
Intranasal Zolmitriptan-Loaded Bilosomes with Extended Nasal Mucociliary Transit Time for Direct Nose to Brain Delivery

Abstract
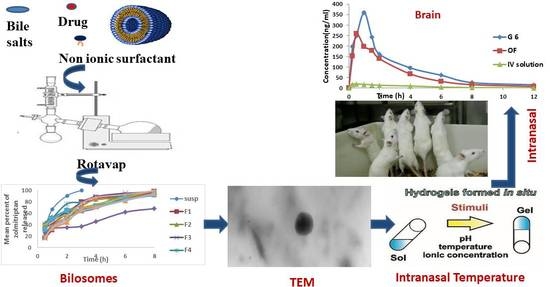
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Optimization of Bilosomes
2.2.1. Preparation of Zolmitriptan-Loaded Niosomes
Evaluation of the Prepared Zolmitriptan-Loaded Niosomes
Assessment of Entrapment Efficiency (EE)
Assessment of Particle Size (PS) and Polydispersity Index (PDI)
Statistical Analysis
2.2.2. Preparation of Zolmitriptan-Loaded Bilosomes
Optimization of Zolmitriptan-Loaded Bilosomes
Characterization of the Prepared Zolmitriptan-Loaded Bilosomes—Assessment of Entrapment Efficiency, Particle Size (PS), Polydispersity Index (PDI) and Zeta Potential (ZP)
Characterization of the Prepared Zolmitriptan-Loaded Bilosomes—In Vitro Release Studies
2.3. Mucoadhesive In Situ Gelling System
2.3.1. Preparation of Mucoadhesive In Situ Gelling System Containing the Optimal Bilosomes
2.3.2. Characterization of the Prepared Mucoadhesive In Situ Gelling System
Assessment of Release Parameters of Zolmitriptan from the Prepared Mucoadhesive In Situ Gelling System
Assessment of Sol to Gel Transition Temperature and Time
Assessment of Rheological Constants
Transmission Electron Microscopy (TEM)
Differential Scanning Calorimetry (DSC)
Determination of Nasal Mucociliary Transit Time
2.4. In Vivo Animal Study
2.4.1. Study Design and Dose Administration
2.4.2. Chromatographic Conditions
2.4.3. Sample Preparation
2.4.4. In Vivo Brain and Systemic-Kinetic Studies
2.4.5. Statistical Analysis
3. Results and Discussion
3.1. Optimization of Bilosomes
3.1.1. Preparation of Zolmitriptan-Loaded Niosomes
Evaluation of the Prepared Zolmitriptan-Loaded Niosomes
3.1.2. Investigation of the Effect Process Variables on the Properties of Zolmitriptan-Loaded Bilosomes
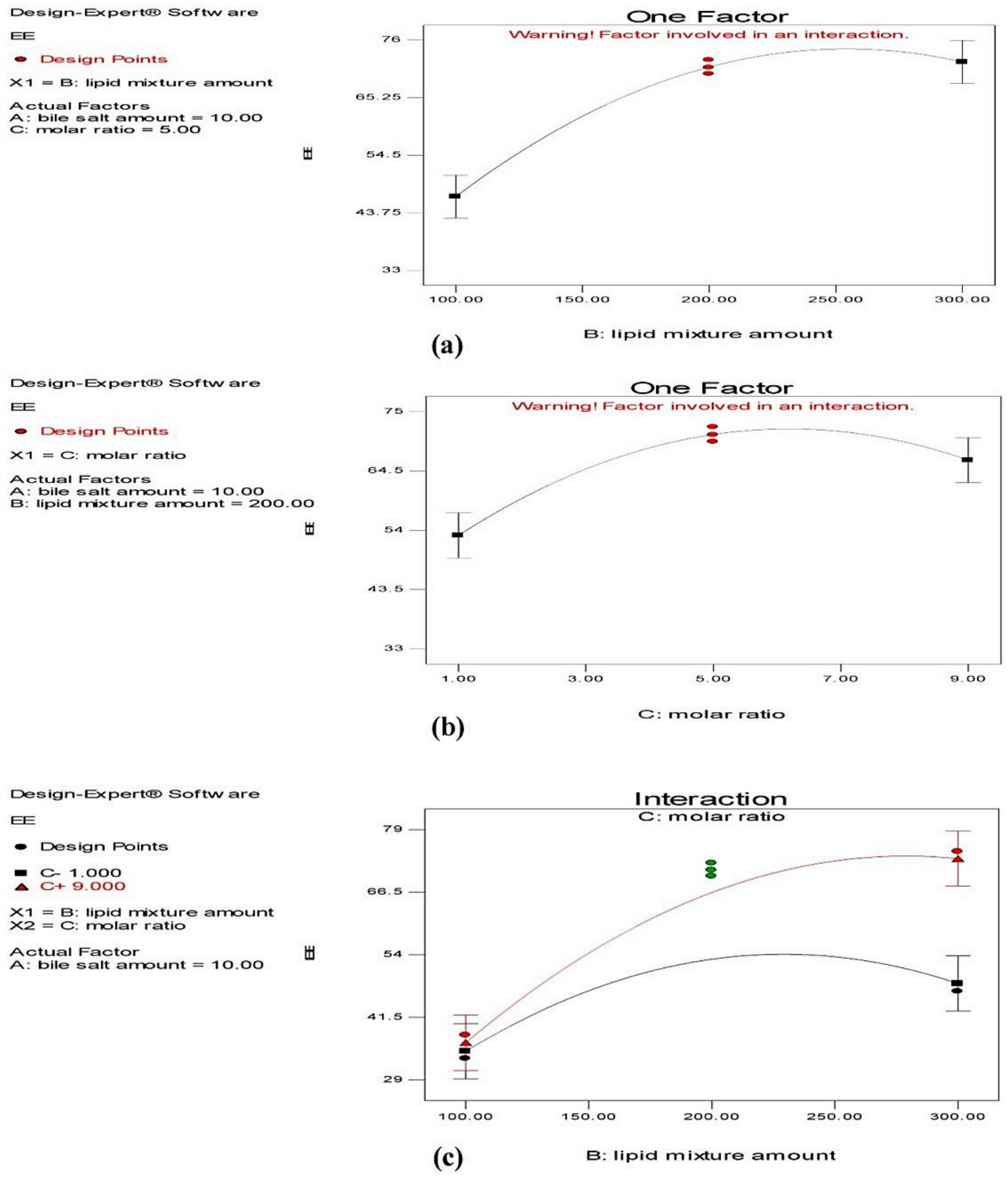
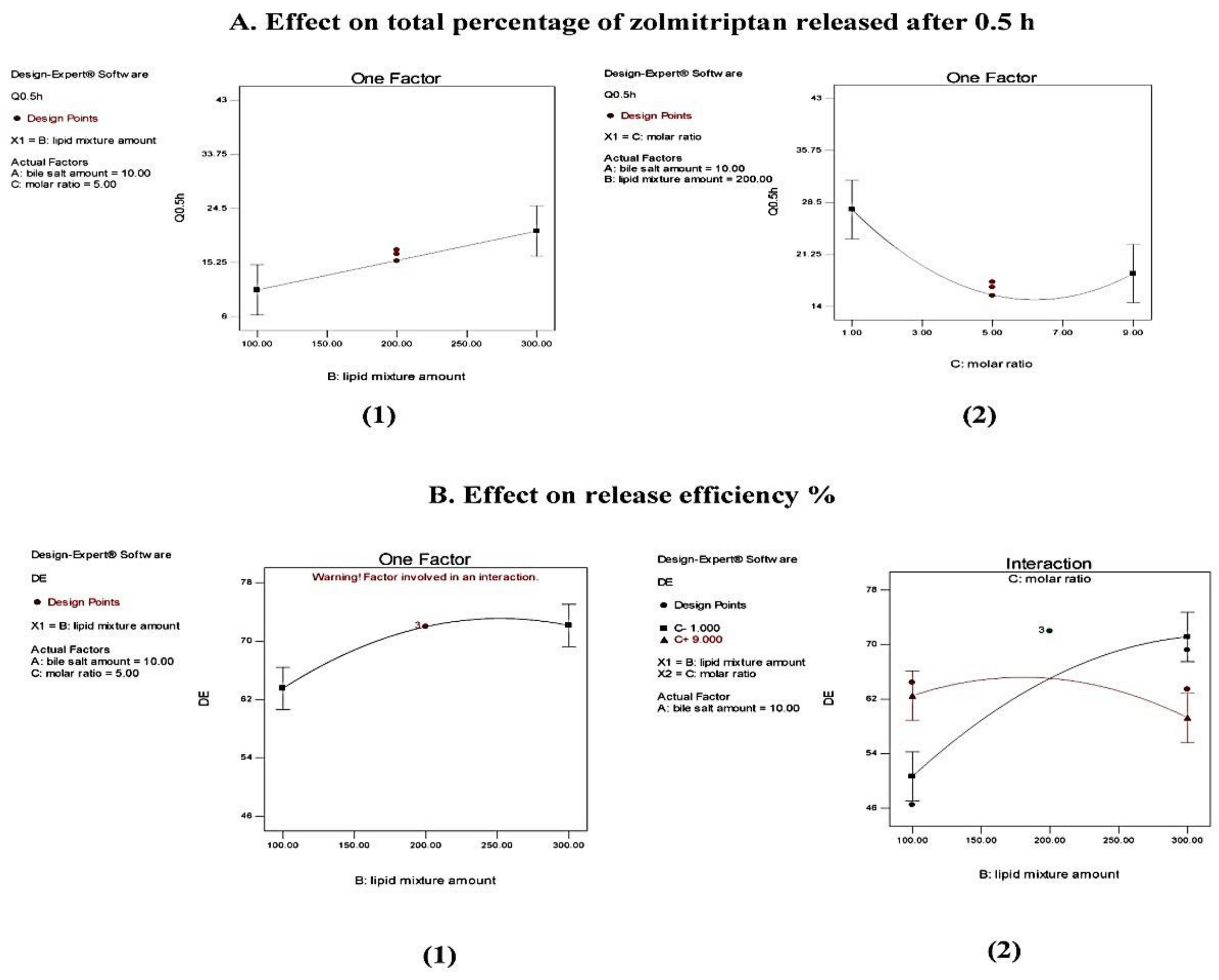
Effect on Entrapment Efficiency (EE)
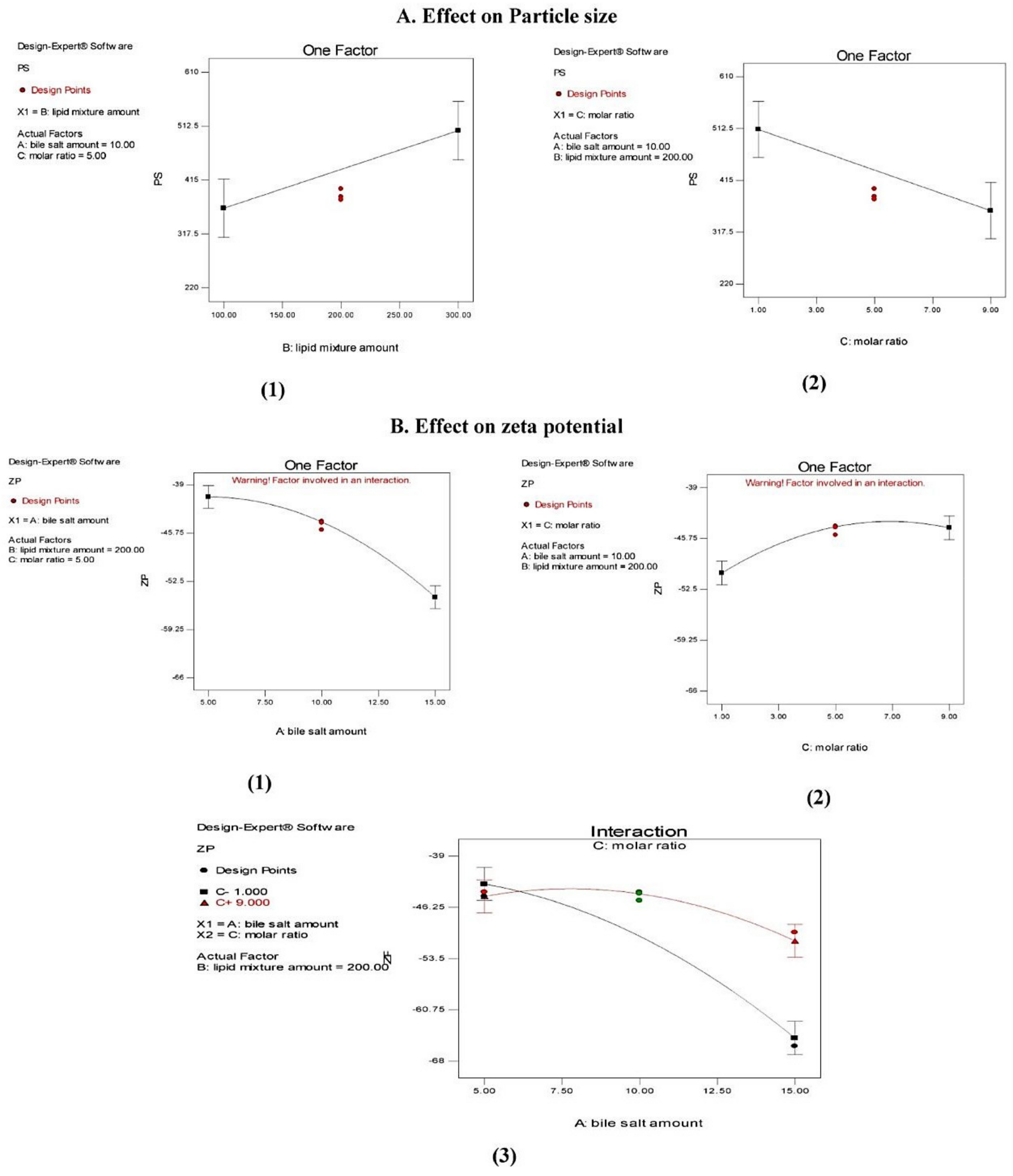
Effect on Particle Size (PS)
Effect on Polydispersity Index (PDI)
Effect on Zeta Potential (ZP)
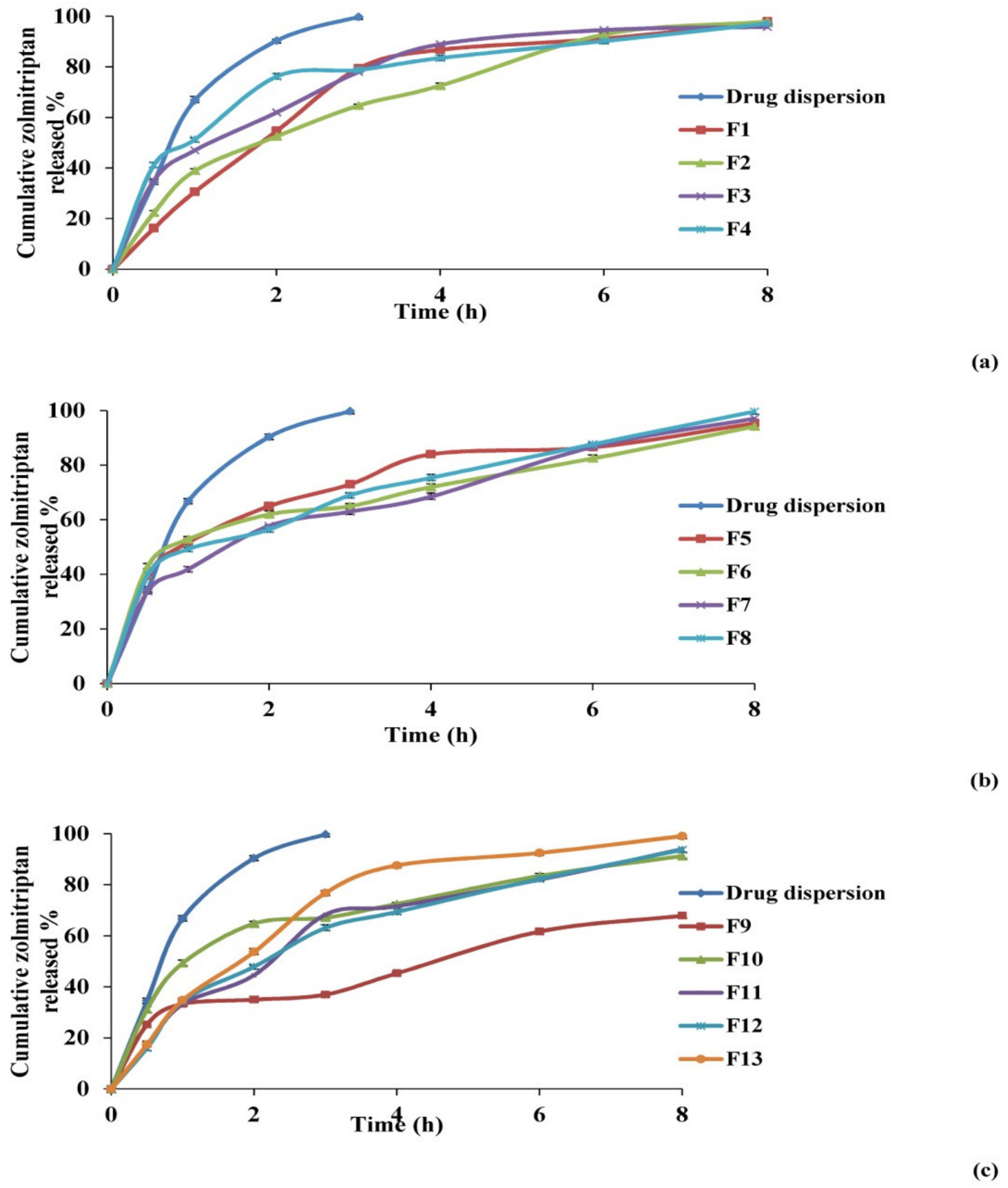
Effect on In Vitro Release Studies
3.1.3. Selection of the Optimized Zolmitriptan-Loaded Bilosomes
3.2. Mucoadhesive In Situ Gelling System
3.2.1. Characterization of the Prepared Mucoadhesive In Situ Gelling System
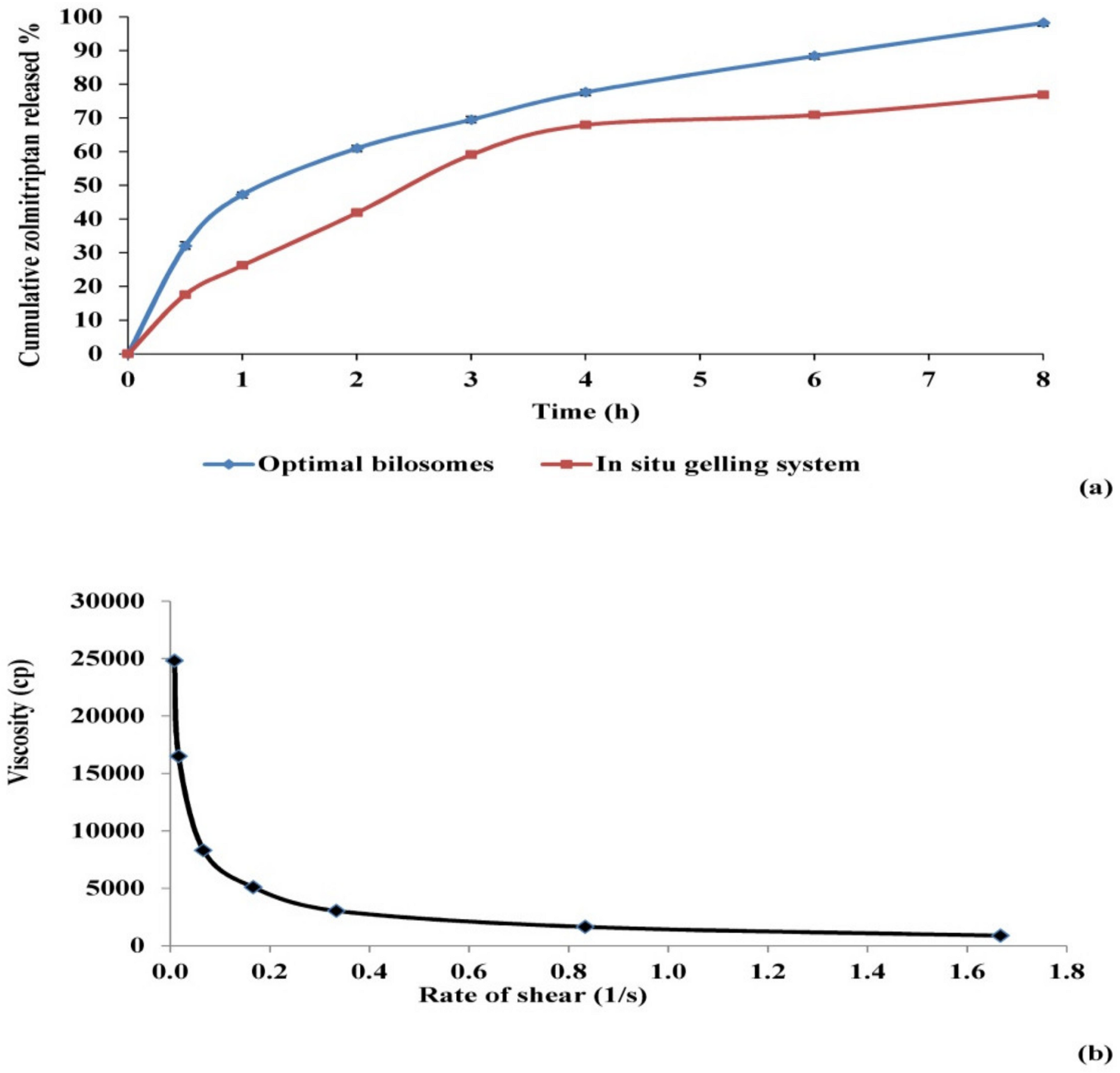
3.2.2. Assessment of Release Parameters of Zolmitriptan from the Prepared Mucoadhesive In Situ Gelling System
3.2.3. Assessment of Sol to Gel Transition Temperature and Time
3.2.4. Assessment of Rheological Constants
3.2.5. Transmission Electron Microscopy (TEM)
3.2.6. Differential Scanning Calorimetry (DSC)
3.2.7. Determination of Nasal Mucociliary Transit Time
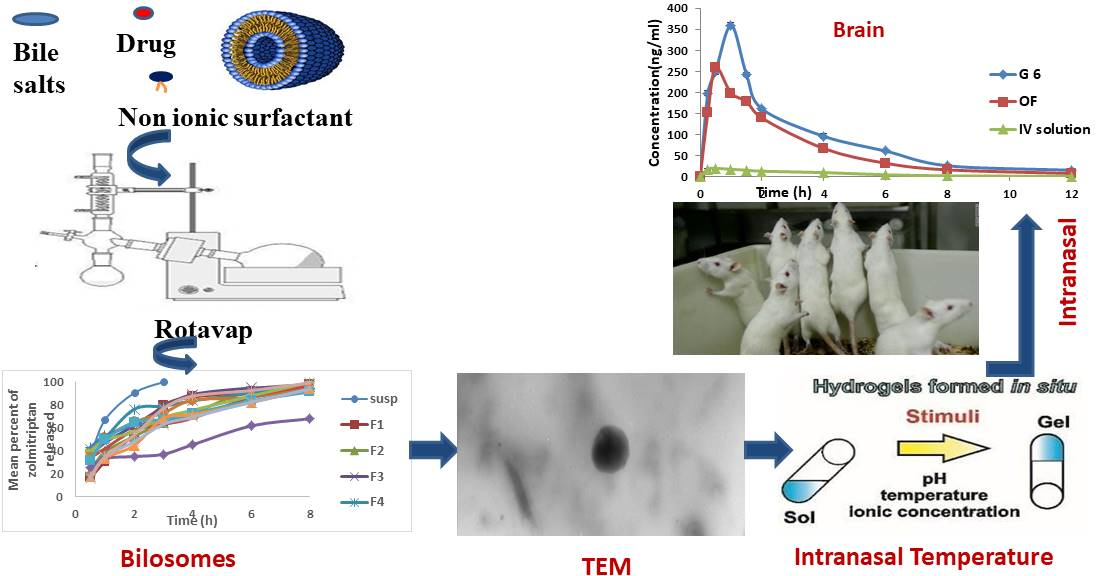
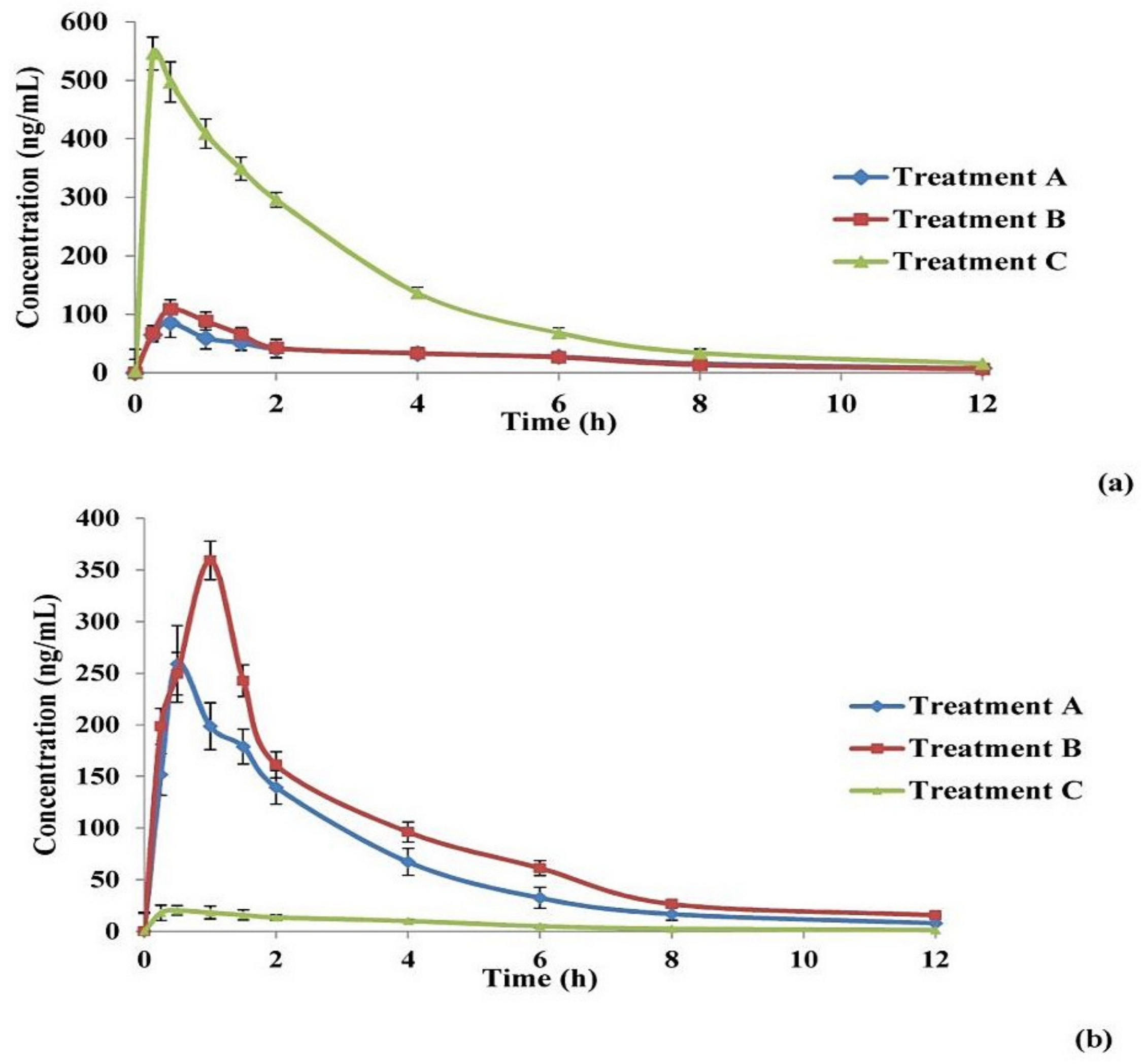
3.3. In Vivo Animals
In Vivo Brain and Systemic–Kinetic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jensen, R.; Stovner, L.J. Epidemiology and Comorbidity of Headache. Lancet Neurol. 2008, 7, 354–361. [Google Scholar] [CrossRef]
- Peterlin, B.L.; Rapoport, A.M. Clinical pharmacology of the serotonin receptor agonist, zolmitriptan. Expert Opin. Drug Metab. Toxicol. 2007, 3, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abera, S.F.; Abyu, G.Y.; Ahmed, M.B.; Aichour, A.N.; Aichour, I.; et al. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [Green Version]
- Weatherall, M.W. The diagnosis and treatment of chronic migraine. Ther. Adv. Chronic Dis. 2015, 6, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.; Rasmussen, B.K.; Fenger, K.; Olesen, J. Migraine without aura and migraine with aura are distinct clinical entities: A study of four hundred and eighty-four male and female migraineurs from the general population. Cephalalgia 1996, 16, 239–245. [Google Scholar] [CrossRef]
- Kurth, T.; Slomke, M.A.; Kase, C.S.; Cook, N.R.; Lee, I.M.; Gaziano, J.M.; Diener, H.C.; Buring, J.E. Migraine, headache, and the risk of stroke in women: A prospective study. Neurology 2005, 64, 1020–1026. [Google Scholar] [CrossRef]
- Prajapati, S.T.; Patel, M.V.; Patel, C.N. Preparation and evaluation of sublingual tablets of zolmitriptan. Int. J. Pharm. Investig. 2014, 4, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, A.A.; Salah, S. Fast relief from migraine attacks using fast-disintegrating sublingual zolmitriptan tablets. Drug Dev. Ind. Pharm. 2012, 38, 762–769. [Google Scholar] [CrossRef]
- Spencer, C.M.; Gunasekara, N.S.; Hills, C. Zolmitriptan. Drugs 1999, 58, 347–374. [Google Scholar] [CrossRef]
- Gizurarson, S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr. Drug Deliv. 2012, 9, 566–582. [Google Scholar] [CrossRef] [Green Version]
- Abd-Elal, R.M.; Shamma, R.N.; Rashed, H.M.; Bendas, E.R. Trans-nasal zolmitriptan novasomes: In-vitro preparation, optimization and in-vivo evaluation of brain targeting efficiency. Drug Deliv. 2016, 23, 3374–3386. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.F.; Scales, H.E.; Shakir, E.; Alexander, J.; Carter, K.C.; Mullen, A.B.; Ferro, V.A. Oral delivery of tetanus toxoid using vesicles containing bile salts (bilosomes) induces significant systemic and mucosal immunity. Methods 2006, 38, 90–95. [Google Scholar] [CrossRef]
- Aburahma, M.H. Bile salts-containing vesicles: Promising pharmaceutical carriers for oral delivery of poorly water-soluble drugs and peptide/protein-based therapeutics or vaccines. Drug Deliv. 2016, 23, 1847–1867. [Google Scholar] [CrossRef]
- Shukla, A.; Singh, B.; Katare, O. Significant systemic and mucosal immune response induced on oral delivery of diphtheria toxoid using nano-bilosomes. Br. J. Pharmacol. 2011, 164, 820–827. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.; Khatri, K.; Gupta, P.N.; Goyal, A.K.; Mehta, A.; Vyas, S.P. Oral immunization against hepatitis B using bile salt stabilized vesicles (bilosomes). J. Pharm. Pharm. Sci. 2008, 11, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Duangjit, S.; Opanasopit, P.; Rojanarata, T.; Ngawhirunpat, T. Evaluation of meloxicam-loaded cationic transfersomes as transdermal drug delivery carriers. Aaps Pharmscitech 2013, 14, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Saifi, Z.; Rizwanullah, M.; Mir, S.R.; Amin, S. Bilosomes nanocarriers for improved oral bioavailability of acyclovir: A complete characterization through in vitro, ex-vivo and in vivo assessment. J. Drug Deliv. Sci. Technol. 2020, 57, 101634. [Google Scholar] [CrossRef]
- Pavlović, N.; Goločorbin-Kon, S.; Ðanić, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile acids and their derivatives as potential modifiers of drug release and pharmacokinetic profiles. Front. Pharmacol. 2018, 9, 1283. [Google Scholar] [CrossRef] [PubMed]
- Abdelalim, L.R.; Abdallah, O.Y.; Elnaggar, Y.S. High efficacy, rapid onset nanobiolosomes of sildenafil as a topical therapy for erectile dysfunction in aged rats. Int. J. Pharm. 2020, 591, 119978. [Google Scholar] [CrossRef]
- Janga, K.Y.; Tatke, A.; Balguri, S.P.; Lamichanne, S.P.; Ibrahim, M.M.; Maria, D.N.; Jablonski, M.M.; Majumdar, S. Ion-sensitive in situ hydrogels of natamycin bilosomes for enhanced and prolonged ocular pharmacotherapy: In vitro permeability, cytotoxicity and in vivo evaluation. Artif. Cells Nanomed. Biotechnol. Int. J. 2018, 46, 1039–1050. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Inthavong, K.; Qiu, D.; Singh, N.; He, F.; Tu, J. Prediction of nasal spray drug absorption influenced by mucociliary clearance. PLoS ONE 2021, 16, e0246007. [Google Scholar] [CrossRef] [PubMed]
- Aderibigbe, B.A. In situ-based gels for nose to brain delivery for the treatment of neurological diseases. Pharmaceutics 2018, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaghoobian, M.; Haeri, A.; Bolourchian, N.; Shahhosseni, S.; Dadashzadeh, S. The impact of surfactant composition and surface charge of niosomes on the oral absorption of repaglinide as a BCS II model drug. Int. J. Nanomed. 2020, 15, 8767–8781. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, M.A.; Tadros, M.I.; Mohamed, M.I.; El-Helaly, S.N. Low-frequency versus high-frequency ultrasound-mediated transdermal delivery of agomelatine-loaded invasomes: Development, optimization and in-vivo pharmacokinetic assessment. Int. J. Nanomed. 2020, 15, 8893–8910. [Google Scholar] [CrossRef]
- Mahmoud, A.A.; Elkasabgy, N.A.; Abdelkhalek, A.A. Design and characterization of emulsified spray dried alginate microparticles as a carrier for the dually acting drug roflumilast. Eur. J. Pharm. Sci. 2018, 122, 64–76. [Google Scholar] [CrossRef]
- Khan, K.; Ka, K.; CT, R. Effect of compaction pressure on the dissolution efficiency of some direct compression systems. Pharmaceutica. Acta Helv. 1972, 47, 594–607. [Google Scholar]
- Peppas, N. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar]
- Korsmeyer, R.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of potassium chloride release from compressed, hydrophilic, polymeric matrices: Effect of entrapped air. J. Pharm. Sci. 1983, 72, 1189–1191. [Google Scholar] [CrossRef]
- Zaki, N.M.; Awad, G.A.; Mortada, N.D.; Abd ElHady, S.S. Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur. J. Pharm. Sci. 2007, 32, 296–307. [Google Scholar] [CrossRef]
- Fatouh, A.M.; Elshafeey, A.H.; Abdelbary, A. Agomelatine-based in situ gels for brain targeting via the nasal route: Statistical optimization, in vitro, and in vivo evaluation. Drug Deliv. 2017, 24, 1077–1085. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Tang, X.; Wang, Y.; Guo, S. Smart gelation of chitosan solution in the presence of NaHCO3 for injectable drug delivery system. Int. J. Pharm. 2011, 414, 6–15. [Google Scholar] [CrossRef]
- Simões, A.; Miranda, M.; Cardoso, C.; Veiga, F.; Vitorino, C. Rheology by design: A regulatory tutorial for analytical method validation. Pharmaceutics 2020, 12, 820. [Google Scholar] [CrossRef]
- Tung, I.-C. Rheological behavior of poloxamer 407 aqueous solutions during sol-gel and dehydration processes. Int. J. Pharm. 1994, 107, 85–90. [Google Scholar] [CrossRef]
- Ohshima, H.; Miyagishima, A.; Kurita, T.; Makino, Y.; Iwao, Y.; Sonobe, T.; Itai, S. Freeze-dried nifedipine-lipid nanoparticles with long-term nano-dispersion stability after reconstitution. Int. J. Pharm. 2009, 377, 180–184. [Google Scholar] [CrossRef]
- Lale, A.; Mason, J.; Jones, N. Mucociliary transport and its assessment: A review. Clin. Otolaryngol. Allied Sci. 1998, 23, 388–396. [Google Scholar] [CrossRef]
- El-Nabarawy, N.A.; Teaima, M.H.; Helal, D.A. Assessment of spanlastic vesicles of zolmitriptan for treating migraine in rats. Drug Des. Dev. Ther. 2019, 13, 3929–3937. [Google Scholar] [CrossRef] [Green Version]
- Dalpiaz, A.; Marchetti, N.; Cavazzini, A.; Pasti, L.; Velaga, S.; Gavini, E.; Beggiato, S.; Ferraro, L. Quantitative determination of zolmitriptan in rat blood and cerebrospinal fluid by reversed phase HPLC–ESI-MS/MS analysis: Application to in vivo preclinical pharmacokinetic study. J. Chromatogr. B 2012, 901, 72–78. [Google Scholar] [CrossRef]
- Chen, X.; Liu, D.; Luan, Y.; Jin, F.; Zhong, D. Determination of zolmitriptan in human plasma by liquid chromatography–tandem mass spectrometry method: Application to a pharmacokinetic study. J. Chromatogr. B 2006, 832, 30–35. [Google Scholar] [CrossRef]
- Vyas, T.K.; Babbar, A.K.; Sharma, R.K.; Misra, A. Intranasal mucoadhesive microemulsions of zolmitriptan: Preliminary studies on brain-targeting. J. Drug Target. 2005, 13, 317–324. [Google Scholar] [CrossRef]
- Mohamed, D.F.; Abdel-Mageed, A.; Abdel-Hamid, F.; Ahmed, M. In-vitro and in-vivo evaluation of niosomal gel containing aceclofenac for sustained drug delivery. Int. J. Pharm. Sci. Res. 2014, 1, 1. [Google Scholar]
- Dinarvand, R.; Moghadam, S.H.; Sheikhi, A.; Atyabi, F. Effect of surfactant HLB and different formulation variables on the properties of poly-D, L-lactide microspheres of naltrexone prepared by double emulsion technique. J. Microencapsul. 2005, 22, 139–151. [Google Scholar] [CrossRef]
- Kazi, K.M.; Mandal, A.S.; Biswas, N.; Guha, A.; Chatterjee, S.; Behera, M.; Kuotsu, K. Niosome: A future of targeted drug delivery systems. J. Adv. Pharm. Technol. Res. 2010, 1, 374–380. [Google Scholar]
- Fouda, N.H.; Abdelrehim, R.T.; Hegazy, D.A.; Habib, B.A. Sustained ocular delivery of Dorzolamide-HCl via proniosomal gel formulation: In-vitro characterization, statistical optimization, and in-vivo pharmacodynamic evaluation in rabbits. Drug Deliv. 2018, 25, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, T.; Sternberg, B.; Florence, A.T. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60 and 80) and a sorbitan triester (Span 85). Int. J. Pharm. 1994, 105, 1–6. [Google Scholar] [CrossRef]
- ElShagea, H.N.; ElKasabgy, N.A.; Fahmy, R.H.; Basalious, E.B. Freeze-dried self-nanoemulsifying self-nanosuspension (snesns): A new approach for the preparation of a highly drug-loaded dosage form. AAPS PharmSciTech 2019, 20, 258. [Google Scholar] [CrossRef]
- Mason, R.L.; Gunst, R.F.; Hess, J.L. Statistical Design and Analysis of Experiments: With Applications to Engineering and Science; John Wiley & Sons: Hoboken, NJ, USA, 2003; Volume 474, pp. 24–28. [Google Scholar]
- Annadurai, G.; Ling, L.Y.; Lee, J.F. Statistical optimization of medium components and growth conditions by response surface methodology to enhance phenol degradation by Pseudomonas putida. J. Hazard. Mater. 2008, 151, 171–178. [Google Scholar] [CrossRef]
- Chauhan, B.; Gupta, R. Application of statistical experimental design for optimization of alkaline protease production from Bacillus sp. RGR-14. Process. Biochem. 2004, 39, 2115–2122. [Google Scholar] [CrossRef]
- Thomas, L.; Viswanad, V. Formulation and optimization of clotrimazole-loaded proniosomal gel using 32 factorial design. Sci. Pharm. 2012, 80, 731–748. [Google Scholar] [CrossRef] [Green Version]
- Mokhtar, M.; Sammour, O.A.; Hammad, M.A.; Megrab, N.A. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int. J. Pharm. 2008, 361, 104–111. [Google Scholar] [CrossRef]
- Moribe, K.; Maruyama, K.; Iwatsuru, M. Encapsulation characteristics of nystatin in liposomes: Effects of cholesterol and polyethylene glycol derivatives. Int. J. Pharm. 1999, 188, 193–202. [Google Scholar] [CrossRef]
- Khalil, R.M.; Abdelbary, G.A.; Basha, M.; Awad, G.E.; El-Hashemy, H.A. Enhancement of lomefloxacin Hcl ocular efficacy via niosomal encapsulation: In vitro characterization and in vivo evaluation. J. Liposome Res. 2017, 27, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Adel, I.M.; ElMeligy, M.F.; Abdelrahim, M.E.; Maged, A.; Abdelkhalek, A.A.; Abdelmoteleb, A.M.; Elkasabgy, N.A. Design and Characterization of Spray-Dried Proliposomes for the Pulmonary Delivery of Curcumin. Int. J. Nanomed. 2021, 16, 2667–2687. [Google Scholar] [CrossRef] [PubMed]
- Uchegbu, I.F.; Florence, A.T. Non-ionic surfactant vesicles (niosomes): Physical and pharmaceutical chemistry. Adv. Colloid Interface Sci. 1995, 58, 1–55. [Google Scholar] [CrossRef]
- Abdelkader, H.; Ismail, S.; Kamal, A.; Alany, R.G. Preparation of niosomes as an ocular delivery system for naltrexone hydrochloride: Physicochemical characterization. Die Pharm. -Int. J. Pharm. Sci. 2010, 65, 811–817. [Google Scholar]
- Salama, A.H.; Abdelkhalek, A.A.; Elkasabgy, N.A. Etoricoxib-loaded bio-adhesive hybridized polylactic acid-based nanoparticles as an intra-articular injection for the treatment of osteoarthritis. Int. J. Pharm. 2020, 578, 119081. [Google Scholar] [CrossRef]
- Abd-Elsalam, W.H.; ElKasabgy, N.A. Mucoadhesive olaminosomes: A novel prolonged release nanocarrier of agomelatine for the treatment of ocular hypertension. Int. J. Pharm. 2019, 560, 235–245. [Google Scholar] [CrossRef]
- Kamel, R.; El-Wakil, N.A.; Abdelkhalek, A.A.; Elkasabgy, N.A. Topical cellulose nanocrystals-stabilized nanoemulgel loaded with ciprofloxacin HCl with enhanced antibacterial activity and tissue regenerative properties. J. Drug Deliv. Sci. Technol. 2021, 64, 102553. [Google Scholar]
- Gagliardi, A.; Paolino, D.; Iannone, M.; Palma, E.; Fresta, M.; Cosco, D. Sodium deoxycholate-decorated zein nanoparticles for a stable colloidal drug delivery system. Int. J. Nanomed. 2018, 13, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Lupo, N.; Steinbring, C.; Friedl, J.D.; Le-Vinh, B.; Bernkop-Schnürch, A. Impact of bile salts and a medium chain fatty acid on the physical properties of self-emulsifying drug delivery systems. Drug Dev. Ind. Pharm. 2021, 47, 22–35. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Tsai, M.J.; Wu, P.C.; Tsai, Y.H.; Wu, Y.H.; Fang, J.Y. Elastic liposomes as carriers for oral delivery and the brain distribution of (+)-catechin. J. Drug Target. 2011, 19, 709–718. [Google Scholar] [CrossRef]
- Govender, T.; Stolnik, S.; Garnett, M.C.; Illum, L.; Davis, S.S. PLGA nanoparticles prepared by nanoprecipitation: Drug loading and release studies of a water soluble drug. J. Control. Release 1999, 57, 171–185. [Google Scholar] [CrossRef]
- Pardakhty, A.; Varshosaz, J.; Rouholamini, A. In vitro study of polyoxyethylene alkyl ether niosomes for delivery of insulin. Int. J. Pharm. 2007, 328, 130–141. [Google Scholar] [CrossRef]
- Williams, M. The Mathematics of Diffusion; Crank, J., Ed.; Clarendon Press: Oxford, UK, 1975; p. 414. [Google Scholar]
- Serra, C.H.d.R.; Chang, K.H.; Dezani, T.M.; Porta, V.; Storpirtis, S. Dissolution efficiency and bioequivalence study using urine data from healthy volunteers: A comparison between two tablet formulations of cephalexin. Braz. J. Pharm. Sci. 2015, 51, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Malakar, J.; Nayak, A.K.; Goswami, S. Use of response surface methodology in the formulation and optimization of bisoprolol fumarate matrix tablets for sustained drug release. ISRN Pharm. 2012, 2012, 35–38. [Google Scholar] [CrossRef] [Green Version]
- Kassem, M.A.; Aboul-Einien, M.H.; El Taweel, M.M. Dry gel containing optimized felodipine-loaded transferosomes: A promising transdermal delivery system to enhance drug bioavailability. AAPS PharmSciTech 2018, 19, 2155–2173. [Google Scholar] [CrossRef]
- Elela, M.M.A.; ElKasabgy, N.A.; Basalious, E.B. Bio-shielding in situ forming gels (BSIFG) loaded with lipospheres for depot injection of quetiapine fumarate: In vitro and in vivo evaluation. AAPS PharmSciTech 2017, 18, 2999–3010. [Google Scholar] [CrossRef]
- Adel, S.; ElKasabgy, N.A. Design of innovated lipid-based floating beads loaded with an antispasmodic drug: In-vitro and in-vivo evaluation. J. Liposome Res. 2014, 24, 136–149. [Google Scholar] [CrossRef]
- Copetti, G.; Grassi, M.; Lapasin, R.; Pricl, S. Synergistic gelation of xanthan gum with locust bean gum: A rheological investigation. Glycoconj. J. 1997, 14, 951–961. [Google Scholar] [CrossRef]
- Owen, D.H.; Peters, J.J.; Katz, D.F. Rheological properties of contraceptive gels. Contraception 2000, 62, 321–326. [Google Scholar] [CrossRef]
- Chang, J.Y.; Oh, Y.K.; Choi, H.G.; Kim, Y.B.; Kim, C.K. Rheological evaluation of thermosensitive and mucoadhesive vaginal gels in physiological conditions. Int. J. Pharm. 2002, 241, 155–163. [Google Scholar] [CrossRef]
- Abdel-Salam, F.S.; Elkheshen, S.A.; Mahmoud, A.A.; Basalious, E.B.; Amer, M.S.; Mostafa, A.A.; Elkasabgy, N.A. In-situ forming chitosan implant-loaded with raloxifene hydrochloride and bioactive glass nanoparticles for treatment of bone injuries: Formulation and biological evaluation in animal model. Int. J. Pharm. 2020, 580, 119213. [Google Scholar] [CrossRef]
- Gizurarson, S. The effect of cilia and the mucociliary clearance on successful drug delivery. Biol. Pharm. Bull. 2015, 38, 497–506. [Google Scholar] [CrossRef] [Green Version]
- El-Mahrouk, G.; Aboul-Einien, M.H.; Elkasabgy, N.A. Formulation and evaluation of meloxicam orally dispersible capsules. Asian J Pharm Sci. 2009, 4, 8–22. [Google Scholar]
- Adel, I.M.; ElMeligy, M.F.; Abdelkhalek, A.A.; Elkasabgy, N.A. Design and characterization of highly porous curcumin loaded freeze-dried wafers for wound healing. Eur. J. Pharm. Sci. 2021, 164, 105888. [Google Scholar] [CrossRef]
- Schipper, N.G.; Verhoef, J.C.; Mercus, F.W. The nasal mucocilliary clearance relevance to nasal drug delivery. Pharm Res. 1991, 8, 807–814. [Google Scholar] [CrossRef]
- EMC. Zomig Tablets 2.5 mg. Available online: https://www.medicines.org.uk/emc/product/1372/smpc#gref (accessed on 20 August 2021).
- Salama, H.A.; Mahmoud, A.A.; Kamel, A.O.; Abdel Hady, M.; Awad, G.A. Brain delivery of olanzapine by intranasal administration of transfersomal vesicles. J. Liposome Res. 2012, 22, 336–345. [Google Scholar] [CrossRef]
- Fahmy, U.A.; Badr-Eldin, S.M.; Ahmed, O.A.; Aldawsari, H.M.; Tima, S.; Asfour, H.Z.; Al-Rabia, M.W.; Negm, A.A.; Sultan, M.H.; Madkhali, O.A.; et al. Intranasal niosomal in situ gel as a promising approach for enhancing flibanserin bioavailability and brain delivery: In vitro optimization and ex vivo/in vivo evaluation. Pharmaceutics 2020, 12, 485. [Google Scholar] [CrossRef]
- Kapoor, M.; Cloyd, J.C.; Siegel, R.A. A review of intranasal formulations for the treatment of seizure emergencies. J. Control. Release 2016, 237, 147–159. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Soane, R.; Hinchcliffe, M.; Davis, S.S.; Illum, L. Clearance characteristics of chitosan based formulations in the sheep nasal cavity. Int. J. Pharm. 2001, 217, 183–191. [Google Scholar] [CrossRef]
- Alsarra, I.A.; Hamed, A.Y.; Alanazi, F.K.; El Maghraby, G.M. Vesicular Systems for Intranasal Drug Delivery, in Drug Delivery to the Central Nervous System; Springer: Berlin/Heidelberg, Germany, 2010; pp. 175–203. [Google Scholar]
- Shepherd, G. Responses of mitral cells to olfactory nerve volleys in the rabbit. J. Physiol. 1963, 168, 89100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seju, U.; Kumar, A.; Sawant, K.K. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: In vitro and in vivo studies. Acta Biomater. 2011, 12, 4169–4176. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gu, P.; Zhang, W.; Cai, C.; He, H.; Tang, X. Evaluation of submicron emulsion as vehicles for rapid-onset intranasal delivery and improvement in brain targeting of zolmitriptan. Drug Deliv. 2011, 18, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gu, P.; Zhang, W.; Qi, N.; Cai, C.; He, H.; Tang, X. Preparation and evaluation of zolmitriptan submicron emulsion for rapid and effective nasal absorption in beagle dogs. Drug Dev. Ind. Pharm. 2011, 37, 1509–1516. [Google Scholar] [CrossRef]
- Khezri, F.A.N.Z.; Lakshmi, C.S.R.; Bukka, R.; Nidhi, M.; Nargund, S.L. Pharmacokinetic study and brain tissue analysis of Zolmitriptan loaded chitosan nanoparticles in rats by LC-MS method. Int. J. Biol. Macromol. 2020, 142, 52–62. [Google Scholar] [CrossRef]
- Al Khateb, K.; Ozhmukhametova, E.K.; Mussin, M.N.; Seilkhanov, S.K.; Rakhypbekov, T.K.; Lau, W.M.; Khutoryanskiy, V.V. In situ gelling systems based on Pluronic F127/Pluronic F68 formulations for ocular drug delivery. Int. J. Pharm. 2016, 502, 70–79. [Google Scholar] [CrossRef]
- Chatterjee, B.; Amalina, N.; Sengupta, P.; Mandal, U.K. Mucoadhesive polymers and their mode of action: A recent update. J. Appl. Pharm. Sci. 2017, 7, 195–203. [Google Scholar]
- Abou-Taleb, H.A.; Khallaf, R.A.; Abdel-Aleem, J.A. Intranasal niosomes of nefopam with improved bioavailability: Preparation, optimization, and in-vivo evaluation. Drug Des. Dev. Ther. 2018, 12, 3501–3516. [Google Scholar] [CrossRef] [Green Version]
- Salem, H.F.; Kharshoum, R.M.; Abou-Taleb, H.A.; Naguib, D.M. Nanosized transferosome-based intranasal in situ gel for brain targeting of resveratrol: Formulation, optimization, in vitro evaluation, and in vivo pharmacokinetic study. AAPS PharmSciTech 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Sherje, A.P.; Londhe, V. Development and evaluation of pH-responsive cyclodextrin-based in situ gel of paliperidone for intranasal delivery. AAPS PharmSciTech 2018, 19, 384–394. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variable (Factor) | Level | ||
|---|---|---|---|
| Low (−1) | Medium (0) | High (+1) | |
| X1: Sodium deoxycholate amount (mg) | 5 | 10 | 15 |
| X2: Cholesterol/Span® 40 amount (mg) | 100 | 200 | 300 |
| X3: Cholesterol: Span® 40 molar ratio (w/w) | 1:1 | 1:5 | 1:9 |
| Dependent variable (Response) | Desirability Constraint | ||
| Y1: Entrapment efficiency (%) | Maximize | ||
| Y2: Particle size (nm) | Minimize | ||
| Y3: Polydispersity index | Minimize | ||
| Y4: Zeta potential (mV) | Minimize | ||
| Y5: Q0.5 h (%) | Maximize | ||
| Y6: Release efficiency (%) | Maximize | ||
| Formulation Code | Composition | ||
|---|---|---|---|
| Sodium Deoxycholate (mg) | Cholesterol/Span® 40 Mixture (mg) | Cholesterol: Span® 40 Molar Ratio | |
| Midpoint Formulations | |||
| F1 | 5 | 100 | 1:5 |
| F2 | 15 | 100 | 1:5 |
| F3 | 5 | 300 | 1:5 |
| F4 | 15 | 300 | 1:5 |
| F5 | 5 | 200 | 1:1 |
| F6 | 15 | 200 | 1:1 |
| F7 | 5 | 200 | 1:9 |
| F8 | 15 | 200 | 1:9 |
| F9 | 10 | 100 | 1:1 |
| F10 | 10 | 300 | 1:1 |
| F11 | 10 | 100 | 1:9 |
| F12 | 10 | 300 | 1:9 |
| Center point formulations | |||
| F13 | 10 | 200 | 1:5 |
| F14 | 10 | 200 | 1:5 |
| F15 | 10 | 200 | 1:5 |
| Non-Ionic Surfactant | EE% | |
|---|---|---|
| Name | HLB | |
| Span® 20 | 8.6 | 14.90 ± 0.70 |
| Span® 40 | 6.7 | 44.72 ± 1.17 |
| Span® 60 | 4.7 | 33.33 ± 1.55 |
| Span® 80 | 4.3 | 24.60 ± 1.35 |
| Tween® 65 | 10.5 | 11.90 ± 1.65 |
| Tween® 80 | 15.0 | 6.50 ± 0.70 |
| Brij® 35 | 16.0 | 9.70 ± 0.70 |
| Brij® O10 | 12.0 | 8.80 ± 0.83 |
| Formulation Code | Responses | |||||
|---|---|---|---|---|---|---|
| EE (%) | PS (nm) | PDI | ZP (mV) | Q0.5 h (%) | RE (%) | |
| F1 | 42.73 ± 1.19 | 343.33 ± 18.53 | 0.34 ± 0.03 | −41.4 ± 1.15 | 16.0 ± 0.64 | 72.0 ± 3.09 |
| F2 | 37.67 ± 1.33 | 230.53 ± 12.62 | 0.49 ± 0.07 | −55.0 ± 1.27 | 22.3 ± 1.00 | 68.6 ± 2.06 |
| F3 | 63.40 ± 0.74 | 448.72 ± 19.32 | 0.33 ± 0.04 | −39.3 ± 0.81 | 34.8 ± 1.60 | 76.1 ± 3.04 |
| F4 | 67.46 ± 0.77 | 602.20 ± 13.21 | 0.35 ± 0.04 | −54.2 ± 1.50 | 39.6 ± 1.41 | 77.3 ± 1.09 |
| F5 | 51.63 ± 1.22 | 560.50 ± 20.55 | 0.26 ± 0.01 | −45.0 ± 1.48 | 38.5 ± 1.24 | 74.1 ± 1.07 |
| F6 | 44.97 ± 1.35 | 527.00 ± 55.27 | 0.35 ± 0.04 | −65.1 ± 2.05 | 42.4 ± 0.75 | 70.0 ± 3.08 |
| F7 | 65.73 ± 1.66 | 409.27 ± 34.68 | 0.35 ± 0.04 | −44.5 ± 0.50 | 32.8 ± 1.00 | 67.8 ± 1.06 |
| F8 | 51.03 ± 1.59 | 460.43 ± 48.91 | 0.28 ± 0.02 | −48.8 ± 0.95 | 38.4 ± 1.22 | 70.8 ± 2.05 |
| F9 | 33.53 ± 1.59 | 531.23 ± 31.97 | 0.16 ± 0.03 | −45.3 ± 0.60 | 24.5 ± 0.96 | 46.5 ± 3.02 |
| F10 | 46.06 ± 1.45 | 597.96 ± 34.41 | 0.35 ± 0.04 | −51.2 ± 0.70 | 31.3 ± 0.55 | 69.2 ± 1.07 |
| F11 | 37.55 ± 0.83 | 356.97 ± 42.54 | 0.16 ± 0.03 | −44.8 ± 1.05 | 18.1 ± 1.00 | 64.4 ± 3.03 |
| F12 | 74.33 ± 1.32 | 352.23 ± 43.37 | 0.29 ± 0.03 | −48.0 ± 0.50 | 15.5 ± 0.68 | 63.5 ± 0.14 |
| F13 | 70.97 ± 1.30 | 384.40 ± 10.05 | 0.017 ± 0.00 | −45.3 ± 0.64 | 15.5 ± 0.96 | 71.9 ± 0.19 |
| F14 | 71.39 ± 1.30 | 379.30 ± 10.05 | 0.019 ± 0.00 | −44.3 ± 0.64 | 16.7 ± 0.96 | 72.1 ± 0.19 |
| F15 | 70.83 ± 1.30 | 398.20 ± 10.05 | 0.012 ± 0.00 | −44.1 ± 0.64 | 17.4 ± 0.96 | 71.8 ± 0.19 |
| Factor | Optimal Level | ||
|---|---|---|---|
| X1: Sodium deoxycholate amount (mg) | 5 | ||
| X2: Cholesterol/Span® 40 amount (mg) | 255 | ||
| X3: Cholesterol: Span® 40 molar ratio (w/w) | 1:7.7 | ||
| Response | Expected value | Observed value | Residual value a |
| Y1: Entrapment efficiency (%) | 71.70 | 70.34 ± 0.10 | 1.36 |
| Y2: Particle size (nm) | 399.27 | 399.80 ± 4.95 | −0.53 |
| Y3: Polydispersity index | 0.33 | 0.33 ± 0.05 | −0.004 |
| Y4: Zeta potential (mv) | −42.50 | −41.90 ±0.19 | −0.60 |
| Y5: Q0.5 h (%) a | 31.87 | 32.20 ± 1.09 | −0.33 |
| Y6: Release Efficiency (%) | 73.86 | 71.76 ± 0.34 | 2.10 |
| In Plasma | |||
| Parameter | Treatment A | Treatment B | Treatment C |
| Cmax(ng/mL) a | 108.58 ± 1.94 | 86.66 ± 4.13 | 535.59 ± 9.04 |
| tmax(h) b | 0.5 | 0.5 | 0.25 |
| AUC0–12 h(ng·h/mL) c | 361.86 ± 6.89 | 340.64 ± 36.48 | 1579.32 ± 33.48 |
| AUC0–∞(ng·h/mL) d | 388.62 ± 4.77 | 370.43 ± 31.26 | 1643.89 ± 35.43 |
| K(h−1) | 0.251 ± 0.010 | 0.231 ± 0.006 | 0.277 ± 0.003 |
| t1/2(h) MRTe | 2.76 ± 0.15 4.08 | 3.00 ± 0.24 4.80 | 2.5 ± 0.38 3.33 |
| Absolute bioavailability (%) * | 23.65 | 22.78 | --- |
| In Brain | |||
| Parameter | Treatment A | Treatment B | Treatment C |
| Cmax(ng/mL) f | 260.43 ± 6.90 | 360.30 ± 7.78 | 21.13 ± 2.09 |
| tmax(h) g | 0.5 | 1 | 0.5 |
| AUC0–12 h(ng·h/mL) h | 768.24 ± 43.69 | 1081.88 ± 43.37 | 91.92 ± 12.20 |
| AUC0–∞(ng·h/mL) i | 801.15 ± 46.94 | 1147.08 ± 51.79 | 97.73 ± 13.86 |
| K(h−1) | 0.275 ± 0.011 | 0.25 ± 0.003 | 0.256 ± 0.003 |
| t1/2(h) | 2.52 ± 0.18 | 2.77 ± 0.16 | 2.70 ± 0.31 |
| MRT(h) j | 3.36 | 3.87 | 3.31 |
| Brain bioavailability (%) ** | 819.75 | 1173.64 | ---- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Taweel, M.M.; Aboul-Einien, M.H.; Kassem, M.A.; Elkasabgy, N.A. Intranasal Zolmitriptan-Loaded Bilosomes with Extended Nasal Mucociliary Transit Time for Direct Nose to Brain Delivery. Pharmaceutics 2021, 13, 1828. https://doi.org/10.3390/pharmaceutics13111828
El Taweel MM, Aboul-Einien MH, Kassem MA, Elkasabgy NA. Intranasal Zolmitriptan-Loaded Bilosomes with Extended Nasal Mucociliary Transit Time for Direct Nose to Brain Delivery. Pharmaceutics. 2021; 13(11):1828. https://doi.org/10.3390/pharmaceutics13111828
Chicago/Turabian StyleEl Taweel, Mai M., Mona H. Aboul-Einien, Mohammed A. Kassem, and Nermeen A. Elkasabgy. 2021. "Intranasal Zolmitriptan-Loaded Bilosomes with Extended Nasal Mucociliary Transit Time for Direct Nose to Brain Delivery" Pharmaceutics 13, no. 11: 1828. https://doi.org/10.3390/pharmaceutics13111828