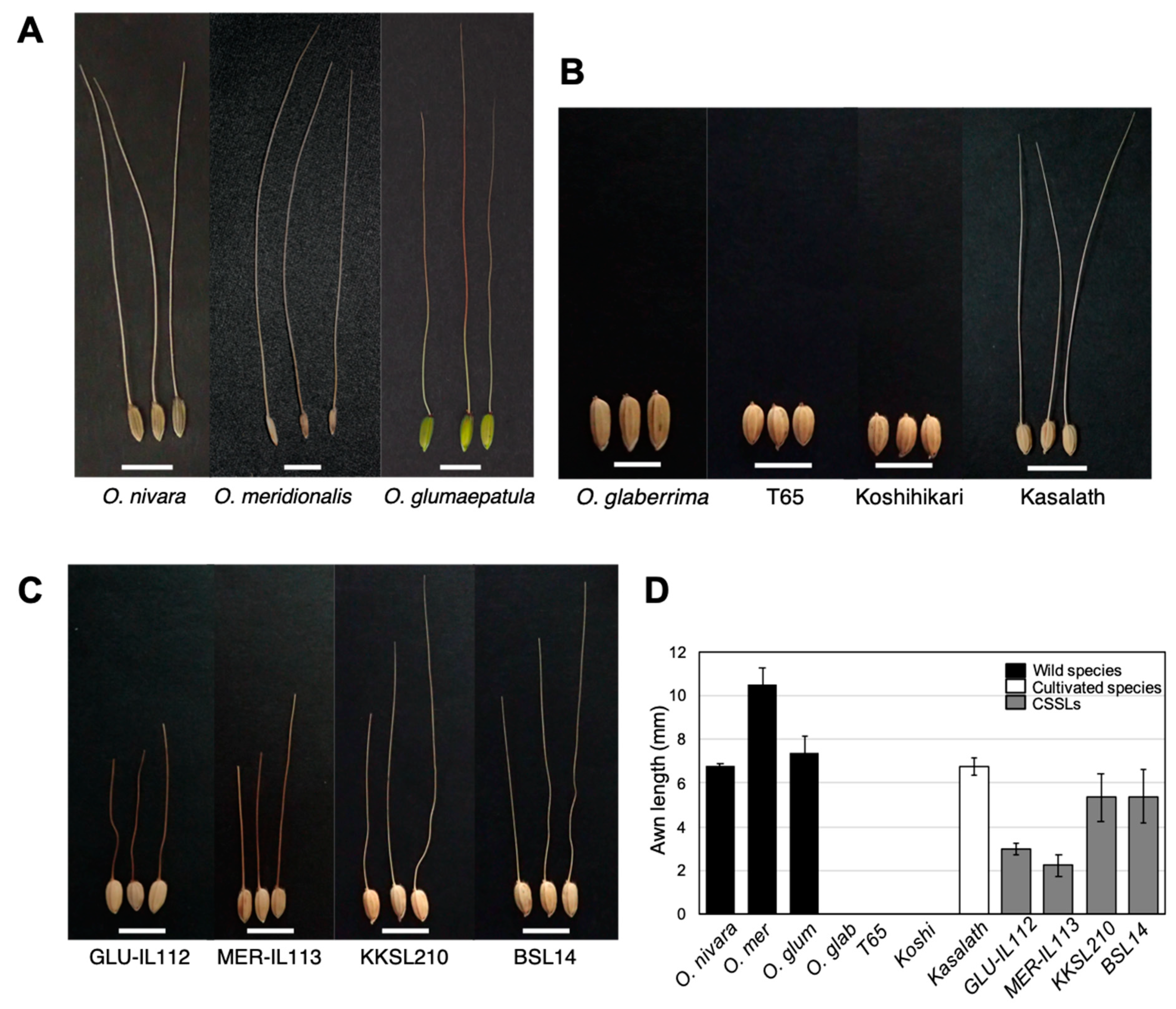
2.2. Awn Phenotype in Wild Rice Species, Cultivated Rice Species, and CSSLs
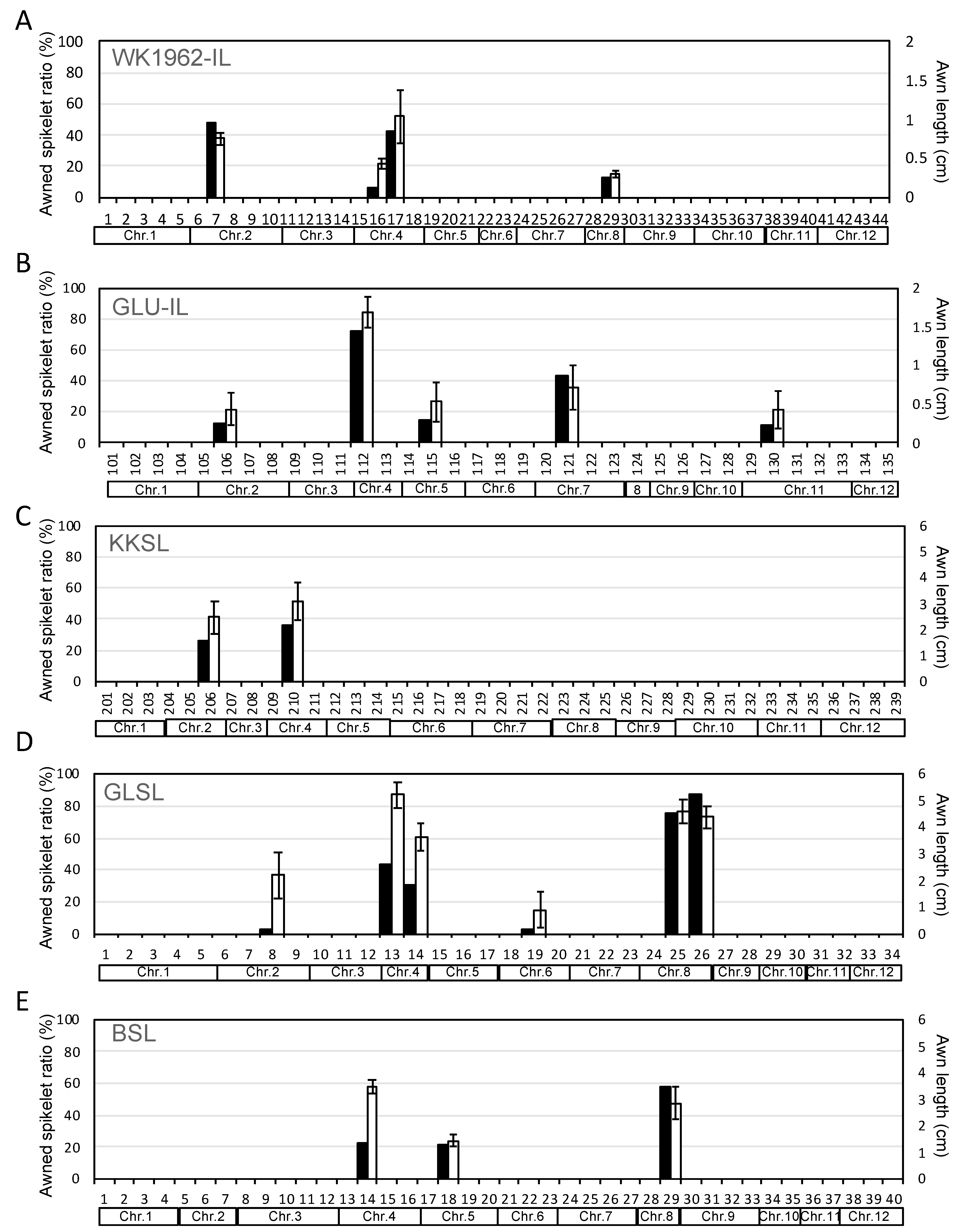
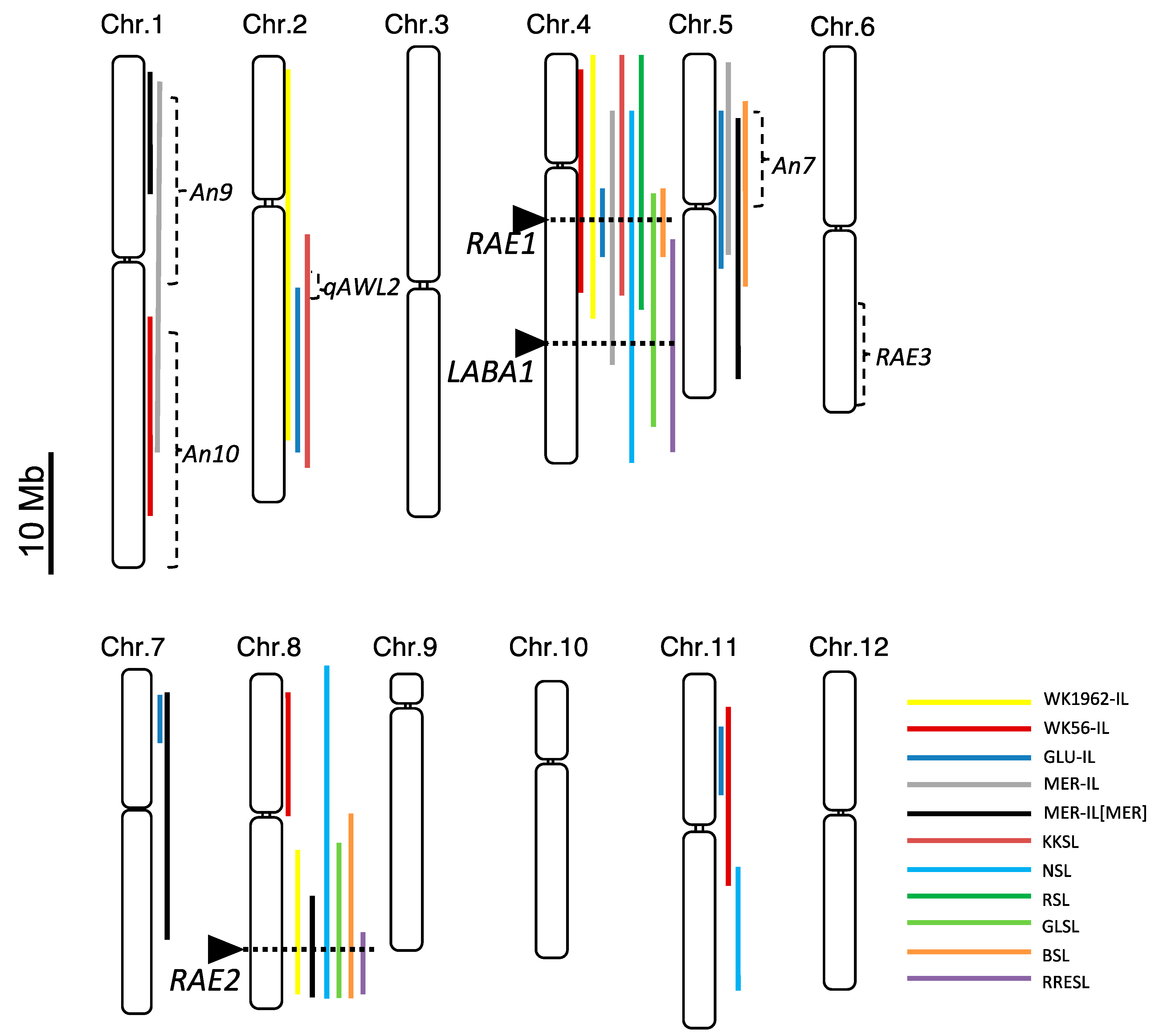
2.3. Identification of the Responsible Loci for Awn Development
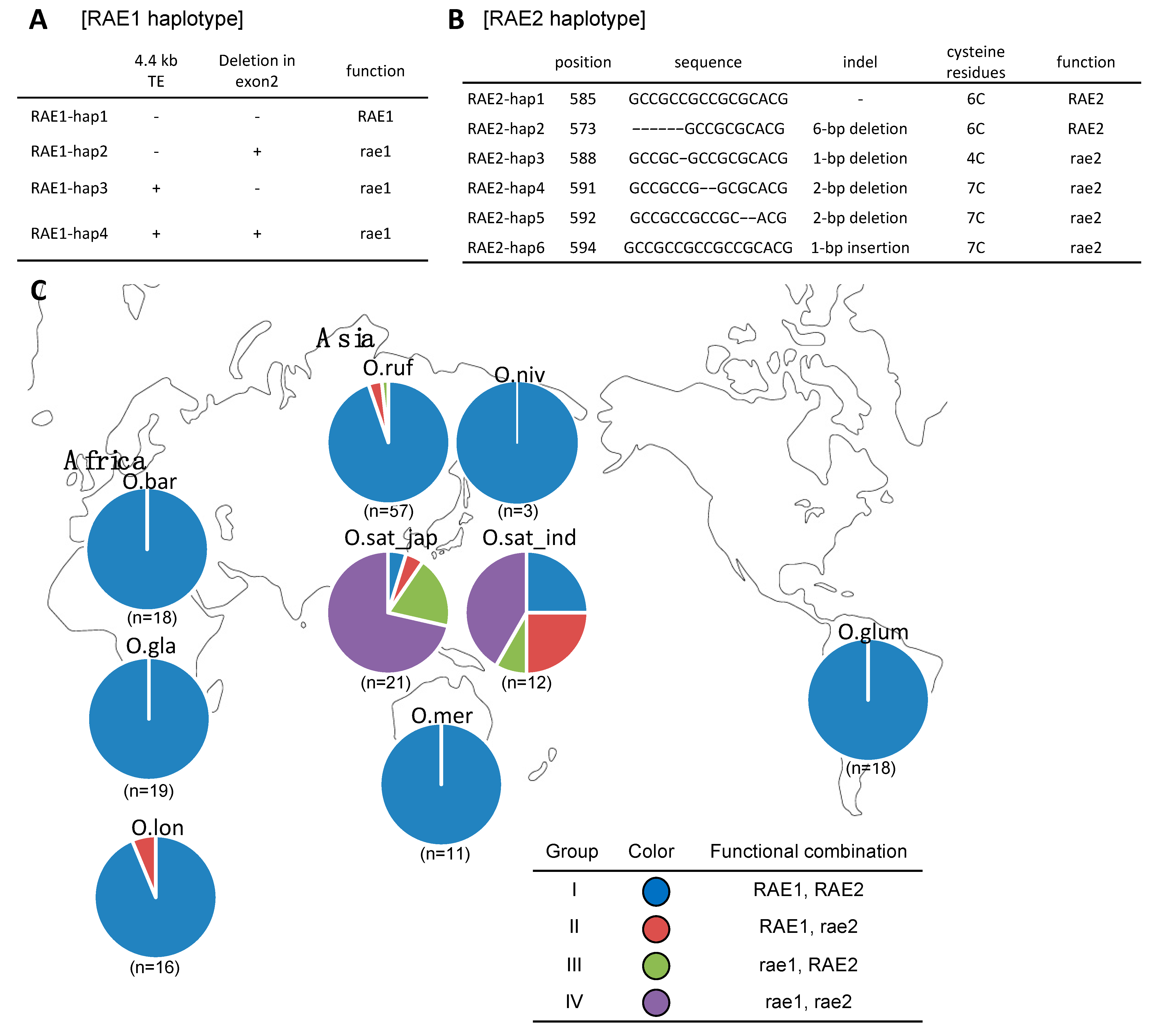
2.4. RAE1 and RAE2 Sequences in Donor and Recurrent Parents
2.5. Awn Development Is Regulated by Different Functional Combinations of RAE1 and RAE2 among the AA Genome Group
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Marker-Assisted Selection (MAS) for Developing RRESL
4.3. Determination of Substituted Segments in RRESL for Making Graphical Genotype
4.4. Growth Conditions and Phenotypic Evaluation
4.5. Chromosomal Localization of Responsible Loci for Awn Development
4.6. Identifying the Functional Mutations of RAE1 and RAE2 in the Donor and Recurrent Parents of CSSL
4.7. Evaluation of Functional RAE1 and RAE2 Allele in AA Genome Rice Species Using SRA Data
4.8. Alignment of TE Inserted Region among AA Genome Species
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khush, G.S. Origin, Dispersal, Cultivation and Variation of Rice. Plant Mol. Biol. 1997, 35, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.A.; Morishima, H.; Kadowaki, K. Diversity in the Oryza Genus. Curr. Opin. Plant Biol. 2003, 6, 139–146. [Google Scholar] [CrossRef]
- Huang, X.; Kurata, N.; Wei, X.; Wang, Z.-X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; et al. A Map of Rice Genome Variation Reveals the Origin of Cultivated Rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, S.; Izawa, T.; Lin, S.Y.; Ebana, K.; Fukuta, Y.; Sasaki, T.; Yano, M. An SNP Caused Loss of Seed Shattering During Rice Domestication. Science 2006, 312, 1392–1396. [Google Scholar] [CrossRef] [Green Version]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The Molecular Genetics of Crop Domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.T.; Thomson, M.J.; Pfeil, B.E.; McCouch, S. Caught Red-Handed: Rc Encodes a Basic Helix-Loop-Helix Protein Conditioning Red Pericarp in Rice. Plant Cell 2006, 18, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Glémin, S.; Bataillon, T. A Comparative View of the Evolution of Grasses under Domestication: Tansley Review. New Phytol. 2009, 183, 273–290. [Google Scholar] [CrossRef]
- Grundbacher, F. The Physiological Function of the Cereal Awn. Bot. Rev. 1963, 29, 366–381. [Google Scholar] [CrossRef]
- Johnson, R.R.; Willmer, C.M.; Moss, D.N. Role of Awns in Photosynthesis, Respiration, and Transpiration of Barley Spikes. Crop Sci. 1975, 15, 217–221. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Li, H.; Zhang, L.; Teng, N.; Lin, Q.; Wang, J.; Kuang, T.; Li, Z.; Li, B.; et al. Awns Play a Dominant Role in Carbohydrate Production during the Grain-Filling Stages in Wheat (Triticum aestivum). Physiol. Plant 2006, 127, 701–709. [Google Scholar] [CrossRef]
- Luo, J.; Liu, H.; Zhou, T.; Gu, B.; Huang, X.; Shangguan, Y.; Zhu, J.; Li, Y.; Zhao, Y.; Wang, Y.; et al. An-1 Encodes a Basic Helix-Loop-Helix Protein That Regulates Awn Development, Grain Size, and Grain Number in Rice. Plant Cell 2013, 25, 3360–3376. [Google Scholar] [CrossRef] [Green Version]
- Gu, B.; Zhou, T.; Luo, J.; Liu, H.; Wang, Y.; Shangguan, Y.; Zhu, J.; Li, Y.; Sang, T.; Wang, Z.; et al. An-2 Encodes a Cytokinin Synthesis Enzyme That Regulates Awn Length and Grain Production in Rice. Mol. Plant 2015, 8, 1635–1650. [Google Scholar] [CrossRef]
- Furuta, T.; Komeda, N.; Asano, K.; Uehara, K.; Gamuyao, R.; Angeles-Shim, R.B.; Nagai, K.; Doi, K.; Wang, D.R.; Yasui, H.; et al. Convergent Loss of Awn in Two Cultivated Rice Species Oryza sativa and Oryza glaberrima Is Caused by Mutations in Different Loci. G3 2015, 5, 2267–2274. [Google Scholar] [CrossRef] [Green Version]
- Hua, L.; Wang, D.R.; Tan, L.; Fu, Y.; Liu, F.; Xiao, L.; Zhu, Z.; Fu, Q.; Sun, X.; Gu, P.; et al. LABA1, a Domestication Gene Associated with Long, Barbed Awns in Wild Rice. Plant Cell 2015, 27, 1875–1888. [Google Scholar] [CrossRef] [Green Version]
- Toriba, T.; Hirano, H.-Y. The DROOPING LEAF and OsETTIN2 Genes Promote Awn Development in Rice. Plant J. 2014, 77, 616–626. [Google Scholar] [CrossRef]
- Bessho-Uehara, K.; Wang, D.R.; Furuta, T.; Minami, A.; Nagai, K.; Gamuyao, R.; Asano, K.; Angeles-Shim, R.B.; Shimizu, Y.; Ayano, M.; et al. Loss of Function at RAE2, a Previously Unidentified EPFL, Is Required for Awnlessness in Cultivated Asian Rice. Proc. Natl. Acad. Sci. USA 2016, 113, 8969–8974. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Hua, L.; Zhu, Z.; Tan, L.; Zhao, X.; Zhang, W.; Liu, F.; Fu, Y.; Cai, H.; Sun, X.; et al. GAD1 Encodes a Secreted Peptide That Regulates Grain Number, Grain Length, and Awn Development in Rice Domestication. Plant Cell 2016, 28, 2453–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, Z.; Sun, X.; Zhu, X.; Li, B.; Li, J.; Guo, H.; Chen, C.; Pan, Y.; Liang, Y.; et al. Natural Alleles of GLA for Grain Length and Awn Development Were Differently Domesticated in Rice Subspecies Japonica and Indica. Plant Biotechnol. J. 2019, 17, 1547–1559. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, W.; Toriba, T.; Ohmori, Y.; Yoshida, A.; Kawai, A.; Mayama-Tsuchida, T.; Ichikawa, H.; Mitsuda, N.; Ohme-Takagi, M.; Hirano, H.-Y. The YABBY Gene TONGARI-BOUSHI1 Is Involved in Lateral Organ Development and Maintenance of Meristem Organization in the Rice Spikelet. Plant Cell 2012, 24, 80–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Zou, T.; He, Z.; Yuan, G.; Luo, T.; Zhu, J.; Liang, Y.; Deng, Q.; Wang, S.; Zheng, A.; et al. Grain Length and AWN 1 Negatively Regulates Grain Size in Rice: GLA1 Mediates Grain Size. J. Integr. Plant Biol. 2019, 61, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Tsuchimoto, S.; Ohtsubo, H.; Ohtsubo, E. Evolutionary Relationships among Rice Species with AA Genome Based on SINE Insertion Analysis. Genes Genet. Syst. 2002, 77, 323–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.L.; Sanchez, P.L.; Yu, S.; Lorieux, M.; Eizenga, G.C. Chromosome Segment Substitution Lines: A Powerful Tool for the Introgression of Valuable Genes from Oryza Wild Species into Cultivated Rice (O. sativa). Rice 2010, 3, 218–234. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Aida, Y.; Nakamura, K.; Tsunematsu, H.; Doi, K.; Yoshimura, A. Reciprocal Chromosome Segment Substitution Series Derived from Japonica and Indica Cross of Rice (Oryza sativa L.). Breed. Sci. 2002, 52, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Ebitani, T.; Takeuchi, Y.; Nonoue, Y.; Yamamoto, T.; Takeuchi, K.; Yano, M. Construction and Evaluation of Chromosome Segment Substitution Lines Carrying Overlapping Chromosome Segments of Indica Rice Cultivar ‘Kasalath’ in a Genetic Background of Japonica Elite Cultivar ‘Koshihikari’. Breed. Sci. 2005, 55, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Ando, T.; Yamamoto, T.; Shimizu, T.; Ma, X.F.; Shomura, A.; Takeuchi, Y.; Lin, S.Y.; Yano, M. Genetic Dissection and Pyramiding of Quantitative Traits for Panicle Architecture by Using Chromosomal Segment Substitution Lines in Rice. Theor. Appl. Genet. 2008, 116, 881–890. [Google Scholar] [CrossRef]
- Abe, T.; Nonoue, Y.; Ono, N.; Omoteno, M.; Kuramata, M.; Fukuoka, S.; Yamamoto, T.; Yano, M.; Ishikawa, S. Detection of QTLs to Reduce Cadmium Content in Rice Grains Using LAC23/Koshihikari Chromosome Segment Substitution Lines. Breed. Sci. 2013, 63, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Liu, F.; Xue, W.; Wang, G.; Ye, S.; Zhu, Z.; Fu, Y.; Wang, X.; Sun, C. Development of Oryza Rufipogon and O. sativa Introgression Lines and Assessment for Yield-Related Quantitative Trait Loci. J. Integr. Plant Biol. 2007, 49, 871–884. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Carabalí, S.; Giraldo, O.; Martínez, C.; Correa, F.; Prado, G.; Tohme, J.; Lorieux, M. Identification of a Rice Stripe Necrosis Virus Resistance Locus and Yield Component QTLs Using Oryza sativa × O. glaberrima Introgression Lines. BMC Plant Biol. 2010, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Shim, R.A.; Angeles, E.R.; Ashikari, M.; Takashi, T. Development and Evaluation of Oryza glaberrima Steud. Chromosome Segment Substitution Lines (CSSLs) in the Background of O. sativa L. Cv. Koshihikari. Breed. Sci. 2010, 60, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Uehara, K.; Angeles-Shim, R.B.; Shim, J.; Ashikari, M.; Takashi, T. Development and Evaluation of Chromosome Segment Substitution Lines (CSSLs) Carrying Chromosome Segments Derived from Oryza rufipogon in the Genetic Background of Oryza sativa L. Breed. Sci. 2014, 63, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessho-Uehara, K.; Furuta, T.; Masuda, K.; Yamada, S.; Angeles-Shim, R.B.; Ashikari, M.; Takashi, T. Construction of Rice Chromosome Segment Substitution Lines Harboring Oryza barthii Genome and Evaluation of Yield-Related Traits. Breed. Sci. 2017, 67, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, Y.; Win, K.T.; Miyazaki, Y.; Ogata, C.; Yasui, H.; Yoshimura, A. Development of Introgression Lines of AA Genome Oryza Species, O. glaberrima, O. rufipogon, and O. nivara, in the Genetic Background of O. sativa L. Cv. Taichung 65. Breed. Sci. 2019, 69, 359–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobrizal, K.; Sanchez, P.L.; Doi, K.; Angeles, E.R.; Khush, G.S.; Yoshimura, A. Development of Oryza glumaepatula Introgression Lines in Rice, Oryza sativa L. Rice Genet. Newsl. 1999, 16, 107–108. [Google Scholar]
- Kurakazu, T. Oryza meridionalis Chromosomal Segment Introgression Lines in Cultivated Rice, O. sativa L. Rice Genet. Newsl. 2001, 18, 81–82. [Google Scholar]
- Furuta, T.; Uehara, K.; Angeles-Shim, R.B.; Shim, J.; Nagai, K.; Ashikari, M.; Takashi, T. Development of Chromosome Segment Substitution Lines Harboring Oryza nivara Genomic Segments in Koshihikari and Evaluation of Yield-Related Traits. Breed. Sci. 2016, 66, 845–850. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, S. Mapping of Genes for Awn in Rice Using Oryza meridionalis Introgression Lines. Rice Genet. Newsl. 2003, 20, 17–18. [Google Scholar]
- International Rice Research Institute. Standard Evaluation System (SES) for Rice, 5th ed.; International Rice Research Institute: Los Banos, Philippines, 2013. [Google Scholar]
- Matsushita, S. Identification of New Alleles of Awnness Genes, An7 and An8, in Rice Using Oryza glumaepatula Introgression Lines. Rice Genet. Newsl. 2003, 20, 19–20. [Google Scholar]
- Amarasinghe, Y.P.J.; Kuwata, R.; Nishimura, A.; Phan, P.D.T.; Ishikawa, R.; Ishii, T. Evaluation of Domestication Loci Associated with Awnlessness in Cultivated Rice, Oryza sativa. Rice 2020, 13, 26. [Google Scholar] [CrossRef]
- Gu, X.Y.; Kianian, S.F.; Hareland, G.A.; Hoffer, B.L.; Foley, M.E. Genetic analysis of adaptive syndromes interrelated with seed dormancy in weedy rice (Oryza sativa). Theor. Appl. Genet. 2005, 110, 1108–1118. [Google Scholar] [CrossRef]
- Fawcett, J.A.; Kado, T.; Sasaki, E.; Takuno, S.; Yoshida, K.; Sugino, R.P.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; et al. QTL Map Meets Population Genomics: An Application to Rice. PLoS ONE 2013, 8, e83720. [Google Scholar] [CrossRef] [Green Version]
- Ikemoto, M.; Otsuka, M.; Thanh, P.T.; Phan, P.D.T.; Ishikawa, R.; Ishii, T. Gene Interaction at Seed-Awning Loci in the Genetic Background of Wild Rice. Genes Genet. Syst. 2017, 92, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Vieira, F.G.; Crawford, J.E.; Chu, C.; Nielsen, R. Asian Wild Rice Is a Hybrid Swarm with Extensive Gene Flow and Feralization from Domesticated Rice. Genome Res. 2017, 27, 1029–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kumar, S.; Agarwal, S. Ranvijay Fast and Memory Efficient Approach for Mapping NGS Reads to a Reference Genome. J. Bioinform. Comput. Biol. 2019, 17, 1950008. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mose, L.E.; Perou, C.M.; Parker, J.S. Improved Indel Detection in DNA and RNA via Realignment with ABRA2. Bioinformatics 2019, 35, 2966–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSSL Name | Line Number | Donor Parent | Recurrent Parent | Reference |
|---|---|---|---|---|
| WK1962-IL | 44 | WK1962 (O. rufipogon) | T65 (O. sativa japonica) | [32] |
| WK56-IL | 34 | WK56 (O. nivara) | [32] | |
| GLU-IL | 35 | WK35 (O. glumaepatula) | [33] | |
| MER-IL | 36 | W1625 (O. meridionalis) | [34] | |
| MER-IL [MER] | 42 | W1625 (O. meridionalis) | [34] | |
| KKSL | 39 | Kasalath (O. sativa)a | Koshihikari (O. sativa japonica) | [24] |
| NSL | 26 | W0054 (O. nivara) | [35] | |
| RSL | 33 | W0106 (O. rufipogon) | [30] | |
| GLSL | 34 | IRGC104038 (O. glaberrima) a | [29] | |
| BSL | 40 | W0009 (O. barthii) | [31] | |
| RRESL | 35 | IRGC105666 (O. glumaepatula) | this study |
| Cultivated/Wild | Line Name | RAE1 TE | RAE1 2nd_exon_G | RAE1 Function | RAE2 Cys no. | RAE2 Function | Awn Phenotype |
|---|---|---|---|---|---|---|---|
| cultivated | Koshihikari (O. sativa japonica) | + | G | rae1 | 4 | rae2 | - |
| cultivated | T65 (O. sativa japonica) | + | G | rae1 | 4 | rae2 | - |
| cultivated | IRGC104038 (O. glaberrima) | - | G | RAE1 | 6 | RAE2 | - |
| cultivated | Kasalath (O. sativa indica, aus) | - | G | RAE1 | 7 | rae2 | + |
| wild | IRGC105715 (O. nivara) | - | G | RAE1 | 6 | RAE2 | + |
| wild | W0054 (O. nivara) | - | G | RAE1 | 6 | RAE2 | + |
| wild | W1962 (O. rufipogon) | - | G | RAE1 | 6 | RAE2 | + |
| wild | W0106 (O. rufipogon) | - | G | RAE1 | 7 | rae2 | + |
| wild | W1625 (O. meridionalis) | - | G | RAE1 | 6 | RAE2 | + |
| wild | WK35 (O. glumaepatula) | - | G | RAE1 | 6 | RAE2 | + |
| wild | IRGC105666 (O. glumaepatula) | - | G | RAE1 | 6 | RAE2 | + |
| wild | W0009 (O. barthii) | - | G | RAE1 | 6 | RAE2 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bessho-Uehara, K.; Yamagata, Y.; Takashi, T.; Makino, T.; Yasui, H.; Yoshimura, A.; Ashikari, M. Exploring the Loci Responsible for Awn Development in Rice through Comparative Analysis of All AA Genome Species. Plants 2021, 10, 725. https://doi.org/10.3390/plants10040725
Bessho-Uehara K, Yamagata Y, Takashi T, Makino T, Yasui H, Yoshimura A, Ashikari M. Exploring the Loci Responsible for Awn Development in Rice through Comparative Analysis of All AA Genome Species. Plants. 2021; 10(4):725. https://doi.org/10.3390/plants10040725
Chicago/Turabian StyleBessho-Uehara, Kanako, Yoshiyuki Yamagata, Tomonori Takashi, Takashi Makino, Hideshi Yasui, Atsushi Yoshimura, and Motoyuki Ashikari. 2021. "Exploring the Loci Responsible for Awn Development in Rice through Comparative Analysis of All AA Genome Species" Plants 10, no. 4: 725. https://doi.org/10.3390/plants10040725