
Doxorubicin-Loaded Core–Shell UiO-66@SiO2 Metal–Organic Frameworks for Targeted Cellular Uptake and Cancer Treatment

 , , , and
, , , and
Abstract
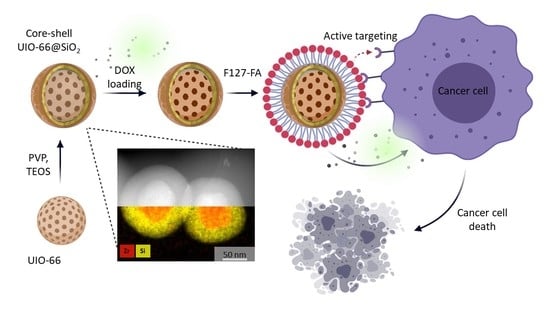
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of UiO-66 and UiO-66-NH2 Nanoparticles
2.3. Synthesis of F127-FA Conjugate
2.4. Modification of the Surface of Nanoparticles with Silica Shells (Silanization of MOFs)
2.5. Loading Nanoparticles with Doxorubicin and Modification with Folic Acid Conjugate
2.6. Cell Cultures
2.6.1. Flow Cytometry
2.6.2. Confocal Laser Scanning Microscopy
2.7. Cytotoxicity Study In Vitro
2.8. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterization of UiO-66 and UiO-66-NH2
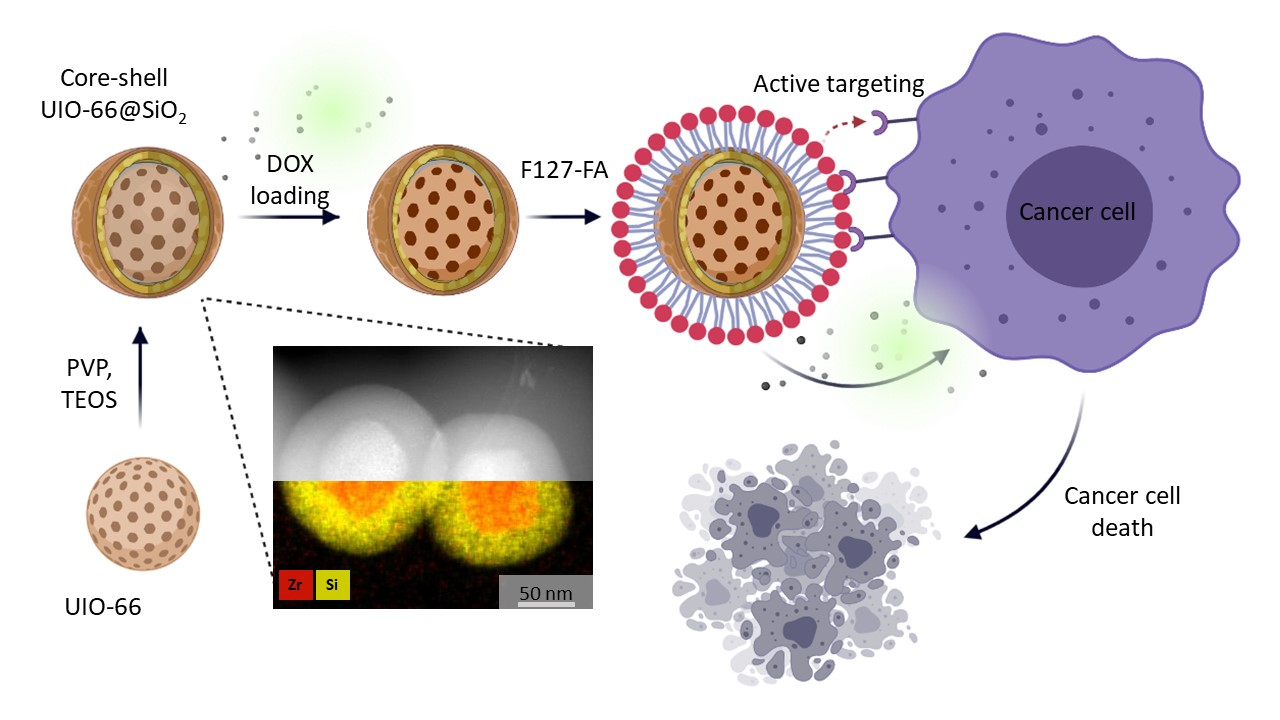
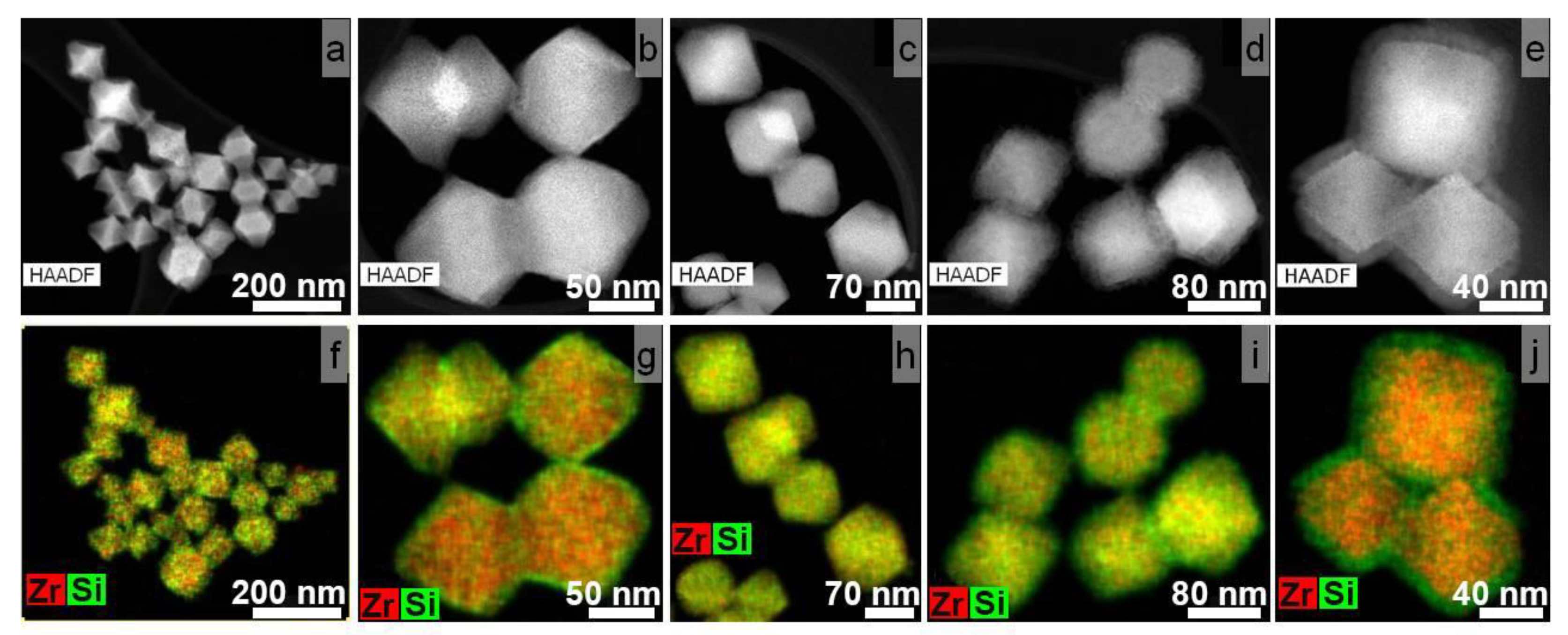
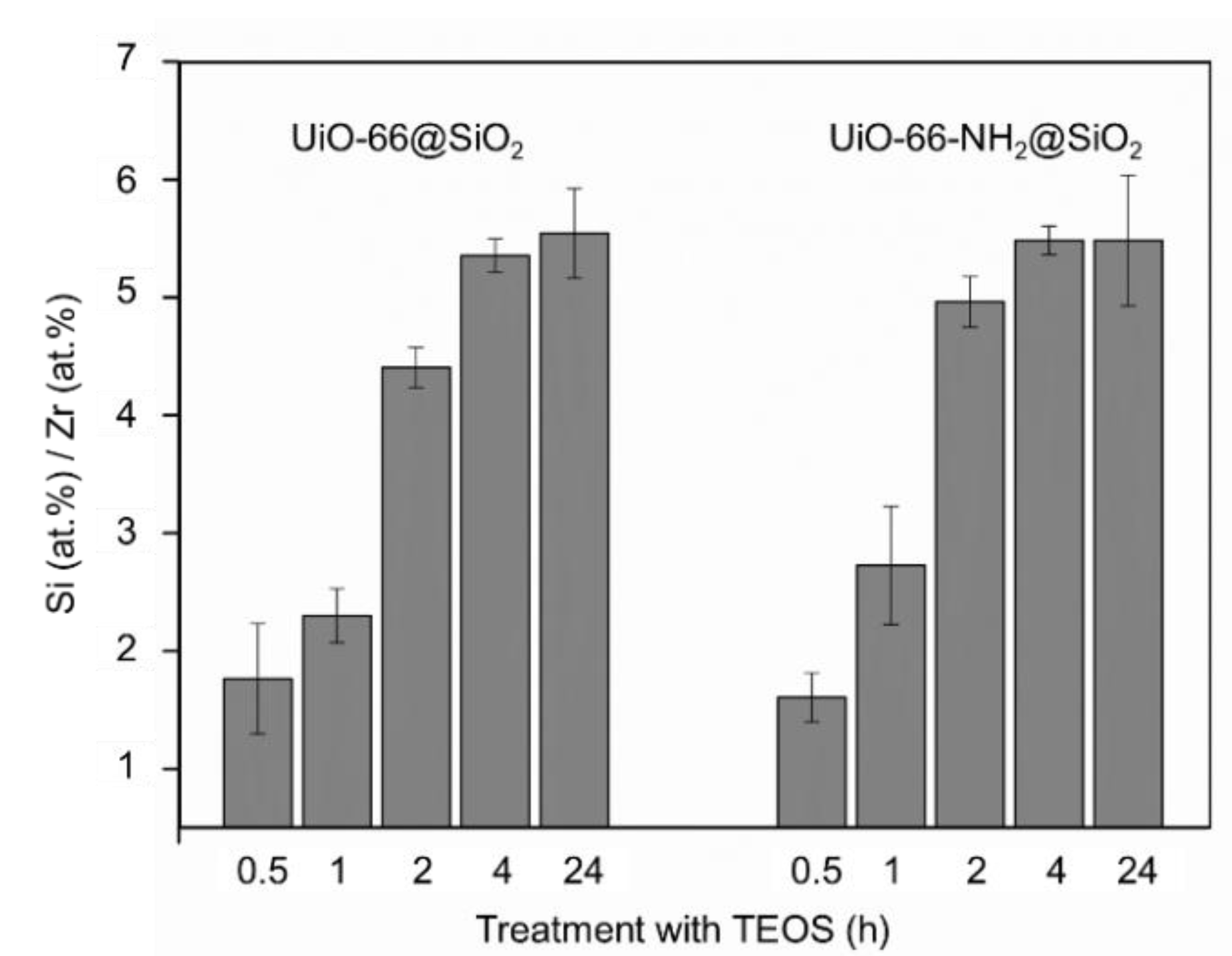
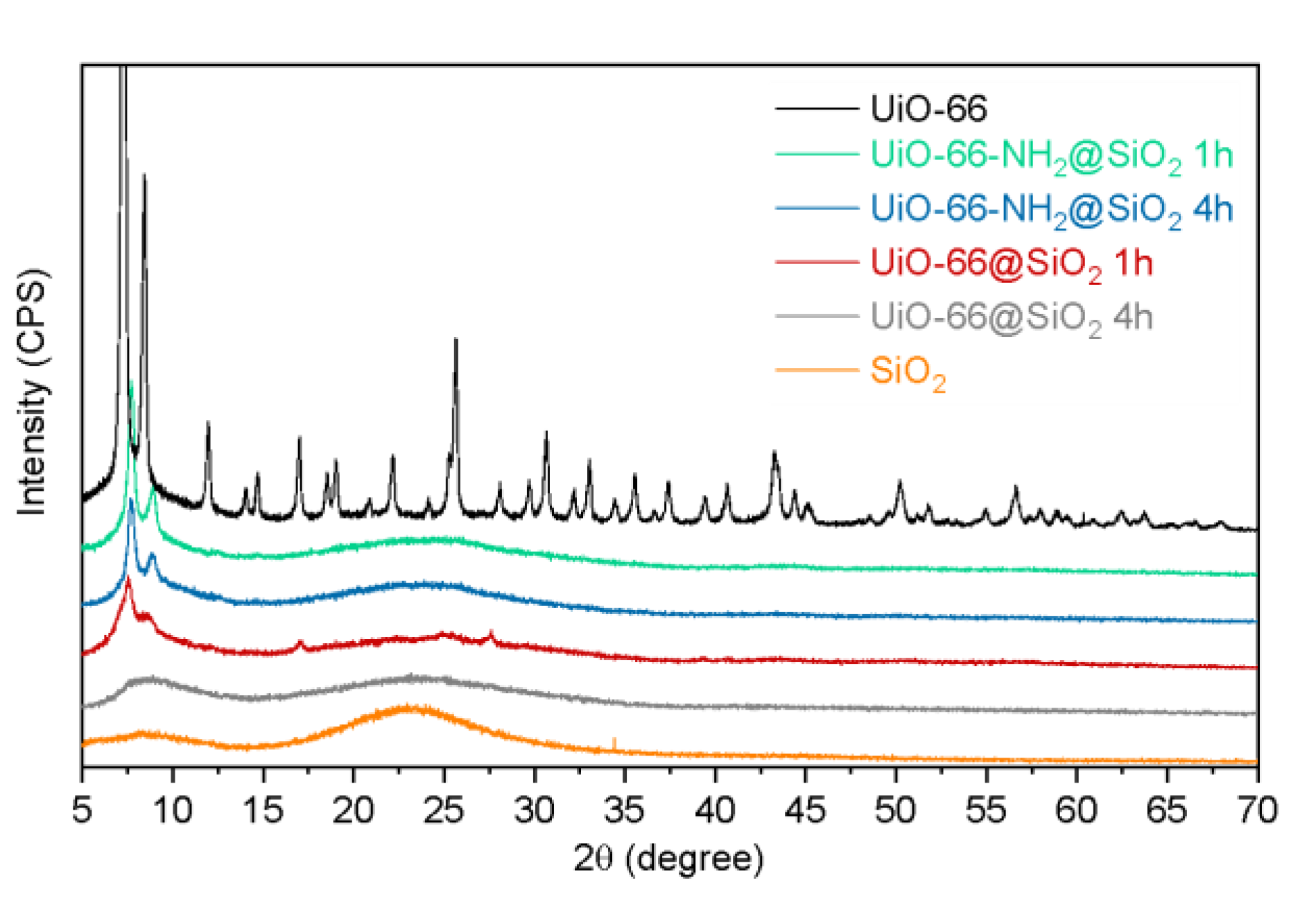
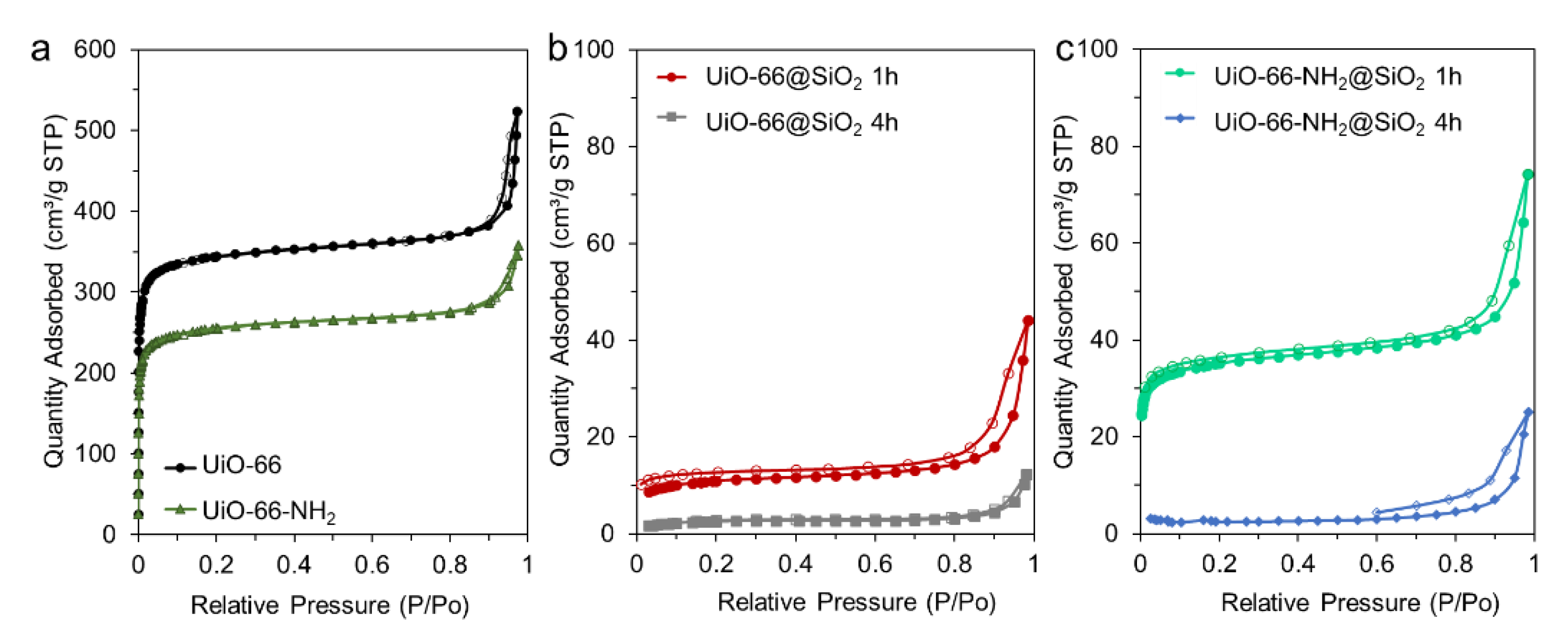
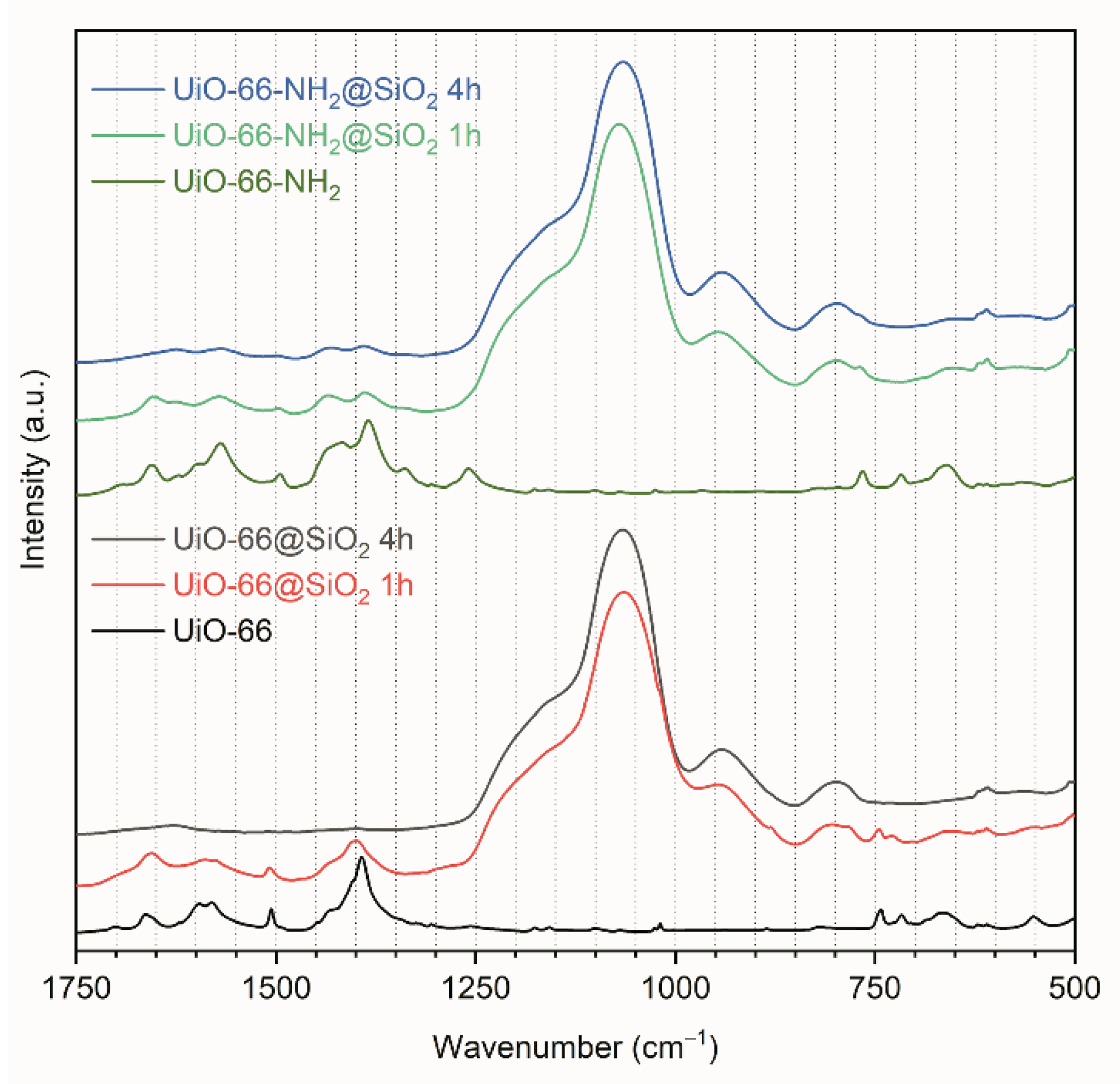
3.2. Synthesis of Core-Shell Structures UiO-66@SiO2
3.3. Colloidal Stability Study in Model Biological Media
3.4. Loading of MOFs with DOX
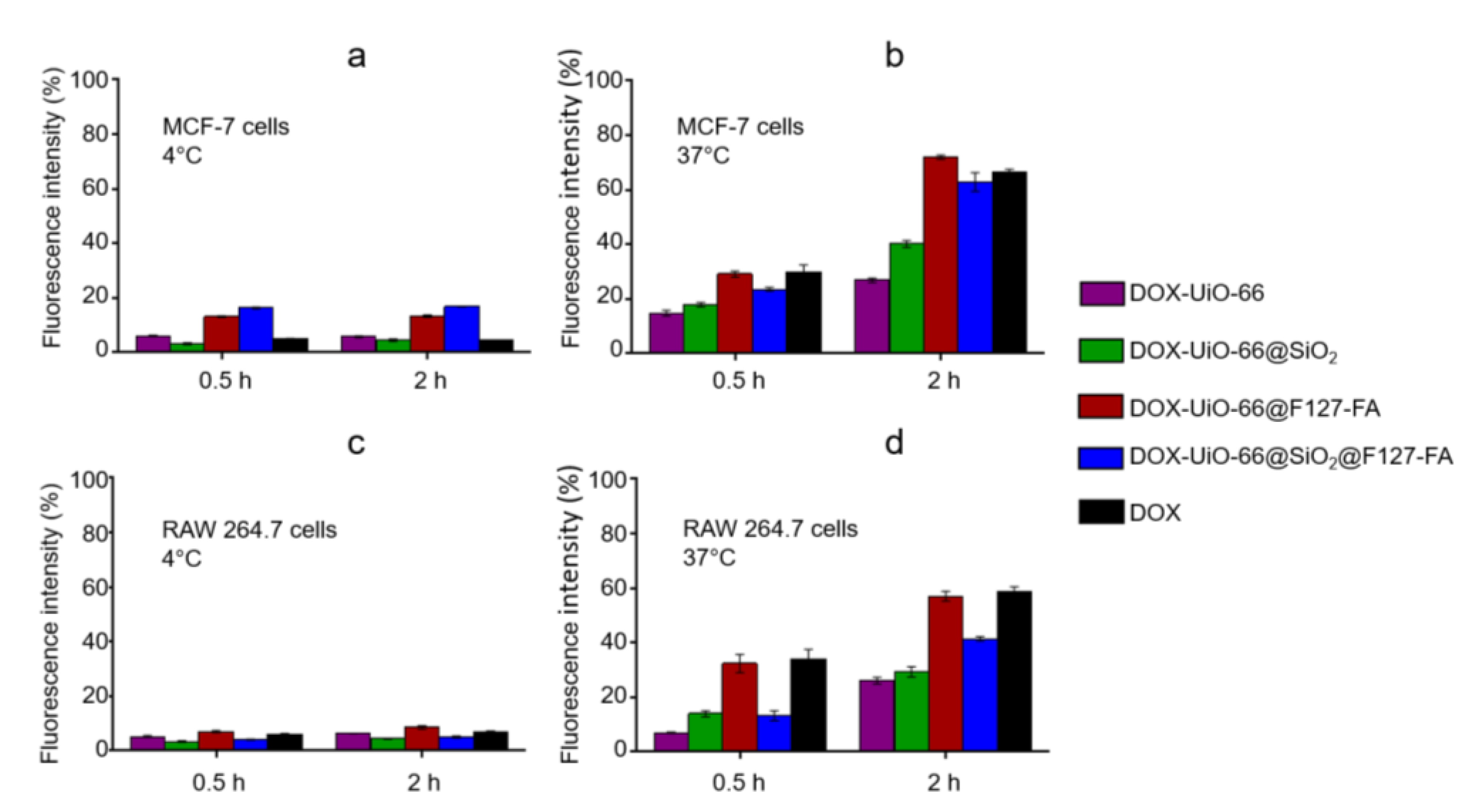
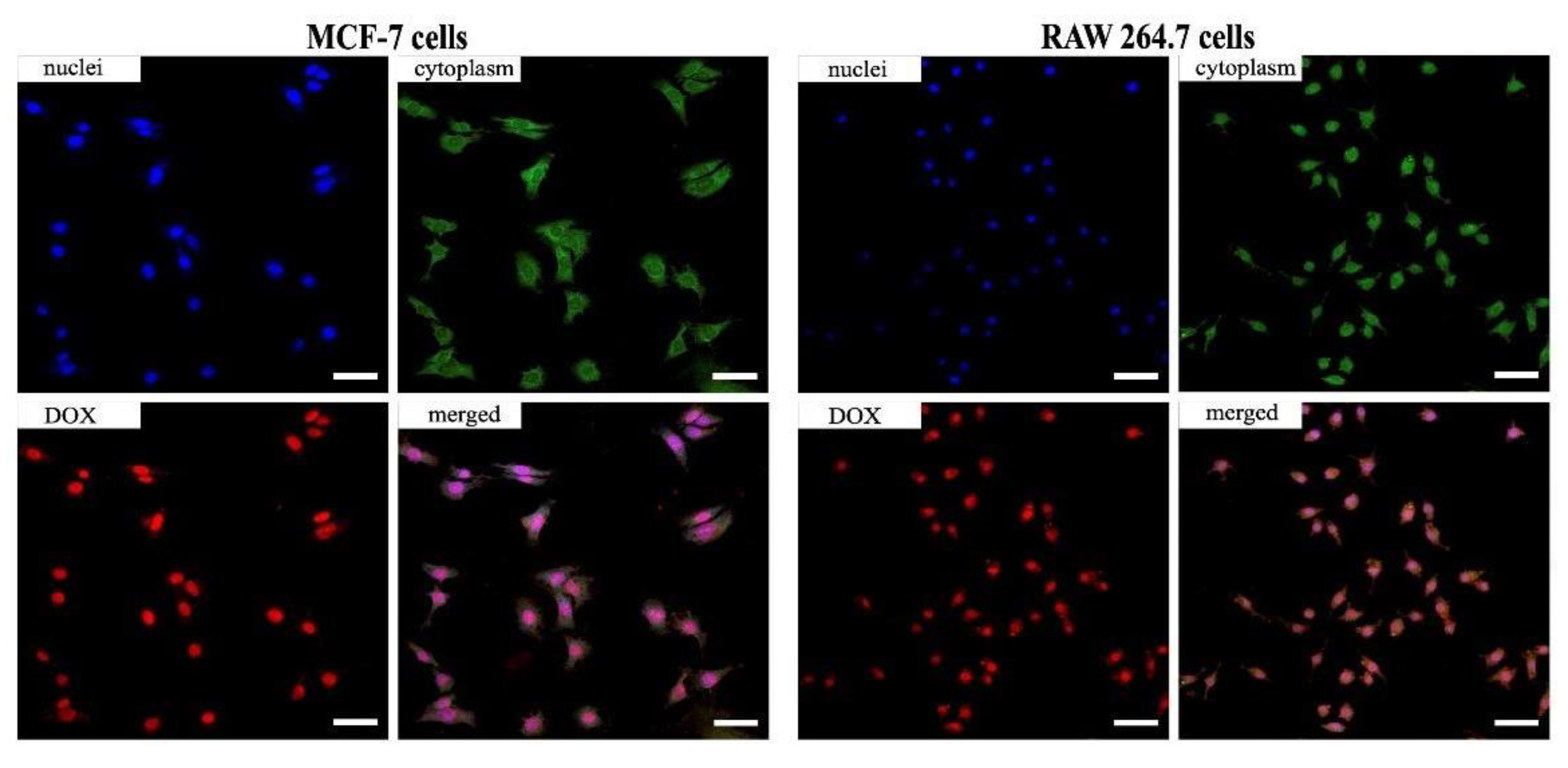
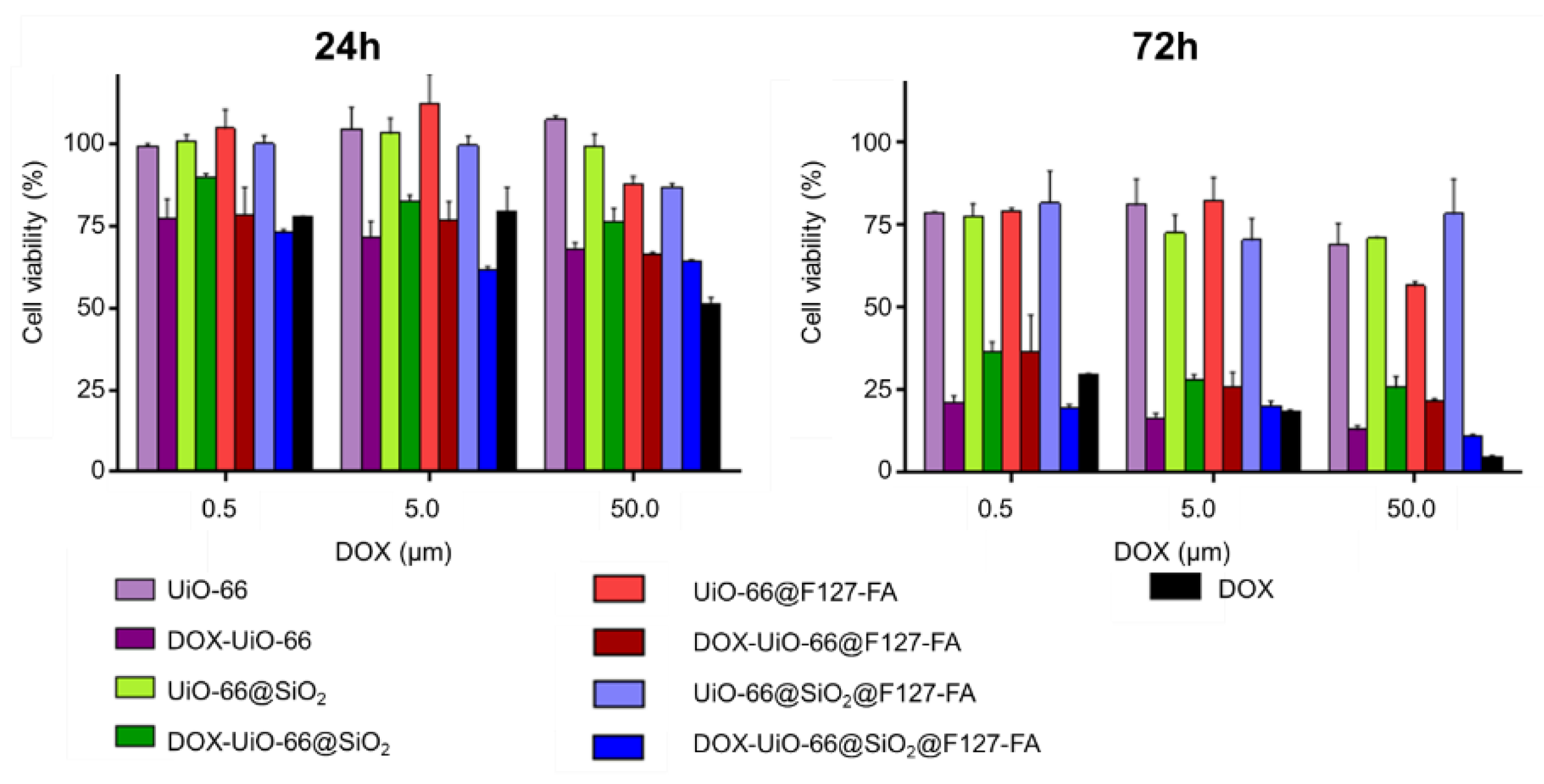
3.5. Evaluation of Cellular Uptake and Cytotoxicity Study In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Safaei, M.; Foroughi, M.M.; Ebrahimpoor, N.; Jahani, S.; Omidi, A.; Khatami, M. A review on metal-organic frameworks: Synthesis and applications. TrAC Trends Anal. Chem. 2019, 118, 401–425. [Google Scholar] [CrossRef]
- Zou, D.; Liu, D. Understanding the modifications and applications of highly stable porous frameworks via UiO-66. Mater. Today Chem. 2019, 12, 139–165. [Google Scholar] [CrossRef]
- Delpiano, G.R.; Tocco, D.; Medda, L.; Magner, E.; Salis, A. Adsorption of Malachite Green and Alizarin Red S Dyes Using Fe-BTC Metal Organic Framework as Adsorbent. Int. J. Mol. Sci. 2021, 22, 788. [Google Scholar] [CrossRef]
- Winarta, J.; Shan, B.; McIntyre, S.M.; Ye, L.; Wang, C.; Liu, J.; Mu, B. A Decade of UiO-66 Research: A Historic Review of Dynamic Structure, Synthesis Mechanisms, and Characterization Techniques of an Archetypal Metal-Organic Framework. Cryst. Growth Des. 2020, 20, 1347–1362. [Google Scholar] [CrossRef]
- Anik, Ü.; Timur, S.; Dursun, Z. Metal organic frameworks in electrochemical and optical sensing platforms: A review. Microchim. Acta 2019, 186, 196. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wu, X.-L.; Xiong, J.; Zong, M.-H.; Lou, W.-Y. Metal-organic frameworks as novel matrices for efficient enzyme immobilization: An update review. Coord. Chem. Rev. 2020, 406, 213149. [Google Scholar] [CrossRef]
- Simon-Yarza, T.; Rojas, S.; Horcajada, P.; Serre, C. The Situation of Metal-Organic Frameworks in Biomedicine; 2017; Volume 4; ISBN 9780081006924. Available online: https://www.researchgate.net/publication/311996625_The_Situation_of_Metal-Organic_Frameworks_in_Biomedicine (accessed on 3 May 2022).
- Ruyra, A.; Yazdi, A.; Espín, J.; Carné-Sánchez, A.; Roher, N.; Lorenzo, J.; Imaz, I.; Maspoch, D. Synthesis, culture medium stability, and in vitro and in vivo zebrafish embryo toxicity of metal-organic framework nanoparticles. Chem.-A Eur. J. 2015, 21, 2508–2518. [Google Scholar] [CrossRef]
- Zhao, H.X.; Zou, Q.; Sun, S.K.; Yu, C.; Zhang, X.; Li, R.J.; Fu, Y.Y. Theranostic metal-organic framework core-shell composites for magnetic resonance imaging and drug delivery. Chem. Sci. 2016, 7, 5294–5301. [Google Scholar] [CrossRef] [Green Version]
- Della Rocca, J.; Liu, D.; Lin, W. Nanoscale Metal–Organic Frameworks for Biomedical Imaging and Drug Delivery. Acc. Chem. Res. 2011, 44, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.M.L.; Rieter, W.J.; Lin, W. Manganese-based nanoscale metal-organic frameworks for magnetic resonance imaging. J. Am. Chem. Soc. 2008, 130, 14358–14359. [Google Scholar] [CrossRef]
- Taylor-Pashow, K.M.L.; Della Rocca, J.; Xie, Z.; Tran, S.; Lin, W. Postsynthetic modifications of iron-carboxylate nanoscale metal-organic frameworks for imaging and drug delivery. J. Am. Chem. Soc. 2009, 131, 14261–14263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieter, W.J.; Pott, K.M.; Taylor, K.M.L.; Lin, W. Nanoscale coordination polymers for platinum-based anticancer drug delivery. J. Am. Chem. Soc. 2008, 130, 11584–11585. [Google Scholar] [CrossRef] [PubMed]
- Abánades Lázaro, I.; Haddad, S.; Sacca, S.; Orellana-Tavra, C.; Fairen-Jimenez, D.; Forgan, R.S. Selective Surface PEGylation of UiO-66 Nanoparticles for Enhanced Stability, Cell Uptake, and pH-Responsive Drug Delivery. Chem 2017, 2, 561–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Moreno, P.; Buzón, P.; Boulaiz, H.; Peula-García, J.M.; Ortega-Vinuesa, J.L.; Luque, I.; Salvati, A.; Marchal, J.A. Balancing the effect of corona on therapeutic efficacy and macrophage uptake of lipid nanocapsules. Biomaterials 2015, 61, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Besheer, A.; Vogel, J.; Glanz, D.; Kressler, J.; Groth, T.; Mäder, K. Characterization of PLGA Nanospheres Stabilized with Amphiphilic Polymers: Hydrophobically Modified Hydroxyethyl Starch vs. Pluronics. Mol. Pharm. 2009, 6, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Tilley, A.J.; Drummond, C.J.; Boyd, B.J. Disposition and association of the steric stabilizer Pluronic® F127 in lyotropic liquid crystalline nanostructured particle dispersions. J. Colloid Interface Sci. 2013, 392, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, W.; Yu, S.; Huang, X.; Lang, X.; He, Q.; Gao, L.; Zhu, H.; Chen, J. A biocompatible Zr-based metal-organic framework UiO-66-PDC as an oral drug carrier for pH-response release. J. Solid State Chem. 2021, 293, 121805. [Google Scholar] [CrossRef]
- Arcuri, C.; Monarca, L.; Ragonese, F.; Mecca, C.; Bruscoli, S.; Giovagnoli, S.; Donato, R.; Bereshchenko, O.; Fioretti, B.; Costantino, F. Probing Internalization Effects and Biocompatibility of Ultrasmall Zirconium Metal-Organic Frameworks UiO-66 NP in U251 Glioblastoma Cancer Cells. Nanomaterials 2018, 8, 867. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Guo, B.; You, H.; Wang, Z.; Leng, Q.; Ding, L.; Wang, Q. Synthesis and surface modification of mesoporous metal-organic framework (UiO-66) for efficient pH-responsive drug delivery and lung cancer treatment. Nanotechnology 2021, 32, 295704. [Google Scholar] [CrossRef]
- Nagarkar, S.S.; Saha, T.; Desai, A.V.; Talukdar, P.; Ghosh, S.K. Metal-organic framework based highly selective fluorescence turn-on probe for hydrogen sulphide. Sci. Rep. 2015, 4, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.; Kang, S.; Park, N.; Park, E.J.; Jin, X.; Kim, K.-D.; Seo, H.O.; Lee, S.M.; Kim, H.J.; Kwon, W.H.; et al. Metal–Organic Framework@Microporous Organic Network: Hydrophobic Adsorbents with a Crystalline Inner Porosity. J. Am. Chem. Soc. 2014, 136, 6786–6789. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.; Ben Yahia, M.; Hall, S.; Miller, S.R.; Chevreau, H.; Elkaïm, E.; Maurin, G.; Horcajada, P.; Serre, C. Rationale of drug encapsulation and release from biocompatible porous metal-organic frameworks. Chem. Mater. 2013, 25, 2767–2776. [Google Scholar] [CrossRef]
- Zhu, X.; Gu, J.; Wang, Y.; Li, B.; Li, Y.; Zhao, W.; Shi, J. Inherent anchorages in UiO-66 nanoparticles for efficient capture of alendronate and its mediated release. Chem. Commun. 2014, 50, 8779–8782. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.; Zhang, W.; Zhang, J.; Luo, G.; Jia, Y.; Deng, M.; Ling, Y. Facile preparation of UiO-66 nanoparticles with tunable sizes in a continuous flow microreactor and its application in drug delivery. Microporous Mesoporous Mater. 2016, 220, 148–154. [Google Scholar] [CrossRef]
- Nasrabadi, M.; Ghasemzadeh, M.A.; Monfared, M.R.Z. The preparation and characterization of UiO-66 metal-organic frameworks for the delivery of the drug ciprofloxacin and an evaluation of their antibacterial activities. New J. Chem. 2019, 43, 16033–16040. [Google Scholar] [CrossRef]
- He, C.; Lu, K.; Liu, D.; Lin, W. Nanoscale metal-organic frameworks for the co-delivery of cisplatin and pooled siRNAs to enhance therapeutic efficacy in drug-resistant ovarian cancer cells. J. Am. Chem. Soc. 2014, 136, 5181–5184. [Google Scholar] [CrossRef]
- Lu, K.; He, C.; Lin, W. Nanoscale Metal–Organic Framework for Highly Effective Photodynamic Therapy of Resistant Head and Neck Cancer. J. Am. Chem. Soc. 2014, 136, 16712–16715. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Cui, R.; Ji, G.; Liu, Z. Size and surface controllable metal-organic frameworks (MOFs) for fluorescence imaging and cancer therapy. Nanoscale 2018, 10, 6205–6211. [Google Scholar] [CrossRef]
- Jarai, B.M.; Stillman, Z.; Attia, L.; Decker, G.E.; Bloch, E.D.; Fromen, C.A. Evaluating UiO-66 Metal-Organic Framework Nanoparticles as Acid-Sensitive Carriers for Pulmonary Drug Delivery Applications. ACS Appl. Mater. Interfaces 2020, 12, 38989–39004. [Google Scholar] [CrossRef]
- Bůžek, D.; Adamec, S.; Lang, K.; Demel, J. Metal–organic frameworks vs. buffers: Case study of UiO-66 stability. Inorg. Chem. Front. 2021, 8, 720–734. [Google Scholar] [CrossRef]
- Moore, T.L.; Rodriguez-Lorenzo, L.; Hirsch, V.; Balog, S.; Urban, D.; Jud, C.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle colloidal stability in cell culture media and impact on cellular interactions. Chem. Soc. Rev. 2015, 44, 6287–6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butova, V.V.; Burachevskaya, O.A.; Ozhogin, I.V.; Borodkin, G.S.; Starikov, A.G.; Bordiga, S.; Damin, A.; Lillerud, K.P.; Soldatov, A.V. UiO-66 type MOFs with mixed-linkers-1,4-Benzenedicarboxylate and 1,4-naphthalenedicarboxylate: Effect of the modulator and post-synthetic exchange. Microporous Mesoporous Mater. 2020, 305, 110324. [Google Scholar] [CrossRef]
- Butova, V.V.; Burachevskaya, O.A.; Muratidi, M.A.; Surzhikova, I.I.; Zolotukhin, P.V.; Medvedev, P.V.; Gorban, I.E.; Kuzharov, A.A.; Soldatov, M.A. Loading of the Model Amino Acid Leucine in UiO-66 and UiO-66-NH2 :Optimization of Metal–Organic Framework Carriers and Evaluation of Host–Guest Interactions. Inorg. Chem. 2021, 60, 5694–5703. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Hiramatsu, D.; Yoshida, K.; Isoda, S.; Kitagawa, S. Sol−Gel Synthesis of Low-Dimensional Silica within Coordination Nanochannels. J. Am. Chem. Soc. 2008, 130, 9216–9217. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Kadowaki, Y.; Kim, C.R.; Fukushima, T.; Hiramatsu, D.; Kitagawa, S. Incarceration of Nanosized Silica into Porous Coordination Polymers: Preparation, Characterization, and Adsorption Property. Chem. Mater. 2011, 23, 1736–1741. [Google Scholar] [CrossRef]
- Xie, R.; Yang, P.; Peng, S.; Cao, Y.; Yao, X.; Guo, S.; Yang, W. A phosphorylcholine-based zwitterionic copolymer coated ZIF-8 nanodrug with a long circulation time and charged conversion for enhanced chemotherapy. J. Mater. Chem. B 2020, 8, 6128–6138. [Google Scholar] [CrossRef]
- Thommes, M. Physical Adsorption Characterization of Nanoporous Materials. Chem. Ing. Tech. 2010, 82, 1059–1073. [Google Scholar] [CrossRef]
- Sorribas, S.; Zornoza, B.; Téllez, C.; Coronas, J. Ordered mesoporous silica–(ZIF-8) core–shell spheres. Chem. Commun. 2012, 48, 9388. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Zheng, H.; Cao, Y.; Che, S. Coordination polymer coated mesoporous silica nanoparticles for ph-responsive drug release. Adv. Mater. 2012, 24, 6433–6437. [Google Scholar] [CrossRef]
- Tang, X.; Kröger, E.; Nielsen, A.; Strelow, C.; Mews, A.; Kipp, T. Ultrathin and Highly Passivating Silica Shells for Luminescent and Water-Soluble CdSe/CdS Nanorods. Langmuir 2017, 33, 5253–5260. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Ando, M.; Murase, N. Highly Luminescent CdSe/CdxZn1–xS Quantum Dots Coated with Thickness-Controlled SiO2 Shell through Silanization. Langmuir 2011, 27, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yang, P.; Matras-Postolek, K.; Wang, J.; Che, Q.; Cao, Y.; Ma, Q. Low toxic and highly luminescent CdSe/CdxZn1−xS quantum dots with thin organic SiO2 coating for application in cell imaging. J. Nanoparticle Res. 2016, 18, 1–11. [Google Scholar] [CrossRef]
- Ovchinnikov, O.; Aslanov, S.; Smirnov, M.; Perepelitsa, A.; Kondratenko, T.; Selyukov, A.; Grevtseva, I. Colloidal Ag2S/SiO2 core/shell quantum dots with IR luminescence. Opt. Mater. Express 2021, 11, 89. [Google Scholar] [CrossRef]
- Cagel, M.; Grotz, E.; Bernabeu, E.; Moretton, M.A.; Chiappetta, D.A. Doxorubicin: Nanotechnological overviews from bench to bedside. Drug Discov. Today 2017, 22, 270–281. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Kabanov, A.V. Pluronic block copolymers: Evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J. Control. Release 2008, 130, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Sonvico, F.; Dubernet, C.; Marsaud, V.; Appel, M.; Chacun, H.; Stella, B.; Renoir, M.; Colombo, P.; Couvreur, P. Establishment of an in vitro model expressing the folate receptor for the investigation of targeted delivery systems. J. Drug Deliv. Sci. Technol. 2005, 15, 407–410. [Google Scholar] [CrossRef]
- Sousa de Almeida, M.; Susnik, E.; Drasler, B.; Taladriz-Blanco, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine. Chem. Soc. Rev. 2021, 50, 5397–5434. [Google Scholar] [CrossRef]
- Colino, C.I.; Lanao, J.M.; Gutierrez-Millan, C. Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Demina, P.A.; Sindeeva, O.A.; Abramova, A.M.; Prikhozhdenko, E.S.; Verkhovskii, R.A.; Lengert, E.V.; Sapelkin, A.V.; Goryacheva, I.Y.; Sukhorukov, G.B. Fluorescent Convertible Capsule Coding Systems for Individual Cell Labeling and Tracking. ACS Appl. Mater. Interfaces 2021, 13, 19701–19709. [Google Scholar] [CrossRef]
- Chen, C.; Ke, J.; Zhou, X.E.; Yi, W.; Brunzelle, J.S.; Li, J.; Yong, E.-L.; Xu, H.E.; Melcher, K. Structural basis for molecular recognition of folic acid by folate receptors. Nature 2013, 500, 486–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsyupka, D.V.; Mordovina, E.A.; Sindeeva, O.A.; Sapelkin, A.V.; Sukhorukov, G.B.; Goryacheva, I.Y. High-fluorescent product of folic acid photodegradation: Optical properties and cell effect. J. Photochem. Photobiol. A Chem. 2021, 407, 113045. [Google Scholar] [CrossRef]
- Low, P.S.; Henne, W.A.; Doorneweerd, D.D. Discovery and Development of Folic-Acid-Based Receptor Targeting for Imaging and Therapy of Cancer and Inflammatory Diseases. Acc. Chem. Res. 2008, 41, 120–129. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MOF Samples | The Thickness of SiO2 Shell after a Specific Duration of Silanization Process, nm | ||||
|---|---|---|---|---|---|
| 0.5 h | 1 h | 2 h | 4 h | 24 h | |
| UiO-66@SiO2 | 8.5 ± 1.6 | 4.5 ± 0.9 | n.a. | n.a. | 9.9 ± 1.2 |
| UiO-66-NH2@SiO2 | 4.5 ± 1.1 | 4.8 ± 1.1 | n.a. | n.a. | 9.9 ± 1.9 |
| Sample | ξ of Nanoparticles in a Different Medium, mV | ||||
|---|---|---|---|---|---|
| DI Water | Saline Solution | MEM | DMEM | RPMI | |
| UiO-66 | 39.7 ± 0.8 | −5.3 ± 3.1 | −15.3 ± 0.9 | −10.2 ± 0.6 | −21.4 ± 0.9 |
| UiO-66/PVP | −14.9 ± 0.9 | −12.2 ± 1.3 | −9.2 ± 0.3 | −10.1 ± 0.7 | −17.8 ± 0.9 |
| UiO-66@SiO2 24 h | −33.3 ± 0.4 | +19.1 ± 1.2 | −18.0 ± 0.5 | −17.1 ± 0.9 | −21.0 ± 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trushina, D.B.; Sapach, A.Y.; Burachevskaia, O.A.; Medvedev, P.V.; Khmelenin, D.N.; Borodina, T.N.; Soldatov, M.A.; Butova, V.V. Doxorubicin-Loaded Core–Shell UiO-66@SiO2 Metal–Organic Frameworks for Targeted Cellular Uptake and Cancer Treatment. Pharmaceutics 2022, 14, 1325. https://doi.org/10.3390/pharmaceutics14071325
Trushina DB, Sapach AY, Burachevskaia OA, Medvedev PV, Khmelenin DN, Borodina TN, Soldatov MA, Butova VV. Doxorubicin-Loaded Core–Shell UiO-66@SiO2 Metal–Organic Frameworks for Targeted Cellular Uptake and Cancer Treatment. Pharmaceutics. 2022; 14(7):1325. https://doi.org/10.3390/pharmaceutics14071325
Chicago/Turabian StyleTrushina, Daria B., Anastasiia Yu. Sapach, Olga A. Burachevskaia, Pavel V. Medvedev, Dmitry N. Khmelenin, Tatiana N. Borodina, Mikhail A. Soldatov, and Vera V. Butova. 2022. "Doxorubicin-Loaded Core–Shell UiO-66@SiO2 Metal–Organic Frameworks for Targeted Cellular Uptake and Cancer Treatment" Pharmaceutics 14, no. 7: 1325. https://doi.org/10.3390/pharmaceutics14071325