Preparation and Evaluation of Amino Acid Conjugates of Celecoxib as Prodrugs to Improve the Pharmacokinetic and Therapeutic Properties of Celecoxib


Abstract
:1. Introduction
2. Materials and Methods
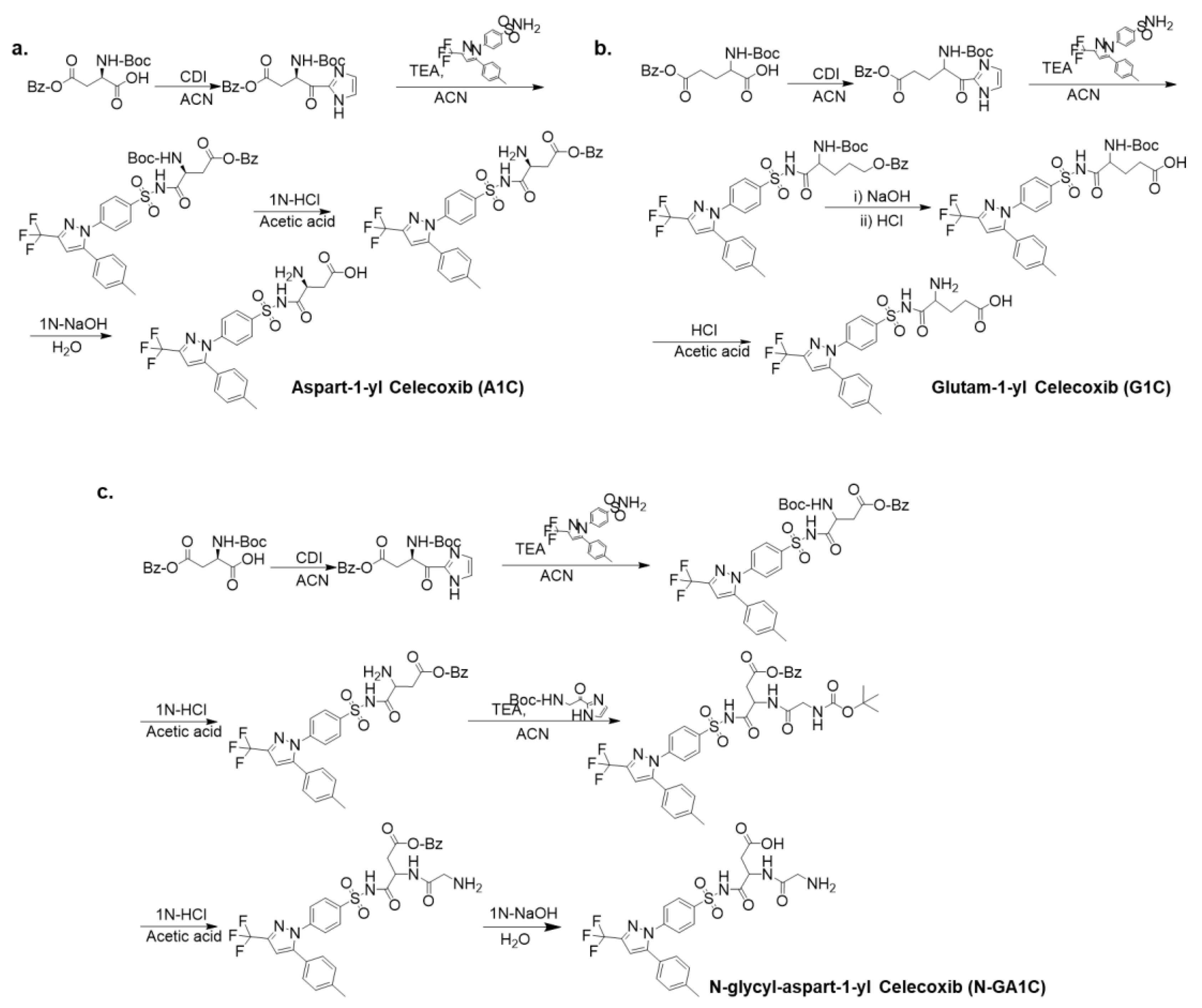
2.1. Synthesis of Aspart-1-yl Celecoxib (A1C) and Glutam-1-yl Celecoxib (G1C)
2.2. Synthesis of N-Glycyl-Aspart-1-yl Celecoxib (N-GA1C)
2.3. HPLC Analysis
2.4. Apparent Distribution Coefficient (Logd6.8) Measurement
2.5. Chemical Stability Study
2.6. Animals
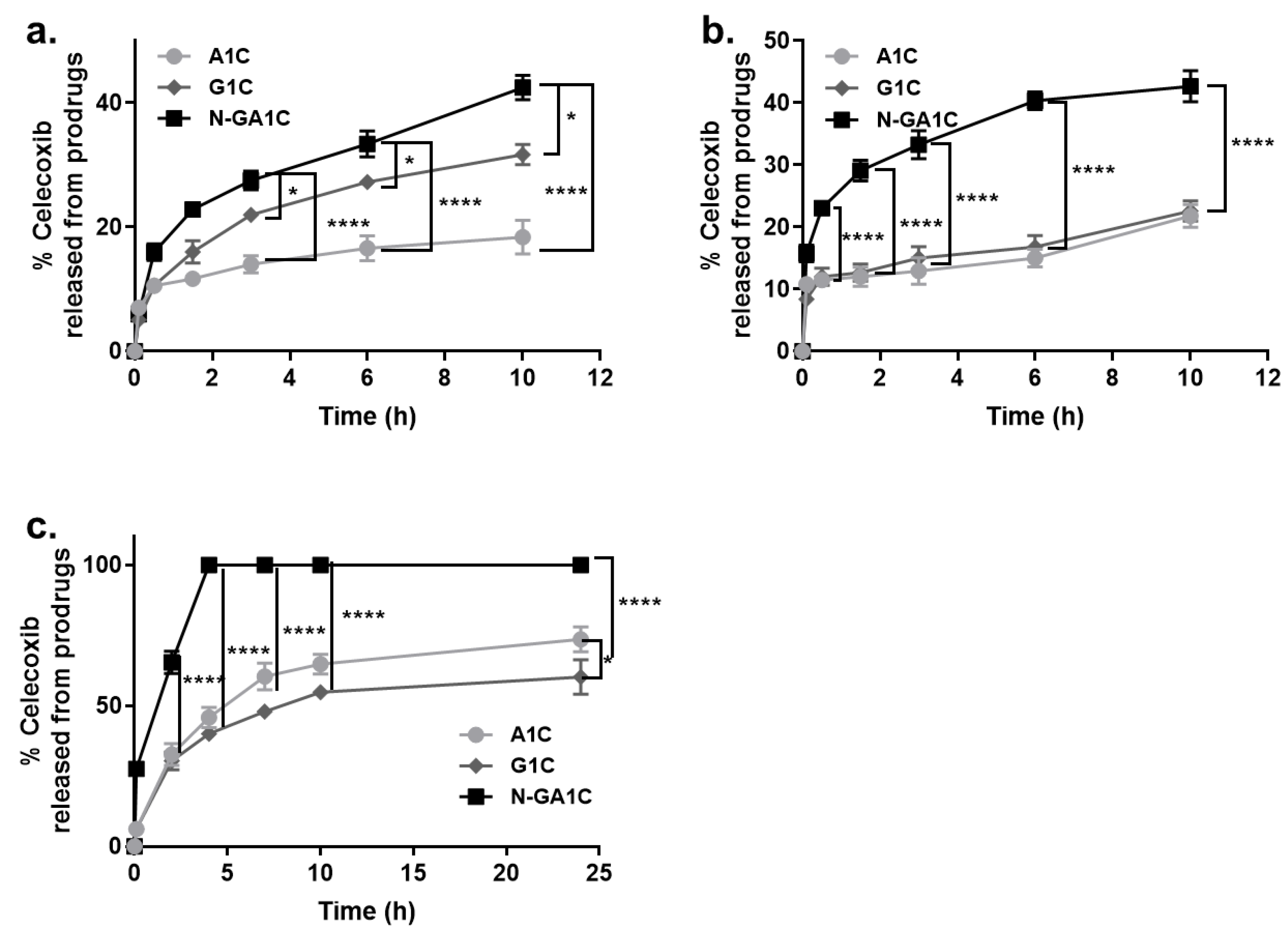
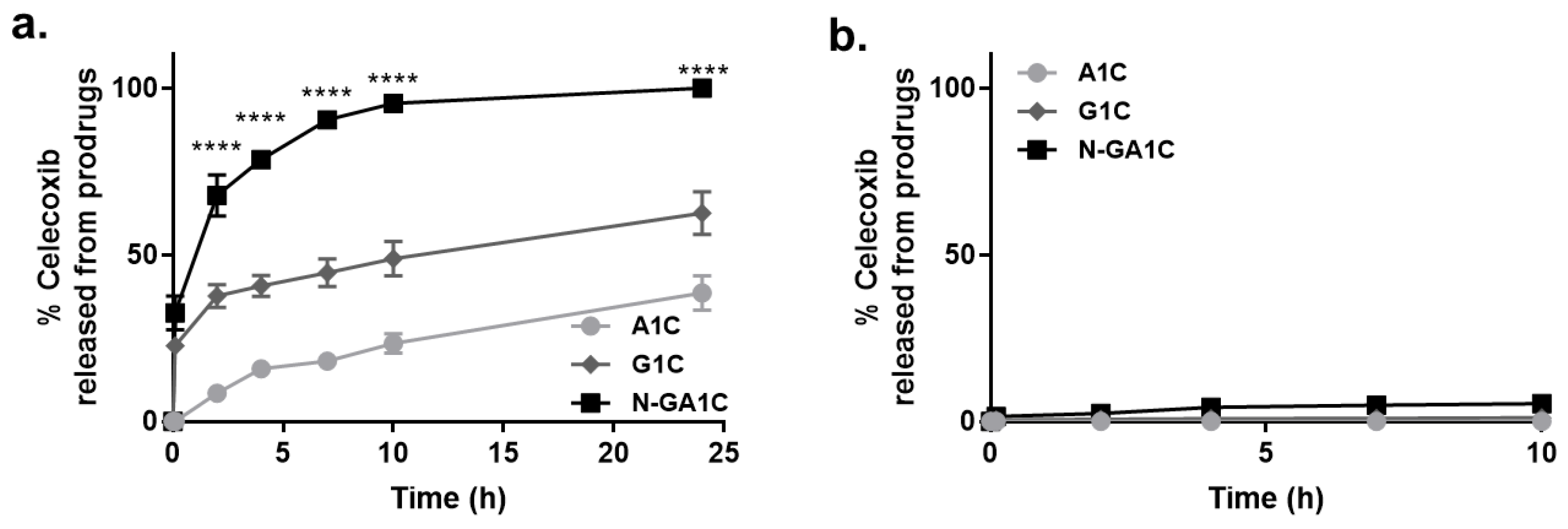
2.7. Drug Release Study
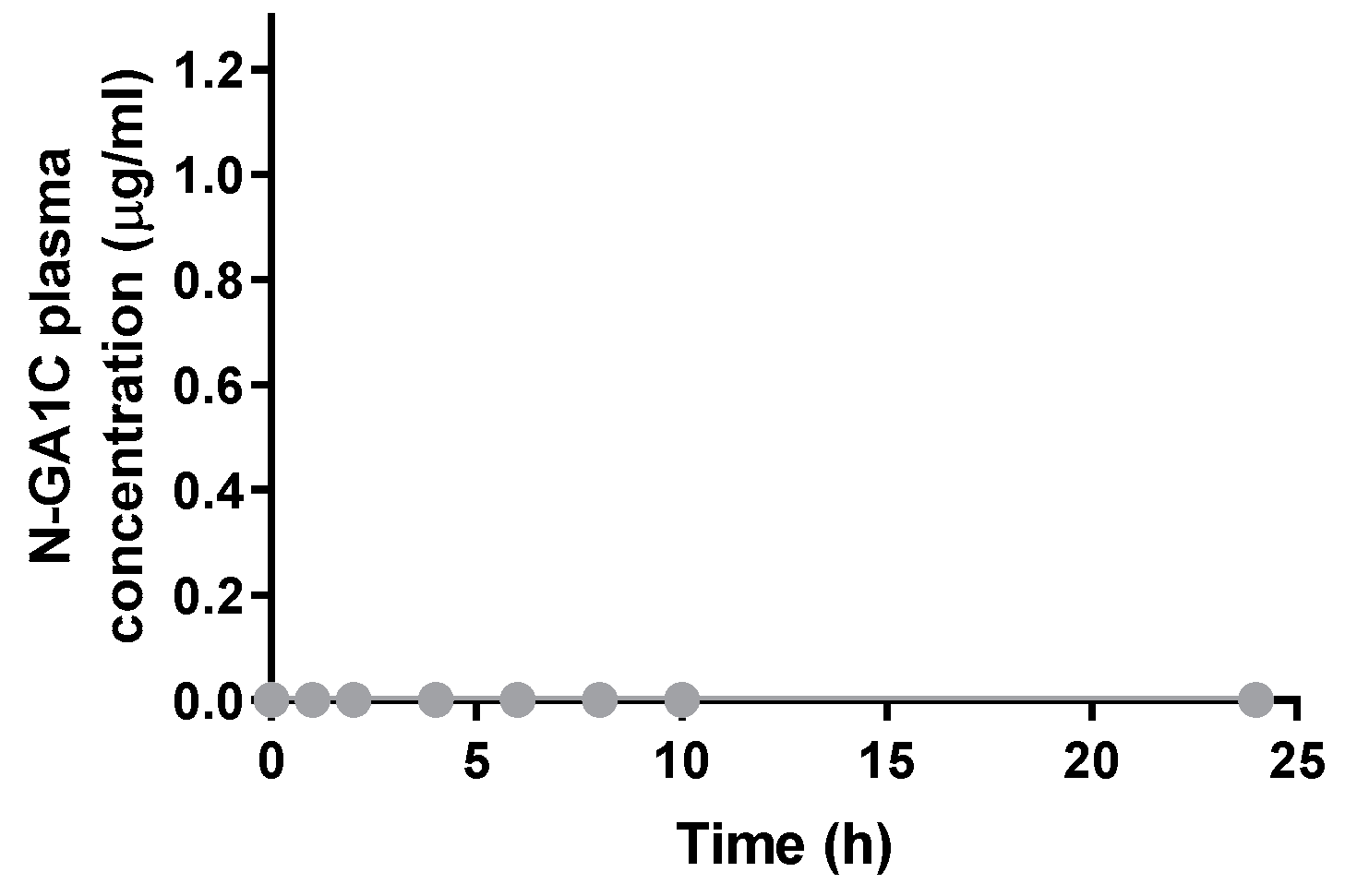
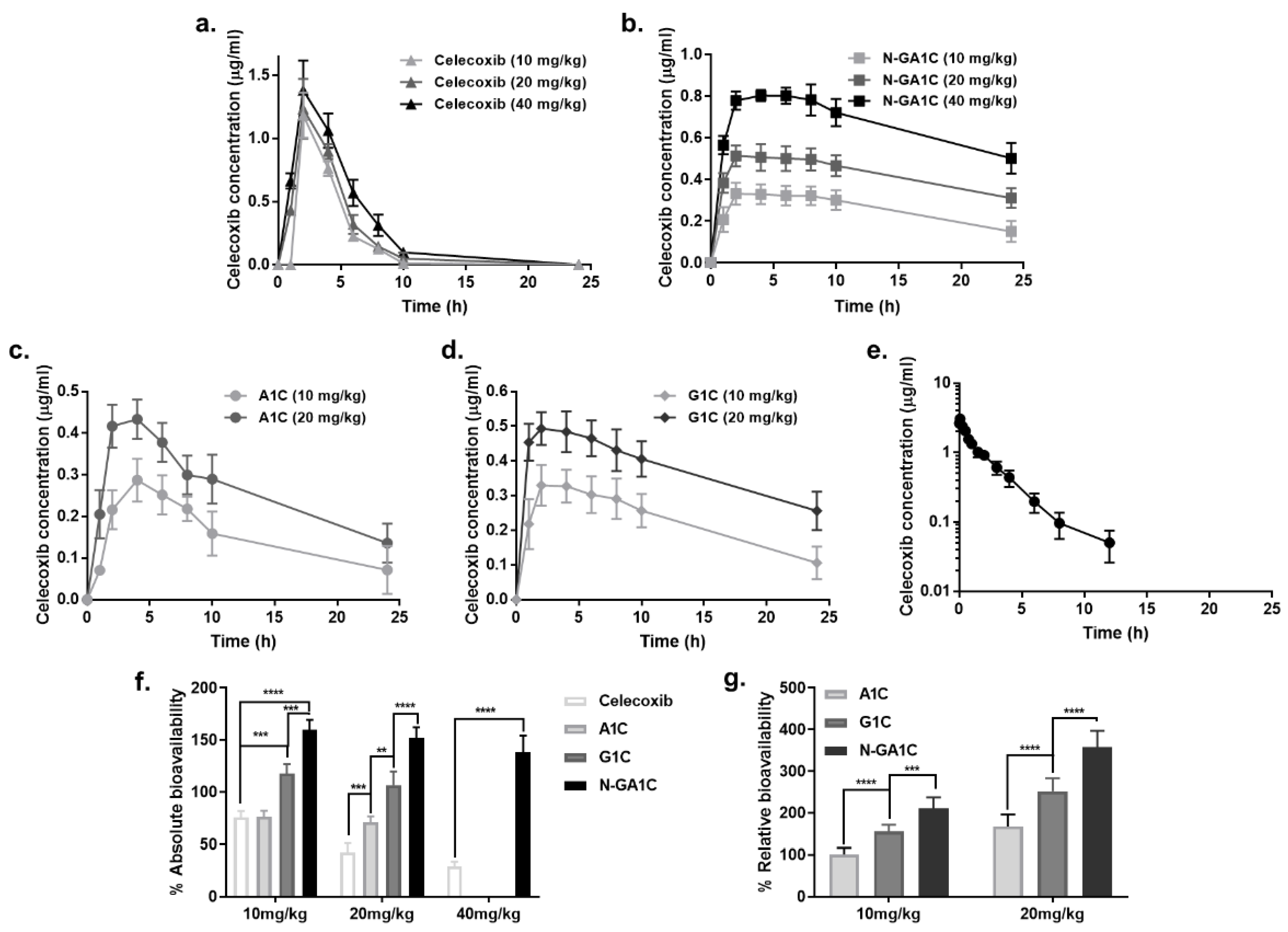
2.8. Pharmacokinetic Study
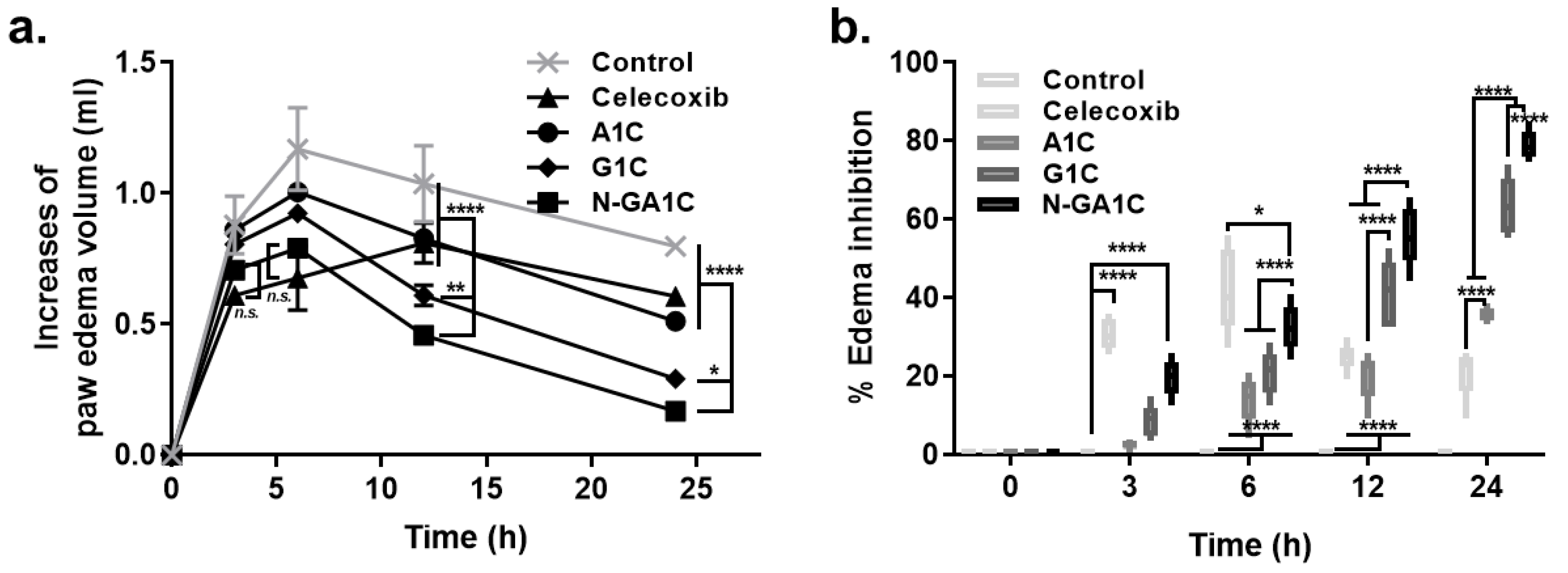
2.9. Carrageenan-Induced Inflammation Study
2.10. Statistical Analysis
3. Results
3.1. Preparation of Celecoxib Derivatives Coupled with Amino Acids
3.2. Celecoxib Derivatives Are Converted to Celecoxib in The Intestinal Contents of Rats
3.3. Systemic Absorption and Exposure of Orally Administered N-GA1C Are Limited
3.4. Celecoxib Derivatives are Metabolized to Celecoxib in the Liver Homogenate of Rats
3.5. Celecoxib Derivatives Improve the PK Properties of Celecoxib
3.6. Oral N-GA1C Prolongs and Enhances the Anti-Inflammatory Activity of Celecoxib
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- FitzGerald, G.A.; Patrono, C. The coxibs, selective inhibitors of cyclooxygenase-2. N. Engl. J. Med. 2001, 345, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Yeomans, N.D.; Solomon, D.H.; Luscher, T.F.; Libby, P.; Husni, M.E.; Graham, D.Y.; Borer, J.S.; Wisniewski, L.M.; Wolski, K.E.; et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016, 375, 2519–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, S.Y.; Ibisogly, A.; Bauer-Brandl, A. Solubility enhancement of BCS Class II drug by solid phospholipid dispersions: Spray drying versus freeze-drying. Int. J. Pharm. 2015, 496, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, K.; Reddy, M.P.; Kaza, R. Design and characterization of microcrystals for enhanced dissolution rate of celecoxib. Curr. Drug Discov. Technol. 2013, 10, 305–314. [Google Scholar] [CrossRef]
- Paulson, S.K.; Vaughn, M.B.; Jessen, S.M.; Lawal, Y.; Gresk, C.J.; Yan, B.; Maziasz, T.J.; Cook, C.S.; Karim, A. Pharmacokinetics of celecoxib after oral administration in dogs and humans: Effect of food and site of absorption. J. Pharm. Exp. 2001, 297, 638–645. [Google Scholar]
- PFIZER. CELEBREX (Celecoxib capsules). FDA Medicat. Guide 1998, 1, 1–22. [Google Scholar]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jung, E.H.; Kim, H.; Yoon, J.-H.; Kim, D.-D.; Jung, Y. Preparation and in vitro evaluation of celecoxib-amino acid conjugates as a colon specific prodrug. J. Pharm. Investig. 2012, 42, 115–120. [Google Scholar] [CrossRef]
- Lee, S.; Lee, Y.; Kim, W.; Nam, J.; Jeong, S.; Yoo, J.W.; Kim, M.S.; Moon, H.R.; Jung, Y. Evaluation of glycine-bearing celecoxib derivatives as a colon-specific mutual prodrug acting on nuclear factor-kappaB, an anti-inflammatory target. Drug Des. Devel. 2015, 9, 4227–4237. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, H.; Kim, W.; Yoon, J.H.; Jeong, S.H.; Jung, Y. Colon-specific delivery of celecoxib is a potential strategy to improve toxicological and pharmacological properties of the selective Cox-2 inhibitor: Implication in treatment of familiar adenomatous polyposis. J. Drug Target. 2012, 20, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, J.; Kim, H.; Kang, S.; Yoon, J.H.; Kim, D.D.; Kim, Y.M.; Jung, Y. N-succinylaspart-1-yl celecoxib is a potential colon-specific prodrug of celecoxib with improved therapeutic properties. J. Pharm. Sci. 2012, 101, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, Y.; Jeong, S.; Nam, J.; Lee, S.; Jung, Y. Colonic delivery of celecoxib is a potential pharmaceutical strategy for repositioning the selective COX-2 inhibitor as an anti-colitic agent. Arch. Pharm. Res. 2015, 38, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, J.; Kim, W.; Nam, J.; Jeong, S.; Lee, S.; Yoo, J.W.; Kim, M.S.; Jung, Y. Celecoxib coupled to dextran via a glutamic acid linker yields a polymeric prodrug suitable for colonic delivery. Drug Des. Devel. 2015, 9, 4105–4113. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, W.; Hong, S.; Park, H.; Yum, S.; Yoon, J.H.; Jung, Y. Colon-targeted celecoxib ameliorates TNBS-induced rat colitis: A potential pharmacologic mechanism and therapeutic advantages. Eur. J. Pharm. 2014, 726, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.J. Carrageenan-induced paw edema in the rat and mouse. Methods Mol. Biol. 2003, 225, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Bateman, K.; Gordon, R.; Mancini, J.; Riendeau, D. Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J. Biol. Chem. 2004, 279, 24866–24872. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IR Spetra (Nujol) (cm−1) | 1H-NMR (DMSO-d6) (δ, ppm) | Elemental Analysis | Log D6.8 | Melting Point | |
|---|---|---|---|---|---|
| Celecoxib | 1720 (C=O, SO2NHCO) | 2.30 (s, 3H), 7.17 (s, 1H), 7.16–7.20 (m, 4H), 7.51 (d, 2H, J = 7.5 Hz), 7.86 (d, 2H, J = 8.0 Hz) | C19H17F3N4O3S (438); calcd: C, 52.05; H, 3.88; N, 12.78; found: C, 52.10; H, 3.89; N, 12.75. | 4.01 | 189 °C–194 °C |
| A1C | 1721 (C=O, SO2NHCO), 1596 (C=O, zwitterionic carboxylate salt) | 1.98 (dd, 1H, J = 10.5, 16.5 Hz), 2.29 (s, 3H), 2.32 (dd, 1H, J = 3.5, 16.0 Hz), 3.34 (m, 1H), 7.13 (s, 1H), 7.15–7.20 (m, 4H), 7.34 (d, 2H, J = 8.5 Hz), 7.80 (d, 2H, J = 9.0 Hz) | C21H19F3N4O5S (496.10) Calcd: C, 50.80; H, 3.86; N, 11.29; Found: C, 50.82; H, 3.91; N, 11.35. | 0.6 | 134 °C–137 °C |
| G1C | 1719 (C=O, SO2NHCO), 1595 (C=O, zwitterionic carboxylate salt) | 1.7 (m, 1H), 2.25 (m, 1H), 2.02 (m, 2H), 2.30 (s, 3H), 3.29 (m, 1H), 7.14 (s, 1H), 7.16–7.21 (m, 4H), 7.59 (d, 2H, J = 8.5 Hz), 7.95 (d, 2H, J = 8.5 Hz) | C22H21F3N4O5S (510.12) Calcd: C, 51.76; H, 4.15; N, 10.98; Found: C, 51.82; H, 4.11; N, 11.20. | 0.81 | 110 °C–115 °C |
| N-GA1C | 1700 (C=O, SO2NHCO), 1668 (C=O, amide), 1596 (C=O, carboxylic) | 2.30 (s, 3H), 2.35 (dd, 1H, J = 7.5, 15.5 Hz), 2.61 (dd, 1H, J = 5.5, 12.5 Hz), 4.02 (t, 2H, J = 5.0 Hz), 4.4 (m, 1H), 7.13 (s, 1H), 7.14–7.21 (m, 4H), 7.51 (d, 2H, J = 8.5 Hz), 7.85 (d, 2H, J = 8.0 Hz) | C23H22F3N5O5S (553.12) Calcd: C, 49.91; H, 4.01; N, 12.56; Found: C, 49.95; H, 4.07; N, 12.50. | −0.13 | 154 °C–158 °C |
| Dose (mg/kg) | Cmax (mg/mL) | Tmax (h) | AUC0–24 (mg/mL)h | AUC0–∞ (mg/mL)h | CL (mL/min/kg) | Vd (L/kg) | BA % (f) | t1/2 (h) | Relative Bioavailability % | |
|---|---|---|---|---|---|---|---|---|---|---|
| Celecoxib (IV) | 10 | - | - | 5.99 ± 0.44 | 6.12 ± 0.52 | 8.01 | 1.04 | - | 1.50 | |
| Celecoxib (Oral) | 10 | 1.18 ± 0.18 | 2 | 4.60 ± 0.45 | 4.64 ± 0.45 | - | - | 75.82% ± 2.66 | - | |
| Celecoxib (Oral) | 20 | 1.23 ± 0.24 | 2 | 5.06 ± 0.49 | 5.19 ± 0.50 | - | - | 42.40% ± 3.94 | - | |
| Celecoxib (Oral) | 40 | 1.38 ± 0.24 | 2 | 6.72 ± 0.58 | 7.12 ± 0.69 | - | - | 29.08% ± 2.02 | - | |
| A1C (Oral) | 10 | 0.29 ± 0.05 | 4 | 3.68 ± 0.39 | 4.68 ± 0.43 | - | - | 76.47% ± 2.48 | 100.85% ± 7.07 | |
| A1C (Oral) | 20 | 0.43 ± 0.05 | 4 | 6.32 ± 0.53 | 8.70 ± 0.72 | - | - | 71.07% ± 2.51 | 167.62%± 12.84 | |
| G1C (Oral) | 10 | 0.31 ± 0.06 | 2 | 5.35 ± 0.44 | 7.22 ± 0.61 | - | - | 117.97% ± 4.032 | 155.59% ± 7.16 | |
| G1C (Oral) | 20 | 0.49 ± 0.05 | 2 | 9.06 ± 0.85 | 13.07 ± 1.11 | - | - | 106.78% ± 5.80 | 251.84%± 14.15 | |
| N-GA1C (Oral) | 10 | 0.33 ± 0.05 | 2 | 6.09 ± 0.49 | 9.78 ± 0.84 | - | - | 159.80% ± 4.31 | 210.76% ± 12.07 | |
| N-GA1C (Oral) | 20 | 0.51 ± 0.05 | 2 | 10.23 ± 0.94 | 18.55 ± 1.19 | - | - | 151.55% ± 4.75 | 357.43% ± 17.74 | |
| N-GA1C (Oral) | 40 | 0.80 ± 0.04 | 4 | 15.75 ± 1.12 | 33.87 ± 3.69 | - | - | 138.35% ± 7.079 | 476.76% ± 21.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.; Kim, J.; Kim, W.; Yoon, I.-S.; Jung, Y. Preparation and Evaluation of Amino Acid Conjugates of Celecoxib as Prodrugs to Improve the Pharmacokinetic and Therapeutic Properties of Celecoxib. Pharmaceutics 2020, 12, 1043. https://doi.org/10.3390/pharmaceutics12111043
Lee Y, Kim J, Kim W, Yoon I-S, Jung Y. Preparation and Evaluation of Amino Acid Conjugates of Celecoxib as Prodrugs to Improve the Pharmacokinetic and Therapeutic Properties of Celecoxib. Pharmaceutics. 2020; 12(11):1043. https://doi.org/10.3390/pharmaceutics12111043
Chicago/Turabian StyleLee, Yonghyun, Jungyun Kim, Wooseong Kim, In-Soo Yoon, and Yunjin Jung. 2020. "Preparation and Evaluation of Amino Acid Conjugates of Celecoxib as Prodrugs to Improve the Pharmacokinetic and Therapeutic Properties of Celecoxib" Pharmaceutics 12, no. 11: 1043. https://doi.org/10.3390/pharmaceutics12111043