Quasi-Irreversible Inhibition of CYP2D6 by Berberine

 , , ,
, , ,
Abstract
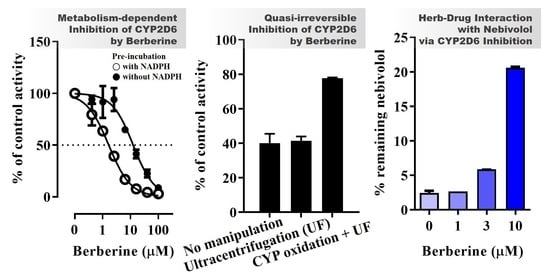
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Direct CYP Inhibition Assay Using Pooled HLM
2.3. In Vitro Metabolism-Dependent CYP2D6 Inhibition Assay Using Pooled HLM and rhCYP2D6
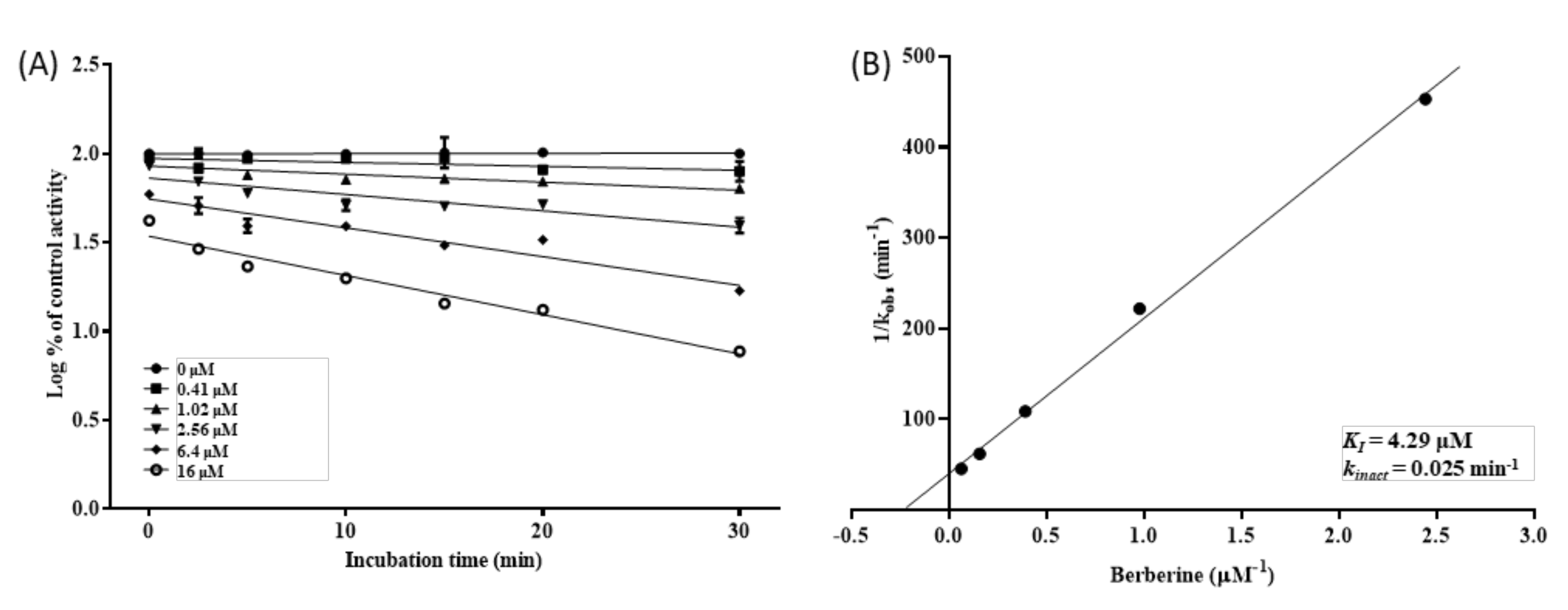
2.4. Determination of Kinetic Parameters for MDI of CYP2D6 by BBR
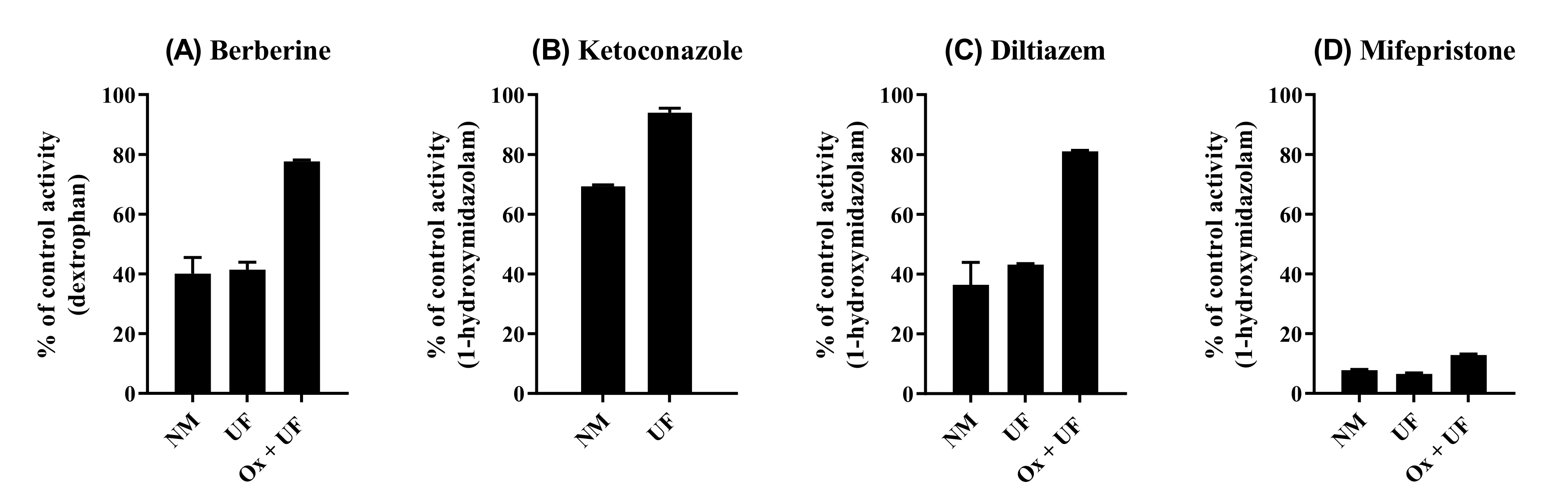
2.5. Reversibility of CYP2D6 Inhibition by BBR
2.6. Metabolic Stability and Metabolite Identification of BBR in Pooled HLM
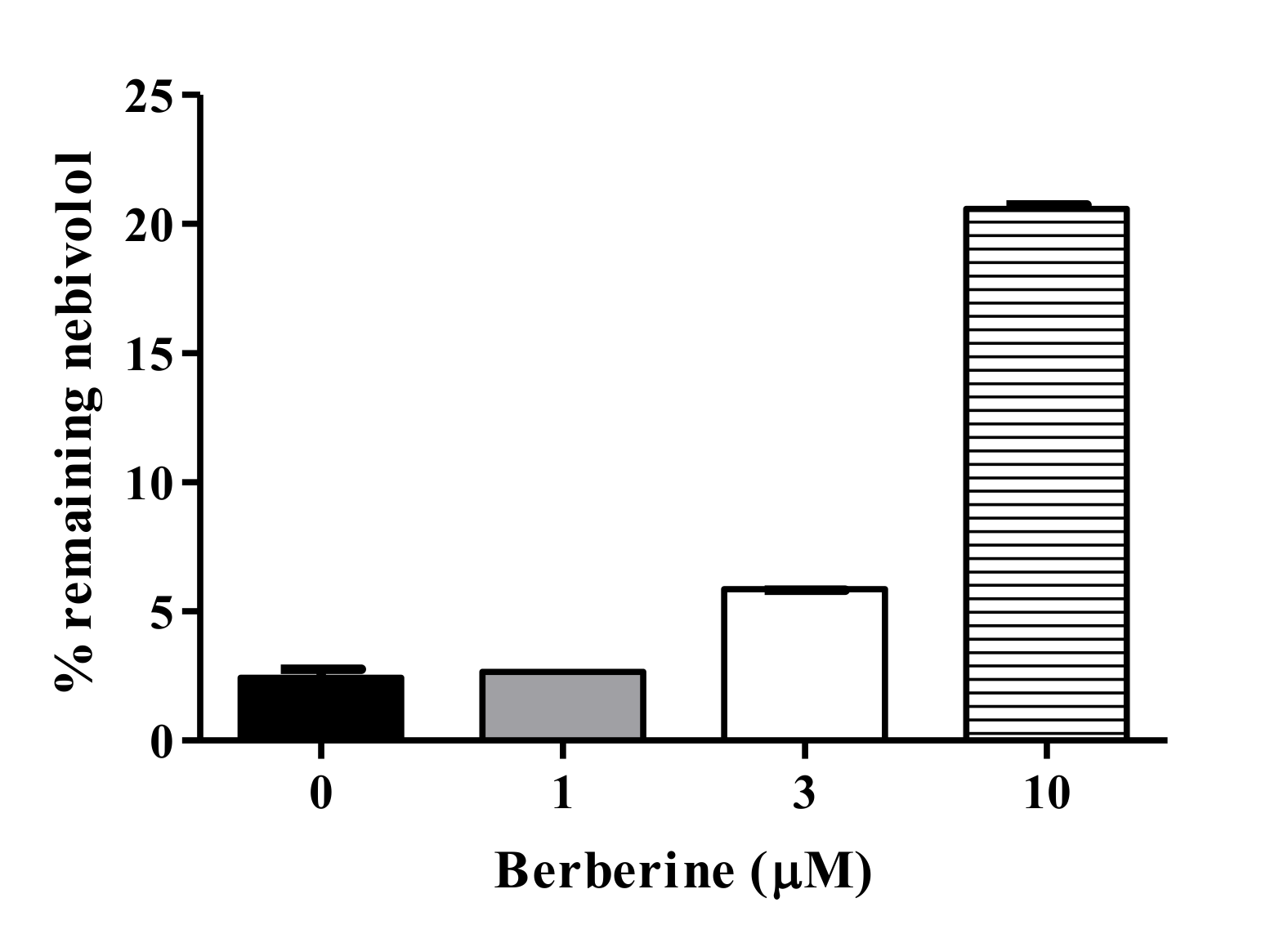
2.7. BBR-Nebivolol Interaction in Pooled HLM
2.8. Data Analysis
3. Results
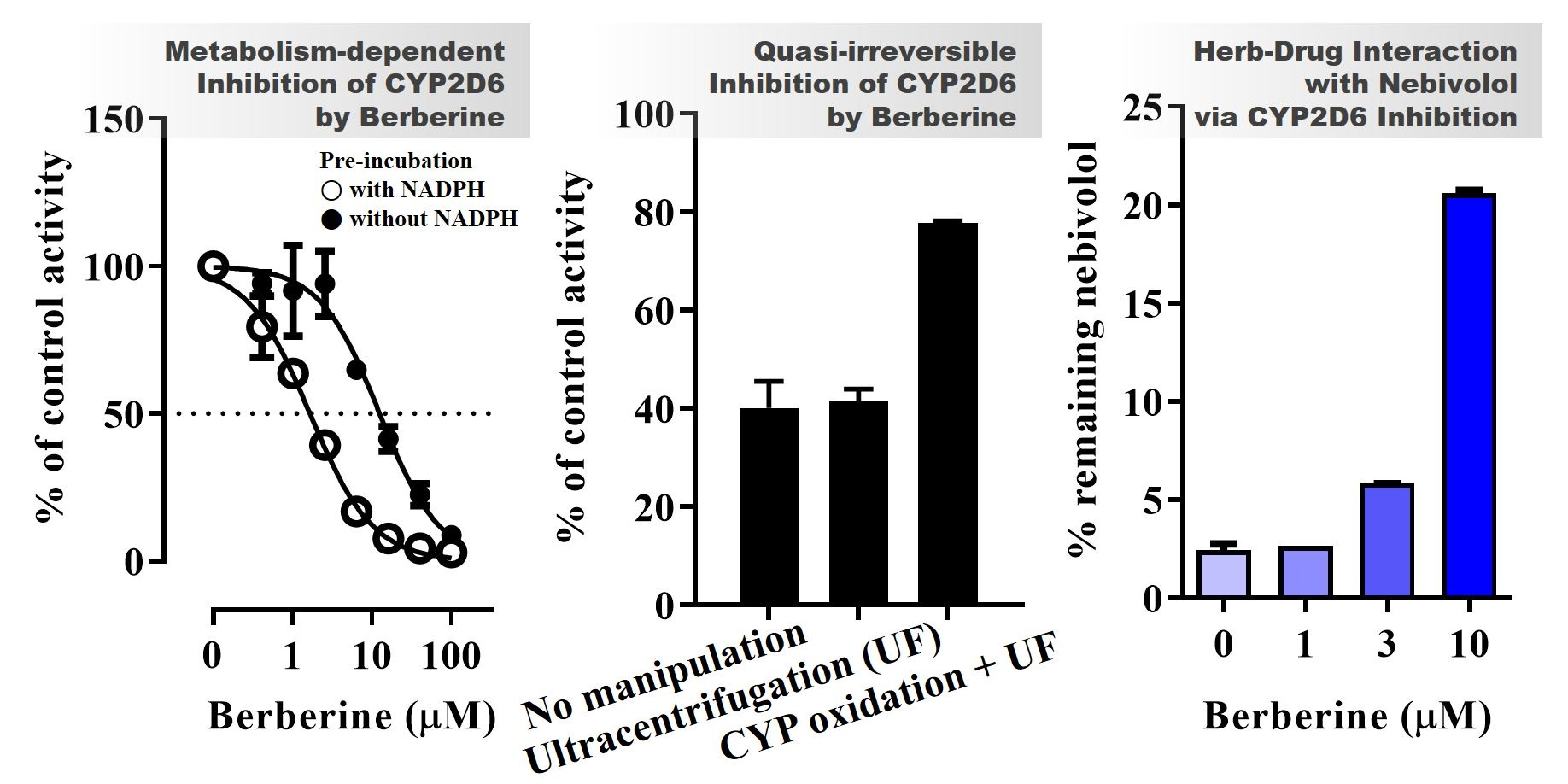
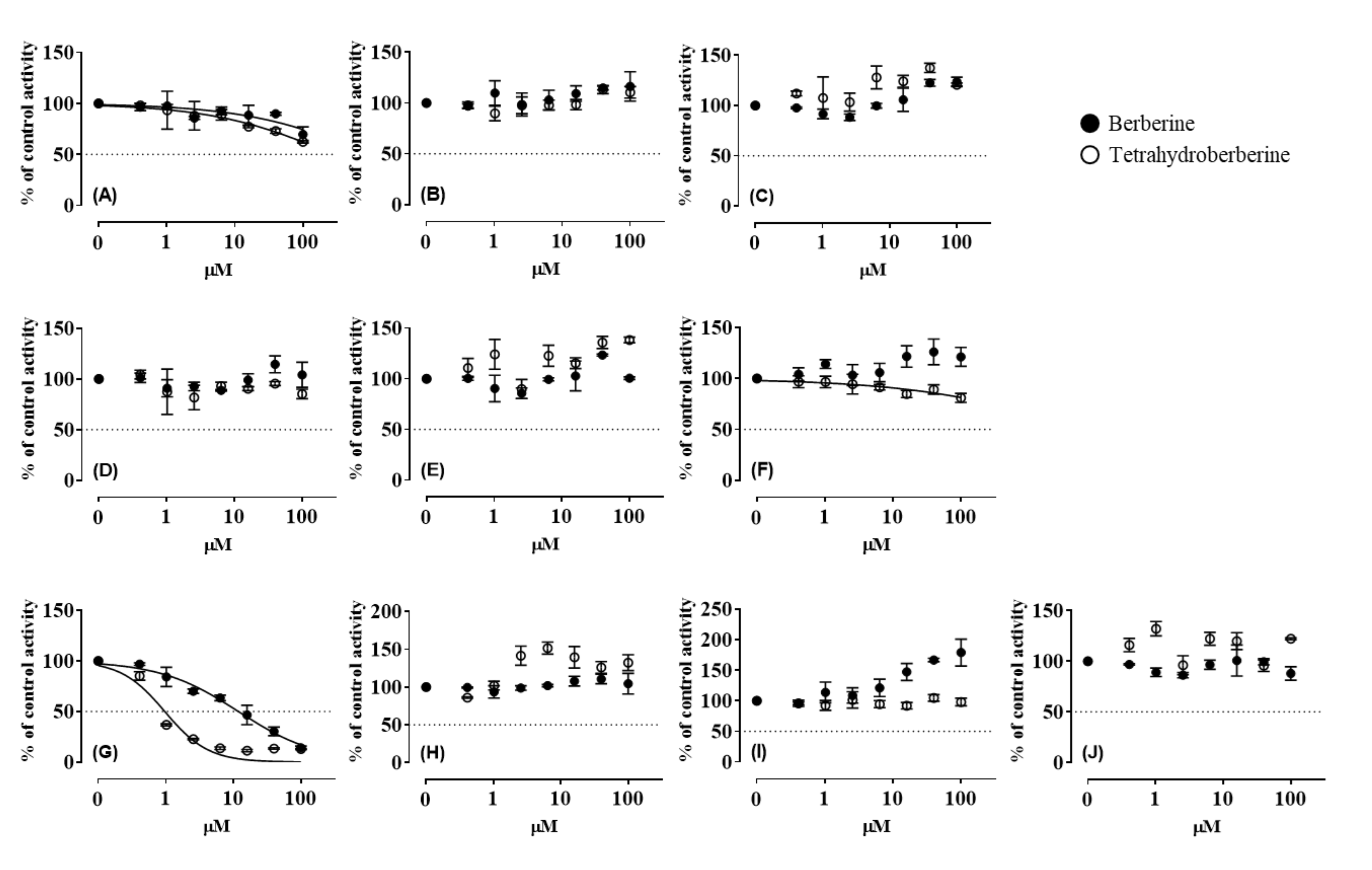
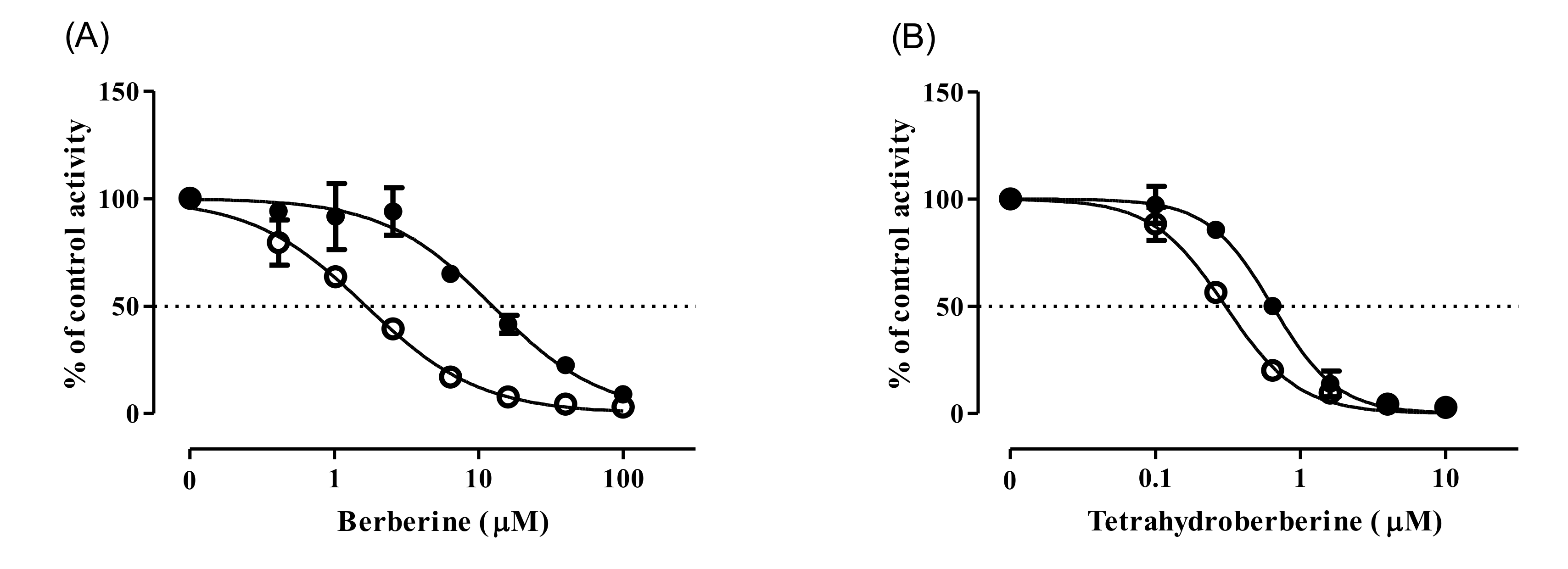
3.1. CYP Inhibition by BBR and THB
3.2. Metabolic Stability and Metabolite Identification of BBR in HLM
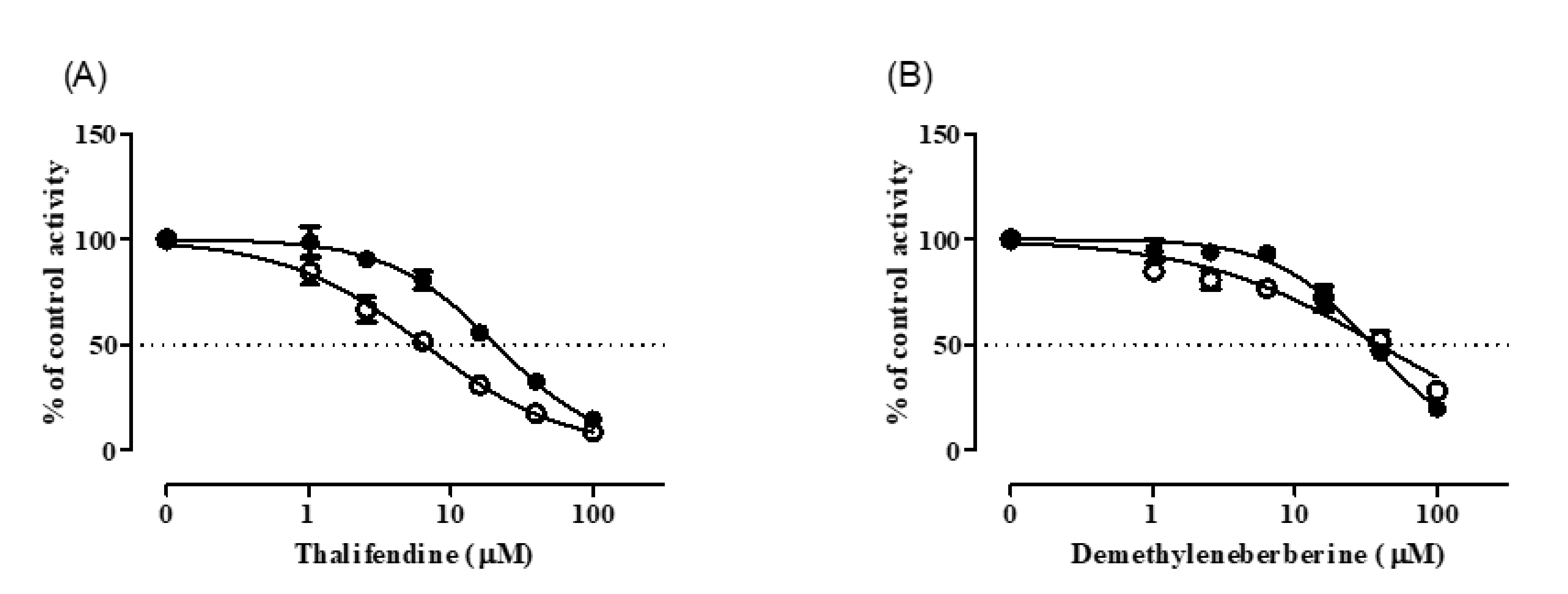
3.3. CYP Inhibition by BRB, TFD, DMB, and JTZ
3.4. Inhibition Kinetics of CYP2D6 by BBR
3.5. Metabolic Interaction between BBR and Nebivolol in HLM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Elmer, G.W.; Lafferty, W.E.; Tyree, P.T.; Lind, B.K. Potential interactions between complementary/alternative products and conventional medicines in a medicare population. Ann. Pharmacother. 2007, 41, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornsson, T.D.; Callaghan, J.T.; Einolf, H.J.; Fischer, V.; Gan, L.; Grimm, S.; Kao, J.; King, S.P.; Miwa, G.; Ni, L. The conduct of In Vitro and In Vivo drug-drug interaction studies: A pharmaceutical research and manufacturers of America (phrma) perspective. Drug Metab. Dispos. 2003, 31, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Dresser, G.K.; Spence, J.D.; Bailey, D.G. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome p450 3a4 inhibition. Clin. Pharmacokinet. 2000, 38, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Hachad, H.; Yao, C.; Ragueneau-Majlessi, I. Relationship between extent of inhibition and inhibitor dose: Literature evaluation based on the metabolism and transport drug interaction database. Curr. Drug Metab. 2003, 4, 371–380. [Google Scholar] [CrossRef]
- Shaikh, A.S.; Thomas, A.B.; Chitlange, S.S. Herb-drug interaction studies of herbs used in treatment of cardiovascular disorders—A narrative review of preclinical and clinical studies. Phytother. Res. 2020, 34, 1008–1026. [Google Scholar] [CrossRef]
- Ernst, E. Harmless herbs? A review of the recent literature. Am. J. Med. 1998, 104, 170–178. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Lee, S.Y.; Oh, S.J.; Lee, K.H.; Jung, Y.S.; Kim, S.K. Assessment of drug-drug interactions caused by metabolism-dependent cytochrome p450 inhibition. Chem. Biol. Interact. 2012, 198, 49–56. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A.Y. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. [Google Scholar]
- VandenBrink, B.M.; Isoherranen, N. The role of metabolites in predicting drug-drug interactions: Focus on irreversible p450 inhibition. Curr. Opin. Drug Discov. Dev. 2010, 13, 66. [Google Scholar]
- Lahiri, S.; Dutta, N. Berberine and chloramphenicol in the treatment of cholera and severe diarrhoea. J. Indian Med. Assoc. 1967, 48, 1–11. [Google Scholar]
- Amin, A.; Subbaiah, T.; Abbasi, K. Berberine sulfate: Antimicrobial activity, bioassay, and mode of action. Can. J. Microbiol. 1969, 15, 1067–1076. [Google Scholar] [CrossRef]
- Sack, R.B.; Froehlich, J.L. Berberine inhibits intestinal secretory response of Vibrio cholerae and Escherichia coli enterotoxins. Infect. Immun. 1982, 35, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.E.; Greenough, W.B., III. Control of diarrheal diseases. Annu. Rev. Public Health 1989, 10, 221–244. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Jin, C.; Xiao, X.-H.; Dong, X.-P. Antimicrobial properties of berberines alkaloids in coptis chinensis franch by microcalorimetry. J. Biochem. Biophys. Methods 2008, 70, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Yao, X.J.; Tan, Y.-H. Effects of berberine on delayed afterdepolarizations in ventricular muscles In Vitro and In Vivo. J. Cardiovasc. Pharmacol. 1994, 23, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.W.; Yao, X.Q.; Chen, Z.Y.; Ko, W.H.; Huang, Y. Cardiovascular actions of berberine. Cardiovasc. Drug Rev. 2001, 19, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kulsum, U.; Chowdhury, S.A.; Fujisawa, S.-I.; Ishihara, M.; Yokoe, I.; Sakagami, H. Tumor-specific cytotoxicity and apoptosis-inducing activity of berberines. Anticancer Res. 2005, 25, 4053–4059. [Google Scholar]
- Piyanuch, R.; Sukhthankar, M.; Wandee, G.; Baek, S.J. Berberine, a natural isoquinoline alkaloid, induces nag-1 and atf3 expression in human colorectal cancer cells. Cancer Lett. 2007, 258, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Serafim, T.L.; Oliveira, P.J.; Sardao, V.A.; Perkins, E.; Parke, D.; Holy, J. Different concentrations of berberine result in distinct cellular localization patterns and cell cycle effects in a melanoma cell line. Cancer Chemother. Pharmacol. 2008, 61, 1007–1018. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-S.; Yang, J.-S.; Lin, H.-J.; Yu, C.-S.; Tan, T.-W.; Lin, Y.-T.; Lin, C.-C.; Lu, H.-F.; Chung, J.-G. Berberine inhibits wehi-3 leukemia cells In Vivo. In Vivo 2007, 21, 407–412. [Google Scholar]
- Kuo, C.-L.; Chi, C.-W.; Liu, T.-Y. The anti-inflammatory potential of berberine In Vitro and In Vivo. Cancer Lett. 2004, 203, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Chen, J.-C.; Hsiang, C.-Y.; Wu, S.-L.; Wu, H.-C.; Ho, T.-Y. Berberine suppresses inflammatory agents-induced interleukin-1β and tumor necrosis factor-α productions via the inhibition of iκb degradation in human lung cells. Pharmacol. Res. 2007, 56, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Minoda, K.; Nagaoka, Y.; Hayashi, T.; Uesato, S. Antiviral activity of berberine and related compounds against human cytomegalovirus. Bioorg. Med. Chem. Lett. 2007, 17, 1562–1564. [Google Scholar] [CrossRef] [PubMed]
- Koppen, L.M.; Whitaker, A.; Rosene, A.; Beckett, R.D. Efficacy of berberine alone and in combination for the treatment of hyperlipidemia: A systematic review. J. Evid. Based Complementary Altern. Med. 2017, 22, 956–968. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wei, J.; Xue, R.; Wu, J.-D.; Zhao, W.; Wang, Z.-Z.; Wang, S.-K.; Zhou, Z.-X.; Song, D.-Q.; Wang, Y.-M. Berberine lowers blood glucose in type 2 diabetes mellitus patients through increasing insulin receptor expression. Metabolism 2010, 59, 285–292. [Google Scholar] [CrossRef]
- Qiu, F.; Zhu, Z.; Kang, N.; Piao, S.; Qin, G.; Yao, X. Isolation and identification of urinary metabolites of berberine in rats and humans. Drug Metab. Dispos. 2008, 36, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ren, G.; Wang, Y.-X.; Kong, W.-J.; Yang, P.; Wang, Y.-M.; Li, Y.-H.; Yi, H.; Li, Z.-R.; Song, D.-Q. Bioactivities of berberine metabolites after transformation through cyp450 isoenzymes. J. Transl. Med. 2011, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Jang, H.; Lee, J.-Y.; Ma, J.Y.; Oh, S.J.; Kim, S.K. Inhibitory effects of hwang-ryun-hae-dok-tang on cytochrome p450 in human liver microsomes. Xenobiotica 2015, 45, 131–138. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.K. Direct and metabolism-dependent cytochrome p450 inhibition assays for evaluating drug–drug interactions. J. Appl. Toxicol. 2013, 33, 100–108. [Google Scholar] [CrossRef]
- Choi, Y.J.; Quan, K.T.; Park, I.; Lee, S.J.; Kang, K.W.; Na, M.; Kim, S.K. Discovery of rubiarbonone c as a selective inhibitor of cytochrome p450 4f enzymes. Arch. Toxicol. 2018, 92, 3325–3336. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Wong, Y.N. Enzyme kinetics for clinically relevant cyp inhibition. Curr. Drug Metab. 2005, 6, 241–257. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.S.; Oh, S.J.; Lee, J.-Y.; Ryu, C.S.; Kim, Y.-M.; Lee, B.H.; Kim, S.K. Metabolic characterization of meso-dihydroguaiaretic acid in liver microsomes and in mice. Food Chem. Toxicol. 2015, 76, 94–102. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, H.; Cha, I.J.; Park, J.S.; Shon, J.H.; Liu, K.H.; Shin, J.G. High-throughput screening of inhibitory potential of nine cytochrome p450 enzymes in vitro using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2651–2658. [Google Scholar] [CrossRef]
- Hu, X.; Lan, T.; Dai, D.; Xu, R.-a.; Yuan, L.; Zhou, Q.; Li, Y.; Cai, J.; Hu, G. Evaluation of 24 cyp2d6 variants on the metabolism of nebivolol In Vitro. Drug Metab. Dispos. 2016, 44, 1828–1831. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hellum, B.H.; Liang, A.; Nilsen, O.G. The In Vitro inhibition of human cyp1a2, cyp2d6 and cyp3a4 by tetrahydropalmatine, neferine and berberine. Phytother. Res. 2012, 26, 277–283. [Google Scholar] [CrossRef]
- Obach, R.S.; Walsky, R.L.; Venkatakrishnan, K. Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug-drug interactions. Drug Metab. Dispos. 2007, 35, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perloff, E.; Mason, A.; Dehal, S.; Blanchard, A.; Morgan, L.; Ho, T.; Dandeneau, A.; Crocker, R.; Chandler, C.; Boily, N. Validation of cytochrome p450 time-dependent inhibition assays: A two-time point ic50 shift approach facilitates k inact assay design. Xenobiotica 2009, 39, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Takakusa, H.; Kimura, T.; Inoue, S.-i.; Kusuhara, H.; Ando, O. Analysis of mechanism-based inhibition of cyp 3a4 by a series of fluoroquinolone antibacterial agents. Drug Metab. Dispos. 2016, 44, 1608–1616. [Google Scholar] [CrossRef] [Green Version]
- Orr, S.T.; Ripp, S.L.; Ballard, T.E.; Henderson, J.L.; Scott, D.O.; Obach, R.S.; Sun, H.; Kalgutkar, A.S. Mechanism-based inactivation (MBI) of cytochrome p450 enzymes: Structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks. J. Med. Chem. 2012, 55, 4896–4933. [Google Scholar] [CrossRef]
- Reinen, J.; Smit, M.; Wenker, M. Evaluation of strategies for the assessment of drug-drug interactions involving cytochrome p450 enzymes. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 737–750. [Google Scholar] [CrossRef]
- Bertelsen, K.M.; Venkatakrishnan, K.; von Moltke, L.L.; Obach, R.S.; Greenblatt, D.J. Apparent mechanism-based inhibition of human cyp2d6 In Vitro by paroxetine: Comparison with fluoxetine and quinidine. Drug Metab. Dispos. 2003, 31, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Fu, S.; Pi, R.; He, F. Neuropharmacological and pharmacokinetic properties of berberine: A review of recent research. J. Pharm. Pharmacol. 2009, 61, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Freston, J.; Chiu, Y.L.; Mulford, D.; Ballard, E., II. Comparative pharmacokinetics and safety of lansoprazole oral capsules and orally disintegrating tablets in healthy subjects. Aliment. Pharmacol. Ther. 2003, 17, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Alolga, R.N.; Fan, Y.; Chen, Z.; Liu, L.-W.; Zhao, Y.-J.; Li, J.; Chen, Y.; Lai, M.-D.; Li, P.; Qi, L.-W. Significant pharmacokinetic differences of berberine are attributable to variations in gut microbiota between Africans and Chinese. Sci. Rep. 2016, 6, 27671. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | BBR (nM) | BRB (nM) | TFD (nM) | DMB (nM) | JTZ (nM) | M1 (m/z = 310) (1000 × cps) |
|---|---|---|---|---|---|---|
| 0 | 1049 ± 16 | 5.4 ± 0.1 | 5.4 ± 0.1 | 6.1 ± 0.1 | 10.4 ± 0.3 | 1.3 ± 0.25 |
| 7.5 | 807 ± 11 | 5.6 ± 0.0 | 20.9 ± 0.4 | 24.1 ± 0.3 | 10.7 ± 0.1 | 28 ± 0.66 |
| 15 | 713 ± 12 | 5.8 ± 0.1 | 33.3 ± 0.6 | 38.1 ± 0.5 | 10.9 ± 0.3 | 80 ± 0.31 |
| 30 | 539 ± 4 | 6.0 ± 0.1 | 44.3 ± 1.0 | 52.6 ± 1.9 | 10.5 ± 0.3 | 220 ± 2.6 |
| 60 | 313 ± 15 | 6.1 ± 0.0 | 50.4 ± 0.7 | 64.3 ± 1.6 | 10.0 ± 0.2 | 540 ± 15 |
| 120 | 147 ± 5 | 6.2 ± 0.0 | 49.3 ± 1.0 | 75.0 ± 1.0 | 9.6 ± 0.2 | 1100 ± 26 |
| IC50 (µM) | CYP Enzyme | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A4 (MDZ) | 3A4 (TST) | |
| BRB | 51 (41–62) | 17 (14–22) | 96 (79–116) | 72 (62–83) | 57 (52–64) | 49 (41–59) | 45 (41–49) | 34 (29–41) | 28 (26–31) | 45 (38–53) |
| TFD | >100 | >100 | >100 | >100 | >100 | >100 | 28 (24–33) | 32 (25–42) | >100 | >100 |
| DMB | >100 | >100 | >100 | >100 | >100 | >100 | 32 (27–37) | 7.7 (6.0–9.8) | 66 (55–78) | 7.3 (5.6–9.4) |
| JTZ | >100 | >100 | >100 | 26 (19–37) | >100 | >100 | 10 (8–12) | 11 (10–13) | >100 | 11 (10–13) |
| TRB | 6.9 (6.5–7.4) | 4.7 (4.1–5.3) | 9.6 (8.7–10.5) | 9.5 (7.9–11.3) | 6.5 (5.8–7.3) | 5.4 (5.0–5.8) | 3.4 (3.2–3.7) | 2.7 (2.5–2.9) | 5.3 (4.7–6.1) | 4.6 (4.1–5.2) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.G.; Lee, H.S.; Jeon, J.S.; Choi, Y.J.; Choi, Y.J.; Yoo, S.-Y.; Kim, E.-y.; Lee, K.; Park, I.; Na, M.; et al. Quasi-Irreversible Inhibition of CYP2D6 by Berberine. Pharmaceutics 2020, 12, 916. https://doi.org/10.3390/pharmaceutics12100916
Kim HG, Lee HS, Jeon JS, Choi YJ, Choi YJ, Yoo S-Y, Kim E-y, Lee K, Park I, Na M, et al. Quasi-Irreversible Inhibition of CYP2D6 by Berberine. Pharmaceutics. 2020; 12(10):916. https://doi.org/10.3390/pharmaceutics12100916
Chicago/Turabian StyleKim, Ha Gyeong, Han Sol Lee, Jang Su Jeon, Young Jae Choi, Yeon Jung Choi, So-Yeol Yoo, Eun-yeong Kim, Kiho Lee, InWha Park, MinKyun Na, and et al. 2020. "Quasi-Irreversible Inhibition of CYP2D6 by Berberine" Pharmaceutics 12, no. 10: 916. https://doi.org/10.3390/pharmaceutics12100916