Beta-Arrestins in the Treatment of Heart Failure Related to Hypertension: A Comprehensive Review

 ,
, 
 , ,
, ,  ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Side Effects of Conventional Antihypertensive Drugs and an Insight on the Process of New Cardiac Drug Discovery
3. Detrimental and Palliative Characteristics: A Paradox of the β-Arrestins Isoform
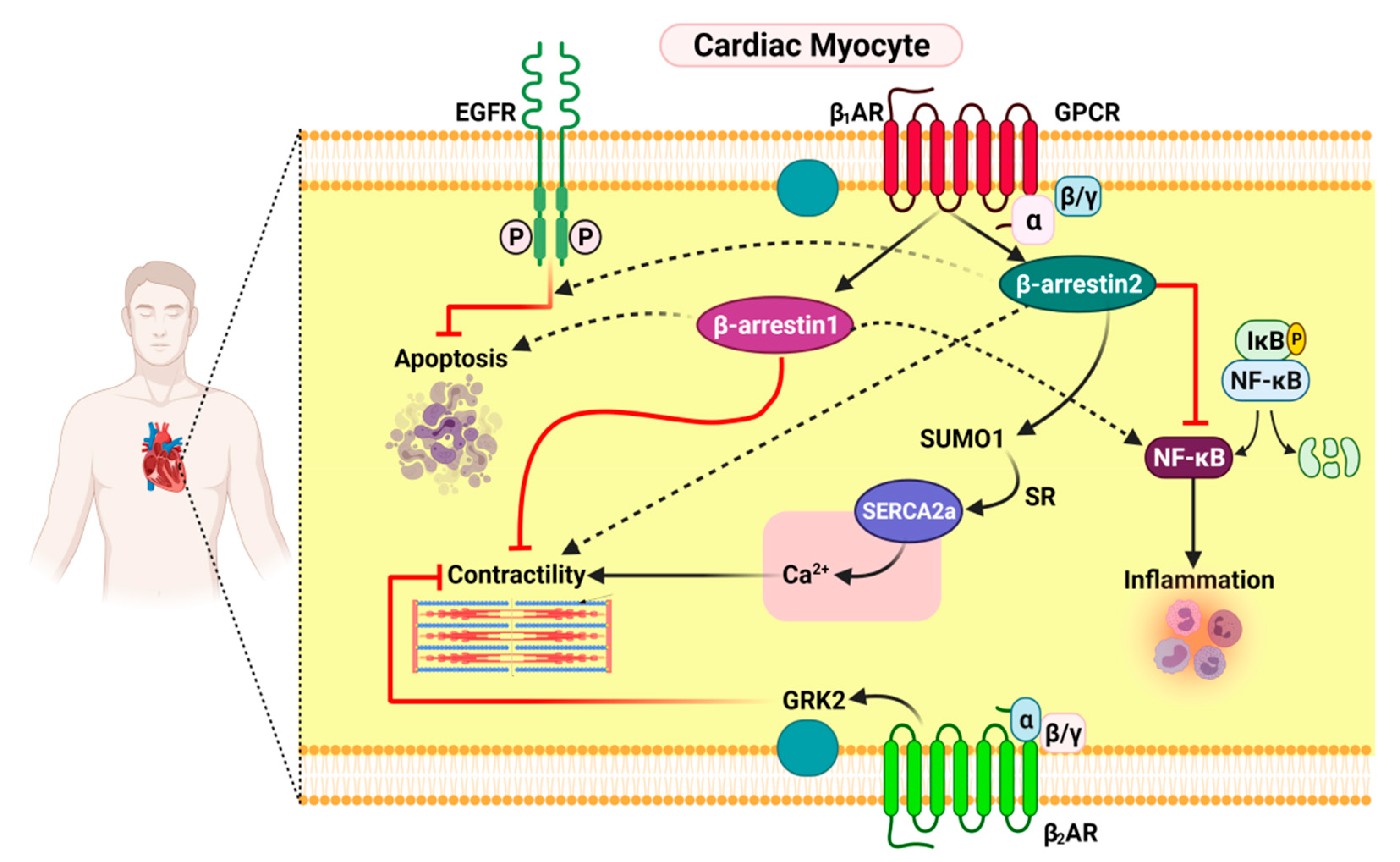
3.1. Functional Distinction between β-Arrestin1 and β-Arrestin2 in Cardiac Myocytes
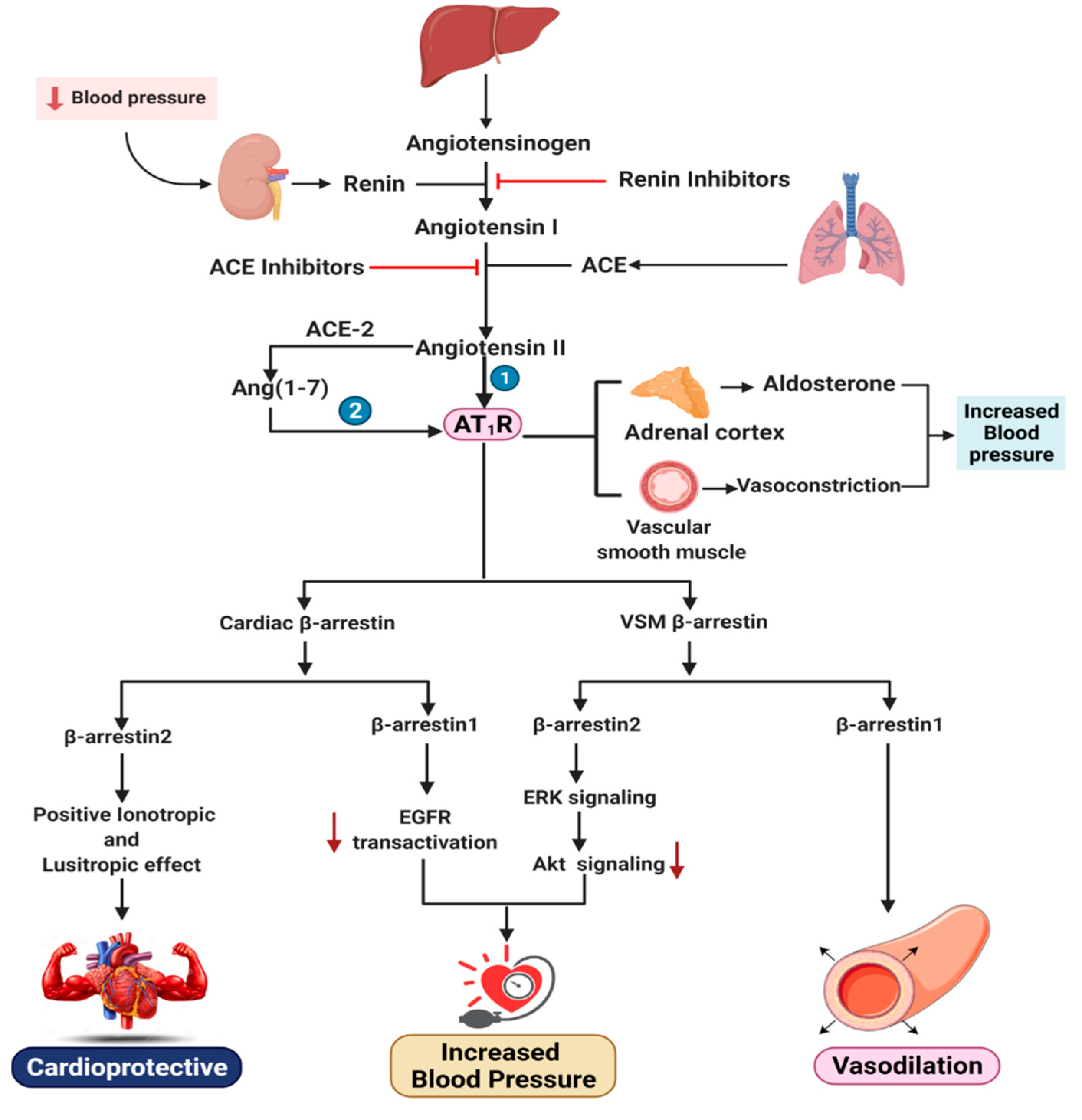
3.2. Functional Distinction between β-Arrestin1 and β-Arrestin2 in Cardiac AT1R Signaling and VSM
4. Effects of β-Arrestin1 and β-Arrestin2 on the Renin–Angiotensin–Aldosterone System
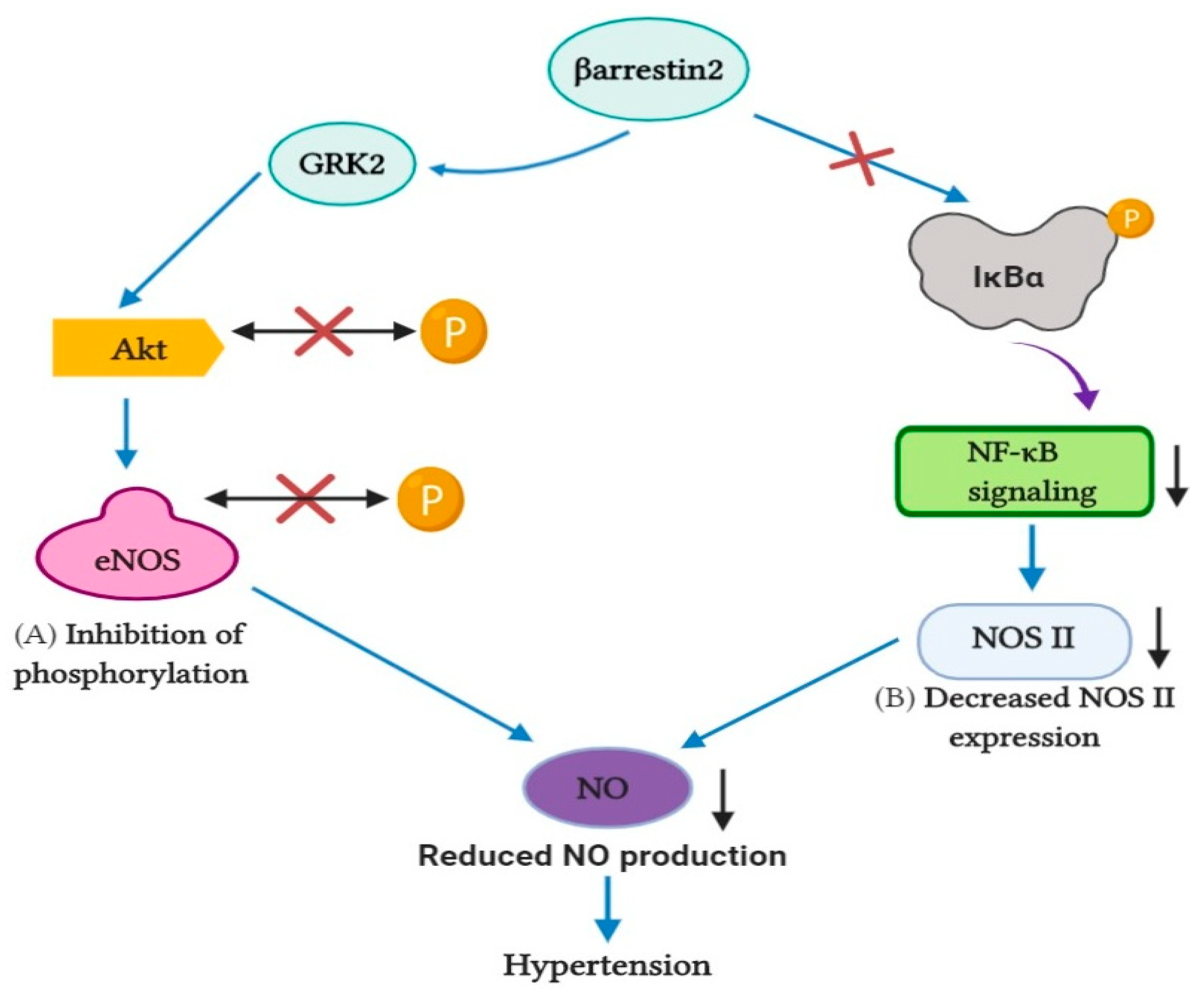
5. Effects of β-Arrestin1 and β-Arrestin2 on Cardiac Beta-Adrenergic Receptors
6. TRV027, a Novel β-Arrestin-Biased Ligand: Progress and Failures
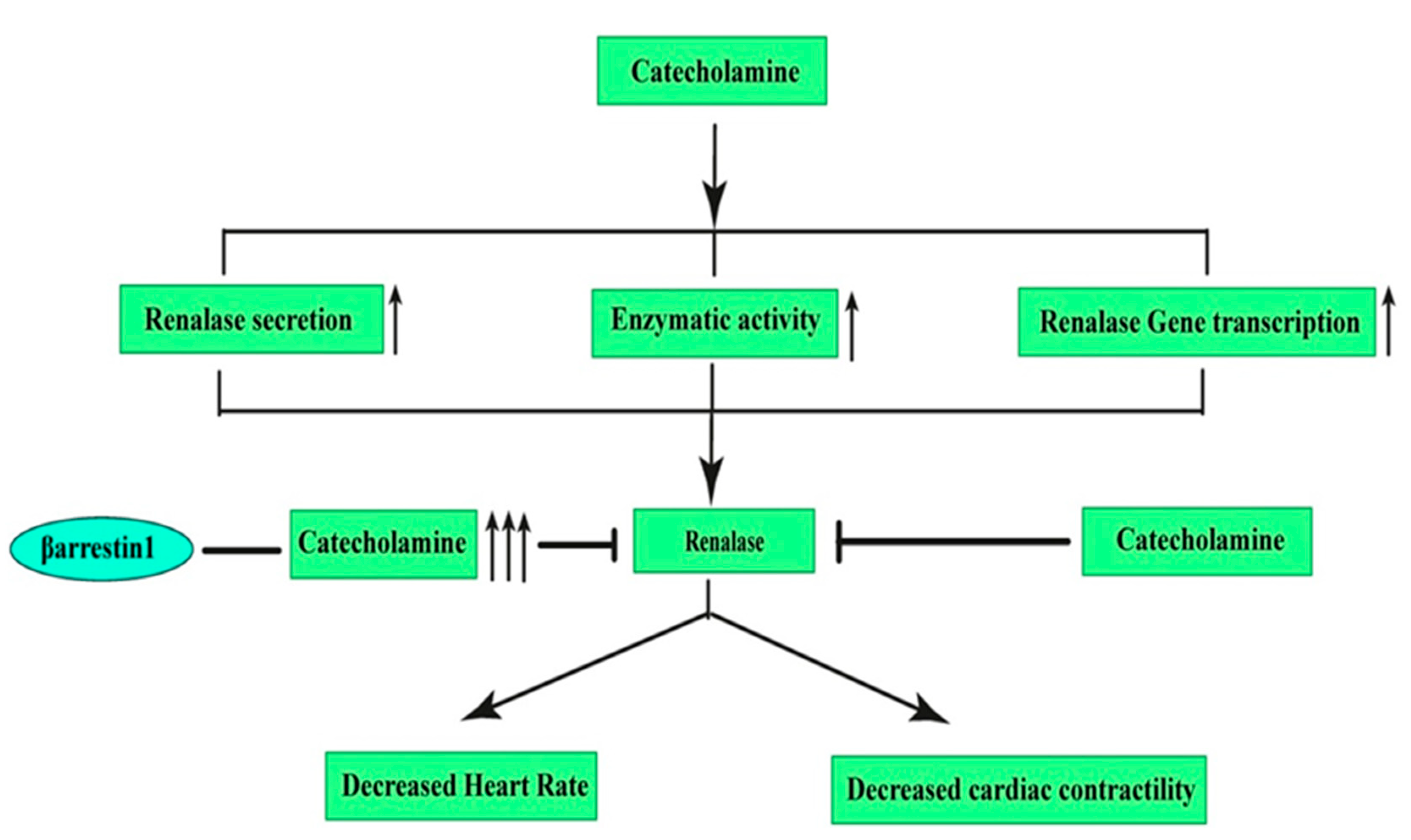
7. Effects of β-Arrestin1 and β-Arrestin2 on Renalase: A Novel Target for Hypertension
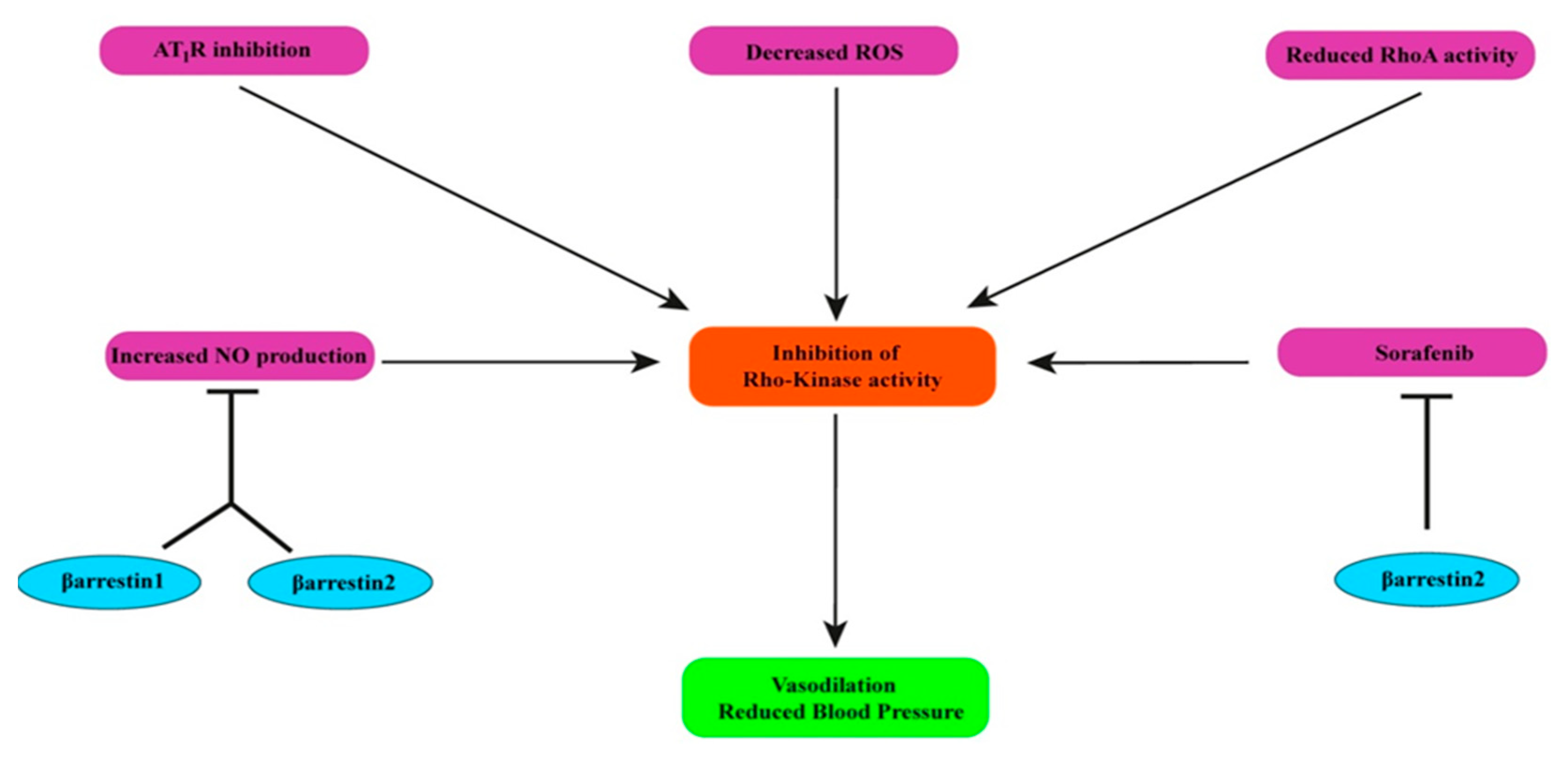
8. Effects of β-Arrestin1 and β-Arrestin2 on Rho-Kinase
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heron, M.P. Deaths: Leading causes for 2013. Natl. Vital. Stat. Rep. 2016, 65, 1–95. [Google Scholar] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; Floyd, J.; Fornage, M.; Gillespie, C.; Isasi, C.R.; et al. Heart disease and stroke statistics-2017 update: A report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Islam, A.K.M.M.; Mohibullah, A.K.M.; Paul, T. Cardiovascular disease in Bangladesh: A review. Bangladesh Heart J. 2016, 31, 80–99. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Uddin, R.; Islam, S.M.S. Clustering patterns of behavioural risk factors for cardiovascular diseases in Bangladeshi adolescents: A population-based study. Health Policy Technol. 2019. [Google Scholar] [CrossRef]
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017. [Google Scholar] [CrossRef]
- Francis, G.S.; Tang, W.H.W. Pathophysiology of congestive heart failure. Rev. Cardiovasc. Med. 2019, 4, 14–20. [Google Scholar]
- Messerli, F.H.; Rimoldi, S.F.; Bangalore, S. The Transition from Hypertension to Heart Failure: Contemporary Update. JACC Heart Fail. 2017, 5, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Pucci, M. Diagnosis and management of hypertension in primary care. Prescriber 2019, 30, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Paulis, L.; Unger, T. Novel therapeutic targets for hypertension. Nat. Rev. Cardiol. 2010, 7, 431–441. [Google Scholar] [CrossRef]
- Violin, J.D.; DeWire, S.M.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, L.P.; Schonegge, A.M.; Bouvier, M. Structural Insight into G Protein-Coupled Receptor Signaling Efficacy and Bias between Gs and β-Arrestin. ACS Pharmacol. Transl. Sci. 2019, 2, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.D.; Perry, S.J.; Regier, D.S.; Prescott, S.M.; Topham, M.K.; Lefkowitz, R.J. Targeting of diacylglycerol degradation to M1 muscarinic receptors by β-arrestins. Science 2007, 315, 663–666. [Google Scholar] [CrossRef]
- Perry, S.J.; Baillie, G.S.; Kohout, T.A.; McPhee, I.; Magiera, M.M.; Ang, K.L.; Miller, W.E.; McLean, A.J.; Conti, M.; Houslay, M.D.; et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 2002, 298, 834–836. [Google Scholar] [CrossRef]
- Girvin, B. Hypertension clinic drug choices: Tips for pharmacist prescribers. Prescriber 2019, 30, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Messerli, F.H. Vasodilatory edema: A common side effect of antihypertensive therapy. Curr. Cardiol. Rep. 2002, 4, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Lithell, H.O.L. Effect of antihypertensive drugs on insulin, glucose, and lipid metabolism. Diabetes Care 1991, 14, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E.D.; Cooper, R.A.; Lewis, E.J. Review of the Overall Experience of Captopril in Hypertension. Arch. Intern. Med. 1984, 144, 1441–1444. [Google Scholar] [CrossRef]
- Ponce, S.P.; Jennings, A.E.; Madias, N.E.; Harrington, J.T. Drug-Induced Hyperkalemia. Medicine 1985, 64, 357–370. [Google Scholar] [CrossRef]
- Davies, R.O.; Irvin, J.D.; Kramsch, D.K.; Walker, J.F.; Moncloa, F. Enalapril worldwide experience. Am. J. Med. 1984, 77, 23–35. [Google Scholar] [CrossRef]
- Jett, G.K. Captopril-induced angioedema. Ann. Emerg. Med. 1984, 13, 489–490. [Google Scholar] [CrossRef]
- Semple, P.F.; Herd, G.W. Cough and Wheeze Caused by Inhibitors of Angiotensin-Converting Enzyme. N. Engl. J. Med. 1986, 314, 61. [Google Scholar] [CrossRef]
- Maning, J.; Negussie, S.; Clark, M.A.; Lymperopoulos, A. Biased agonism/antagonism at the AngII-AT1 receptor: Implications for adrenal aldosterone production and cardiovascular therapy. Pharmacol. Res. 2017, 125, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A. Beta-arrestin Biased Agonism/Antagonism at Cardiovascular Seven Transmembrane-spanning Receptors. Curr. Pharm. Des. 2012, 18, 192–198. [Google Scholar] [CrossRef]
- Thomas, W.G.; Thekkumkara, T.J.; Baker, K.M. Cardiac effects of AII: AT1A Receptor Signaling, Desensitization, and Internalization. Adv. Exp. Med. Biol. 1996, 396, 59–69. [Google Scholar] [CrossRef]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483. [Google Scholar] [CrossRef] [Green Version]
- Kenakin, T. New concepts in drug discovery: Collateral efficacy and permissive antagonism. Nat. Rev. Drug Discov. 2005, 4, 919–927. [Google Scholar] [CrossRef]
- Fisher, A.; Heldman, E.; Gurwitz, D.; Haring, R.; Barak, D.; Meshulam, H.; Marciano, D.; Brandeis, R.; Pittel, Z.; Segal, M. Selective signaling via unique M1 muscarinic agonists. Ann. N. Y. Acad. Sci. 1993, 695, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; Lefkowitz, R.J. β-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007, 8, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; Crombie, A.L.; Soergel, D.G.; Lark, M.W. Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharmacol. Sci. 2014, 35, 308–316. [Google Scholar] [CrossRef]
- Fan, H.; Bitto, A.; Zingarelli, B.; Luttrell, L.M.; Borg, K.; Halushka, P.V.; Cook, J.A. Beta-arrestin 2 negatively regulates sepsis-induced inflammation. Immunology 2010, 130, 344–351. [Google Scholar] [CrossRef]
- Boerrigter, G.; Whalen, E.J.; Lark, M.; Burnett, J.C. Cardiorenal Actions of TRV120027, a Novel, β-Arrestin-Biased Ligand at the Angiotensin II Type I Receptor, in Healthy and Heart Failure Canines: A Novel Therapeutic Strategy for Acute Heart Failure. J. Card. Fail. 2010, 16, S73. [Google Scholar] [CrossRef]
- Kim, K.S.; Abraham, D.; Williams, B.; Violin, J.D.; Mao, L.; Rockman, H.A. β-arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 2012, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monasky, M.M.; Taglieri, D.M.; Henze, M.; Warren, C.M.; Utter, M.S.; Soergel, D.G.; Violin, J.D.; John Solaro, R. The β-arrestin-biased ligand TRV120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am. J. Physiol. Heart Circ. Physiol. 2013, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCrink, K.A.; Maning, J.; Vu, A.; Jafferjee, M.; Marrero, C.; Brill, A.; Bathgate-Siryk, A.; Dabul, S.; Koch, W.J.; Lymperopoulos, A. Cardiac βarrestin2 Improves Contractility and Adverse Remodeling in Heart Failure, But Is Underexpressed in Humans. J. Am. Coll. Cardiol. 2017, 70, 2948–2949. [Google Scholar] [CrossRef]
- Turu, G.; Balla, A.; Hunyady, L. The Role of β-Arrestin Proteins in Organization of Signaling and Regulation of the AT1 Angiotensin Receptor. Front. Endocrinol. 2019, 10, 519. [Google Scholar] [CrossRef]
- Zhai, P.; Yamamoto, M.; Galeotti, J.; Liu, J.; Masurekar, M.; Thaisz, J.; Irie, K.; Holle, E.; Yu, X.; Kupershmidt, S.; et al. Cardiac-specific overexpression of AT1 receptor mutant lacking G αq/Gαi causes hypertrophy and bradycardia in transgenic mice. J. Clin. Investig. 2005, 115, 3045–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gareri, C.; Rockman, H.A. G-protein-coupled receptors in heart disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Rockman, H.A.; Choi, D.J.; Akhter, S.A.; Jaber, M.; Giros, B.; Lefkowitz, R.J.; Caron, M.G.; Koch, W.J. Control of myocardial contractile function by the level of beta-adrenergic receptor kinase 1 in gene-targeted mice. J. Biol. Chem. 1998, 273, 18180–18184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjalmarson, A.; Goldstein, S.; Fagerberg, B.; Wedel, H.; Waagstein, F.; Kjekshus, J.; Wikstrand, J.; El Allaf, D.; Vítovec, J.; Aldershvile, J.; et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: The Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-HF). MERIT-HF Study Group. JAMA 2000, 283, 1295–1302. [Google Scholar] [CrossRef]
- Investigators, C.; Commentary, S. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): A randomised trial. Lancet 1999, 353, 9–13. [Google Scholar]
- Kveiborg, B.; Major-Petersen, A.; Christiansen, B.; Torp-Pedersen, C. Carvedilol in the treatment of chronic heart failure: Lessons from the Carvedilol Or Metoprolol European Trial. Vasc Health Risk Manag. 2007, 3, 31–37. [Google Scholar]
- Wisler, J.W. A unique mechanism of beta-blocker action: Carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.M.; Tilley, D.G.; Chen, J.; Salazar, N.C.; Whalen, E.J.; Violin, J.D.; Rockman, H.A. β-Blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc. Natl. Acad. Sci. USA 2008, 105, 14555–14560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, R.; Schilling, J.; Song, J.; Carter, R.L.; Du, Y.; Yoo, S.M.; Traynham, C.J.; Koch, W.J.; Cheung, J.Y.; Tilley, D.G.; et al. Β-Arrestin-Biased Signaling Through the Β2-Adrenergic Receptor Promotes Cardiomyocyte Contraction. Proc. Natl. Acad. Sci. USA 2016, 113, E4107–E4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Fowler, M.B.; Roecker, E.B.; Coats, A.J.S.; Katus, H.A.; Krum, H.; Mohacsi, P.; Rouleau, J.L.; Tendera, M.; Staiger, C.; et al. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: Results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 2002, 106, 2194–2199. [Google Scholar] [CrossRef] [Green Version]
- Lymperopoulos, A.; Desimine, L.; McCrink, K.A.; Maning, J.; Wertz, S.L.; Markan, U.; Pasupuleti, S.; Brill, A.; Parker, B.M. Positive cardiac inotropy by carvedilol via unique beta-arrestin2-dependent SERCA2a stimulation. Eur. Heart J. 2018, 39, 644. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Soltys, S.; Koch, W.J. An adrenal β-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5825–5830. [Google Scholar] [CrossRef] [Green Version]
- Ferraino, K.E.; Cora, N.; Pollard, C.M.; Sizova, A.; Maning, J.; Lymperopoulos, A. Adrenal angiotensin II type 1 receptor biased signaling: The case for “biased” inverse agonism for effective aldosterone suppression. Cell. Signal. 2021, 82. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Koch, W.J. Adrenal beta-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J. Am. Coll. Cardiol. 2011, 57, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Dabul, S.; Bathgate-Siryk, A.; Valero, T.R.; Jafferjee, M.; Sturchler, E.; Mcdonald, P.; Koch, W.J.; Lymperopoulos, A. Suppression of adrenal ßarrestin1-dependent aldosterone production by ARBs: Head-to-head comparison. Sci. Rep. 2015, 5, 8116. [Google Scholar] [CrossRef] [Green Version]
- Lymperopoulos, A.; Sturchler, E.; Bathgate-Siryk, A.; Dabul, S.; Garcia, D.; Walklett, K.; Rengo, G.; McDonald, P.; Koch, W.J. Different potencies of angiotensin receptor blockers at suppressing adrenal β-arrestin1-dependent post-myocardial infarction hyperaldosteronism. J. Am. Coll. Cardiol. 2014, 64, 2805–2806. [Google Scholar] [CrossRef] [Green Version]
- Appear, B. Knockout Mice; Stimulation, normal but demonstrate altered cardiac responses to beta-adrenergic Beta-arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to beta-adrenergic stimulation. Circ. Res. 1997, 81, 1021–1026. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, L.; Song, Y.; Zhang, M.; Shan, D.; Liu, Y.; Fang, M.; Lv, F.; Xiao, R.P.; Zhang, Y. β-arrestin 2 mediates cardiac ischemia-reperfusion injury via inhibiting GPCR-independent cell survival signalling. Cardiovasc. Res. 2017, 113, 1615–1626. [Google Scholar] [CrossRef]
- Schaff, M.; Receveur, N.; Bourdon, C.; Ohlmann, P.; Lanza, F.; Gachet, C.; Mangin, P.H. β-arrestin-1 participates in thrombosis and regulates integrin α IIbβ 3 signalling without affecting p2y receptors desensitisation and function. Thromb. Haemost. 2012, 107, 735–748. [Google Scholar] [CrossRef] [Green Version]
- Walters, R.W.; Shukla, A.K.; Kovacs, J.J.; Violin, J.D.; DeWire, S.M.; Lam, C.M.; Chen, J.R.; Muehlbauer, M.J.; Whalen, E.J.; Lefkowitz, R.J. β-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J. Clin. Investig. 2009, 119, 1312–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watari, K.; Nakaya, M.; Nishida, M.; Kim, K.M.; Kurose, H. β-arrestin2 in Infiltrated Macrophages Inhibits Excessive Inflammation after Myocardial Infarction. PLoS ONE 2013, 8, e68351. [Google Scholar] [CrossRef]
- Eckhart, A.D.; Ozaki, T.; Tevaearai, H.; Rockman, H.A.; Koch, W.J. Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates β-adrenergic receptor signaling and increases resting blood pressure. Mol. Pharmacol. 2002, 61, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–606. [Google Scholar] [CrossRef]
- Lee, S.U.; In, H.J.; Kwon, M.S.; Park, B.O.; Jo, M.; Kim, M.O.; Cho, S.; Lee, S.; Lee, H.J.; Kwak, Y.S.; et al. β-Arrestin 2 mediates G protein-coupled receptor 43 signals to nuclear factor-κB. Biol. Pharm. Bull. 2013, 36, 1754–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noma, T.; Lemaire, A.; Naga Prasad, S.V.; Barki-Harrington, L.; Tilley, D.G.; Chen, J.; Le Corvoisier, P.; Violin, J.D.; Wei, H.; Lefkowitz, R.J.; et al. β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Investig. 2007, 117, 2445–2448. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, K.; Whalen, E.J.; Violin, J.D.; Stiber, J.A.; Rosenberg, P.B.; Premont, R.T.; Coffman, T.M.; Rockman, H.A.; Lefkowitz, R.J. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 16284–16289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zidar, D.A.; Violin, J.D.; Whalen, E.J.; Lefkowitz, R.J. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 9649–9654. [Google Scholar] [CrossRef] [Green Version]
- Aplin, M.; Christensen, G.L.; Schneider, M.; Heydorn, A.; Gammeltoft, S.; Kjølbye, A.L.; Sheikh, S.P.; Hansen, J.L. Differential extracellular signal-regulated kinases 1 and 2 activation by the angiotensin type 1 receptor supports distinct phenotypes of cardiac myocytes. Basic Clin. Pharmacol. Toxicol. 2007, 100, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 2007, 13, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Gao, E.; Ebert, S.N.; Dorn, G.W.; Koch, W.J. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J. Biol. Chem. 2010, 285, 16378–16386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenal adrenoceptors in heart failure: Fine-tuning cardiac stimulation. Trends Mol. Med. 2007, 13, 503–511. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Soltys, S.; Koch, W.J. Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol. Ther. 2008, 16, 302–307. [Google Scholar] [CrossRef]
- Kim, J.; Zhang, L.; Peppel, K.; Wu, J.-H.; Zidar, D.A.; Brian, L.; DeWire, S.M.; Exum, S.T.; Lefkowitz, R.J.; Freedman, N.J. β-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ. Res. 2008, 103, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Charles, R.; Namkung, Y.; Cotton, M.; Laporte, S.A.; Claing, A. β-Arrestin-mediated angiotensin II signaling controls the activation of ARF6 protein and endocytosis in migration of vascular smooth muscle cells. J. Biol. Chem. 2016, 291, 3967–3981. [Google Scholar] [CrossRef] [Green Version]
- Morinelli, T.A.; Walker, L.P.; Velez, J.C.Q.; Ullian, M.E. Clathrin-dependent internalization of the angiotensin II AT1A receptor links receptor internalization to COX-2 protein expression in rat aortic vascular smooth muscle cells. Eur. J. Pharmacol. 2015, 748, 143–148. [Google Scholar] [CrossRef]
- Morinelli, T.A.; Lee, M.H.; Kendall, R.T.; Luttrell, L.M.; Walker, L.P.; Ullian, M.E. Angiotensin II activates NF-κB through AT1A receptor recruitment of β-arrestin in cultured rat vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2013, 304. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.; Kim, J.; Hara, M.R.; Ren, X.R.; Lefkowitz, R.J. β-arrestin-2 mediates anti-apoptotic signaling through regulation of BAD phosphorylation. J. Biol. Chem. 2009, 284, 8855–8865. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.T.; Lee, M.-H.; Pleasant, D.L.; Robinson, K.; Kuppuswamy, D.; McDermott, P.J.; Luttrell, L.M. Arrestin-dependent angiotensin AT1 receptor signaling regulates Akt and mTor-mediated protein synthesis. J. Biol. Chem. 2014, 289, 26155–26166. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.C.; Lee, M.H.; Appleton, K.M.; El-Shewy, H.M.; Morinelli, T.A.; Peterson, Y.K.; Luttrell, L.M.; Jaffa, A.A. The arrestin-selective angiotensin AT1 receptor agonist [Sar1,Ile4,Ile8]-AngII negatively regulates bradykinin B2 receptor signaling via AT1-B2 receptor heterodimers. J. Biol. Chem. 2013, 288, 18872–18884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, F.T.; Lu, T.L.; Fu, H.W. Opposing effects of β-arrestin1 and β-arrestin2 on activation and degradation of Src induced by protease-activated receptor 1. Cell. Signal. 2006, 18, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lau, C.S.; Mathur, M.; Wang, P.; DeFea, K.A. Differential effects of β-arrestins on the internalization, desensitization and ERK1/2 activation downstream of protease activated receptor-2. Am. J. Physiol. Cell Physiol. 2007, 293. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, W.B.; Friedman, P.A. Β-Arrestin-Dependent Parathyroid Hormone-Stimulated Extracellular Signal-Regulated Kinase Activation and Parathyroid Hormone Type 1 Receptor Internalization. Endocrinology 2007, 148, 4073–4079. [Google Scholar] [CrossRef]
- Fan, H.; Luttrell, L.M.; Tempel, G.E.; Senn, J.J.; Halushka, P.V.; Cook, J.A. β-Arrestins 1 and 2 differentially regulate LPS-induced signaling and pro-inflammatory gene expression. Mol. Immunol. 2007, 44, 3092–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atlas, S.A. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 2007, 8, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.M.; Chappell, M.C. A new myocardial conversion of angiotensin I. Curr. Opin. Cardiol. 1994, 9, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.G.A.G.; Souza, L.L.; Becari, C.; Duarte, D.A.; Camacho, F.R.B.; Oliveira, J.A.C.; Gomes, M.D.; Oliveira, E.B.; Salgado, M.C.O.; Garcia-Cairasco, N.; et al. Angiotensin II-independent angiotensin-(1-7) formation in rat hippocampus: Involvement of thimet oligopeptidase. Hypertension 2013, 62, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.; Brosnihan, K.B.; Jacobsen, D.W.; DiCorleto, P.E.; Ferrario, C.M. Production of angiotensin-(1-7) by human vascular endothelium. Hypertension 1992, 19, II56–II61. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A. Angiotensin-(1-7). Hypertension 2014, 63, 1138–1147. [Google Scholar] [CrossRef]
- Porrello, E.R.; Delbridge, L.M.D.; Thomas, W.G. The angiotensin II type 2 (AT2) receptor: An enigmatic seven transmembrane receptor. Front. Biosci. 2009, 14, 958–972. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.M. Angiotension-(1-7) and antihypertensive mechanisms. J. Nephrol. 1998, 11, 278–283. [Google Scholar]
- Santos, R.A.S.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.R.; Machado, R.P.; De Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, D.; Waitches, G.; Birchmeier, C.; Fasano, O.; Wigler, M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 1986, 45, 711–719. [Google Scholar] [CrossRef]
- Rabin, M.; Birnbaum, D.; Young, D.; Birchmeier, C.; Wigler, M.; Ruddle, F.H. Human ros1 and mas1 oncogenes located in regions of chromosome 6 associated with tumor-specific rearrangements. Oncogene Res. 1987, 1, 169–178. [Google Scholar] [PubMed]
- Peña Silva, R.A.; Kung, D.K.; Mitchell, I.J.; Alenina, N.; Bader, M.; Santos, R.A.S.; Faraci, F.M.; Heistad, D.D.; Hasan, D.M. Angiotensin 1-7 reduces mortality and rupture of intracranial aneurysms in mice. Hypertension 2014, 64, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, K.; Saad, Y.; Karnik, S.S. Interaction of Phe8 of angiotensin II with Lys199 and His256 of AT1 receptor in agonist activation. J. Biol. Chem. 1995, 270, 28511–28514. [Google Scholar] [CrossRef] [Green Version]
- Holloway, A.C.; Qian, H.; Pipolo, L.; Ziogas, J.; Miura, S.I.; Karnik, S.; Southwell, B.R.; Lew, M.J.; Thomas, W.G. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol. Pharmacol. 2002, 61, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Ahn, S.; Barnes, W.G.; Lefkowitz, R.J. Stable interaction between β-arrestin 2 and angiotensin type 1A receptor is required for β-arrestin 2-mediated activation of extracellular signal-regulated kinases 1 and 2. J. Biol. Chem. 2004, 279, 48255–48261. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, L.B.; Parreiras, E.; Silva, L.T.; Bruder-Nascimento, T.; Duarte, D.A.; Simões, S.C.; Costa, R.M.; Rodríguez, D.Y.; Ferreira, P.A.B.; Silva, C.A.A.; et al. Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Szakadáti, G.; Tóth, A.D.; Oláh, I.; Erdélyi, L.S.; Balla, T.; Várnai, P.; Hunyady, L.; Balla, A. Investigation of the fate of type I angiotensin receptor after biased activation. Mol. Pharmacol. 2015, 87, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Tóth, A.D.; Turu, G.; Hunyady, L.; Balla, A. Novel mechanisms of G-protein-coupled receptors functions: AT1 angiotensin receptor acts as a signaling hub and focal point of receptor cross-talk. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassier, E.; Gallay, N.; Bourquard, T.; Claeysen, S.; Bockaert, J.; Crepieux, P.; Poupon, A.; Reiter, E.; Marin, P.; Vandermoere, F. Phosphorylation of β-arrestin2 at Thr383 by MEK underlies β-arrestin-dependent activation of Erk1/2 by GPCRs. Elife 2017, 6, e23777. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.X.; Chen, J.Y.; Wang, Q.T.; Sun, W.Y.; Liu, L.H.; Zhang, L.L.; Wei, W. Expression and function of β-arrestin 2 stimulated by IL-1β in human fibroblast-like synoviocytes and the effect of paeoniflorin. Int. Immunopharmacol. 2012, 12, 701–706. [Google Scholar] [CrossRef]
- Yang, P.; Kuc, R.E.; Brame, A.L.; Dyson, A.; Singer, M.; Glen, R.C.; Cheriyan, J.; Wilkinson, I.B.; Davenport, A.P.; Maguire, J.J. [Pyr1]apelin-13(1-12) is a biologically active ACE2 metabolite of the endogenous cardiovascular peptide [Pyr1]apelin-13. Front. Neurosci. 2017, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Funder, J.W. Mineralocorticoid receptors and hypertension. J. Steroid Biochem. Mol. Biol. 1995, 53, 53–55. [Google Scholar] [CrossRef]
- Rocha, R.; Chander, P.N.; Zuckerman, A.; Stier, C.T. Role of aldosterone in renal vascular injury in stroke-prone hypertensive rats. Hypertension 1999, 33, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, J.; Sathish, S.; Mayilvanan, C.; Balasubramanian, K. Excess aldosterone-induced changes in insulin signaling molecules and glucose oxidation in gastrocnemius muscle of adult male rat. Mol. Cell. Biochem. 2013, 372, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Aukszi, B. Angiotensin receptor blocker drugs and inhibition of adrenal beta-arrestin-1-dependent aldosterone production: Implications for heart failure therapy. World J. Cardiol. 2017, 9, 200. [Google Scholar] [CrossRef]
- Petrofski, J.A.; Koch, W.J. The β-adrenergic receptor kinase in heart failure. J. Mol. Cell. Cardiol. 2003, 35, 1167–1174. [Google Scholar] [CrossRef]
- Lymperopoulos, A. Chapter Two—Arrestins in the Cardiovascular System: An Update. In Progress in Molecular Biology and Translational Science; Teplow, D.B., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 159, pp. 27–57. [Google Scholar] [CrossRef]
- Zhabyeyev, P.; Zhang, H.; Oudit, G.Y. Is β-Arrestin 2 a Magic Bullet for Heart Failure Treatment? Hypertension 2017, 70, 887–889. [Google Scholar] [CrossRef]
- McCrink, K.A.; Maning, J.; Vu, A.; Jafferjee, M.; Marrero, C.; Brill, A.; Bathgate-Siryk, A.; Dabul, S.; Koch, W.J.; Lymperopoulos, A. β-Arrestin2 Improves Post--Myocardial Infarction Heart Failure via Sarco (endo) plasmic Reticulum Ca2+-ATPase--Dependent Positive Inotropy in Cardiomyocytes. Hypertension 2017, 70, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Gros, R.; Chorazyczewski, J.; Meek, M.D.; Benovic, J.L.; Ferguson, S.S.G.; Feldman, R.D. G-Protein-Coupled Receptor Kinase Activity in Hypertension:Increased Vascular and Lymphocyte G-Protein Receptor Kinase-2 Protein Expression. Hypertension 2000, 35, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Komici, K.; Bencivenga, L.; D’amico, M.L.; Gambino, G.; Liccardo, D.; Ferrara, N.; Rengo, G. GRK2 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2018, 22, 75–83. [Google Scholar] [CrossRef]
- Taguchi, K.; Kobayashi, T.; Takenouchi, Y.; Matsumoto, T.; Kamata, K. Angiotensin II causes endothelial dysfunction via the GRK2/Akt/eNOS pathway in aortas from a murine type 2 diabetic model. Pharmacol. Res. 2011, 64, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, R.; Roed, S.N.; Heding, A.; Elling, C.E. Beta-arrestin2 as a competitor for GRK2 interaction with the GLP-1 receptor upon receptor activation. Pharmacology 2011, 88, 174–181. [Google Scholar] [CrossRef]
- Kizaki, T.; Izawa, T.; Sakurai, T.; Haga, S.; Taniguchi, N.; Tajiri, H.; Watanabe, K.; Day, N.K.; Toba, K.; Ohno, H. β2-Adrenergic receptor regulates Toll-like receptor-4-induced nuclear factor-κB activation through β-arrestin 2. Immunology 2008, 124, 348–356. [Google Scholar] [CrossRef]
- Tan, S.; Li, L.; Chen, T.; Chen, X.; Tao, L.; Lin, X.; Tao, J.; Huang, X.; Jiang, J.; Liu, H.; et al. β-Arrestin-1 protects against endoplasmic reticulum stress/p53-upregulated modulator of apoptosis-mediated apoptosis via repressing p-p65/inducible nitric oxide synthase in portal hypertensive gastropathy. Free Radic. Biol. Med. 2015, 87, 69–83. [Google Scholar] [CrossRef]
- Boerrigter, G.; Lark, M.W.; Whalen, E.J.; Soergel, D.G.; Violin, J.D.; Burnett, J.C. Cardiorenal actions of TRV120027, a novel ß-arrestin-biased ligand at the angiotensin II type i receptor, in healthy and heart failure canines: A novel therapeutic strategy for acute heart failure. Circ. Heart Fail. 2011, 4, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Soergel, D.G.; Subach, R.A.; Cowan, C.L.; Violin, J.D.; Lark, M.W. First clinical experience with TRV027: Pharmacokinetics and pharmacodynamics in healthy volunteers. J. Clin. Pharmacol. 2013, 53, 892–899. [Google Scholar] [CrossRef]
- Soergel, D.; Subach, R.A.; James, I.E.; Cowan, C.L.; Gowen, M.; Lark, M. Trvo27, a Beta-Arrestin Biased Ligand At the Angiotensin 2 Type 1 Receptor, Produces Rapid, Reversible Changes in Hemodynamics in Patients With Stable Systolic Heart Failure. J. Am. Coll. Cardiol. 2013, 61, E683. [Google Scholar] [CrossRef] [Green Version]
- Pang, P.S.; Butler, J.; Collins, S.P.; Cotter, G.; Davison, B.A.; Ezekowitz, J.A.; Filippatos, G.; Levy, P.D.; Metra, M.; Ponikowski, P.; et al. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: A randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). Eur. Heart J. 2017, 38, 2364–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lymperopoulos, A.; Wertz, S.L.; Pollard, C.M.; Desimine, V.L.; Maning, J.; McCrink, K.A. Not all arrestins are created equal: Therapeutic implications of the functional diversity of the β arrestins in the heart. World J. Cardiol. 2019, 11, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; Butler, J.; Collins, S.P.; Cotter, G.; Davison, B.A.; Ezekowitz, J.A.; Filippatos, G.; Levy, P.D.; Metra, M.; Ponikowski, P.; et al. heart failure therapeutics on thebasisofabiased ligand of theangiotensin-2 type 1 receptor. Rationale and design of the blast-ahf study (biased ligand of the angiotensin receptor study in acute heart failure). JACC Heart Fail. 2015, 3, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, G.; Wang, P.; Velazquez, H.; Yao, X.; Li, Y.; Wu, Y.; Peixoto, A.; Crowley, S.; Desir, G. V Renalase is a novel, soluble monoamine oxidase that regulates cardiac function and blood pressure. J. Clin. Investig. 2005, 115, 1275–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desir, G.V.; Tang, L.; Wang, P.; Li, G.; Sampaio-Maia, B.; Quelhas-Santos, J.; Pestana, M.; Velazquez, H. Renalase Lowers Ambulatory Blood Pressure by Metabolizing Circulating Adrenaline. J. Am. Heart Assoc. 2012, 1. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Xu, J.; Velazquez, H.; Wang, P.; Li, G.; Liu, D.; Sampaio-Maia, B.; Quelhas-Santos, J.; Russell, K.; Russell, R.; et al. Renalase deficiency aggravates ischemic myocardial damage. Kidney Int. 2011, 79, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Xu, J.; Wang, P.; Velazquez, H.; Li, Y.; Wu, Y.; Desir, G. V Catecholamines regulate the activity, secretion, and synthesis of renalase. Circulation 2008, 117, 1277. [Google Scholar] [CrossRef] [PubMed]
- Bathgate-Siryk, A.; Dabul, S.; Pandya, K.; Walklett, K.; Rengo, G.; Cannavo, A.; De Lucia, C.; Liccardo, D.; Gao, E.; Leosco, D.; et al. Negative impact of β-arrestin-1 on post-myocardial infarction heart failure via cardiac and adrenal-dependent neurohormonal mechanisms. Hypertension 2014, 64, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Wirth, A. Rho kinase and hypertension. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Griendling, K.K. Reactive oxygen species in hypertension; An update. Am. J. Hypertens. 2004, 17, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Schiffrin, E.L. Reactive oxygen species in vascular biology: Implications in hypertension. Histochem. Cell Biol. 2004, 122, 339–352. [Google Scholar] [CrossRef]
- De Godoy, M.A.F.; Patel, C.A.; Waldman, S.A.; Katsuki, M.; Regan, R.F.; Rattan, S. H-ras inhibits RhoA/ROCK leading to a decrease in the basal tone in the internal anal sphincter. Gastroenterology 2007, 132, 1401–1409. [Google Scholar] [CrossRef]
- Hennenberg, M.; Trebicka, J.; Stark, C.; Kohistani, A.Z.; Heller, J.; Sauerbruch, T. Sorafenib targets dysregulated Rho kinase expression and portal hypertension in rats with secondary biliary cirrhosis. Br. J. Pharmacol. 2009, 157, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Hennenberg, M.; Trebicka, J.; Sauerbruch, T.; Heller, J. Mechanisms of extrahepatic vasodilation in portal hypertension. Gut 2008, 57, 1300–1314. [Google Scholar] [CrossRef] [PubMed]
- Hennenberg, M.; Trebicka, J.; Biecker, E.; Schepke, M.; Sauerbruch, T.; Heller, J. Vascular dysfunction in human and rat cirrhosis: Role of receptor-desensitizing and calcium-sensitizing proteins. Hepatology 2007, 45, 495–506. [Google Scholar] [CrossRef]
- Bubici, C.; Papa, S.; Pham, C.G.; Zazzeroni, F.; Franzoso, G. The NF-kB-mediated control of ROS and JNK signaling. Histol. Histopathol. 2006, 21, 69–80. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakib, A.; Eva, T.A.; Sami, S.A.; Mitra, S.; Nafiz, I.H.; Das, A.; Tareq, A.M.; Nainu, F.; Dhama, K.; Emran, T.B.; et al. Beta-Arrestins in the Treatment of Heart Failure Related to Hypertension: A Comprehensive Review. Pharmaceutics 2021, 13, 838. https://doi.org/10.3390/pharmaceutics13060838
Rakib A, Eva TA, Sami SA, Mitra S, Nafiz IH, Das A, Tareq AM, Nainu F, Dhama K, Emran TB, et al. Beta-Arrestins in the Treatment of Heart Failure Related to Hypertension: A Comprehensive Review. Pharmaceutics. 2021; 13(6):838. https://doi.org/10.3390/pharmaceutics13060838
Chicago/Turabian StyleRakib, Ahmed, Taslima Akter Eva, Saad Ahmed Sami, Saikat Mitra, Iqbal Hossain Nafiz, Ayan Das, Abu Montakim Tareq, Firzan Nainu, Kuldeep Dhama, Talha Bin Emran, and et al. 2021. "Beta-Arrestins in the Treatment of Heart Failure Related to Hypertension: A Comprehensive Review" Pharmaceutics 13, no. 6: 838. https://doi.org/10.3390/pharmaceutics13060838