
Polymer Pro-Drug Nanoparticles for Sustained Release of Cytotoxic Drugs Evaluated in Patient-Derived Glioblastoma Cell Lines and In Situ Gelling Formulations

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
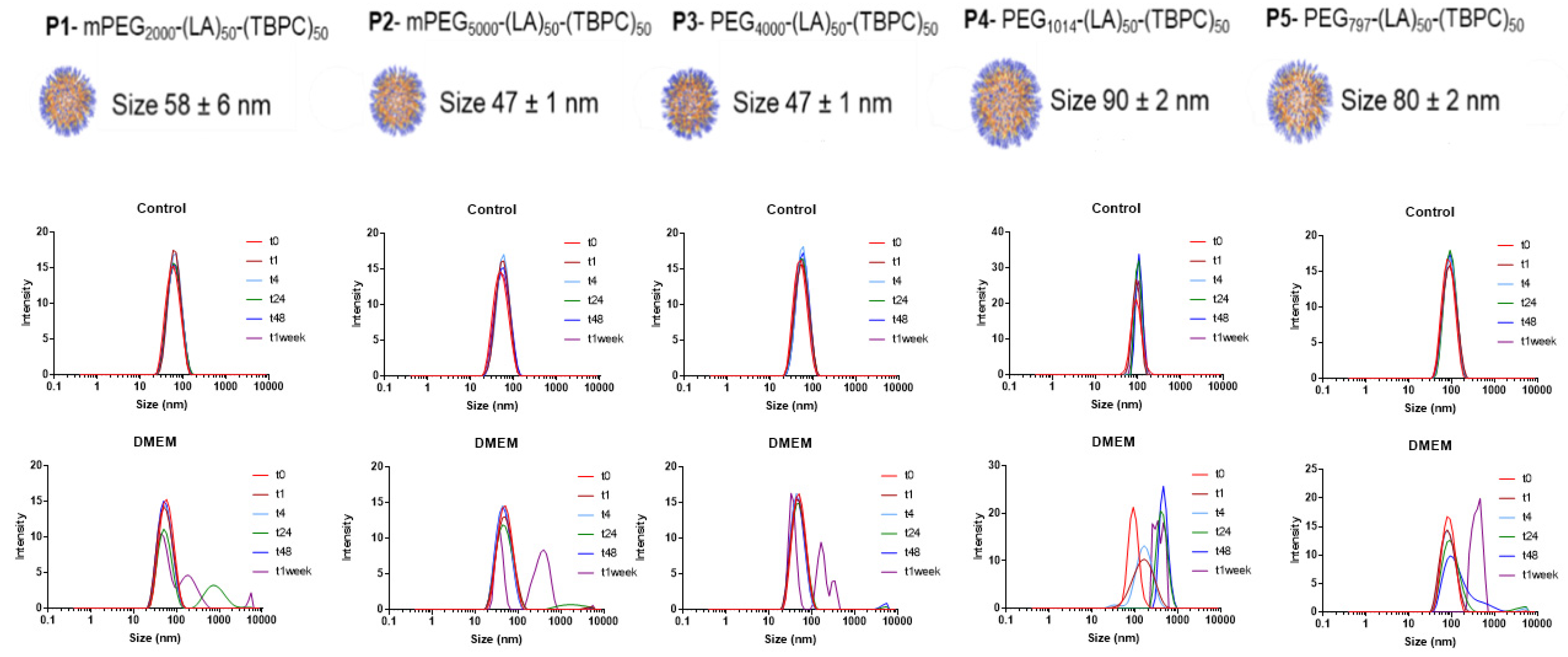
2.1. Nanoparticle Stability Studies
2.2. Cell Culture
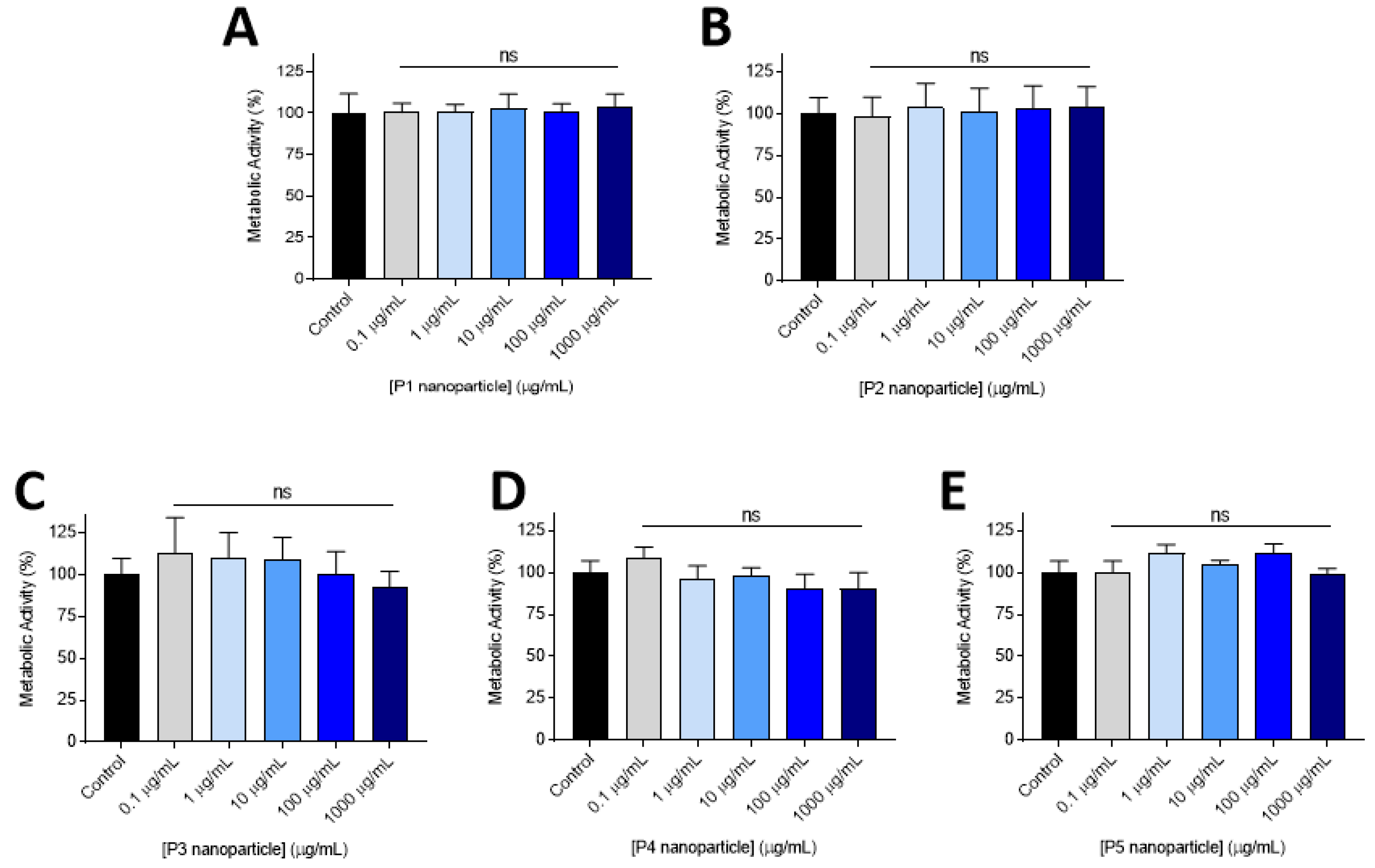
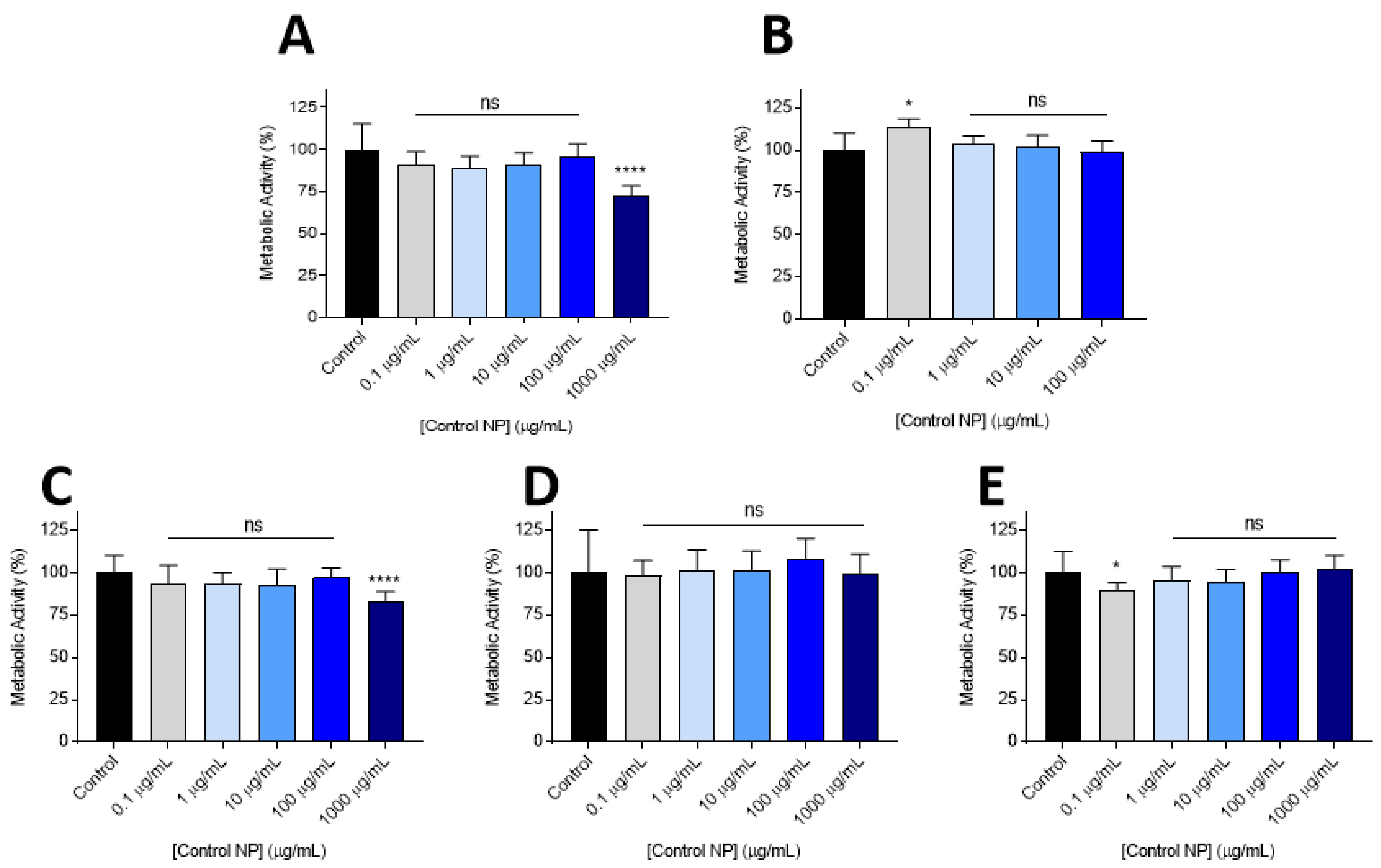
2.3. Cytotoxicity Experiments
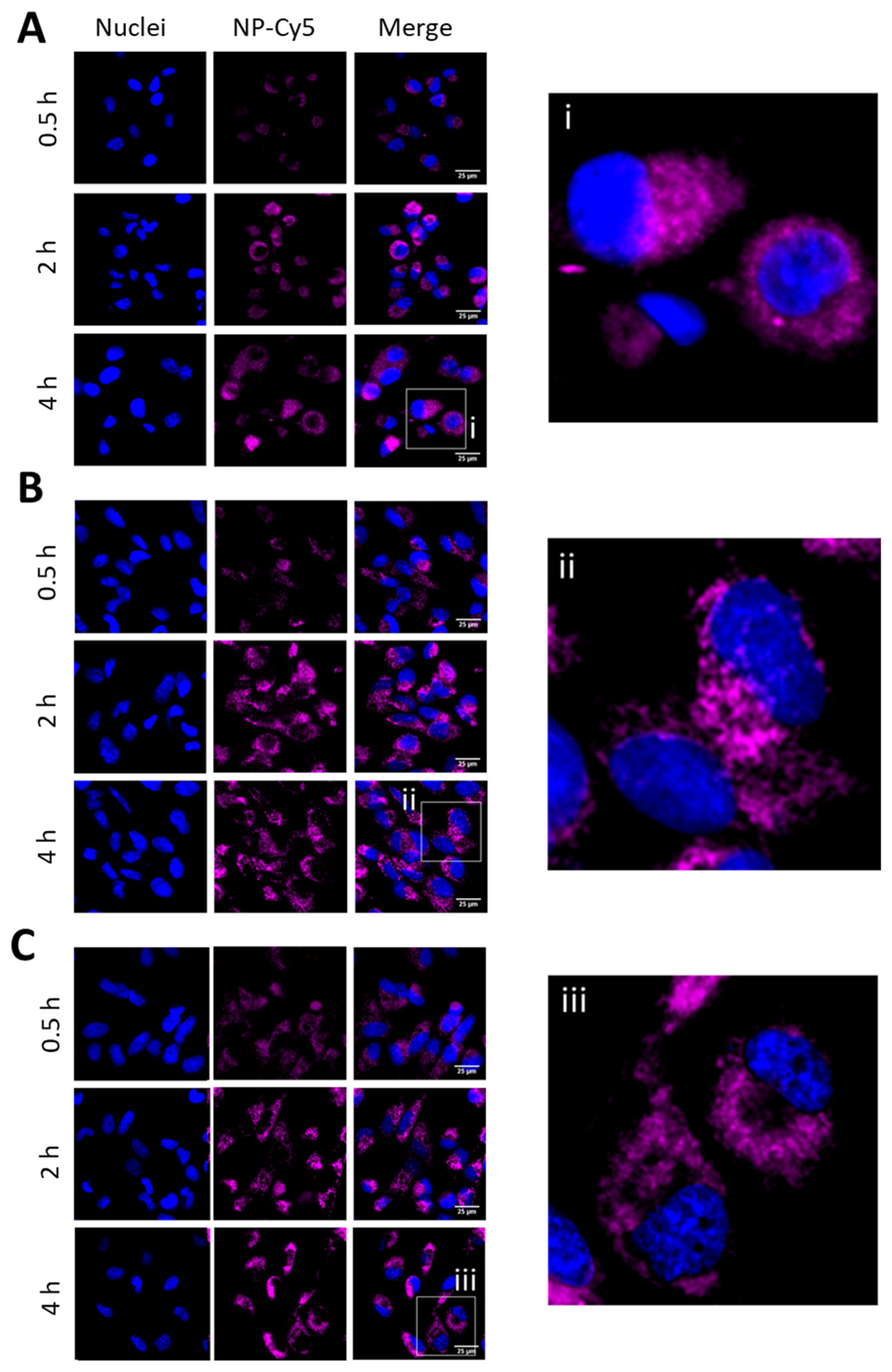
2.4. Fluorescent Live-Cell Microscopy
2.5. Fluorometric-Based Uptake Assessment
2.6. Preparation of PLGA/PEG Matrix
2.7. Release Studies
2.7.1. DOX Release from DOX-NPs
2.7.2. DOX-NP Release from PLGA/PEG Paste
2.8. Statistical Analysis
3. Results
3.1. Nanoparticle Stability
3.2. Cytocompatability
3.3. Lead Nanoparticle Cytocombability Screening
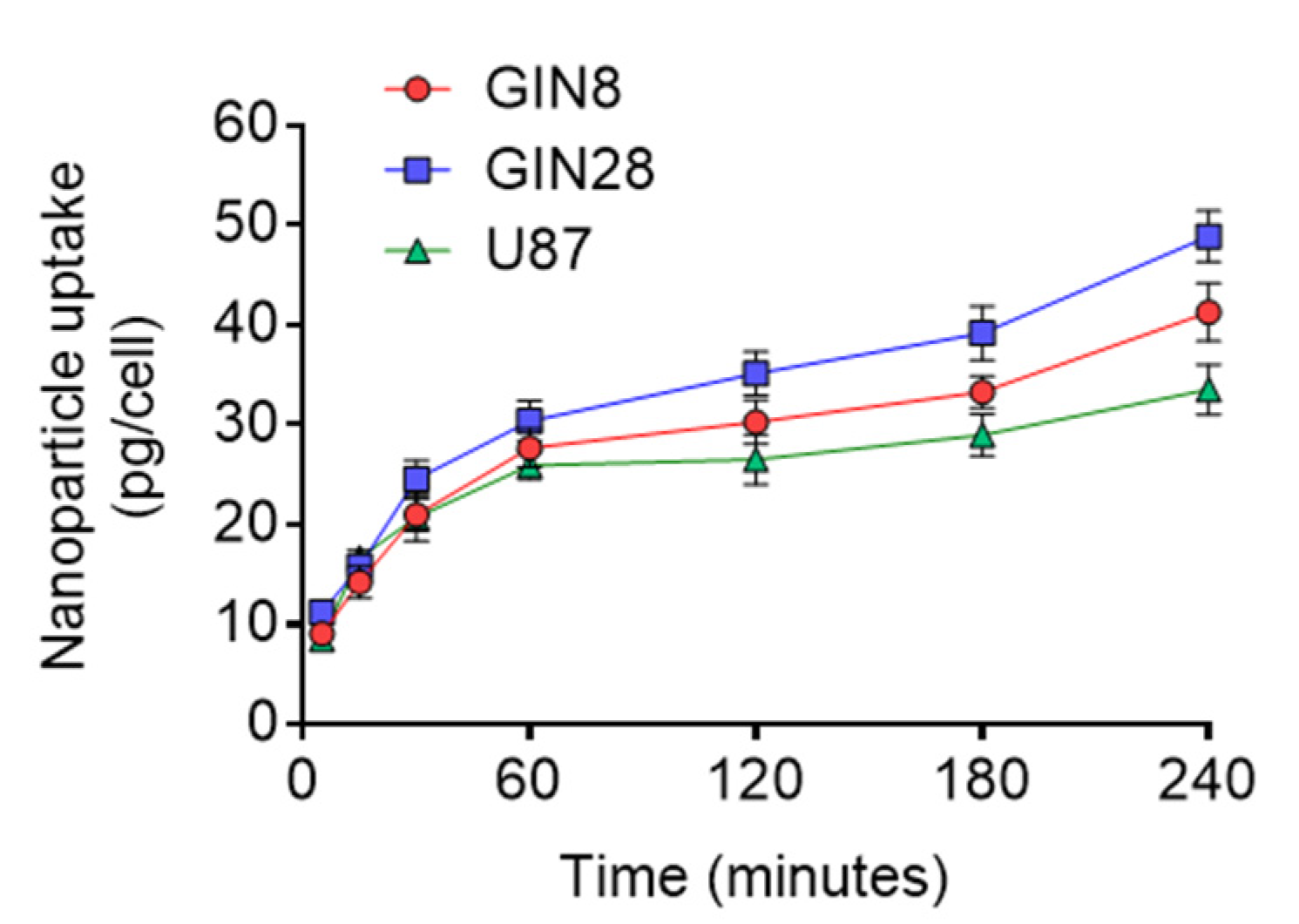
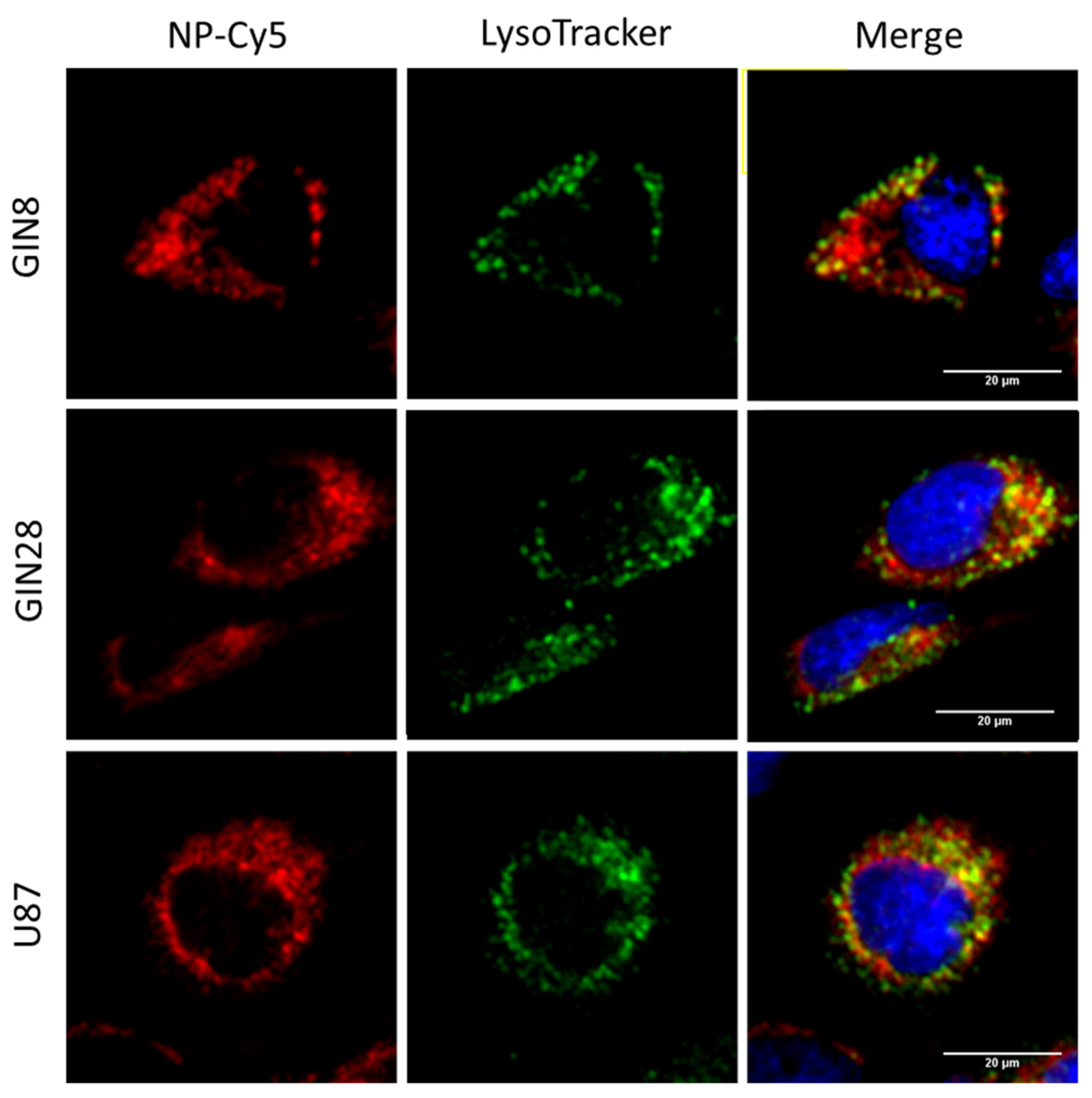
3.4. Assays to Evaluate Cellular Internalisation of NPs
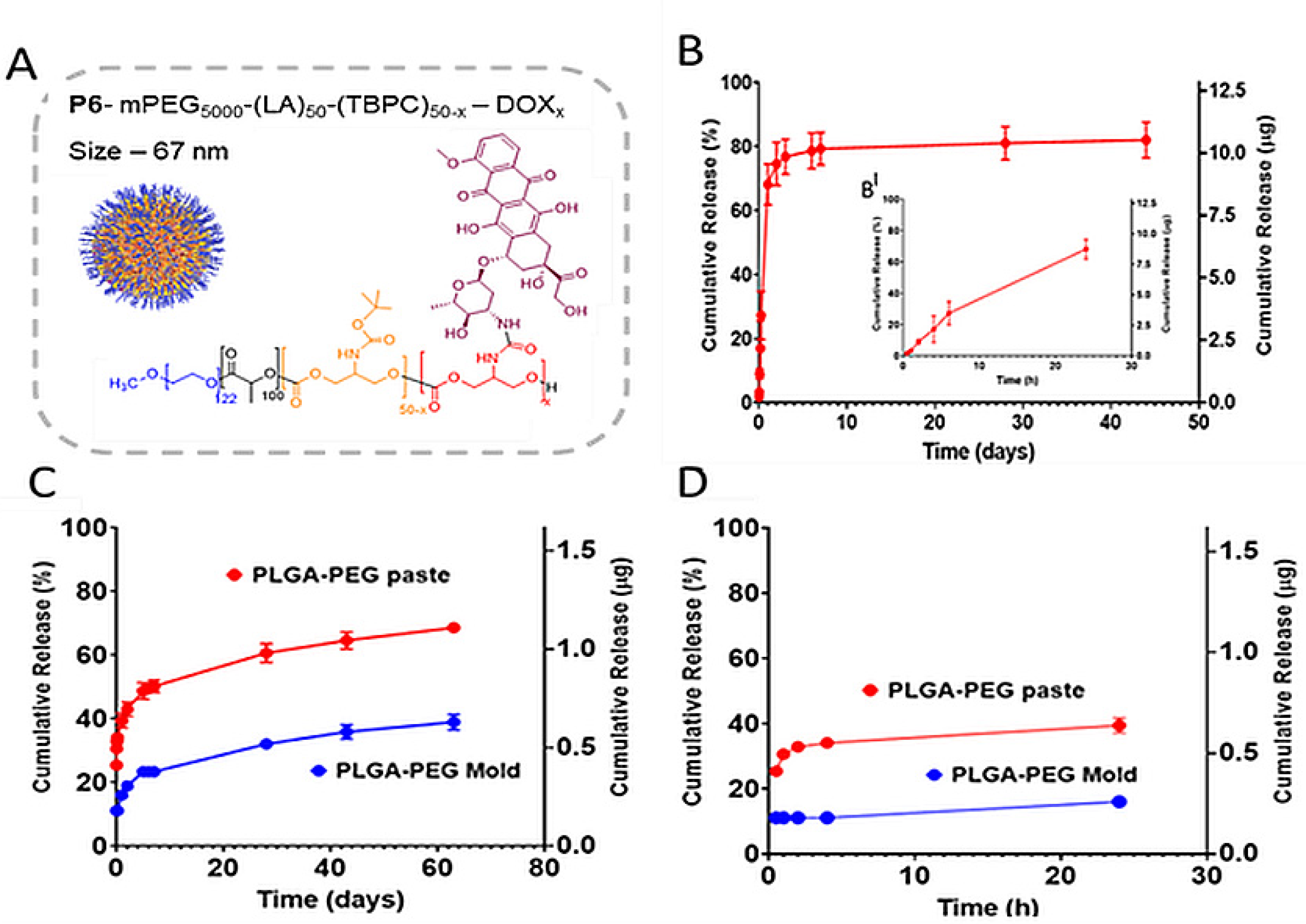
3.5. Drug Release Studies
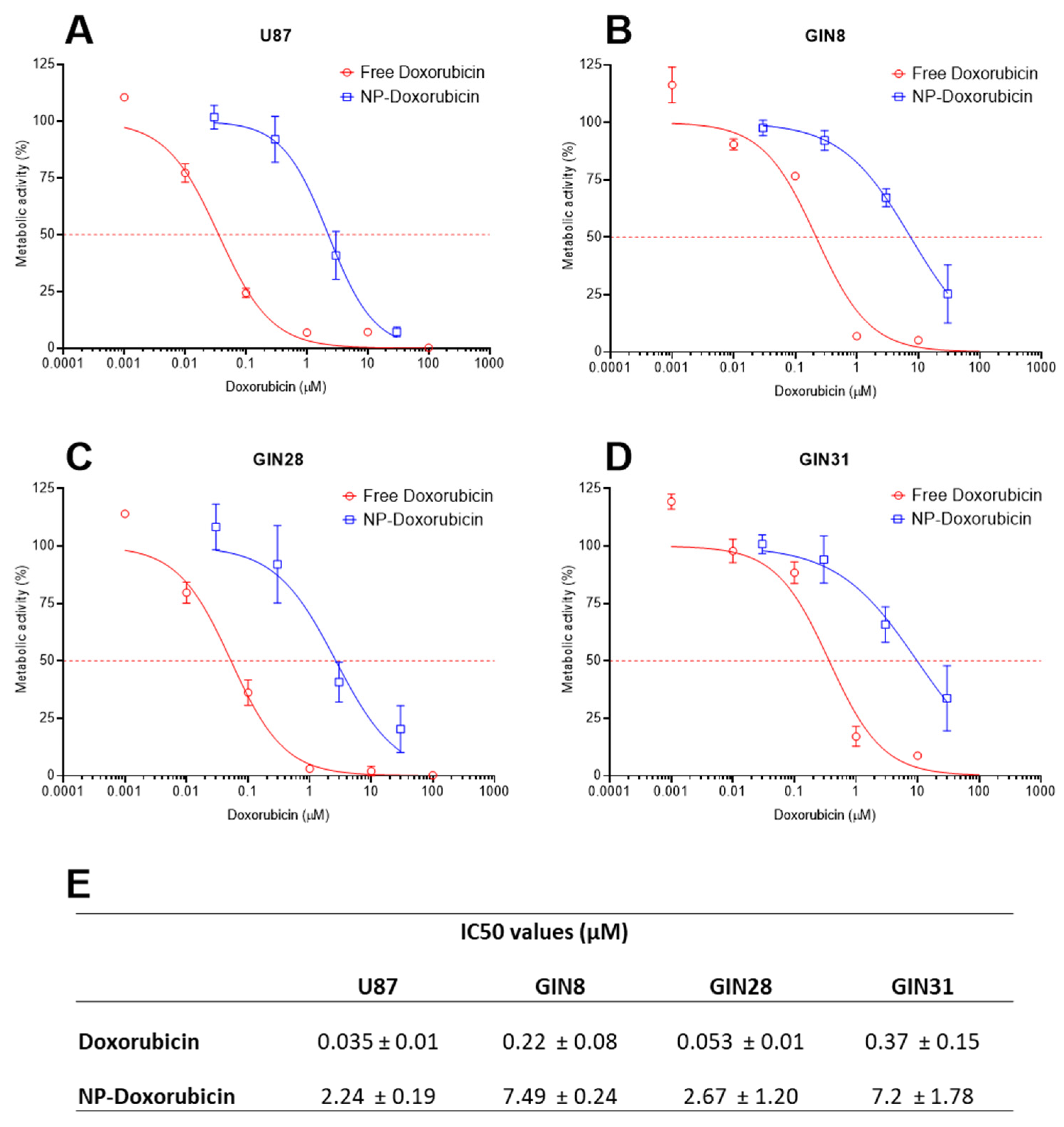
3.6. Effects of DOX-Loaded Nanoparticles in Glioblastoma Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Glas, M.; Rath, B.H.; Simon, M.; Reinartz, R.; Schramme, A.; Trageser, D.; Eisenreich, R.; Leinhaas, A.; Keller, M.; Schildhaus, H.-U.; et al. Residual tumor cells are unique cellular targets in glioblastoma. Ann. Neurol. 2010, 68, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.J.; Rahman, C.V.; Clarke, P.A.; Ritchie, A.A.; Gould, T.W.; Ward, J.H.; Shakesheff, K.M.; Grundy, R.G.; Rahman, R. Surgical delivery of drug releasing poly(lactic-co-glycolic acid)/poly(ethylene glycol) paste with in vivo effects against glio-blastoma. Ann. Royal Coll. Surg. Engl. 2014, 96, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Rahman, C.V.; Smith, S.J.; Morgan, P.S.; Langmack, K.A.; Clarke, P.A.; Ritchie, A.A.; MacArthur, D.C.; Rose, F.R.; Shakesheff, K.M.; Grundy, R.G.; et al. Adjuvant Chemotherapy for Brain Tumors Delivered via a Novel Intra-Cavity Moldable Polymer Matrix. PLoS ONE 2013, 8, e77435. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.J.; Tyler, B.M.; Gould, T.; Veal, G.J.; Gorelick, N.; Rowlinson, J.; Serra, R.; Ritchie, A.; Berry, P.; Otto, A.; et al. Overall Survival in Malignant Glioma Is Significantly Prolonged by Neurosurgical Delivery of Etoposide and Temozolomide from a Thermo-Responsive Biodegradable Paste. Clin. Cancer Res. 2019, 25, 5094–5106. [Google Scholar] [CrossRef] [Green Version]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [Green Version]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Almers, W. Exocytosis. Annu. Rev. Physiol. 1990, 52, 607–624. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, R.; Meldolesi, J. News about non-secretory exocytosis: Mechanisms, properties, and functions. J. Mol. Cell Biol. 2019, 11, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Oh, N. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 2014, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Liu, Y.; Chen, Y.; Zhao, W.; Zhai, Q.; Rathod, S.; Huang, Y.; Tang, S.; Kwon, Y.T.; Fernandez, C.; et al. Doxorubicin delivered by a redox-responsive dasatinib-containing polymeric prodrug carrier for combination therapy. J. Control. Release 2017, 258, 43–55. [Google Scholar] [CrossRef]
- Saravanakumar, G.; Kim, J.; Kim, W.J. Reactive-Oxygen-Species-Responsive Drug Delivery Systems: Promises and Challenges. Adv. Sci. 2017, 4, 1600124. [Google Scholar] [CrossRef]
- Taresco, V.; Abelha, T.F.; Cavanagh, R.J.; Vasey, C.E.; Anane-Adjei, A.B.; Pearce, A.K.; Monteiro, P.F.; Spriggs, K.A.; Clarke, P.; Ritchie, A.; et al. Functionalized Block Co-Polymer Pro-Drug Nanoparticles with Anti-Cancer Efficacy in 3D Spheroids and in an Orthotopic Triple Negative Breast Cancer Model. Adv. Ther. 2020. [Google Scholar] [CrossRef]
- Vasey, C.E.; Pearce, A.K.; Sodano, F.; Cavanagh, R.; Abelha, T.F.; Crucitti, V.C.; Anane-Adjei, A.B.; Ashford, M.; Gellert, P.; Taresco, V.; et al. Amphiphilic tri- and tetra-block co-polymers combining versatile functionality with facile assembly into cytocompatible nanoparticles. Biomater. Sci. 2019, 7, 3832–3845. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, S.; Taniguchi, K.; Yokomizo, A.; Kuwano, T.; Ono, M.; Mori, T.; Hori, S.; Kohno, K.; Kuwano, M. Possible in-volvement of multidrug-resistance-associated protein (MRP) gene expression in spontaneous drug resistance to vincristine, etoposide and adriamycin in human glioma cells. Int. J. Cancer 1994, 58, 860–864. [Google Scholar] [CrossRef]
- Pang, Z.; Feng, L.; Hua, R.; Chen, J.; Gao, H.; Pan, S.; Jiang, X.; Zhang, P. Lactoferrin-Conjugated Biodegradable Polymersome Holding Doxorubicin and Tetrandrine for Chemotherapy of Glioma Rats. Mol. Pharm. 2010, 7, 1995–2005. [Google Scholar] [CrossRef]
- Beier, C.P.; Schmid, C.; Gorlia, T.; Kleinletzenberger, C.; Beier, D.; Grauer, O.; Steinbrecher, A.; Hirschmann, B.; Brawanski, A.; Dietmaier, C.; et al. RNOP-09: Pegylated liposomal doxorubicine and prolonged temozolomide in addition to radiotherapy in newly diagnosed glioblastoma—A phase II study. BMC Cancer 2009, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Maksimenko, O.; Malinovskaya, J.; Shipulo, E.; Osipova, N.; Razzhivina, V.; Arantseva, D.; Yarovaya, O.; Mostovaya, U.; Khalansky, A.; Fedoseeva, V.; et al. Doxorubicin-loaded PLGA nanoparticles for the chemotherapy of glioblastoma: Towards the pharmaceutical development. Int. J. Pharm. 2019, 572, 118733. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Yathindranath, V.; Thliveris, J.A.; Kopec, B.M.; Siahaan, T.J.; Miller, D.W. Doxorubicin-loaded iron oxide nanoparticles for glioblastoma therapy: A combinational approach for enhanced delivery of nanoparticles. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Miao, T.; Su, M.; Khan, N.U.; Ju, X.; Chen, H.; Liu, F.; Han, L. PSMA-targeted nanoparticles for specific penetration of blood-brain tumor barrier and combined therapy of brain metastases. J. Control. Release 2020. [Google Scholar] [CrossRef]
- Xu, H.; Han, Y.; Zhao, G.; Zhang, L.; Zhao, Z.; Wang, Z.; Zhao, L.; Hua, L.; Naveena, K.; Lu, J.; et al. Hypox-ia-Responsive Lipid-Polymer Nanoparticle-Combined Imaging-Guided Surgery and Multitherapy Strategies for Glioma. ACS Appl. Mater. Interfaces 2020, 12, 52319–52328. [Google Scholar] [CrossRef]
- Lu, H.; Utama, R.H.; Kitiyotsawat, U.; Babiuch, K.; Jiang, Y.; Barner-Kowollik, C. Enhanced transcellular penetration and drug delivery by crosslinked polymeric micelles into pancreatic multicellular tumor spheroids. Biomater. Sci. 2015, 3, 1085–1095. [Google Scholar] [CrossRef]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef]
- Petrecca, K.; Guiot, M.-C.; Panet-Raymond, V.; Souhami, L. Failure pattern following complete resection plus radiotherapy and temozolomide is at the resection margin in patients with glioblastoma. J. Neuro-Oncol. 2013, 111, 19–23. [Google Scholar] [CrossRef]
- Smith, S.; Diksin, M.; Chhaya, S.; Sairam, S.; Estevez-Cebrero, M.A.; Rahman, R. The Invasive Region of Glioblastoma Defined by 5ALA Guided Surgery Has an Altered Cancer Stem Cell Marker Profile Compared to Central Tumour. Int. J. Mol. Sci. 2017, 18, 2452. [Google Scholar] [CrossRef] [Green Version]
- Rahman, C.V.; Kuhn, G.; White, L.J.; Kirby, G.T.S.; Varghese, O.P.; McLaren, J.S.; Cox, H.C.; Rose, F.R.A.J.; Müller, R.; Hilborn, J.; et al. PLGA/PEG-hydrogel composite scaffolds with controllable mechanical properties. J. Biomed. Mater. Res. Part B: Appl. Biomater. 2013, 101, 648–655. [Google Scholar] [CrossRef]
- Dhillon, A.; Schneider, P.; Kuhn, G.; Reinwald, Y.; White, L.J.; Levchuk, A.; Rose, F.R.A.J.; Müller, R.; Shakesheff, K.M.; Rahman, C.V. Analysis of sintered polymer scaffolds using concomitant synchrotron computed tomography and in situ mechanical testing. J. Mater. Sci. Mater. Med. 2011, 22, 2599–2605. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.; Minut, R.I.; McCrorie, P.; Vasey, C.; Larder, R.R.; Krumins, E.; Marlow, M.; Rahman, R.; Alexander, C.; Taresco, V.; et al. Role of self-assembly conditions and amphiphilic balance on nanoparticle formation of PEG-PDLLA copolymers in aqueous environments. J. Polym. Sci. Part A: Polym. Chem. 2019, 57, 1801–1810. [Google Scholar] [CrossRef] [Green Version]
- Nance, E.A.; Woodworth, G.F.; Sailor, K.A.; Shih, T.-Y.; Xu, Q.; Swaminathan, G.; Xiang, D.; Eberhart, C.; Hanes, J. A Dense Poly(Ethylene Glycol) Coating Improves Penetration of Large Polymeric Nanoparticles Within Brain Tissue. Sci. Transl. Med. 2012, 4, 149ra119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nance, E.; Zhang, C.; Shih, T.-Y.; Xu, Q.; Schuster, B.S.; Hanes, J. Brain-Penetrating Nanoparticles Improve Paclitaxel Efficacy in Malignant Glioma Following Local Administration. ACS Nano 2014, 8, 10655–10664. [Google Scholar] [CrossRef]
- Sasso, L.; Purdie, L.; Grabowska, A.M.; Jones, A.T.; Alexander, C. Time and cell-dependent effects of endocytosis inhibitors on the internalization of biomolecule markers and nanomaterials. J. Interdiscip. Nanomed. 2018, 3, 67–81. [Google Scholar] [CrossRef]
- Herd, H.; Daum, N.; Jones, A.T.; Huwer, H.; Ghandehari, H.; Lehr, C.-M. Nanoparticle Geometry and Surface Orientation Influence Mode of Cellular Uptake. ACS Nano 2013, 7, 1961–1973. [Google Scholar] [CrossRef] [Green Version]
- Sodano, F.; Cavanagh, R.J.; Pearce, A.K.; Lazzarato, L.; Rolando, B.; Fraix, A.; Abelha, T.F.; Vasey, C.E.; Alexander, C.; Taresco, V.; et al. Enhancing doxorubicin anticancer activity with a novel polymeric platform photoreleasing nitric oxide. Biomater. Sci. 2020, 8, 1329–1344. [Google Scholar] [CrossRef]
- Hutchby, M.; Houlden, C.E.; Gair Ford, J.; Tyler, S.N.G.; Gagné, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Hindered ureas as masked isocyanates: Facile carbamoylation of nucleophiles under neutral conditions. Angew. Chem. Int. Ed. 2009, 48, 8721–8724. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zhang, Y.; Cheng, J. Dynamic urea bond for the design of reversible and self-healing polymers. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gangarapu, S.; Escorihuela, J.; Fei, G.; Zuilhof, H.; Xia, H. Dynamic covalent urea bonds and their potential for development of self-healing polymer materials. J. Mater. Chem. A 2019, 7, 15933–15943. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
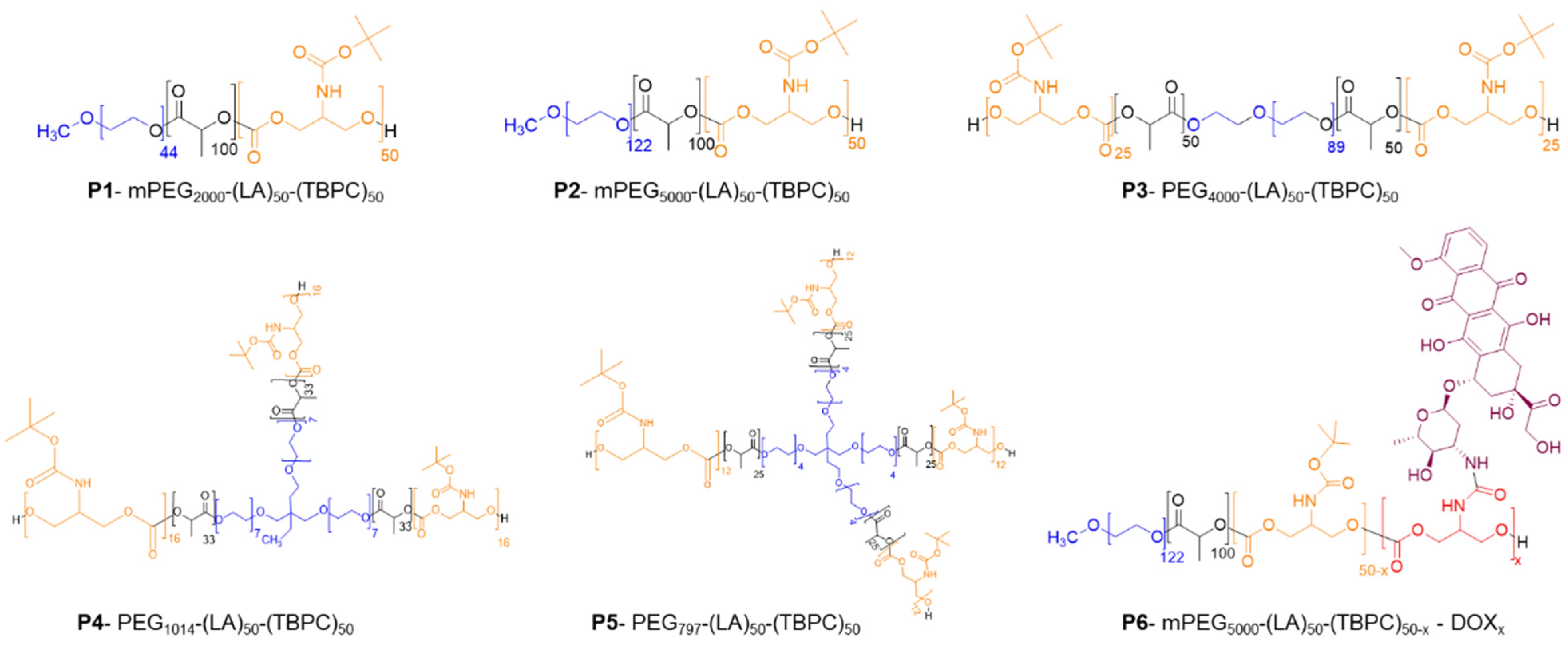
| Polymer | PEG Length (Da) | Final Polymer Arms | Mn (NMR) | Mn 1 (GPC) | Ð |
|---|---|---|---|---|---|
| P1. mPEG2000-(LA)50-(TBPC)50 | 2000 | 1 | 20,500 | 20,400 | 1.3 |
| P2. mPEG5000-(LA)50-(TBPC)50 | 5000 | 1 | 23,000 | 24,000 | 1.2 |
| P3. PEG4000-(LA)50-(TBPC)50 | 4000 | 2 | 21,000 | 20,000 | 1.1 |
| P4. PEG1014-(LA)50-(TBPC)50 | 1014 | 3 | 19,000 | 16,400 | 1.2 |
| P5. PEG797-(LA)50-(TBPC)50 | 797 | 4 | 21,000 | 16,300 | 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasey, C.E.; Cavanagh, R.J.; Taresco, V.; Moloney, C.; Smith, S.; Rahman, R.; Alexander, C. Polymer Pro-Drug Nanoparticles for Sustained Release of Cytotoxic Drugs Evaluated in Patient-Derived Glioblastoma Cell Lines and In Situ Gelling Formulations. Pharmaceutics 2021, 13, 208. https://doi.org/10.3390/pharmaceutics13020208
Vasey CE, Cavanagh RJ, Taresco V, Moloney C, Smith S, Rahman R, Alexander C. Polymer Pro-Drug Nanoparticles for Sustained Release of Cytotoxic Drugs Evaluated in Patient-Derived Glioblastoma Cell Lines and In Situ Gelling Formulations. Pharmaceutics. 2021; 13(2):208. https://doi.org/10.3390/pharmaceutics13020208
Chicago/Turabian StyleVasey, Catherine E., Robert J. Cavanagh, Vincenzo Taresco, Cara Moloney, Stuart Smith, Ruman Rahman, and Cameron Alexander. 2021. "Polymer Pro-Drug Nanoparticles for Sustained Release of Cytotoxic Drugs Evaluated in Patient-Derived Glioblastoma Cell Lines and In Situ Gelling Formulations" Pharmaceutics 13, no. 2: 208. https://doi.org/10.3390/pharmaceutics13020208