Automated Real-Time Tumor Pharmacokinetic Profiling in 3D Models: A Novel Approach for Personalized Medicine

 , , , and
, , , and
Abstract
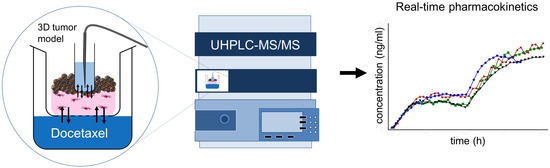
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
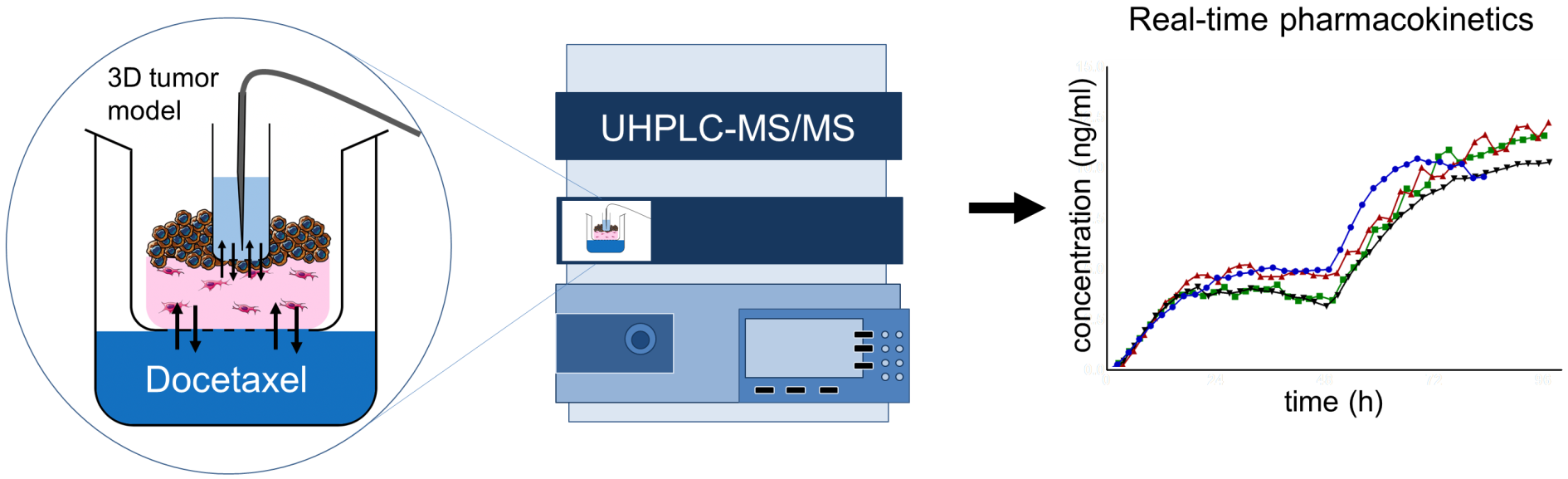
2.3. Sample Port Integration into Tumor Oral Mucosa (TOM) Models
2.4. Docetaxel Treatment of TOM Models
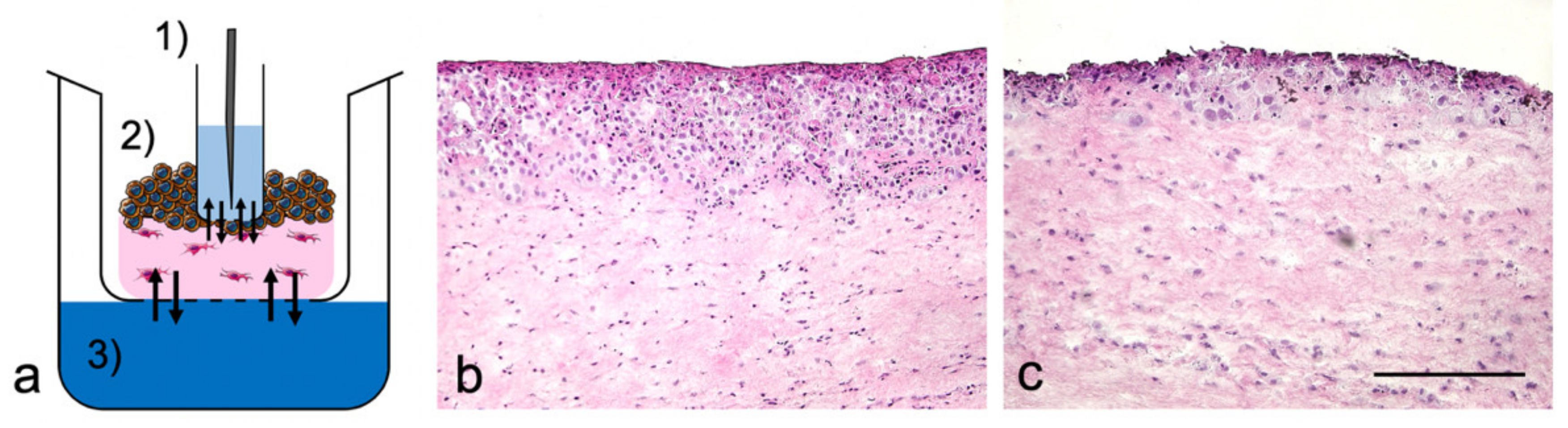
2.5. Morphological Analysis
2.6. UHPLC-MS/MS Analyses
2.7. Validation
2.8. Sample Preparation for the Identification of Degradation Products
2.9. Pharmacokinetic Analysis
3. Results
3.1. TOM Models with Sampling Port
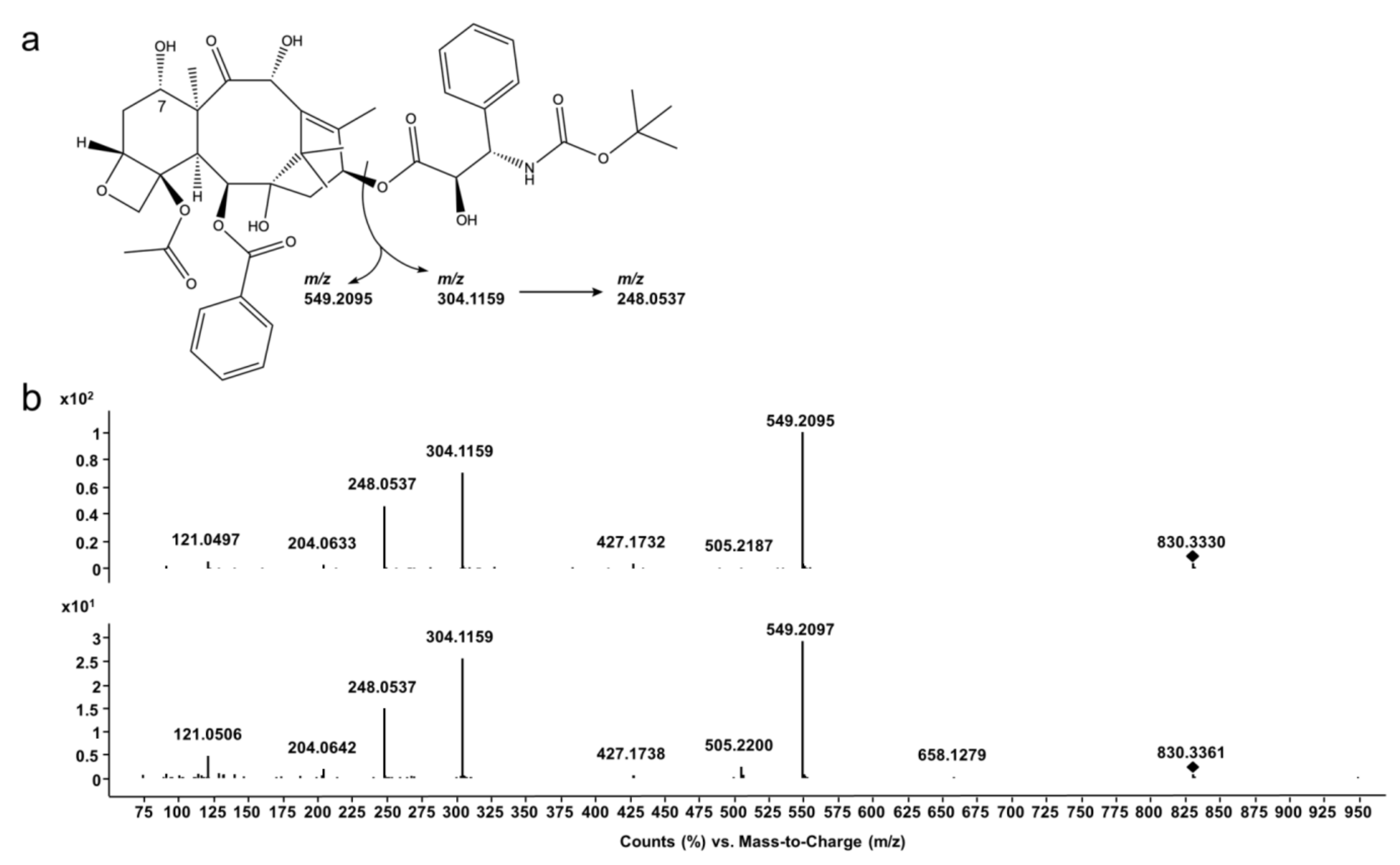
3.2. Docetaxel Epimerization and Degradation Products
3.3. Validation
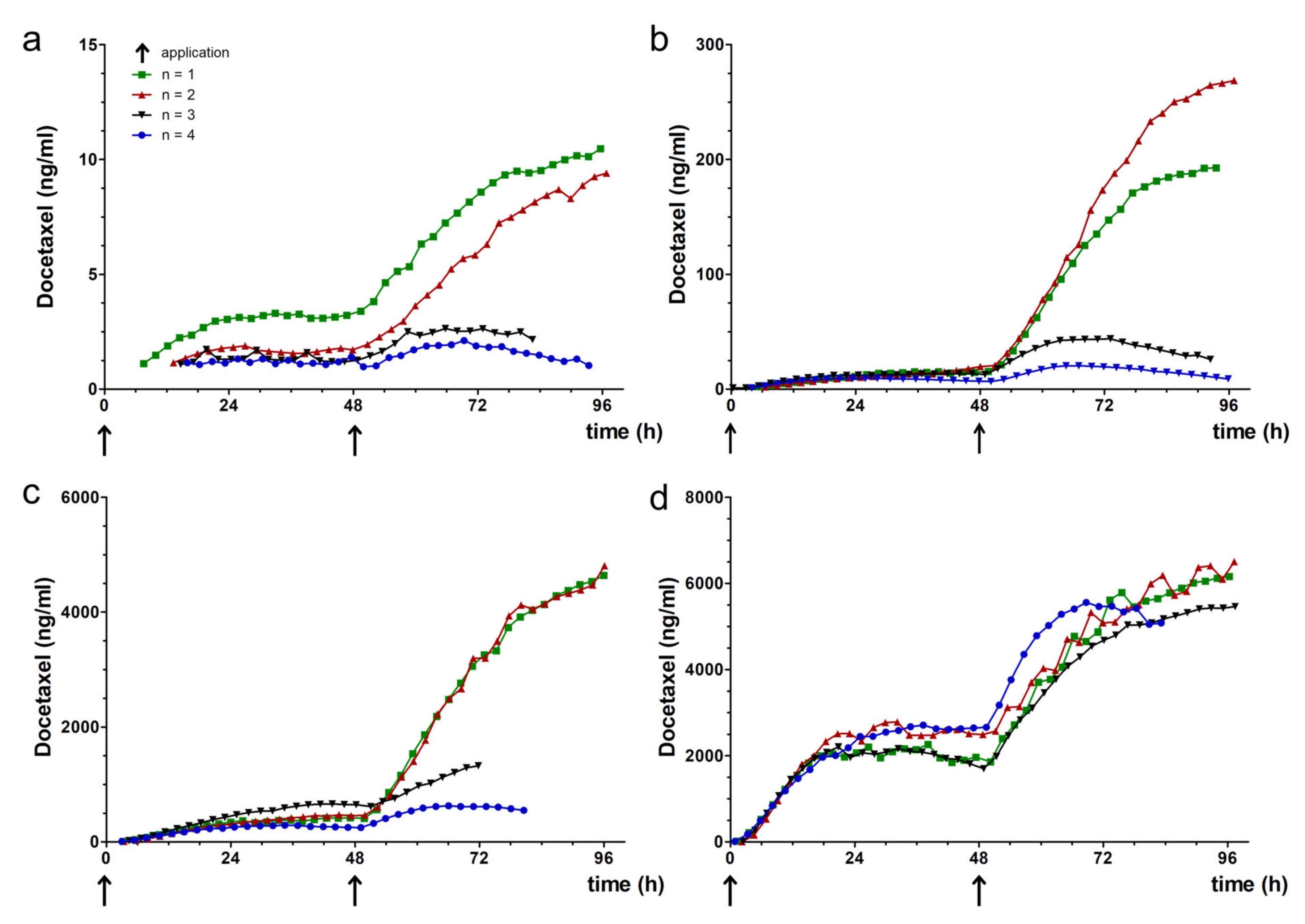
3.4. Docetaxel Pharmacokinetics in TOM Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Horvath, C.J.; Milton, M.N. The TeGenero incident and the Duff Report conclusions: A series of unfortunate events or an avoidable event? Toxicol. Pathol. 2009, 37, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Yu, Y.; Zheng, N.; Yang, Y.; Paholak, H.J.; Yu, L.X.; Sun, D. Applications of Human Pharmacokinetic Prediction in First-in-Human Dose Estimation. AAPS J. 2012, 14, 262–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, E.L.; Bento, A.F.; Cavalli, J.; Oliveira, S.K.; Schwanke, R.C.; Siqueira, J.M.; Freitas, C.S.; Marcon, R.; Calixto, J.B. Non-clinical studies in the process of new drug development - Part II: Good laboratory practice, metabolism, pharmacokinetics, safety and dose translation to clinical studies. Braz. J. Med. Biol. Res. 2016, 49, e5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, L.; Rubin, E.H.; Mehrotra, N.; Pinheiro, J.; Fernandes, L.L.; Roy, A.; Bailey, S.; de Alwis, D.P. Rendering the 3 + 3 Design to Rest: More Efficient Approaches to Oncology Dose-Finding Trials in the Era of Targeted Therapy. Clin. Cancer Res. 2016, 22, 2623–2629. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.H.; Wientjes, M.G.; Lu, D.; Au, J.L. Drug delivery and transport to solid tumors. Pharm. Res. 2003, 20, 1337–1350. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef]
- Saleem, A.; Price, P.M. Early tumor drug pharmacokinetics is influenced by tumor perfusion but not plasma drug exposure. Clin. Cancer Res. 2008, 14, 8184–8190. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.H.; Wientjes, M.G.; Au, J.L. Determinants of paclitaxel uptake, accumulation and retention in solid tumors. Invest. New Drugs 2001, 19, 113–123. [Google Scholar] [CrossRef]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef] [Green Version]
- Schütte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, J.; Pi, Z.; Zheng, Z.; Xing, J.; Song, F.; Liu, S.; Liu, Z. Application of online microdialysis coupled with liquid chromatography-tandem mass spectrometry method in assessing neuroprotective effect of Rhizoma coptidis on diabetic rats. Anal. Methods 2015, 7, 45–52. [Google Scholar] [CrossRef]
- Graf, C.; Rühl, E. Imaging Techniques for Probing Nanoparticles in Cells and Skin. In Biological Responses to Nanoscale Particles; Springer: Berlin, Germany, 2019; pp. 213–239. [Google Scholar]
- Rheinwald, J.G.; Beckett, M.A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981, 41, 1657–1663. [Google Scholar] [PubMed]
- Gronbach, L.; Wolff, C.; Klinghammer, K.; Stellmacherc, J.; Jurmeisterd, P.; Alexievc, U.; Schäfer-Kortinga, M.; Tinhofer, I.; Keilholz, U.; Zoschke, C. A multilayered epithelial mucosa model of head neck squamous cell carcinoma for analysis of tumor-microenvironment interactions and drug development. Clin. Cancer Res. (in revision).
- EMA. Guideline on Bioanalytical Method Validation. 2011. Available online: ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 2 February 2020).
- EMA. ICH Topic Q 2 (R1) Validation of Analytical Procedures: Text and Methodology. 1995. Available online: ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_en.pdf (accessed on 2 February 2020).
- R Development Core Team. R: A language and environment for statistical computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Rao, B.M.; Chakraborty, A.; Srinivasu, M.K.; Devi, M.L.; Kumar, P.R.; Chandrasekhar, K.B.; Srinivasan, A.K.; Prasad, A.S.; Ramanatham, J. A stability-indicating HPLC assay method for docetaxel. J. Pharm. Biomed. Anal. 2006, 41, 676–681. [Google Scholar] [CrossRef]
- Kumar, D.; Tomar, R.S.; Deolia, S.K.; Mitra, M.; Mukherjee, R.; Burman, A.C. Isolation and characterization of degradation impurities in docetaxel drug substance and its formulation. J. Pharm. Biomed. 2007, 43, 1228–1235. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Fujikawa, A.; Ito, H.; Tanaka, N.; Furugen, A.; Miyamori, K.; Takahashi, N.; Ogura, J.; Kobayashi, M.; Yamada, T.; et al. A rapid and sensitive LC/ESI–MS/MS method for quantitative analysis of docetaxel in human plasma and its application to a pharmacokinetic study. J. Chromatogr. B Biomed. Appl. 2012, 893–894, 157–161. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Houghton, P. A proposal regarding reporting of in vitro testing results. Clin. Cancer Res. 2013, 19, 2828–2833. [Google Scholar] [CrossRef] [Green Version]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- EMA. European Public Assessment Report “TAXOTERE”, Annex l - Summary of Product. 2019. Available online: ema.europa.eu/documents/product-information/taxotere-epar-product-information_en.pdf (accessed on 2 February 2020).
- Mohsin, S.; Arellano, I.H.; Choudhury, N.R.; Garg, S. Docetaxel epimerization in silicone films: a case of drug excipient incompatibility. Drug Test. Anal. 2014, 6, 1076–1084. [Google Scholar] [CrossRef]
- Vuilhorgne, M.; Gaillard, C.; Sanderink, G.J.; Royer, I.; Monsarrat, B.; Dubois, J.; Wright, M. Metabolism of Taxoid Drugs. In Taxane Anticancer Agents; American Chemical Society: Washington, DC, USA, 1994; Volume 583, pp. 98–110. [Google Scholar]
- Tian, J.; Stella, V.J. Degradation of paclitaxel and related compounds in aqueous solutions I: Epimerization. J. Pharm. Sci. 2008, 97, 1224–1235. [Google Scholar] [CrossRef]
- Song, D.; Hsu, L.F.; Au, J.L. Binding of taxol to plastic and glass containers and protein under in vitro conditions. J. Pharm. Sci. 1996, 85, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Schuck, V.J.; Rinas, I.; Derendorf, H. In vitro microdialysis sampling of docetaxel. J. Pharm. Biomed. Anal. 2004, 36, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K.Y.; Church, M.K.; Clough, G.F.; Quist, S.R.; Schmelz, M.; Skov, P.S.; Anderson, C.D.; Tannert, L.K.; Giménez-Arnau, A.M.; Frischbutter, S.; et al. Skin microdialysis: methods, applications and future opportunities—an EAACI position paper. Clin. Transl. Allergy 2019, 9, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqua, O.E.D. PKPD and Disease Modeling: Concepts and Applications to Oncology. In Clinical Trial Simulations: Applications and Trends; Kimko, H.H.C., Peck, C.C., Eds.; Springer: New York, NY, USA, 2011; pp. 281–306. [Google Scholar] [CrossRef]
- Patteet, L.; Maudens, K.E.; Stove, C.P.; Lambert, W.E.; Morrens, M.; Sabbe, B.; Neels, H. The use of dried blood spots for quantification of 15 antipsychotics and 7 metabolites with ultra-high performance liquid chromatography - tandem mass spectrometry. Drug Test Anal. 2015, 7, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Aller, M.; Gurny, R.; Veuthey, J.L.; Guillarme, D. Coupling ultra high-pressure liquid chromatography with mass spectrometry: Constraints and possible applications. J. Chromatogr. A 2013, 1292, 2–18. [Google Scholar] [CrossRef]
- Novakova, L.; Pavlik, J.; Chrenkova, L.; Martinec, O.; Cerveny, L. Current antiviral drugs and their analysis in biological materials - Part II: Antivirals against hepatitis and HIV viruses. J. Pharm. Biomed. Anal. 2018, 147, 378–399. [Google Scholar] [CrossRef]
- Gorgens, C.; Guddat, S.; Schanzer, W.; Thevis, M. Screening and confirmation of myo-inositol trispyrophosphate (ITPP) in human urine by hydrophilic interaction liquid chromatography high resolution / high accuracy mass spectrometry for doping control purposes. Drug Test. Anal. 2014, 6, 1102–1107. [Google Scholar] [CrossRef]
- Ambrosio, G.; Joseph, J.F.; Wuest, B.; Mazzarino, M.; de la Torre, X.; Diel, P.; Botre, F.; Parr, M.K. Detection and quantitation of ecdysterone in human serum by liquid chromatography coupled to tandem mass spectrometry. Steroids 2020, 157, 108603. [Google Scholar] [CrossRef]
- Holder, B.R.; McNaney, C.A.; Luchetti, D.; Schaeffer, E.; Drexler, D.M. Bioanalysis of acetylcarnitine in cerebrospinal fluid by HILIC-mass spectrometry. Biomed. Chromatogr. 2015, 29, 1375–1379. [Google Scholar] [CrossRef]
- Kong, T.Y.; Kim, J.H.; Kim, J.Y.; In, M.K.; Choi, K.H.; Kim, H.S.; Lee, H.S. Rapid analysis of drugs of abuse and their metabolites in human urine using dilute and shoot liquid chromatography-tandem mass spectrometry. Arch. Pharm. Res. 2017, 40, 180–196. [Google Scholar] [CrossRef]
- Bourgogne, E.; Wagner, M. [Sample preparation and bioanalysis in mass spectrometry]. Ann. Biol. Clin. (Paris) 2015, 73, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Parr, M.K.; Ambrosio, G.; Wuest, B.; Mazzarino, M.; de la Torre, X.; Sibilia, F.; Joseph, J.F.; Diel, P.; Botrè, F. Targeting the Administration of Ecdysterone in Doping Control Samples. bioRxiv 2019. [Google Scholar] [CrossRef]
- Plock, N.; Kloft, C. Microdialysis--theoretical background and recent implementation in applied life-sciences. Eur. J. Pharm. Sci. 2005, 25, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, J.J.; Hillebrand, M.J.; Thijssen, B.; Rosing, H.; Schinkel, A.H.; Schellens, J.H.; Beijnen, J.H. A sensitive combined assay for the quantification of paclitaxel, docetaxel and ritonavir in human plasma using liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 2984–2990. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Precursor Ion (m/z) | Product Ion (m/z) | Collision Energy | Cell Accelerator Voltage | Polarity |
|---|---|---|---|---|
| 830.3 | 549.1 | 25 | 4 | Positive |
| 830.3 | 304.1 | 20 | 2 | Positive |
| Degradation Product | Formula | RT (min) | m/z | Exact Mass | Adduct | Mass Accuracy (ppm) |
|---|---|---|---|---|---|---|
| Carbamate | C38H45NO12 | 4.30 | 708.3010 | 708.3015 | [M + H]+ | 0.65 |
| 10DABIII | C29H36O10 | 4.55 | 545.2378 | 545.2381 | [M + H]+ | 0.60 |
| Epi-carbamate | C38H45NO12 | 4.72 | 708.3004 | 708.3015 | [M + H]+ | 1.47 |
| Epi-10DABIII | C29H36O10 | 5.31 | 567.2196 | 567.2201 | [M + Na]+ | 0.76 |
| Oxo-10DABIII | C29H34O10 | 5.40 | 565.2041 | 565.2044 | [M + Na]+ | 0.56 |
| Epi-oxo-10DABIII | C29H34O10 | 5.84 | 565.2040 | 565.2044 | [M + Na]+ | 0.67 |
| Docetaxel | C43H53NO14 | 7.95 | 830.3374 | 830.3358 | [M + Na]+ | −1.9 |
| Epi-Docetaxel | C43H53NO14 | 9.08 | 830.3377 | 830.3358 | [M + Na]+ | −2.26 |
| Oxo-Docetaxel | C43H51NO14 | 9.94 | 828.3200 | 828.3202 | [M + Na]+ | 0.19 |
| Epi-oxo-Docetaxel | C43H51NO14 | 11.07 | 828.3192 | 828.3202 | [M + Na]+ | 1.16 |
| QC | Within-day (n = 5) | Between-day (n = 5) | |||||
|---|---|---|---|---|---|---|---|
| Expected c (ng/mL) | Mean Calculated c (ng/mL) | CV (%) | RE (%) | Mean Calculated c (ng/mL) | CV (%) | RE (%) | |
| LLOQ | 1.00 | 1.06 | 7.26 | 5.71 | 1.01 | 11.40 | 1.62 |
| LQC | 3.00 | 2.76 | 2.27 | −7.87 | 2.67 | 3.37 | −10.94 |
| MQC | 3000 | 3027 | 7.83 | 0.89 | 3254 | 9.48 | 8.48 |
| HQC | 7500 | 6982 | 7.79 | −6.91 | 7676 | 9.57 | 2.35 |
| c | AUC (0–48 h) | Cmax (0–48 h) | tmax (0–48 h) | |||
| (ng/mL) | Mean (h × ng/mL) | CV (%) | Mean (ng/mL) | CV (%) | tmax (h) | CV (%) |
| 7 | 66.3 | 44.6 | 1.9 | 194.5 | 43.9 | 14.5 |
| 70 | 444.4 | 12.5 | 14.4 | 25.3 | 39.5 | 20.7 |
| 700 | 13,324 | 26.0 | 461 | 29.0 | 41 | 9.9 |
| 7000 | 85,658 | 8.3 | 2492 | 10.4 | 32 | 21.4 |
| c | AUC (48–96 h) | Cmax (48–96 h) | tmax (48–96 h) | |||
| (ng/mL) | Mean (h × ng/mL) | CV (%) | Mean (ng/mL) | CV (%) | tmax (h) | CV (%) |
| 7 | 151.4 | 84.4 | 6.0 | 65.1 | 82.8 | 16.2 |
| 70 | 3915.1 | 75.0 | 131.4 | 78.5 | 82.7 | 15.5 |
| 700 | 78,890 | 75.1 | 2850 | 66.3 | 83 | 16.6 |
| 7000 | 211,171 | 12.2 | 5920 | 7.3 | 90 | 13.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, J.F.; Gronbach, L.; García-Miller, J.; Cruz, L.M.; Wuest, B.; Keilholz, U.; Zoschke, C.; Parr, M.K. Automated Real-Time Tumor Pharmacokinetic Profiling in 3D Models: A Novel Approach for Personalized Medicine. Pharmaceutics 2020, 12, 413. https://doi.org/10.3390/pharmaceutics12050413
Joseph JF, Gronbach L, García-Miller J, Cruz LM, Wuest B, Keilholz U, Zoschke C, Parr MK. Automated Real-Time Tumor Pharmacokinetic Profiling in 3D Models: A Novel Approach for Personalized Medicine. Pharmaceutics. 2020; 12(5):413. https://doi.org/10.3390/pharmaceutics12050413
Chicago/Turabian StyleJoseph, Jan F., Leonie Gronbach, Jill García-Miller, Leticia M. Cruz, Bernhard Wuest, Ulrich Keilholz, Christian Zoschke, and Maria K. Parr. 2020. "Automated Real-Time Tumor Pharmacokinetic Profiling in 3D Models: A Novel Approach for Personalized Medicine" Pharmaceutics 12, no. 5: 413. https://doi.org/10.3390/pharmaceutics12050413