
Spin Freezing and Its Impact on Pore Size, Tortuosity and Solid State


 , , , ,
, , , ,  and
and 
Abstract
:
1. Introduction
2. Objectives
3. Methods and Materials
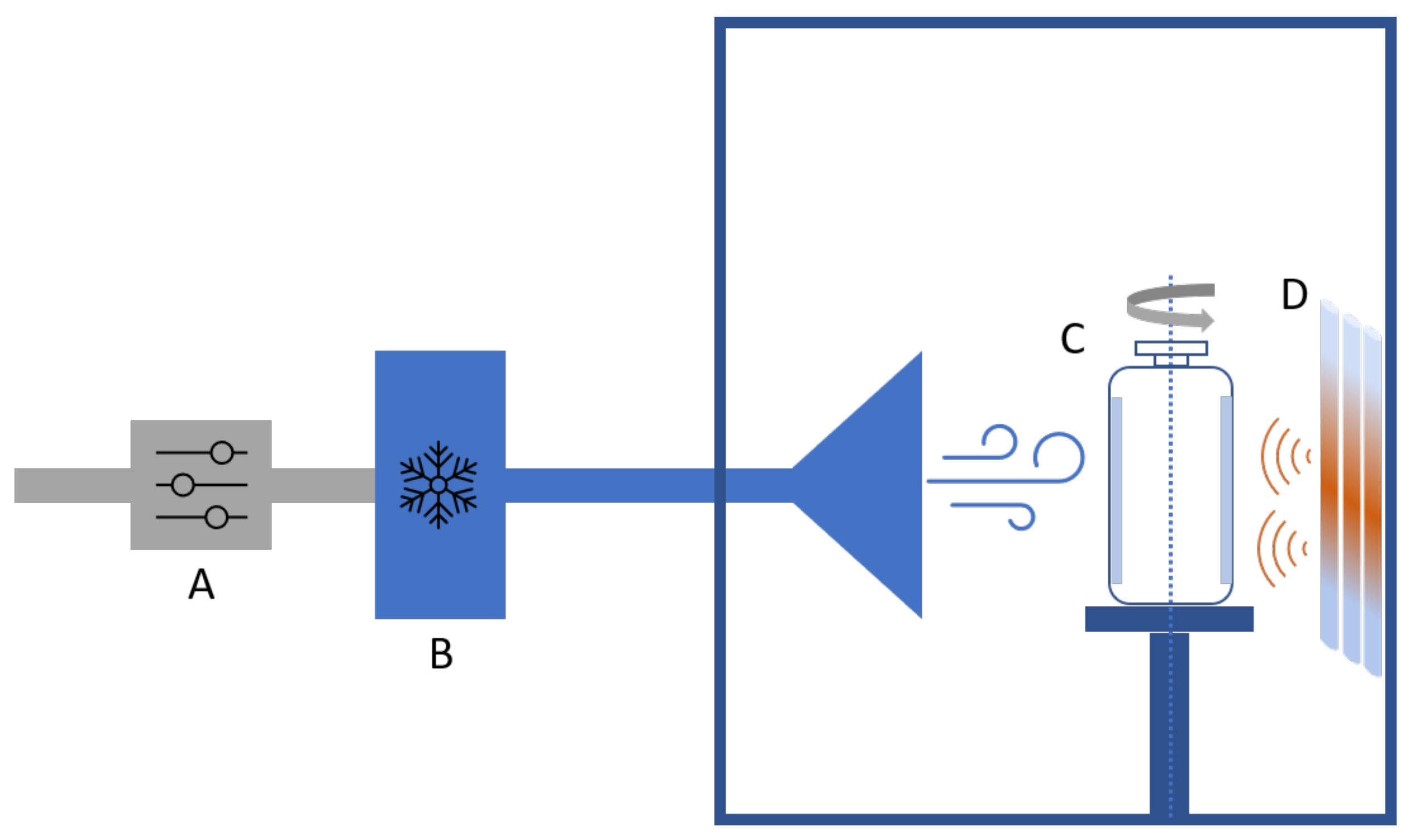
3.1. Spin Freezing and Spin Freeze-Drying
3.2. Determination of Collapse Temperature
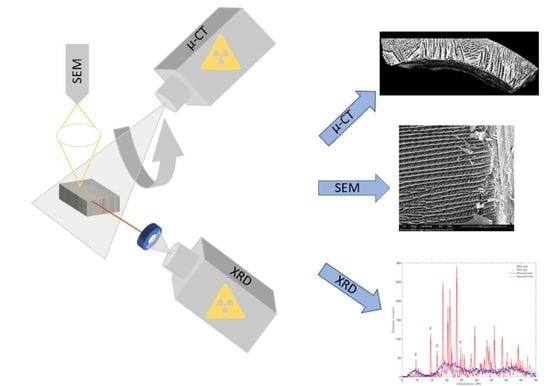
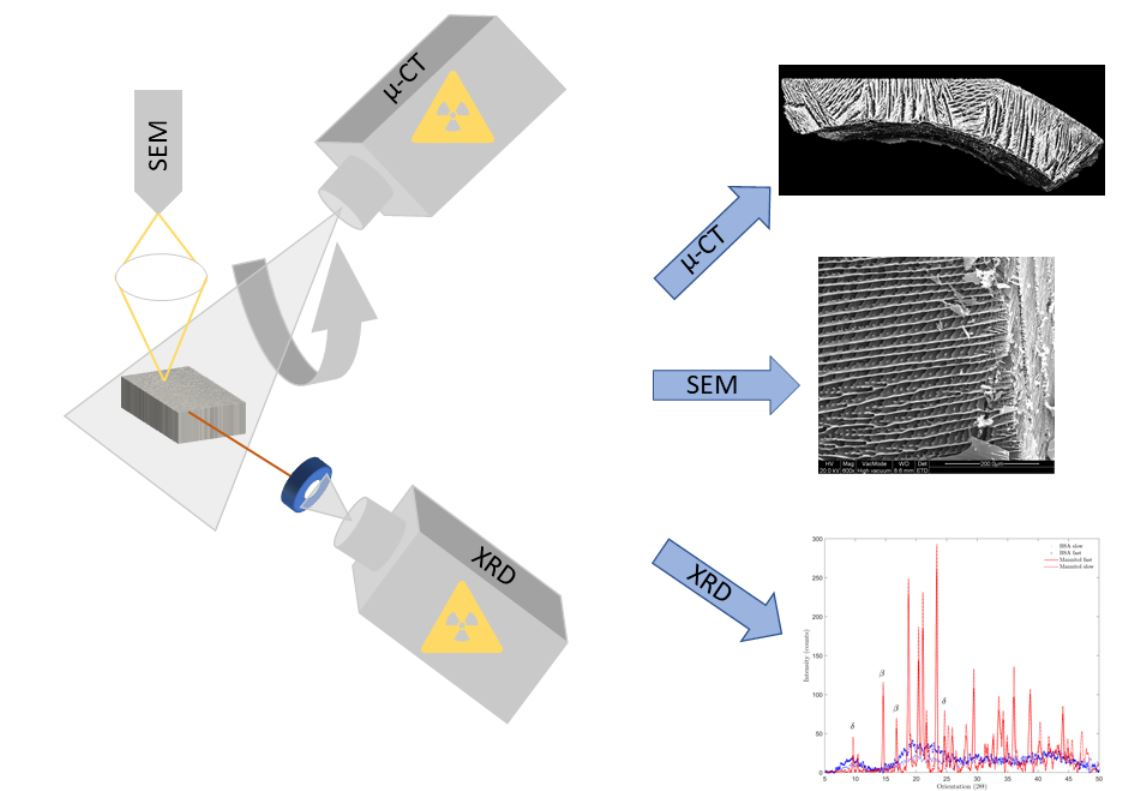
3.3. High Resolution X-ray Micro-Computed Tomography
3.4. Scanning Electron Microscopy
3.5. Determination of Dried Product Mass Transfer Resistance
3.6. Evaluation of the Primary Drying Time
3.7. X-ray Diffraction
4. Results and Discussion
4.1. Determination of Collapse Temperature
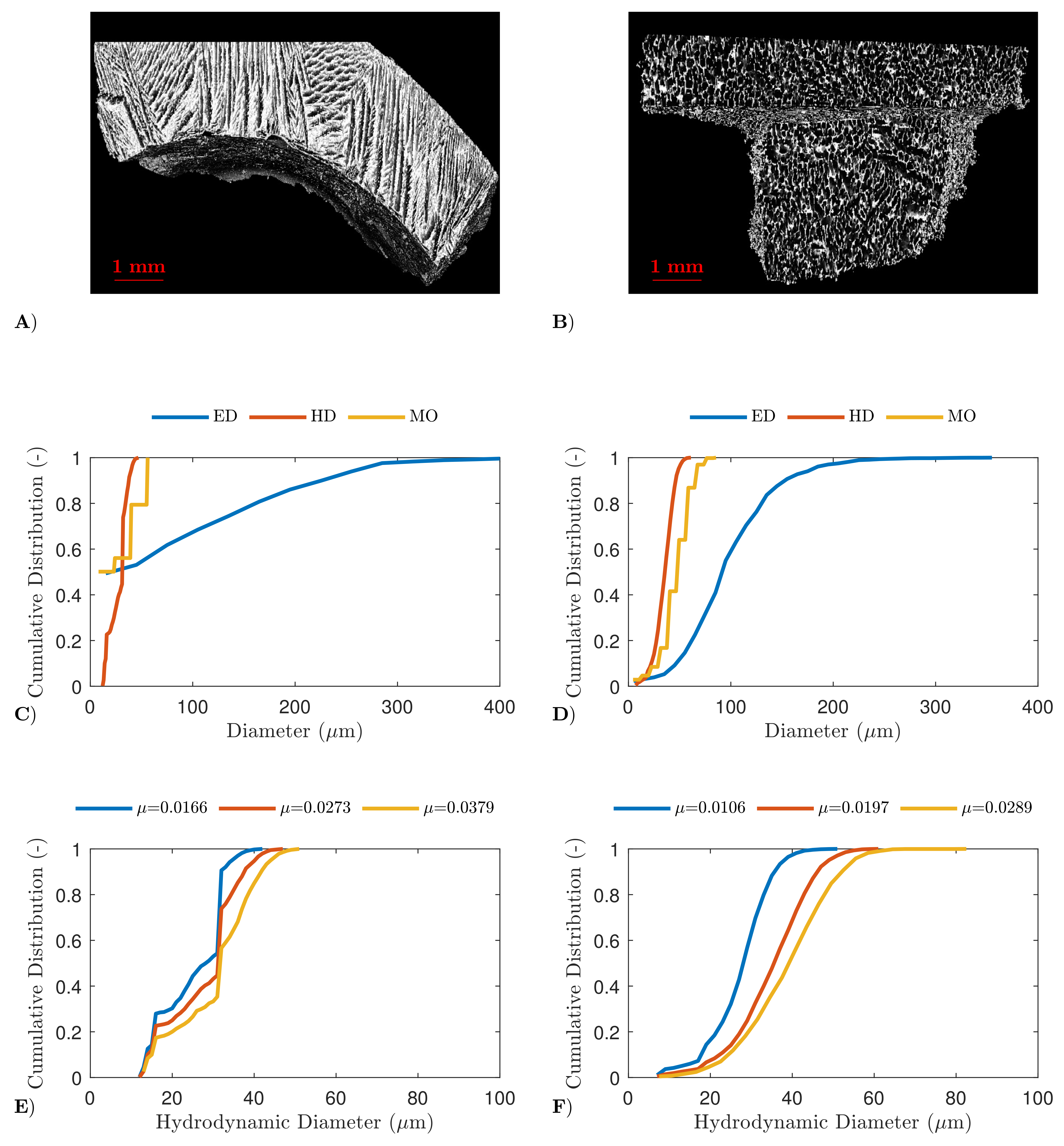
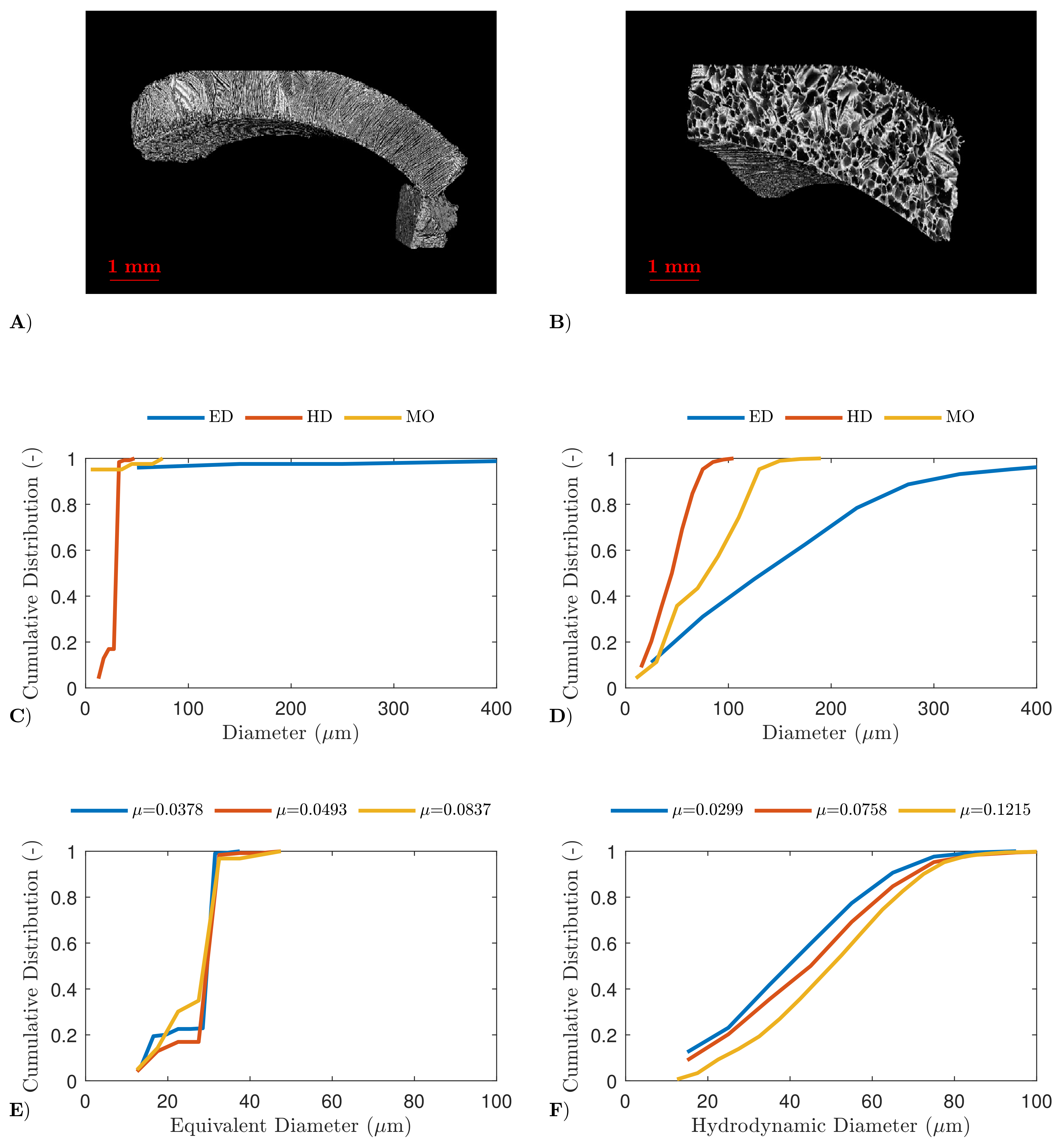
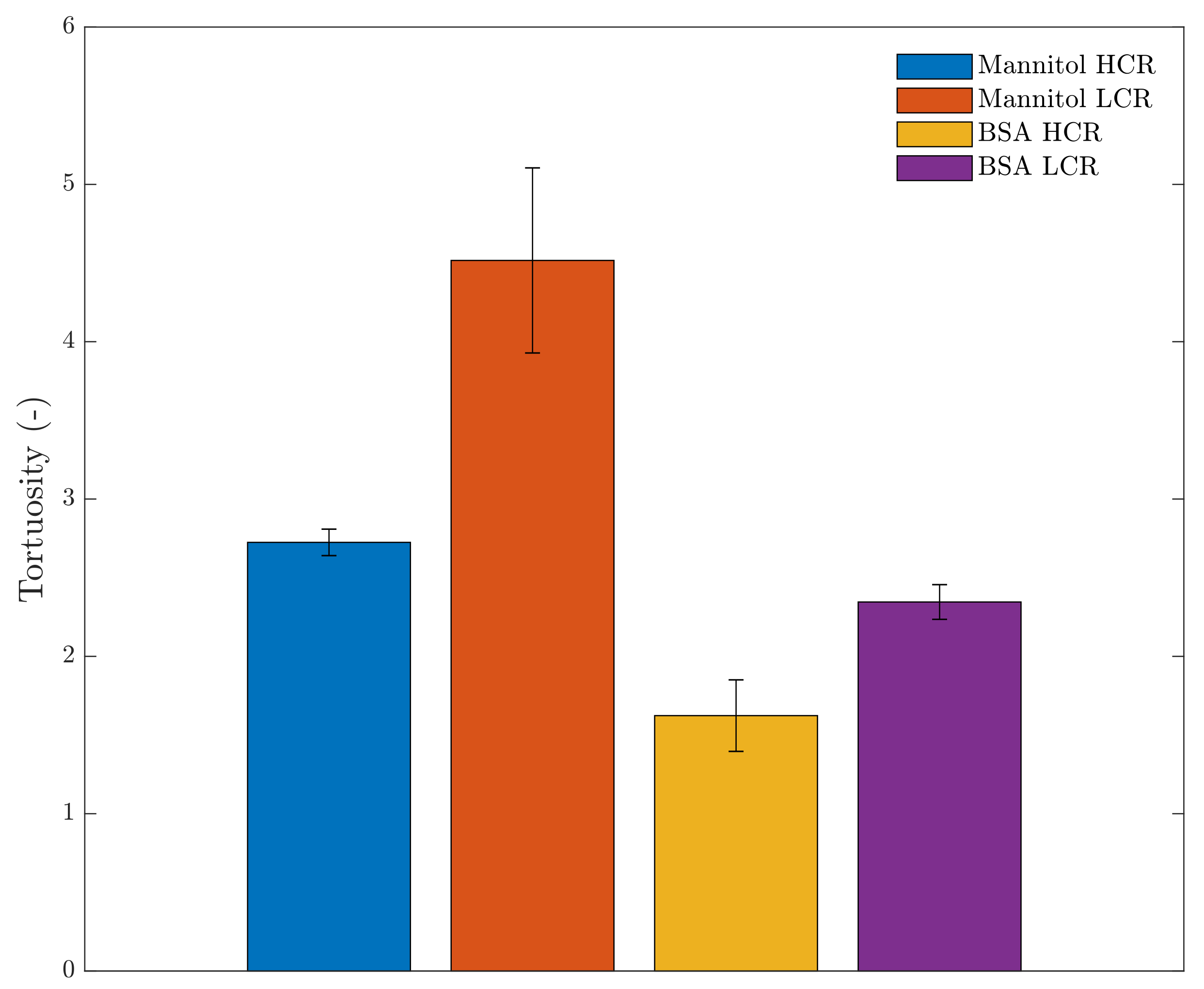
4.2. High-Resolution X-ray Computed Tomography
4.3. Scanning Electron Microscopy
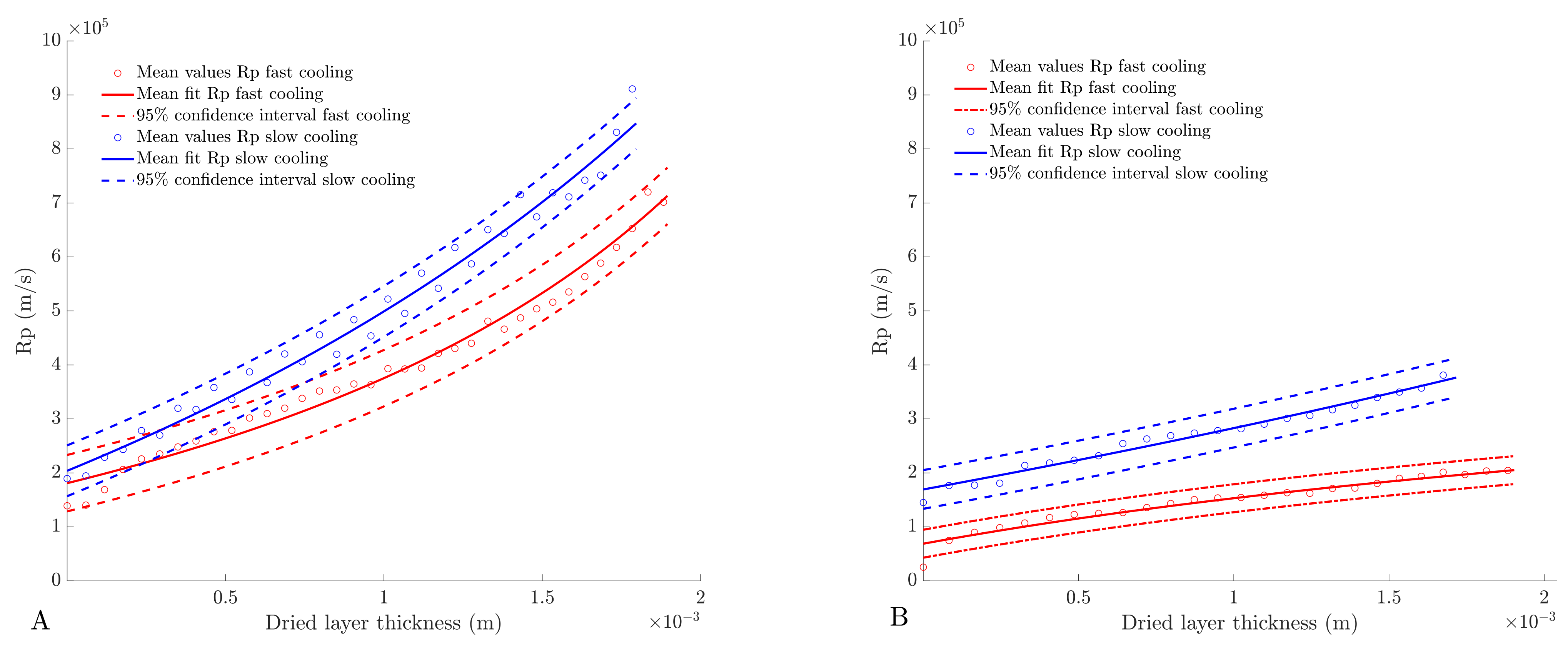
4.4. Determination of Dried Product Mass Transfer Resistance
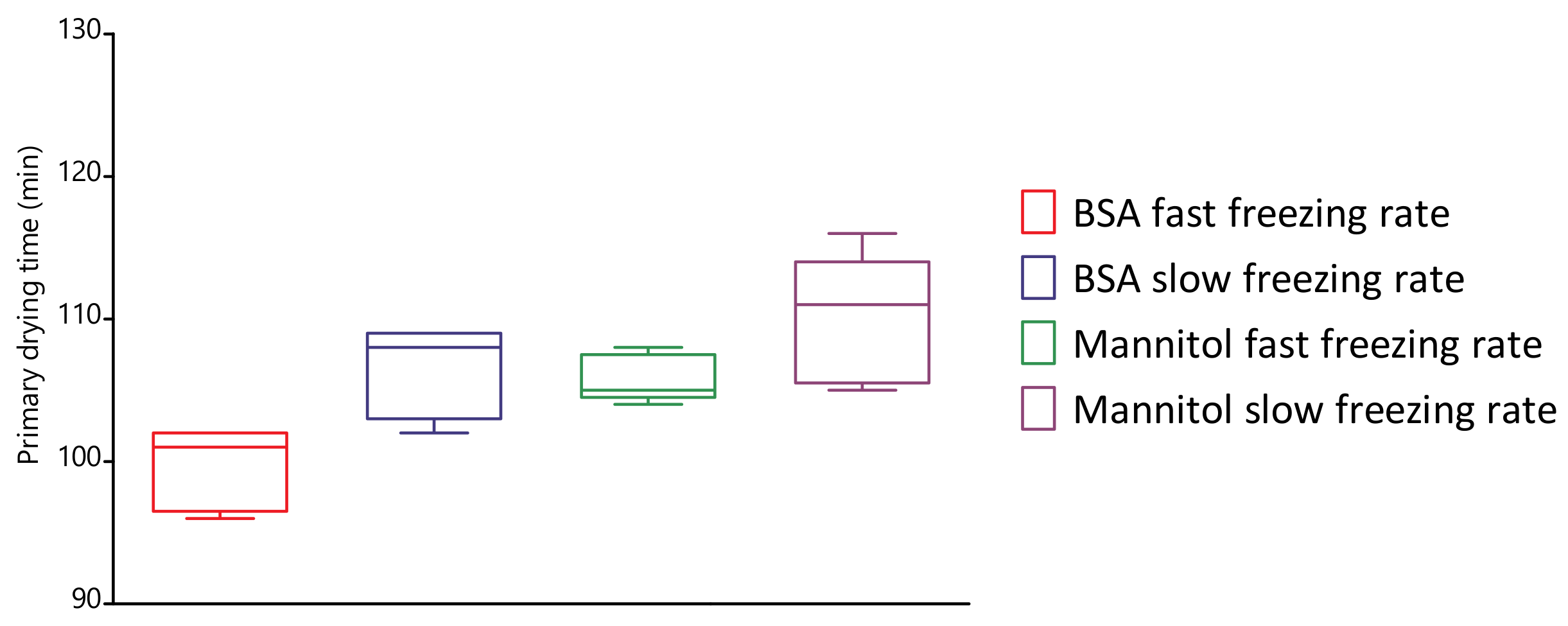
4.5. Evaluation of Primary Drying Times
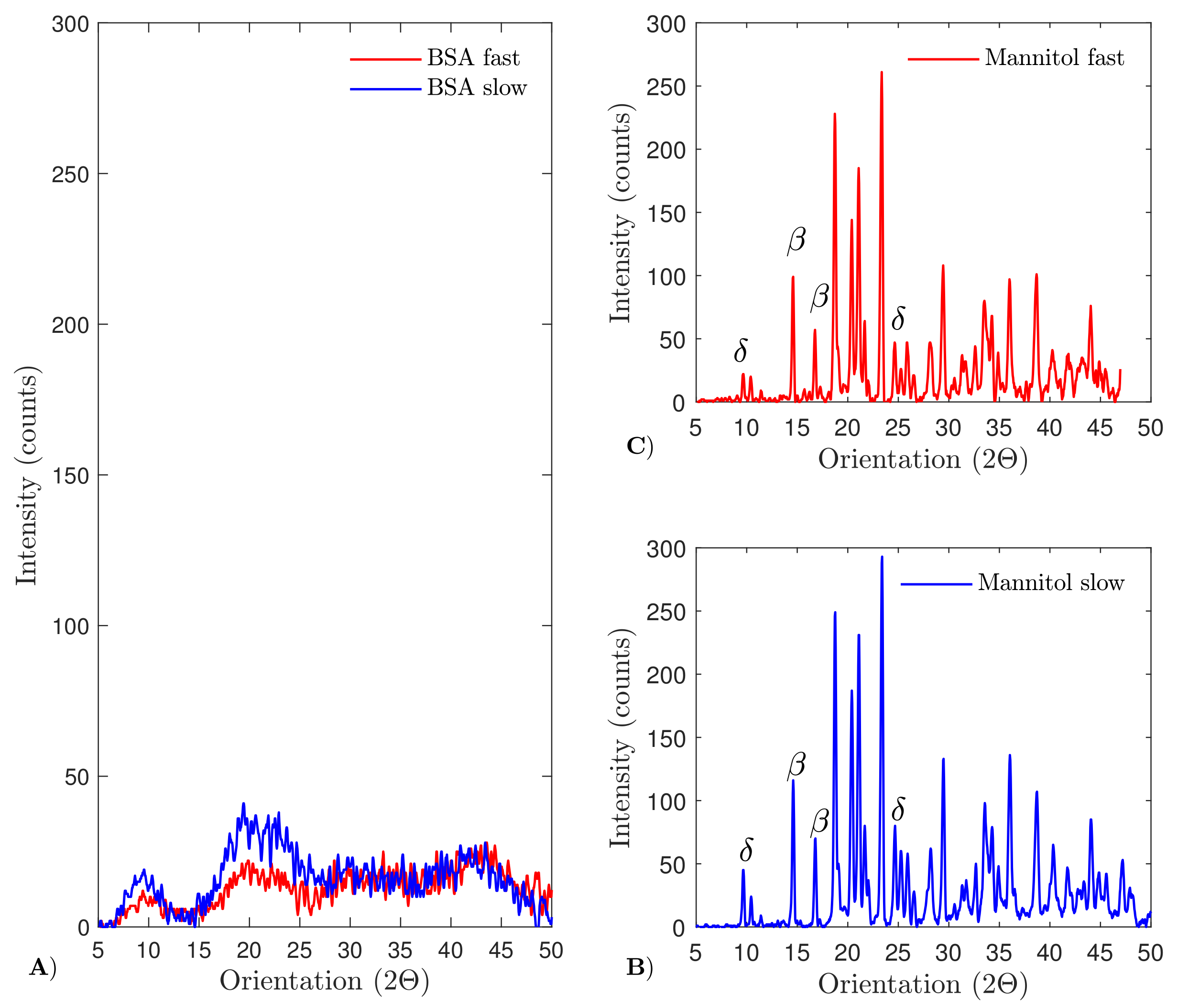
4.6. X-ray Diffraction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mullard, A. 2020 FDA drug approvals. Nat. Rev. Drug Discov. 2021, 20, 85–90. [Google Scholar] [CrossRef]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.R.; Chou, R.C. Therapeutic monoclonal antibodies and derivatives: Historical perspectives and future directions. Biotechnol. Adv. 2016, 34, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Frokjaer, S.; Otzen, D.E. Protein drug stability: A formulation challenge. Nat. Rev. Drug Discov. 2005, 4, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L. How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Pikal, M.J. Freeze Drying. In Encyclopedia of Pharmaceutical Technology; Taylor & Francis: Abingdon, UK, 2002; pp. 1299–1326. [Google Scholar] [CrossRef]
- Tang, X.; Pikal, M.J. Design of Freeze-Drying Processes for Pharmaceuticals: Practical Advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef]
- Emami, F.; Vatanara, A.; Park, E.J.; Na, D.H. Drying technologies for the stability and bioavailability of biopharmaceuticals. Pharmaceutics 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsiccio, A.; Pisano, R. Clarifying the role of cryo- and lyo-protectants in the biopreservation of proteins. Phys. Chem. Chem. Phys. 2018, 20, 8267–8277. [Google Scholar] [CrossRef]
- Arsiccio, A.; Giorsello, P.; Marenco, L.; Pisano, R. Considerations on Protein Stability During Freezing and Its Impact on the Freeze-Drying Cycle: A Design Space Approach. J. Pharm. Sci. 2020, 109, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Shepherd, D.; Sun, J.; Ouellette, D.; Grant, K.L.; Tang, X.; Pikal, M.J. Mechanism of protein stabilization by sugars during freeze-drying and storage: Native structure preservation, specific interaction, and/or immobilization in a glassy matrix? J. Pharm. Sci. 2005, 94, 1427–1444. [Google Scholar] [CrossRef]
- Bhatnagar, B.S.; Bogner, R.H.; Pikal, M.J. Protein stability during freezing: Separation of stresses and mechanisms of protein stabilization. Pharm. Dev. Technol. 2007, 12, 505–523. [Google Scholar] [CrossRef]
- Jiang, S.; Nail, S.L. Effect of process conditions on recovery of protein activity after freezing and freeze-drying. Eur. J. Pharm. Biopharm. 1998, 45, 249–257. [Google Scholar] [CrossRef]
- Searles, J.A.; Carpenter, J.F.; Randolph, T.W. The ice nucleation temperature determines the primary drying rate of lyophilization for samples frozen on a temperature-controlled shelf. J. Pharm. Sci. 2001, 90, 860–871. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Crowe, J.H. The mechanism of cryoprotection of proteins by solutes. Cryobiology 1988, 25, 244–255. [Google Scholar] [CrossRef]
- Gitter, J.H.; Geidobler, R.; Presser, I.; Winter, G. Microwave-assisted freeze-drying of monoclonal antibodies: Product quality aspects and storage stability. Pharmaceutics 2019, 11, 674. [Google Scholar] [CrossRef] [Green Version]
- Renteria Gamiz, A.G.; Dewulf, J.; De Soete, W.; Heirman, B.; Dahlin, P.; Jurisch, C.; Krebser, U.; De Meester, S. Freeze drying in the biopharmaceutical industry: An environmental sustainability assessment. Food Bioprod. Process. 2019, 117, 213–223. [Google Scholar] [CrossRef]
- De Meyer, L.; Van Bockstal, P.J.; Corver, J.; Vervaet, C.; Remon, J.P.; De Beer, T. Evaluation of spin freezing versus conventional freezing as part of a continuous pharmaceutical freeze-drying concept for unit doses. Int. J. Pharm. 2015, 496, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Van Bockstal, P.J. Mechanistic modelling of infrared mediated energy transfer during the primary drying step of continuous freeze-drying process. Eur. J. Pharm. Biopharm. 2016, 114, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Pisano, R.; Arsiccio, A.; Capozzi, L.C.; Trout, B.L. Achieving continuous manufacturing in lyophilization: Technologies and approaches. Eur. J. Pharm. Biopharm. 2019, 142, 265–279. [Google Scholar] [CrossRef]
- Hosokawa Micron, B.V. Active Freeze Drying; Hosokawa Micron B.V.: Doetinchem, The Netherlands, 2012. [Google Scholar]
- Leys, L.; Vanbillemont, B.; Van Bockstal, P.; Lammens, J.; Nuytten, G.; Corver, J.; Vervaet, C.; De Beer, T. A primary drying model-based comparison of conventional batch freeze-drying to continuous spin-freeze-drying for unit doses. Eur. J. Pharm. Biopharm. 2020, 157, 97–107. [Google Scholar] [CrossRef]
- Van Bockstal, P.J.; Corver, J.; De Meyer, L.; Vervaet, C.; De Beer, T. Thermal Imaging as a Noncontact Inline Process Analytical Tool for Product Temperature Monitoring during Continuous Freeze-Drying of Unit Doses. Anal. Chem. 2018, 90, 13591–13599. [Google Scholar] [CrossRef] [Green Version]
- Brouckaert, D.; De Meyer, L.; Vanbillemont, B.; Van Bockstal, P.J.; Lammens, J.; Mortier, S.; Corver, J.; Vervaet, C.; Nopens, I.; De Beer, T. Potential of Near-Infrared Chemical Imaging as Process Analytical Technology Tool for Continuous Freeze-Drying. Anal. Chem. 2018, 90, 4354–4362. [Google Scholar] [CrossRef]
- Vanbillemont, B.; Lammens, J.; Goethals, W.; Vervaet, C.; Boone, M.N.; De Beer, T. 4D micro-computed X-ray tomography as a tool to determine critical process and product information of spin freeze-dried unit doses. Pharmaceutics 2020, 12, 430. [Google Scholar] [CrossRef]
- De Meyer, L.; Lammens, J.; Mortier, S.; Vanbillemont, B.; Van Bockstal, P.; Corver, J.; Nopens, I.; Vervaet, C.; De Beer, T. Modelling the primary drying step for the determination of the optimal dynamic heating pad temperature in a continuous pharmaceutical freeze-drying process for unit doses. Int. J. Pharm. 2017, 532, 185–193. [Google Scholar] [CrossRef] [Green Version]
- De Meyer, L.; Lammens, J.; Vanbillemont, B.; Van Bockstal, P.J.; Corver, J.; Vervaet, C.; Friess, W.; De Beer, T. Dual chamber cartridges in a continuous pharmaceutical freeze-drying concept: Determination of the optimal dynamic infrared heater temperature during primary drying. Int. J. Pharm. 2019, 570, 118631. [Google Scholar] [CrossRef]
- Kasper, J.C.; Friess, W. The freezing step in lyophilization: Physico-chemical fundamentals, freezing methods and consequences on process performance and quality attributes of biopharmaceuticals. Eur. J. Pharm. Biopharm. 2011, 78, 248–263. [Google Scholar] [CrossRef]
- Capozzi, L.C.; Pisano, R. Looking inside the ‘black box’: Freezing engineering to ensure the quality of freeze-dried biopharmaceuticals. Eur. J. Pharm. Biopharm. 2018, 129, 58–65. [Google Scholar] [CrossRef]
- Nunes, C.; Suryanarayanan, R.; Botez, C.E.; Stephens, P.W. Characterization and crystal structure of D-mannitol hemihydrate. J. Pharm. Sci. 2004, 93, 2800–2809. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Suryanarayanan, R.; Rinella, J.V.; Bogner, R.H. Mechanisms by which crystalline mannitol improves the reconstitution time of high concentration lyophilized protein formulations. Eur. J. Pharm. Biopharm. 2018, 131, 70–81. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Patel, S.M.; Bogner, R.H. Reconstitution Time for Highly Concentrated Lyophilized Proteins: Role of Formulation and Protein. J. Pharm. Sci. 2020, 109, 2975–2985. [Google Scholar] [CrossRef]
- Sane, P.; Bogner, R.H.; Bhatnagar, B.; Tchessalov, S. Reconstitution of Highly Concentrated Lyophilized Proteins: Part 1 Amorphous Formulations. J. Pharm. Sci. 2020, 109, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Beech, K.E.; Biddlecombe, J.G.; Van Der Walle, C.F.; Stevens, L.A.; Rigby, S.P.; Burley, J.C.; Allen, S. Insights into the influence of the cooling profile on the reconstitution times of amorphous lyophilized protein formulations. Eur. J. Pharm. Biopharm. 2015, 96, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masschaele, B.; Dierick, M.; Loo, D.V.; Boone, M.N.; Brabant, L.; Pauwels, E.; Cnudde, V.; Hoorebeke, L.V. HECTOR: A 240 kV micro-CT setup optimized for research. J. Phys. Conf. Ser. 2013, 463, 012012. [Google Scholar] [CrossRef]
- Vlassenbroeck, J.; Dierick, M.; Masschaele, B.; Cnudde, V.; Van Hoorebeke, L.; Jacobs, P. Software tools for quantification of X-ray microtomography at the UGCT. Nucl. Instrum. Methods Phys. Res. Sect. A 2007, 580, 442–445. [Google Scholar] [CrossRef]
- Brabant, L.; Vlassenbroeck, J.; De Witte, Y.; Cnudde, V.; Boone, M.N.; Dewanckele, J.; Van Hoorebeke, L. Three-Dimensional Analysis of High-Resolution X-Ray Computed Tomography Data with Morpho. Microsc. Microanal. 2011, 17, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Goshima, H.; Do, G.; Nakagawa, K. Impact of Ice Morphology on Design Space of Pharmaceutical Freeze-Drying. J. Pharm. Sci. 2016, 105, 1920–1933. [Google Scholar] [CrossRef]
- Ullrich, S.; Seyferth, S.; Lee, G. Measurement of shrinkage and cracking in lyophilized amorphous cakes. Part II: Kinetics. Pharm. Res. 2015, 32, 2503–2515. [Google Scholar] [CrossRef]
- Kuu, W.Y.; Hardwick, L.M.; Akers, M.J. Rapid determination of dry layer mass transfer resistance for various pharmaceutical formulations during primary drying using product temperature profiles. Int. J. Pharm. 2006, 313, 99–113. [Google Scholar] [CrossRef]
- Pisano, R.; Barresi, A.A.; Capozzi, L.C.; Novajra, G.; Oddone, I.; Vitale-Brovarone, C. Characterization of the mass transfer of lyophilized products based on X-ray micro-computed tomography images. Dry. Technol. 2017, 35, 933–938. [Google Scholar] [CrossRef]
- Patel, S.M.; Nail, S.L.; Pikal, M.J.; Geidobler, R.; Winter, G.; Hawe, A.; Davagnino, J.; Rambhatla Gupta, S. Lyophilized Drug Product Cake Appearance: What Is Acceptable? J. Pharm. Sci. 2017, 106, 1706–1721. [Google Scholar] [CrossRef]
- Franks, F. Freeze-drying of bioproducts: Putting principles into practice. Eur. J. Pharm. Biopharm. 1998, 45, 221–229. [Google Scholar] [CrossRef]
- Vanbillemont, B.; Carpenter, J.F.; Probst, C.; De Beer, T. The Impact of Formulation Composition and Process Settings of Traditional Batch Versus Continuous Freeze-Drying On Protein Aggregation. J. Pharm. Sci. 2020, 109, 3308–3318. [Google Scholar] [CrossRef]
- Searles, J.A.; Carpenter, J.F.; Randolph, T.W. Annealing to optimize the primary drying rate, reduce freezing-induced drying rate heterogeneity, and determine T’g pharmaceutical lyophilization. J. Pharm. Sci. 2001, 90, 872–887. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Cooling Rate (°C/min) | Freezing Rate (W) | Chamber Pressure (Pa) | Total Drying Time (h) |
|---|---|---|---|---|
| Mannitol fast | 50 | 17 | 10 | 5 |
| Mannitol slow | 4 | 1.4 | ||
| BSA fast | 50 | 17 | ||
| BSA slow | 4 | 1.4 |
| Sample | Voxel Size (µm) | Projections (-) | Analysed Volume (mm) |
|---|---|---|---|
| Mannitol fast | 8 | 2401 | 4.6 |
| Mannitol slow | 4.5 | 2601 | 4.55 |
| BSA fast | 8 | 2401 | 4.6 |
| BSA slow | 8 | 2401 | 4.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lammens, J.; Goudarzi, N.M.; Leys, L.; Nuytten, G.; Van Bockstal, P.-J.; Vervaet, C.; Boone, M.N.; De Beer, T. Spin Freezing and Its Impact on Pore Size, Tortuosity and Solid State. Pharmaceutics 2021, 13, 2126. https://doi.org/10.3390/pharmaceutics13122126
Lammens J, Goudarzi NM, Leys L, Nuytten G, Van Bockstal P-J, Vervaet C, Boone MN, De Beer T. Spin Freezing and Its Impact on Pore Size, Tortuosity and Solid State. Pharmaceutics. 2021; 13(12):2126. https://doi.org/10.3390/pharmaceutics13122126
Chicago/Turabian StyleLammens, Joris, Niloofar Moazami Goudarzi, Laurens Leys, Gust Nuytten, Pieter-Jan Van Bockstal, Chris Vervaet, Matthieu N. Boone, and Thomas De Beer. 2021. "Spin Freezing and Its Impact on Pore Size, Tortuosity and Solid State" Pharmaceutics 13, no. 12: 2126. https://doi.org/10.3390/pharmaceutics13122126