1. Introduction
An in vitro–in vivo correlation (IVIVC) can be defined as a mathematical relationship between an in vitro characteristic of the drug product and an in vivo characteristic of the same drug product. Usually, the in vitro characteristic is the in vitro dissolution rate or the dissolved fractions versus time and the in vivo parameter is absorption rate or oral fractions absorbed versus time [
1].
While a drug product is developed, IVIVCs are useful tools as they serve to predict in vivo behavior and, consequently, can be used to guide formulation development. Thus, this avoids in vivo failures and, if adequately validated, they can be used to obtain a biowaiver based on the in vitro dissolution tests [
1].
From a regulatory point of view, four types of IVIVCs can be defined, but only a level A IVIVC can substitute human bioequivalence studies. Specifically, in a level A IVIVC, a point by point relationship is established between the complete dissolution profile and the complete absorption profile. Active pharmaceutical ingredients (API) belonging to the class II Biopharmaceutic Classification System (BCS) can obtain a dissolution based biowaiver through a validated Level A IVIVC [
2,
3]. A recent study reported a so-called in vitro–in vivo correlation for candesartan cilexetil products, but the dissolution results were not reported and both formulations demonstrated bioequivalence [
4]. A positive correlation between dissolution rate of a proniosomal formulation and its oral bioavailability in rats versus the pure drug was demonstrated using as dissolution media acetate buffer (pH 4.5) and phosphate buffer (pH 6.8) added with tween 80 at 0.2%
w/
v [
5]. Nevertheless, a level A IVIVC was not attempted.
Candesartan cilexetil is a prodrug of candesartan that was designed to increase its bioavailability and it is hydrolyzed to candesartan during absorption. Nevertheless, a recent study challenged this hypothesis by proposing the superior solubility and permeability of candesartan versus the prodrug [
6].
From a pharmacokinetic point of view, the prodrug still provides a low oral bioavailability of candesartan (14%). Candesartan cilexetil low solubility, combined with its efflux transport by the intestinal P-glycoprotein and its vulnerability to enzymatic degradation in small intestine contribute to the observed low oral bioavailability [
7,
8,
9]. Recently, an improvement of candesartan cilexetil oral bioavailability in rabbits has been demonstrated by using naringin as a P-gp inhibitor [
10]. After absorption, candesartan is mainly excreted unchanged in urine and feces (by biliary excretion). Tmax is reached around 3–4 h [
7]. Its protein binding is high (99%) with a distribution volume of 0.1 L/kg [
7,
11] and a half-life of 9 h [
7].
Candesartan cilexetil is a BCS class II drug (low solubility, high permeability) with a molecular weight of 610.7 g/mol, low solubility (intrinsic solubility: 0.0595 mg/L) and that behaves like a weak acid (pka1: 3.50 and pka2: 5.85). Dose number is higher than 1 in the pH range from 1.2 to 6.8 [
12].
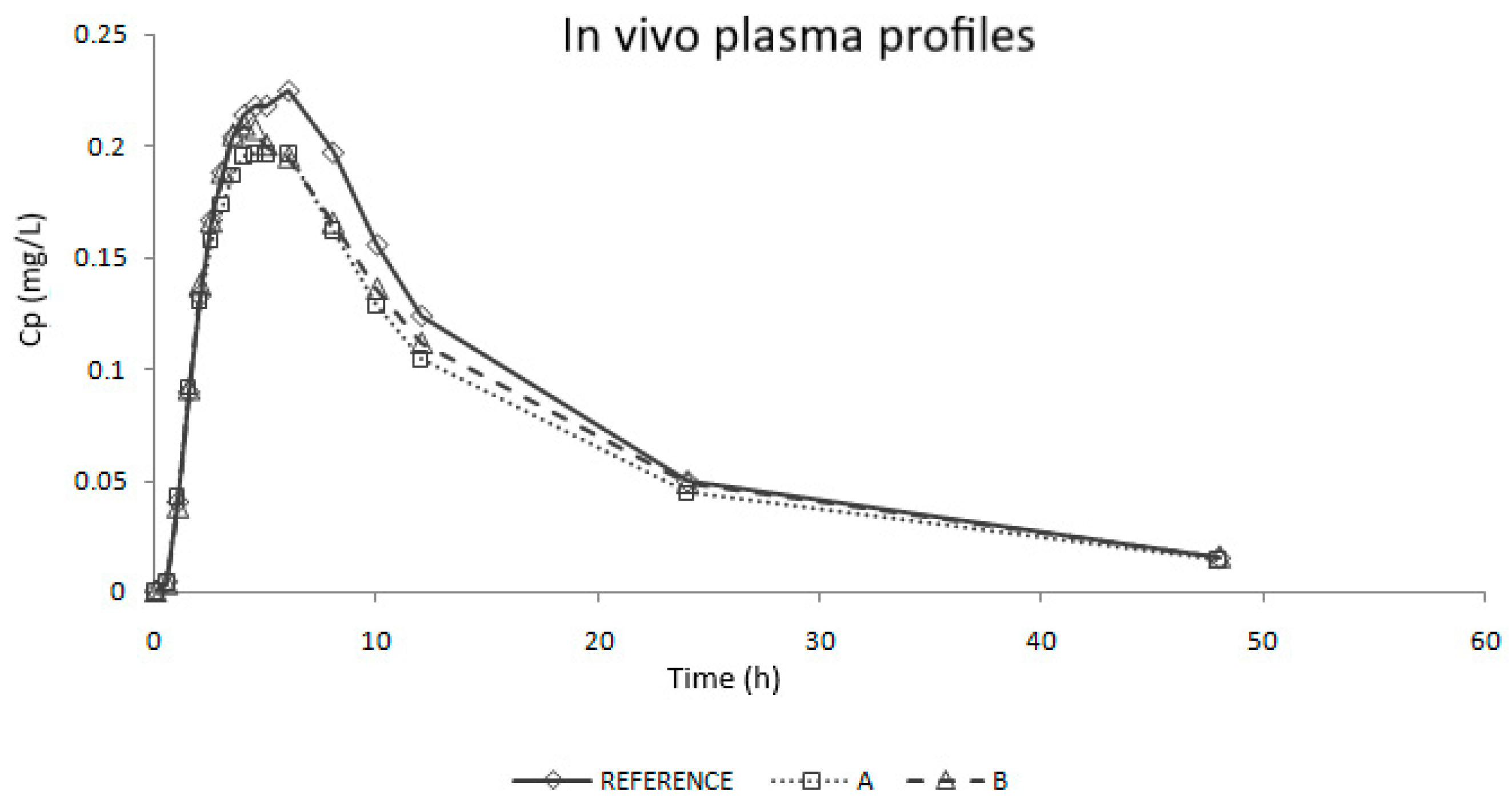
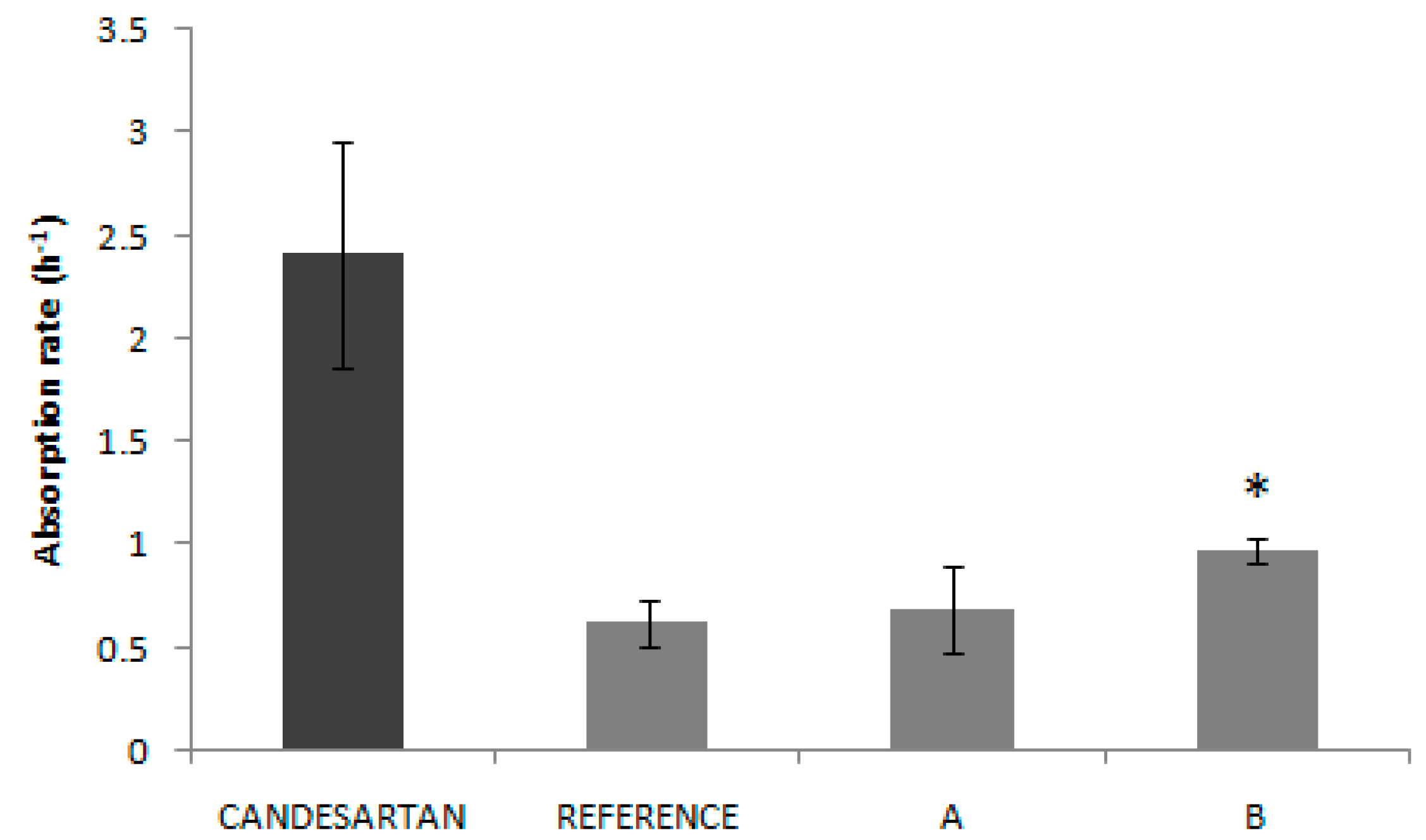
Two generic immediate release products of candesartan cilexetil (Product A and Product B) were developed and succeeded in their corresponding BE study, as their 90% confidence intervals were between 80% and 125% for Cmax and AUC, but their rate of absorption was lower than that of the reference as the 90% CI of Cmax did not include the 100% value. Since the Cmax differences were statistically significant, both datasets can be used to explore the relationship between dissolution rate and absorption rate. We have recently showed how it is possible to combine data from separate Bioequivalence studies to construct an IVIVC by normalizing the data sets with the reference plasma levels ratios in the different studies [
13,
14].
The aims of this work were (1) to establish and validate a level A IVIVC and (2) to obtain a biopredictive in vitro dissolution test for these three candesartan cilexetil products (Atacand®, Astrazeneca SA, Madrid, Spain as Reference, Product A and Product B) to be used as developments tool to reduce the failure rate in future bioequivalence studies.
Prediction of plasma levels using as input in vitro dissolution results could be done with other commercial PBPK modelling packages [
15,
16], but we intended to develop a method that could be easily implemented in basic modelling software or even with excel worksheets. Regarding the design of the dissolution method, we checked the standard conditions already published for candesartan and explore the suggested alternatives in recent reports about biorelevant dissolution conditions selection based on the biopharmaceutical properties of the drug [
17,
18].
4. Discussion
An in vitro dissolution test that is able to predict the in vivo behavior of two IR generic products of candesartan cilexetil has been developed based on an IVIVC. This exercise could be conducted by regulatory agencies with all generic products and their corresponding bioequivalence studies submitted for marketing authorization, as well as those failed bioequivalence studies that have to be reported to regulatory authorities. As the validity of IVIVCs is limited to the design-space of the products included in the IVIVC, such a general IVIVC would have an extremely wide validity and would provide a general dissolution test for all products or a large number of products containing the same drug. This dissolution method could be employed to support variations without the need of in vivo bioequivalence studies if within the design-space and to reduce the failure rate of future generics when conducting their own bioequivalence study.
This approach contrasts with the present approach in the US Food and Drug Administration where, in principle, generic products have to exhibit a similar dissolution profile to that of the reference listed product in the dissolution methods presently recommended by the Division of Bioequivalence, which is generally based on the method of the reference product, but which might not be applicable or meaningful for the generic formulation [
29,
30].
Furthermore, we have performed solubility and permeability experiments with the formulations, a methodology not frequently employed by pharmaceutical companies developing generic products but that could be informative on the impact of excipients on these relevant biopharmaceutical attributes.
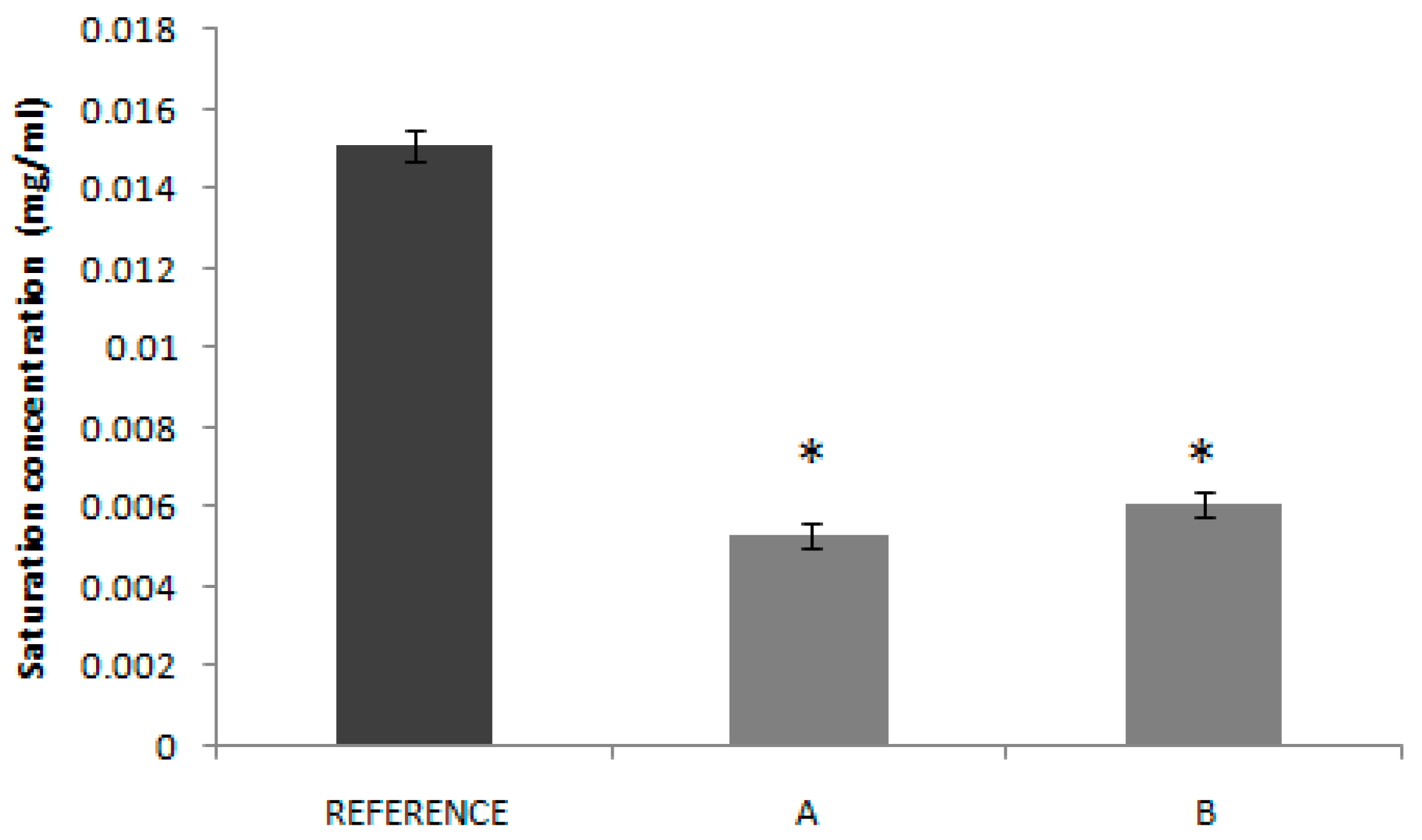
The solubility tests confirm the low solubility classification of candesartan cilexetil, as all the Do values in the API experimental solubility study were higher than 1.00. In
Figure 2, it can be seen that saturation solubility of candesartan cilexetil in products A and B is lower than in the case of the Reference with a statistically significant difference. This fact could explain, at least partially, the lower bioavailability (in absorption rate) of test products, since the rank order of the saturation solubility agrees with the rank order of the Cmax T/R ratios of the BE studies. It is speculated that the dissolution, as the limiting factor of BCS class II drugs absorption, is affected by the solubility of the drug in the product formulations. In fact, several technological innovations developed to increase candesartan solubility have demonstrated to increase drug bioavailability. The formation of co-crystals improved the drug bioavailability in Wistar rats [
31] and nanosized suspensions with higher saturation solubility showed improved bioavailability in a murine model [
32]. Candesartan loaded proniosomes showed faster dissolution and higher bioavailability after oral administration in rats [
5].
Candesartan cilexetil permeability value (5.97·10
−5 cm/s) was higher than metoprolol permeability (2.00·10
−5 cm/s). [
13] This confirms the high permeability of the API candesartan cilexetil and, taking into account the solubility results, it allows us to confirm candesartan cilexetil as BCS class II (low solubility, high permeability) candesartan. The permeability studies with the products show that in all these products, Reference, Product A and Product B, candesartan cilexetil permeability is lower than that of the free API (1.53·10
−5, 1.71·10
−5 and 2.40·10
−5 cm/s). This result suggests that the excipients of these products affect to the permeation process. Nevertheless, even if excipients could be affecting the permeation process, the permeability rank does not correspond to the trend of in vivo Cmax across products, thus, excipient effects alone cannot explain the observed differences. The permeability continues to be sufficiently high and dissolution, not permeability, is known to be the limiting factor for drug absorption in BCS class II drugs.
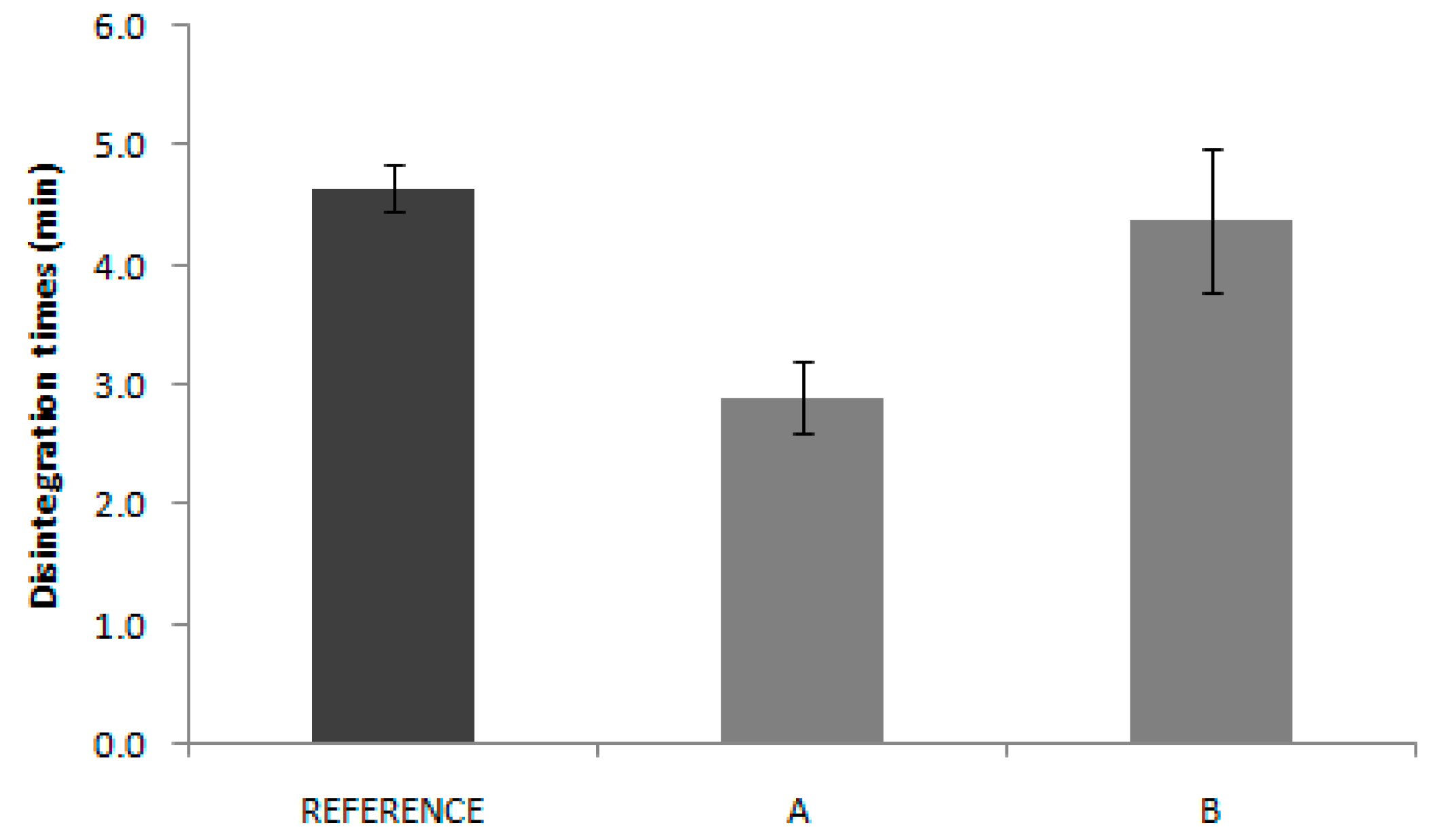
In the same way, disintegration test results are not reflected in the in vivo behavior. Reference disintegration time was 4.6 min, while Product A and B presented disintegrations times of 2.9 and 4.4 min, respectively. Some authors have proposed the use of the disintegration test as the quality control test, which eventually could be used as a biopredictive tool with adequate experimental conditions, but that only applies to highly soluble drugs where the disintegration is the limiting factor for dissolution. [
13] In the case of candesartan products, the pharmacopoeia conditions applied are not informative about the in vivo outcome.
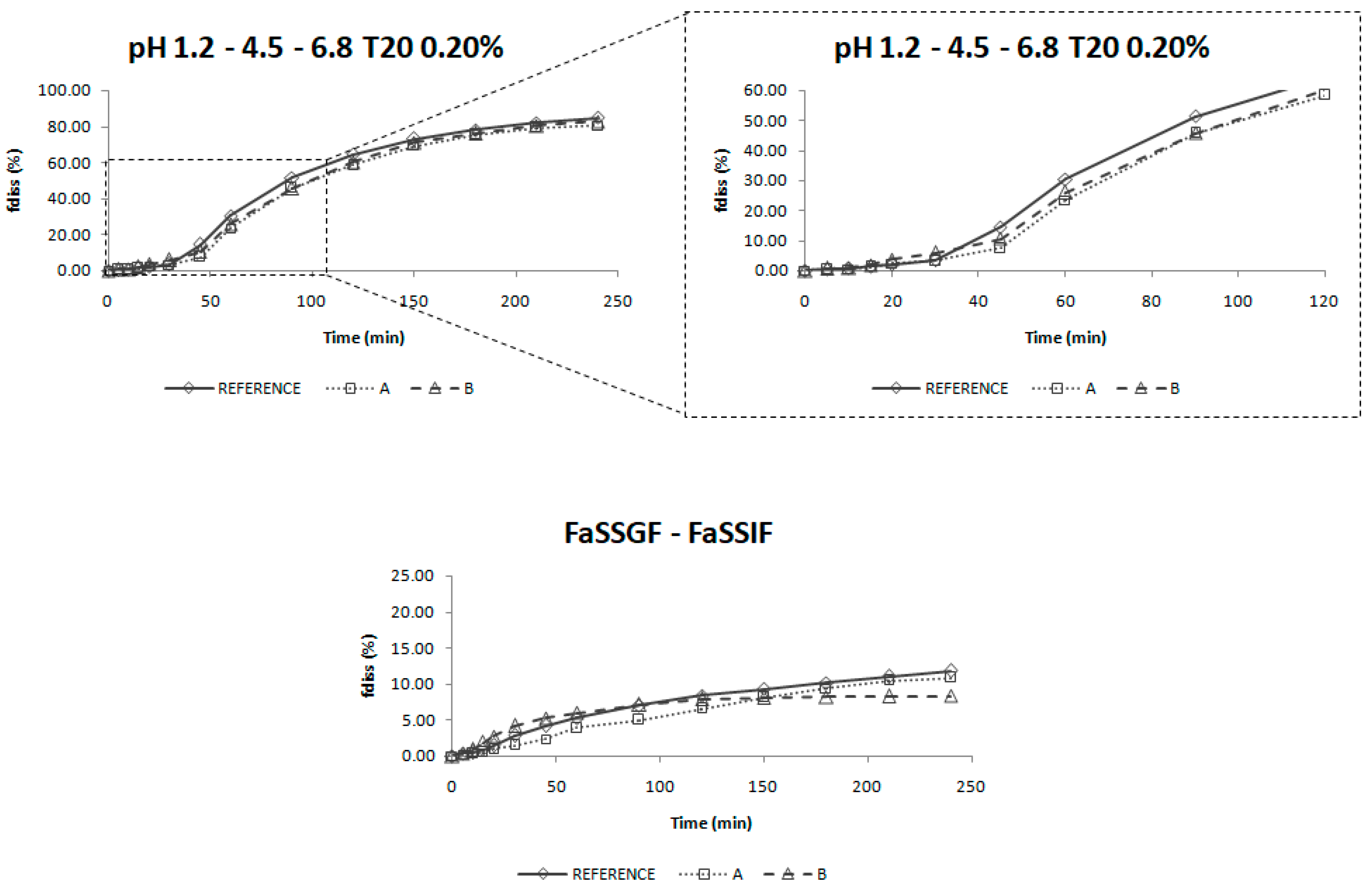
The dissolution profiles obtained in USP IV apparatus with a three-step pH change from 1.2 to 4.5 and 6.8 with 0.2% of Tween 20 were selected as biopredictive.
The incorporation of the surfactant in the dissolution media helps to reflect the surface tension of human gastrointestinal fluids which is found to be around 35–50 mN/m. The surface tension provided by Tween 20 at 0.2% is around 34 mN/m. [
33] Surface tension values in this range have demonstrated to be biorelevant for low solubility drugs. [
34] Moreover, the addition of surfactants helps to achieve sink conditions in the dissolution media [
35], which would reflect the in vivo conditions for a high permeability drug for which the absorption process maintain those sink conditions in the luminal fluids.
Nevertheless, the incorporation of a surfactant in the dissolution media is not enough to reflect the in vivo behavior, as the experiments performed in the USP II apparatus did not provide meaningful results. The change of media pH is also important to reflect candesartan cilexetil in vivo dissolution. As candesartan cilexetil solubility increases with pH, this leads to changes in its dissolution rate in vivo which has to be reflected in vitro in order to be biopredictive.
These dissolution conditions were able to order in vitro dissolution profiles in the same rank order as the in vivo ones and the f
2 values gave the same conclusion as the bioequivalence study. According to Taupitz and Klein, dissolution media modified with synthetic surfactants can be discriminatory to evaluate dissolution differences between formulations of drugs with low solubility, being the amount of surfactant a critical aspect to obtain predictive conditions [
36].
This in vitro dissolution method could be of application for other non-complex immediate release candesartan cilexetil formulations. Complex formulations with lipids for which digestion by intestinal enzymes could affect release or polymer formulations, which could help supersaturation processes may need a different in vitro dissolution method [
17,
37].
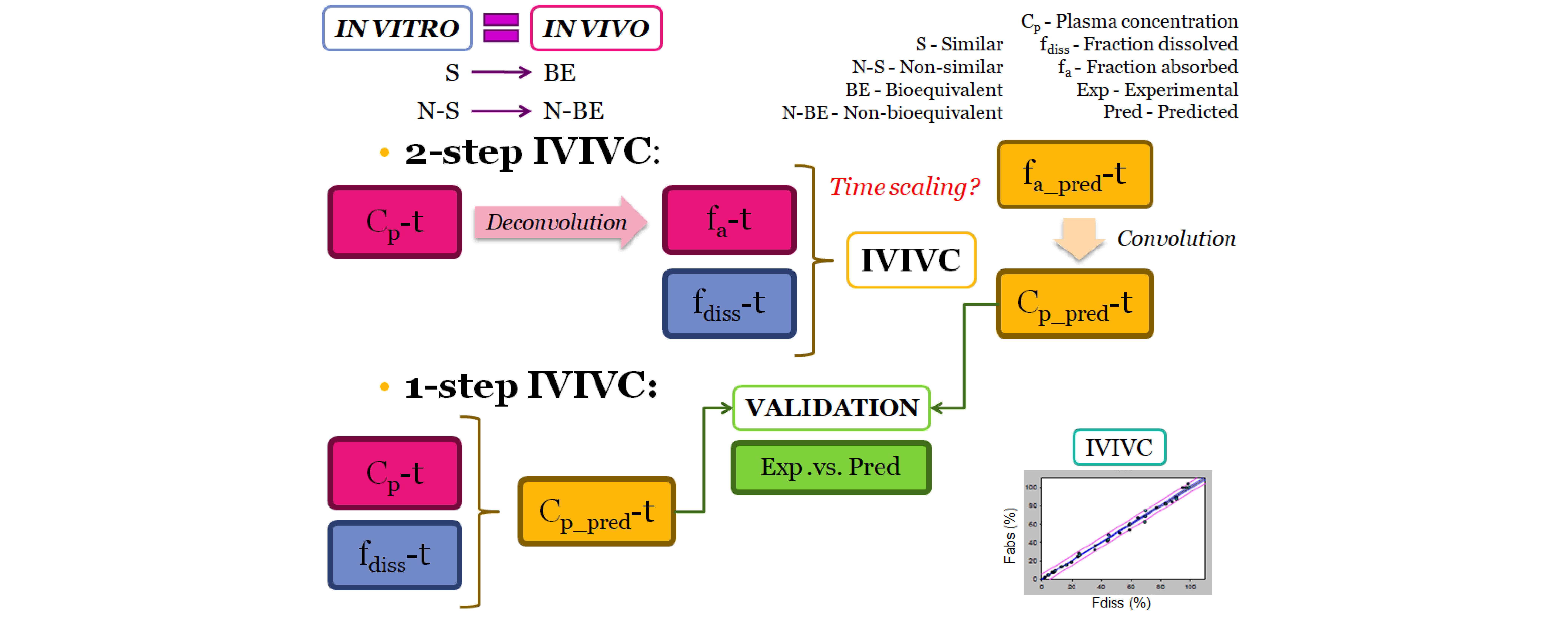
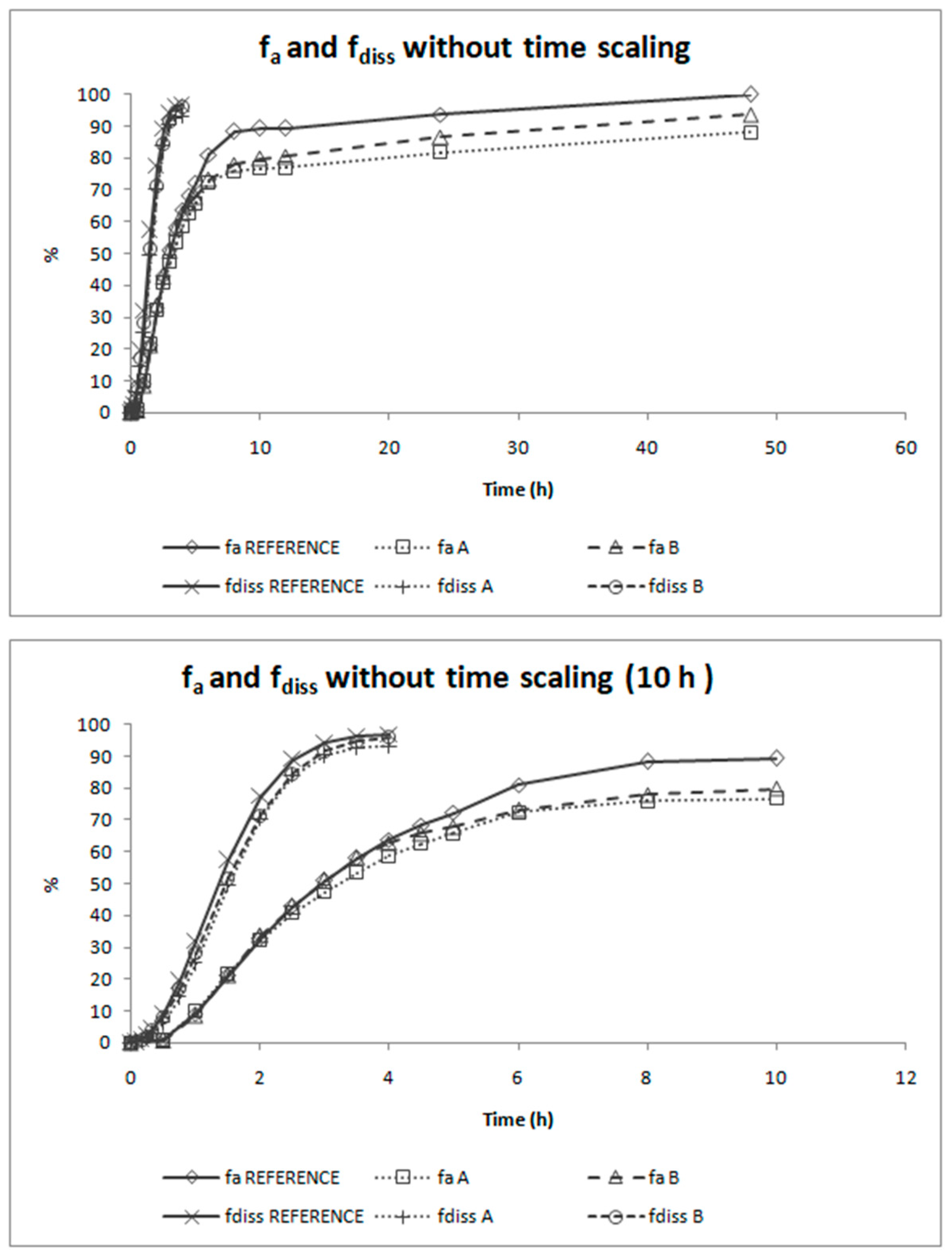
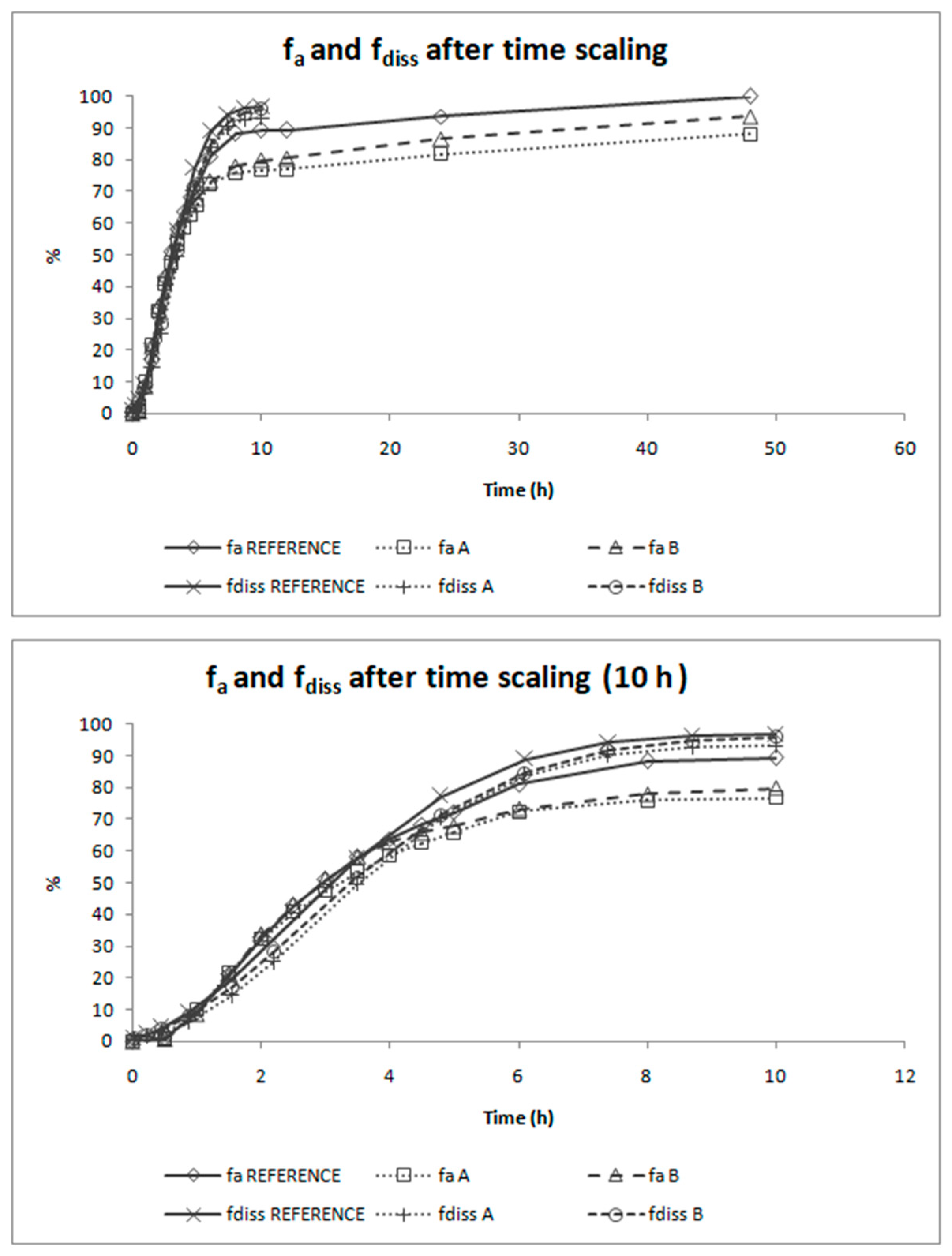
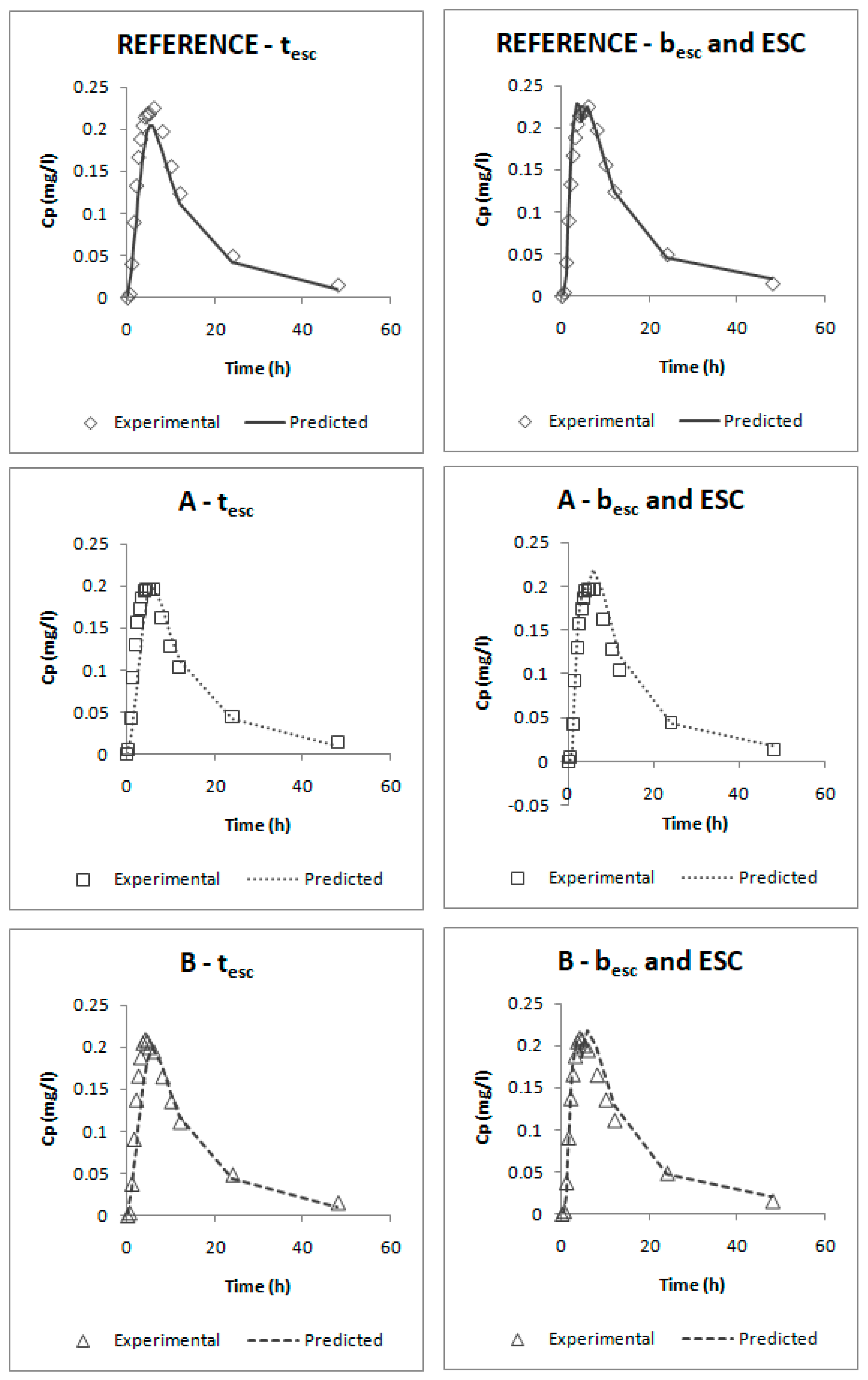
The deconvolution method used to obtain fractions absorbed from the in vivo data of candesartan cilexetil was the Loo-Riegelman method as candesartan follows a two-compartment disposition model. As Loo-Rielgeman mass balance lead to absorption profiles, which always reach 100%, but the dissolution profiles never reached complete dissolution it was necessary to scale in magnitude the in vitro profiles for comparing in vitro and in vivo data, as in
Figure 6.
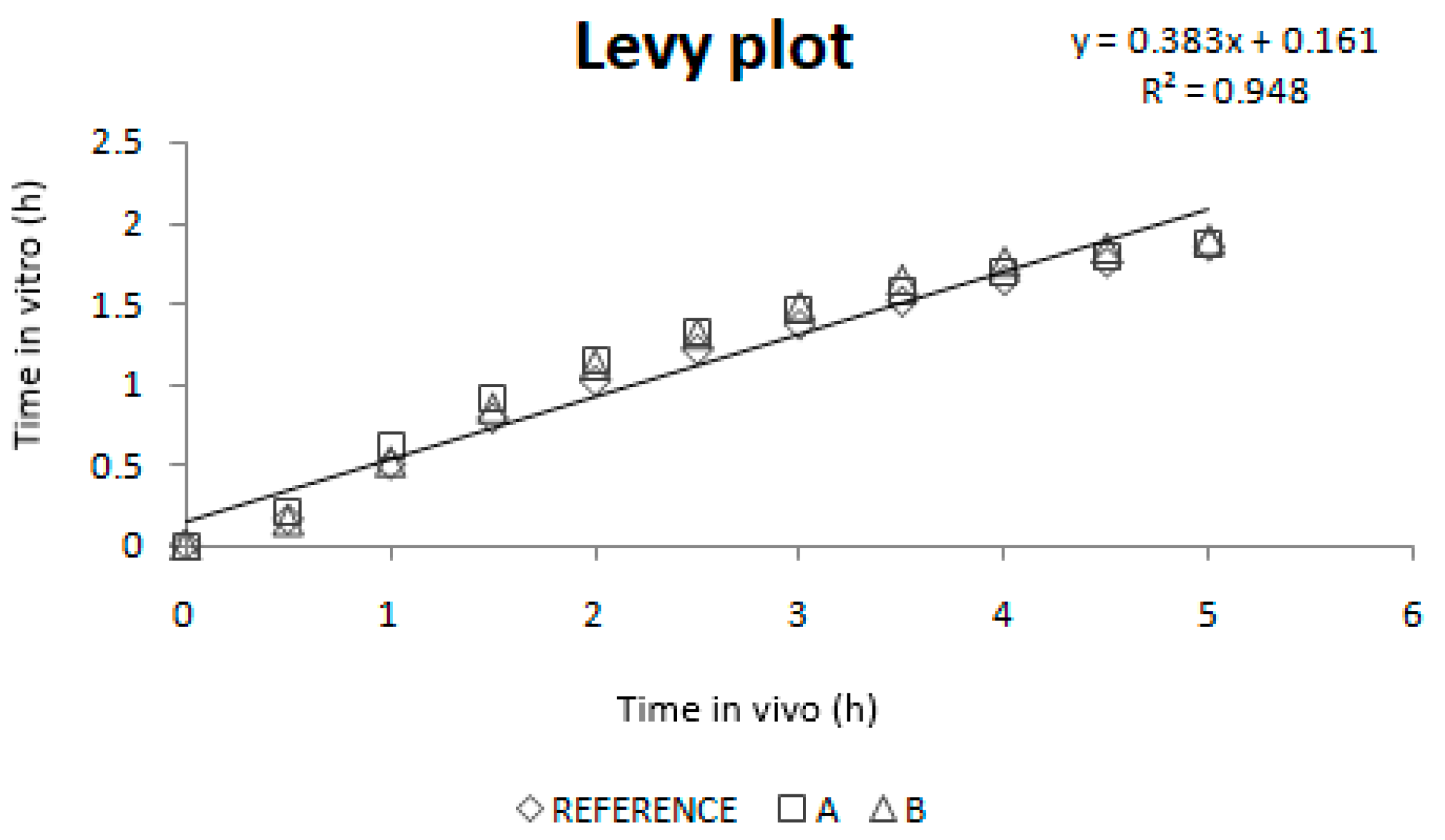
As dissolution and absorption processes did not happen at the same speed, since dissolution was much faster than absorption, a Levy Plot was constructed and the dissolution process was scaled with its equation (IRF). Once the time was scaled, the profiles overlapped and the IVIVCs could be obtained.
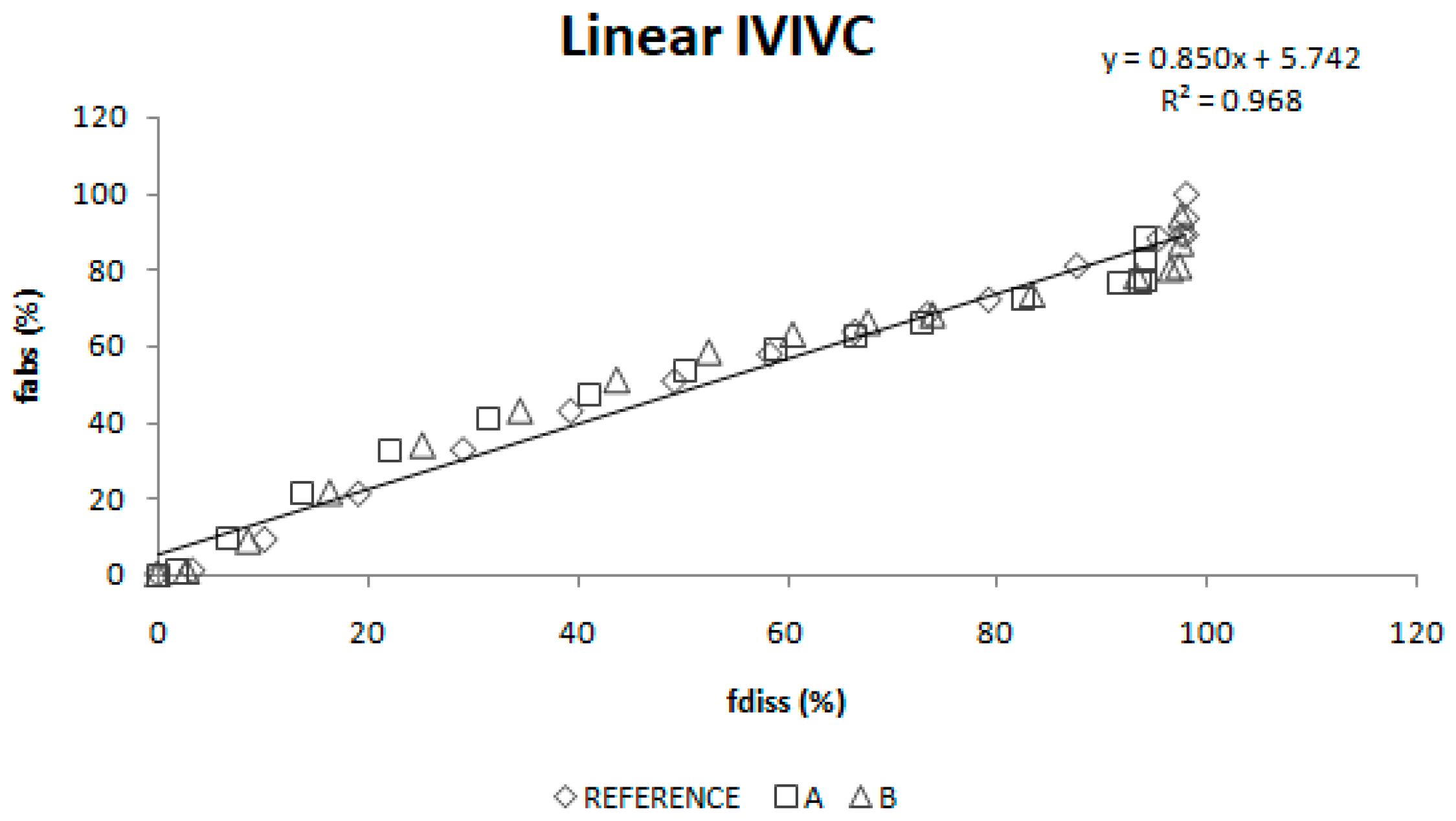
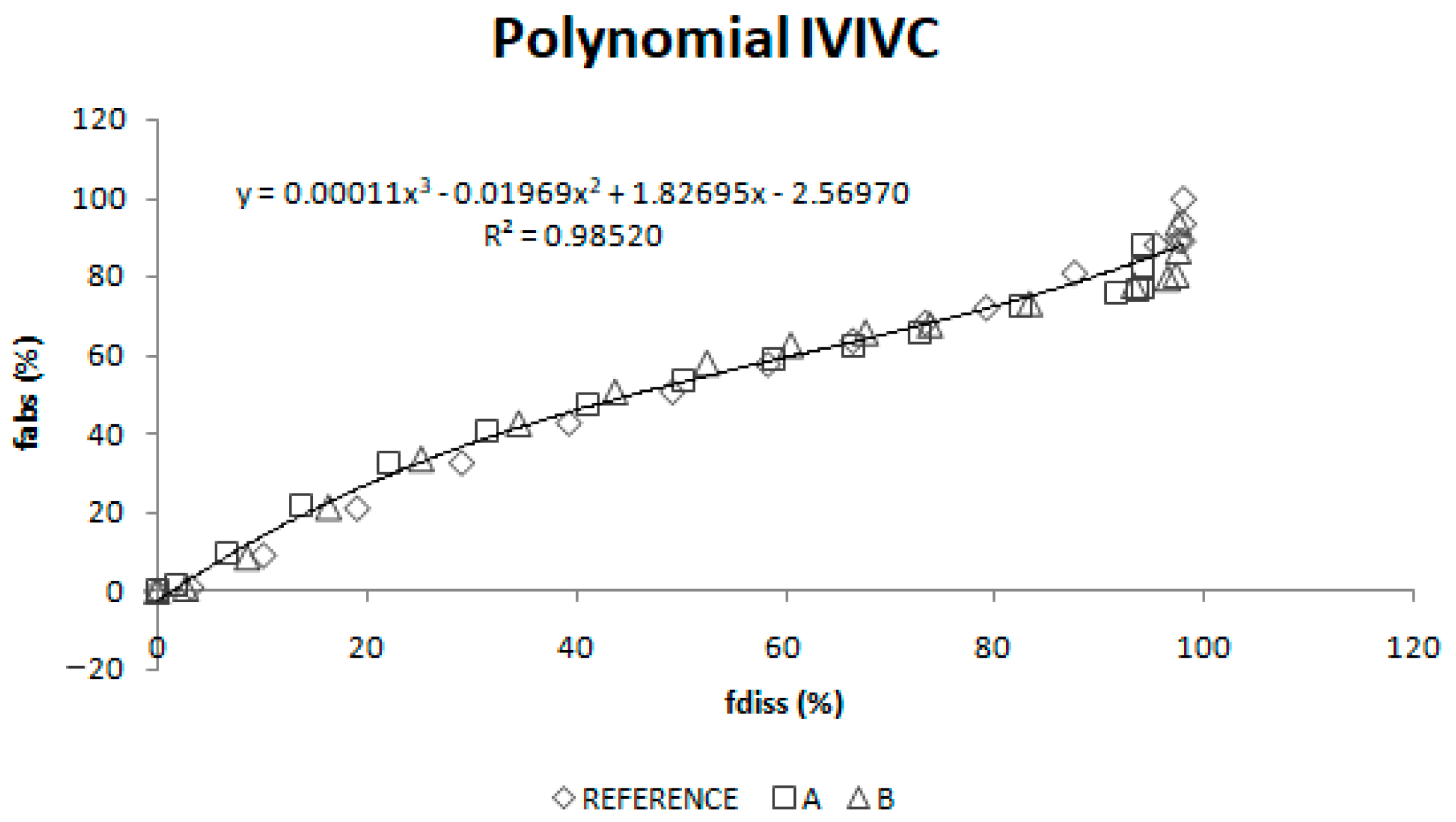
Two different two-step level A correlations were obtained, a linear one and a polynomial one, both with a coefficient of determination (r2) higher than 0.960. Polynomial IVIVC presented a higher r2 and an improved statistically significance on the sum of squared residuals (Fcal = 17.14 and Ftab = 3.19; p = 0.05). However, in this case, the most relevant aspect is the in vivo predictability, apart from the statistical evaluation of the correlation equation itself.
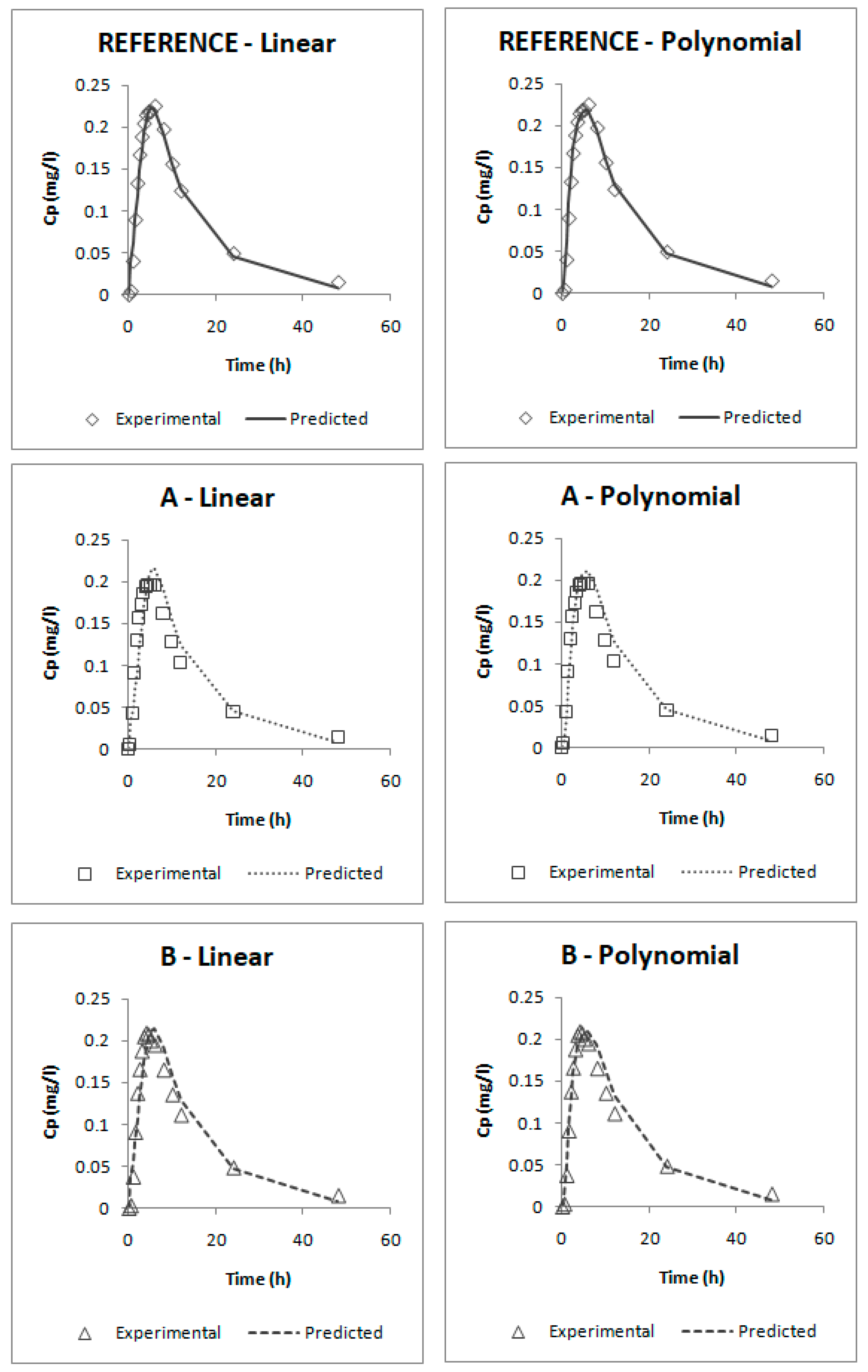
According to the internal validation results (
Table 5), all the prediction errors (PE%) are below the pre-established limits. The prediction error for each parameter and each product are below 15% and the mean percentage of prediction error for each parameter is lower than 10%, thus, both IVIVCs are valid and biopredictive.
Furthermore, as the prediction errors of both correlations are practically the same, their predictability can be considered similar. Applying the parsimony law, the simplest one should be selected, which is the linear IVIVC.
The one-step correlations can be also considered valid and biopredictive as, according to their internal validation (
Table 6), all the PE% are below the pre-established limits (the PE% for each parameter and each product are below 15% and the mean PE% for each parameter is lower than 10%).
Comparing both models, it can be seen that the one that uses the parameters besc and ESC gives lower errors; thus, this one would be preferred. This model was obtained by empirically dividing profiles in five segments:
In the first one, it was assumed that until t = 0.5 h, dissolution is faster than absorption; this happens when the drug is dissolved in the stomach and it cannot be absorbed.
From t = 0.5 h to 2 h, absorption would be faster than dissolution, which could correspond with an absorption window, similarly to that observed for valsartan with similar physicochemical characteristics. [
38,
39] In that period, dissolution is the limiting factor. These in vivo times correspond to the in vitro times 0.150 h and 1.100 h that, as can be observed on
Figure 5 (upper plot), correspond to the period in which dissolution rate showed the highest differences between reference and test products.
After that moment, from 2 h to 4.5 h and from 4.5 h to 10 h, absorption would be again slower than dissolution; this means that we should have a big amount of drug in the intestine that cannot be absorbed quickly. Looking at the predicted profiles at those times, a double peak in the highest plasma levels can be seen; this double peak simulates an enterohepatic circulation that returns part of the drug to the intestine, which, as has been said, cannot be absorbed.
The assumption of an enterohepatic cycle during candesartan disposition is supported by the data reported previously in some animal models [
40], and the enterohepatic cycle has been demonstrated in other drugs of the same family (Angiotensin II AT1-Receptor Antagonists) [
41].
The last stretch from t = 10 h until the end of the profile would correspond with the elimination phase. At those times, dissolution has almost finished.
From a regulatory point of view, all the correlations could be used as substitutes of human bioequivalence studies. Nevertheless, this work has some limitations: (1) As in the human bioequivalence study, the three products are bioequivalent, and the development of a biopredictivein vitro dissolution test is facilitated, since there cannot be a high discrimination capacity between products. (2) In the same way, the similarity between products makes the limit of applicability of the obtained IVIVCs very narrow, since extrapolation outside the observed in vitro dissolution rates is not allowed. (3) In addition, as no pharmacokinetic studies in which candesartan cilexetil was administered intravenously in humans were available, the calculation of the microconstants (kel, k12 and k21), necessary for obtaining the absorption profiles by the Loo-Riegelman method, was approximated from the data of an extravasal administration.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}