Recombinant Peptide Production Platform Coupled with Site-Specific Albumin Conjugation Enables a Convenient Production of Long-Acting Therapeutic Peptide


Abstract
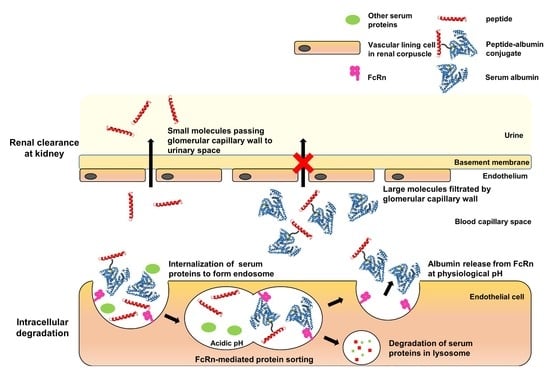
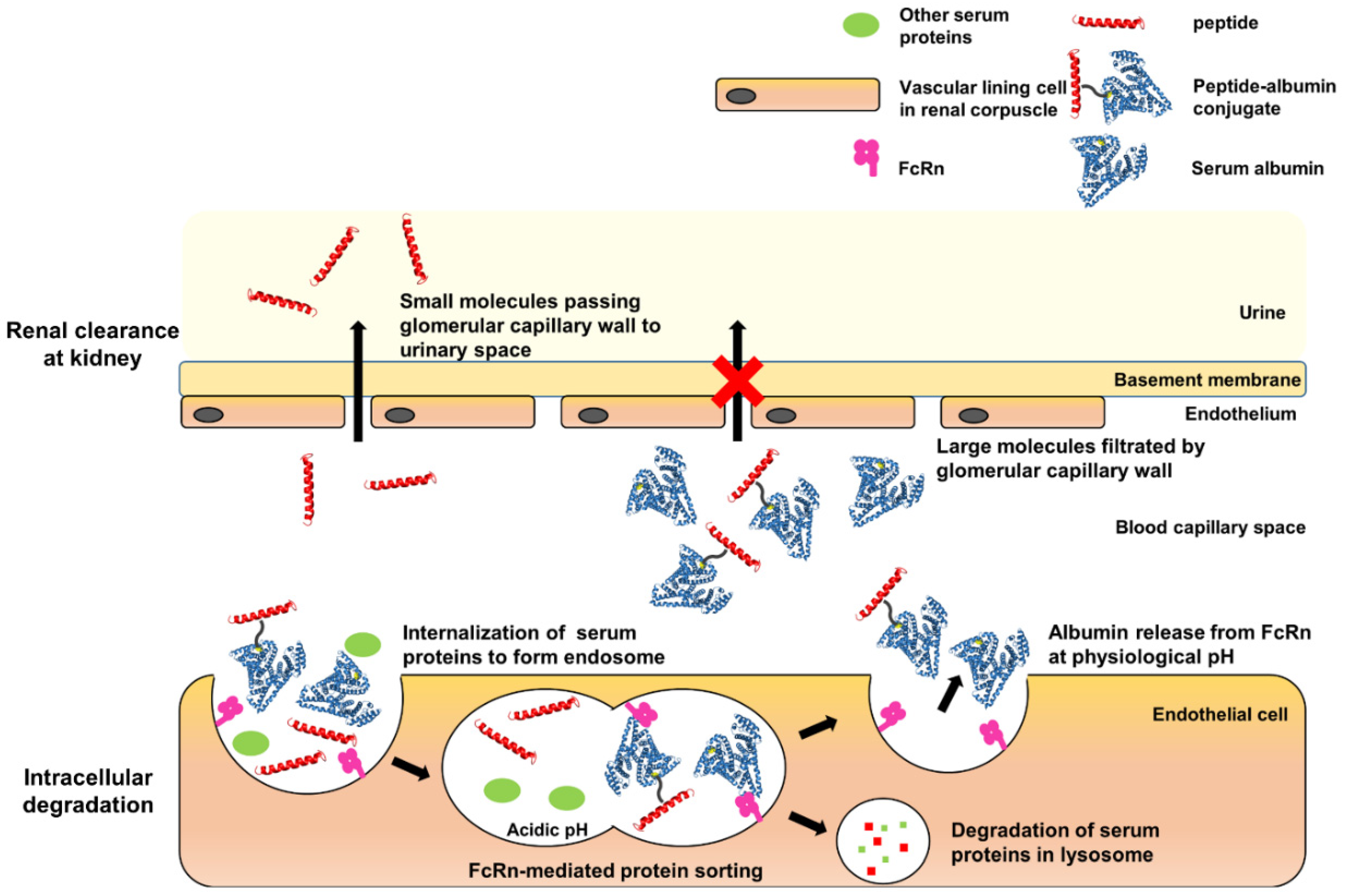
:
{kind=link}
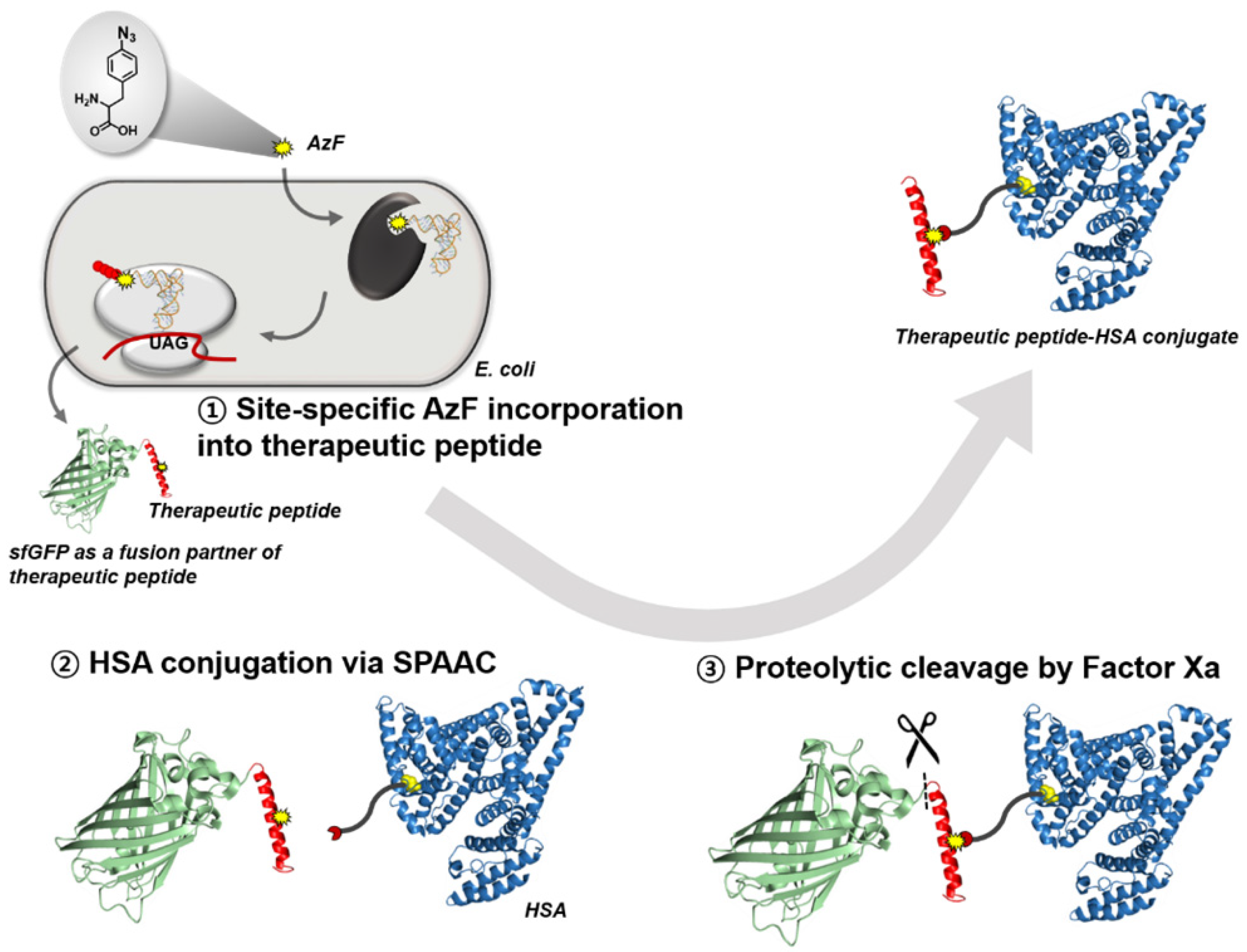{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Expression and Purification of sfGFP and GLP-1 Fusion Protein
2.3. Preparation of HSA Conjugate of GLP-1
2.4. Mass Spectrometric Analysis
2.5. Labeling of sfGFP-GLP1_AzF by SPAAC
2.6. In Vitro Activity ASSAY
2.7. In Vitro GLP-1-Albumin Enzyme-Linked Immune Sorbent Assay
2.8. Pharmacokinetic Studies of GLP-1-HSA Conjugates
2.9. In Vivo Intraperitoneal Glucose Tolerance Test (IPGTT)
3. Results and Discussion
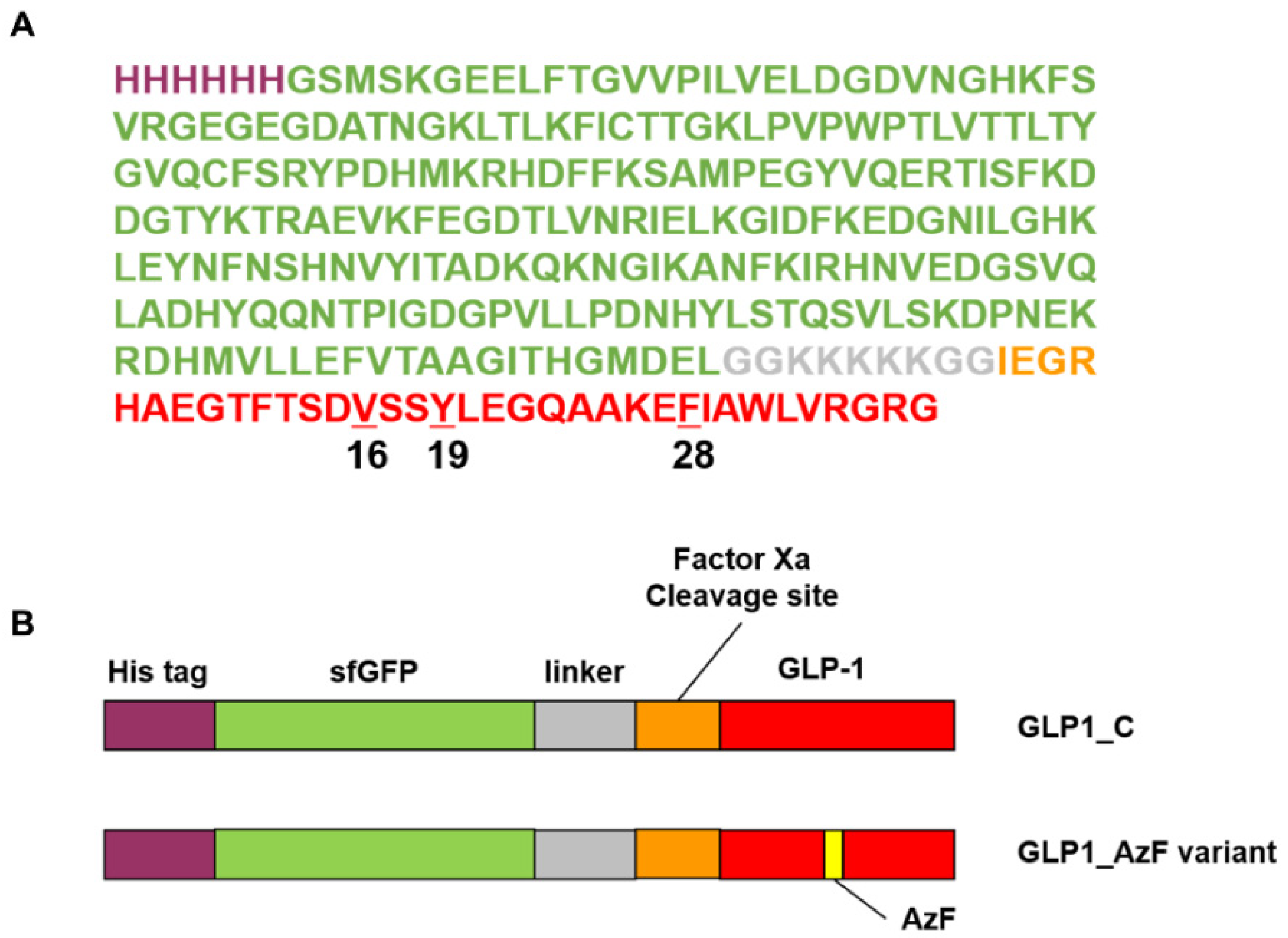
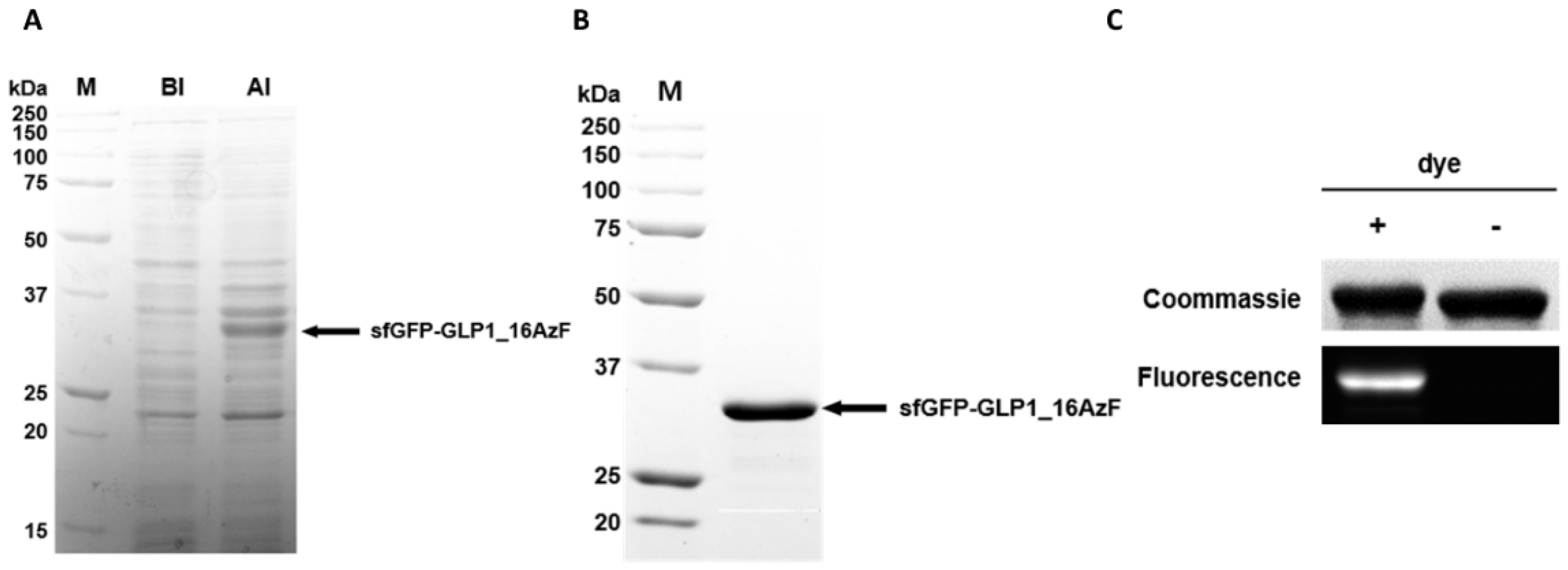
3.1. Site-Specific Incorporation of AzF into V16, Y19, or F28 Site of sfGFP-GLP1 Fusion Protein
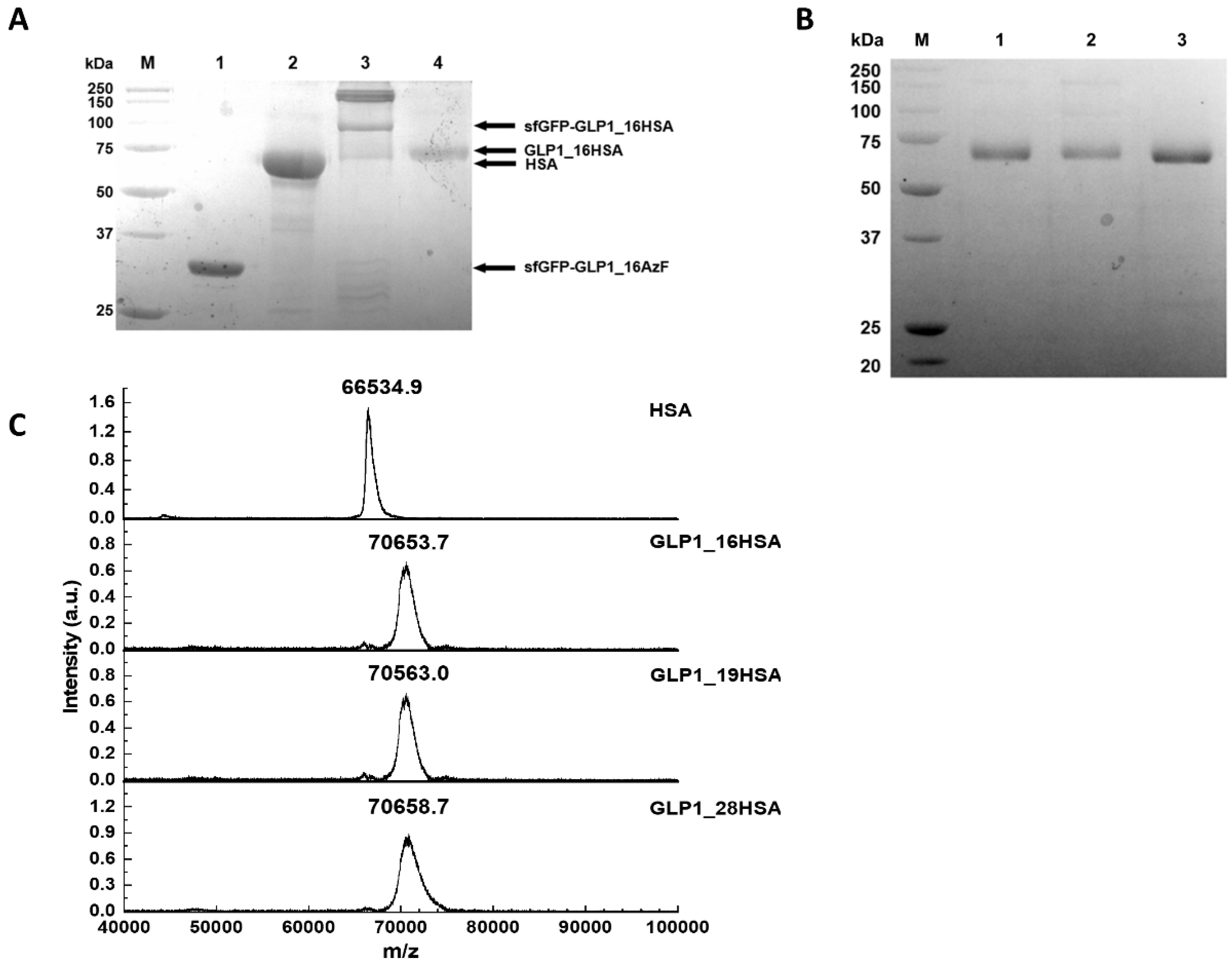
3.2. Preparation of Site-Specifically Albuminconjugated GLP1_HSA Variants
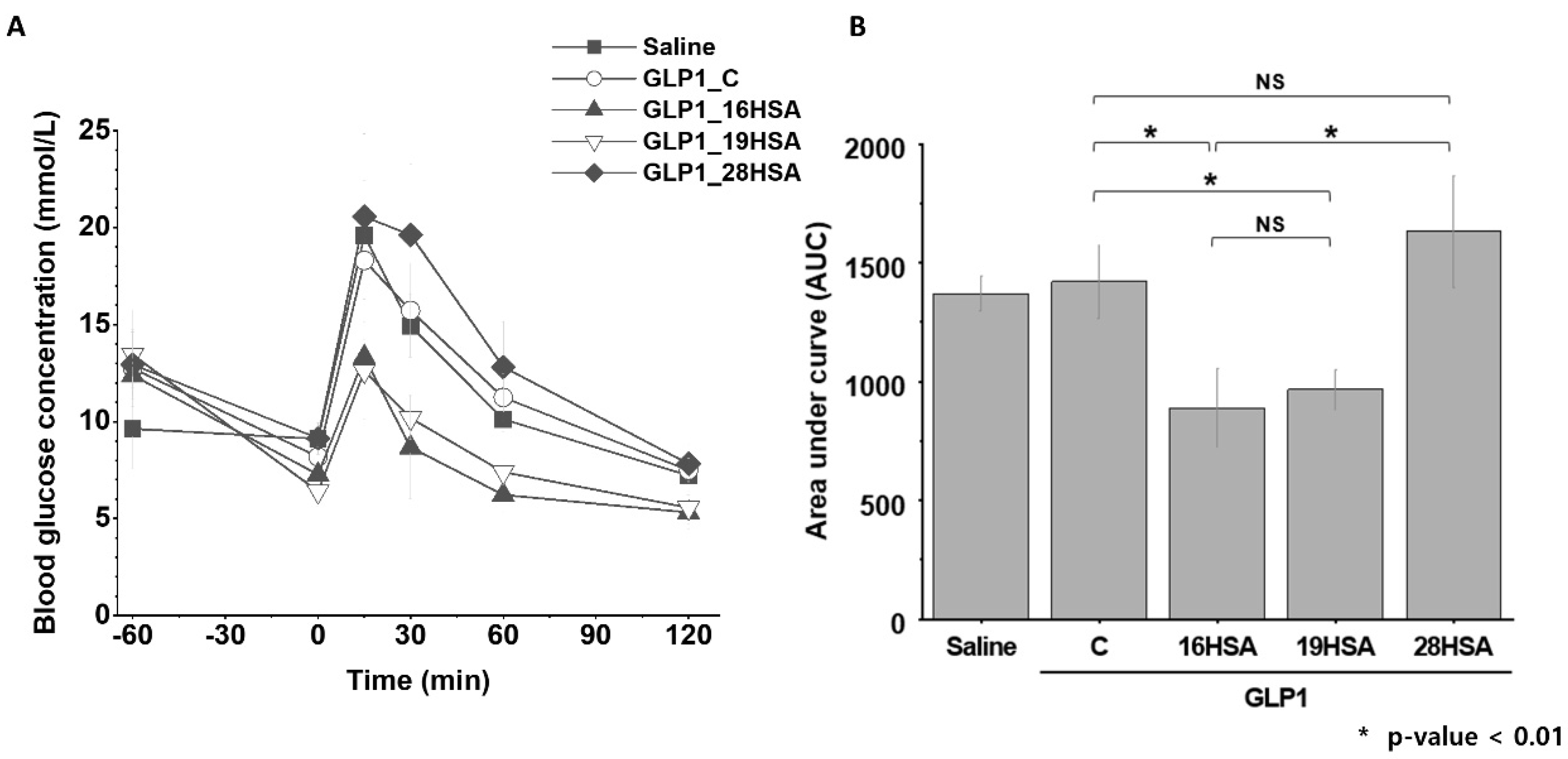
3.3. In Vivo Study
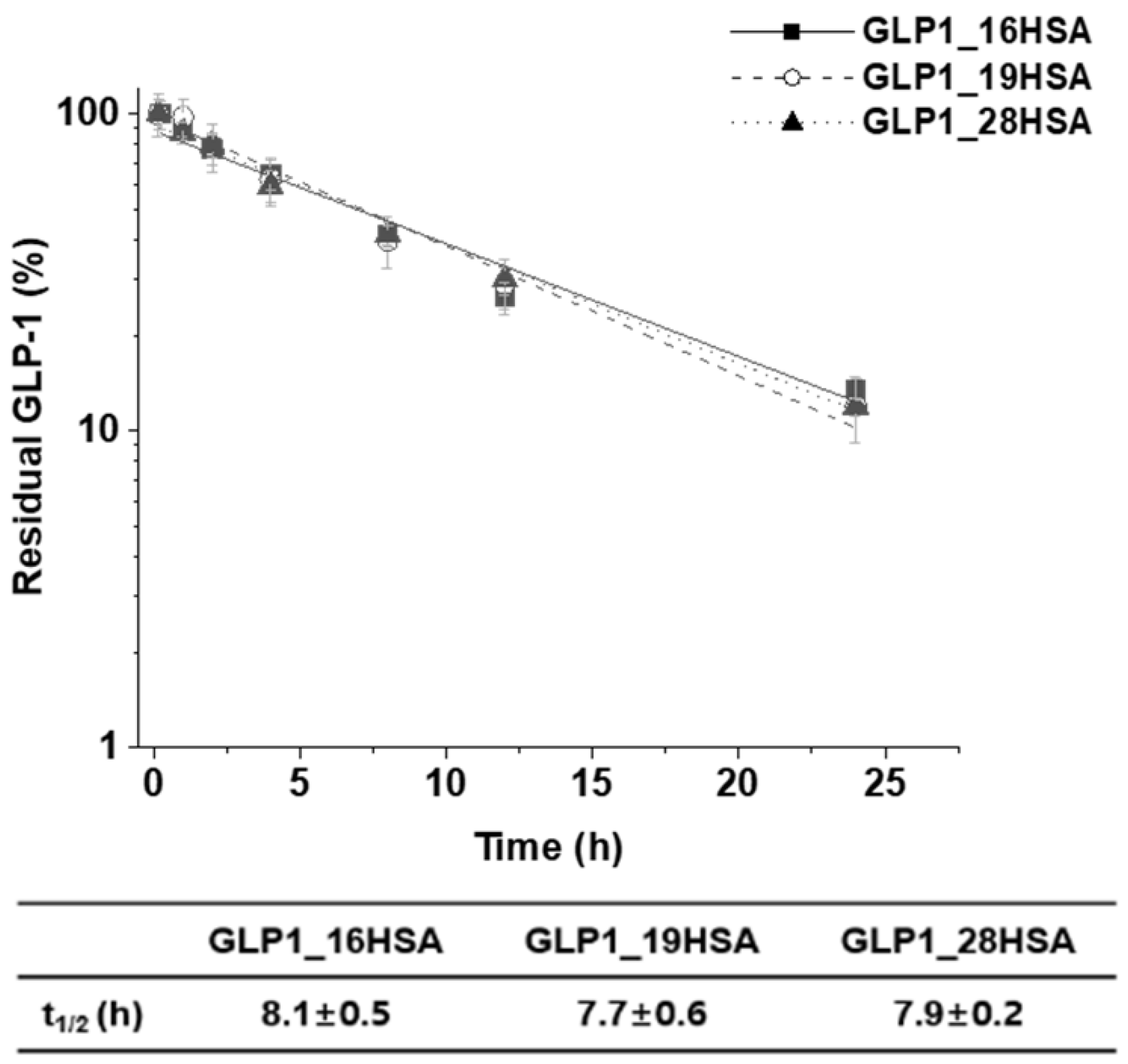
3.4. In Vitro Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, A.C.L.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Transparency Market Research, Peptide Therapeutics Market. Available online: https://www.transparencymarketresearch.com/peptide-therapeutics-market.html (accessed on 10 March 2020).
- Gaglione, R.; Pane, K.; Dell’Olmo, E.; Cafaro, V.; Pizzo, E.; Olivieri, G.; Notomista, E.; Arciello, A. Cost-effective production of recombinant peptides in Escherichia coli. New Biotechnol. 2019, 51, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar]
- Andersen, D.C.; Krummen, L. Recombinant protein expression for therapeutic applications. Curr. Opin. Biotechnol. 2002, 13, 117–123. [Google Scholar] [CrossRef]
- Sanchez-Garcia, L.; Martín, L.; Mangues, R.; Ferrer-Miralles, N.; Vázquez, E.; Villaverde, A. Recombinant pharmaceuticals from microbial cells: A 2015 update. Microb. Cell Fact. 2016, 15, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.; Robinson, D.K. Industrial choices for protein production by large-scale cell culture. Curr. Opin. Biotechnol. 2001, 12, 180–187. [Google Scholar] [CrossRef]
- Costa, S.; Almeida, A.; Castro, A.; Domingues, L. Fusion tags for protein solubility, purification, and immunogenicity in Escherichia coli: The novel Fh8 system. Front. Microbiol. 2014, 5, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Pédelacq, J.D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef]
- Wu, X.; Wu, D.; Lu, Z.; Chen, W.; Hu, X.; Ding, Y. A novel method for high-level production of TEV protease by superfolder GFP tag. J. Biomed. Biotechnol. 2010, 2009, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lövgren, J.; Tian, S.; Lundwall, Å.; Karp, M.; Lilja, H. Production and activation of recombinant hK2 with propeptide mutations resulting in high expression levels. Eur. J. Biochem. 1999, 266, 1050–1055. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, L.B.; Rasmussen, L.K.; Petersen, T.E.; Etzerodt, M.; Fedosov, S.N. Kinetic and structural characterization of a two-domain streptokinase: Dissection of domain functionality. Biochemistry 2000, 39, 6440–6448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, G.J.P.; Murray, B.; Zhou, J.; Frazer, I.H. Expression, purification and immunological characterization of the transforming protein E7, from cervical cancer-associated human papillomavirus type 16. Clin. Exp. Immunol. 1999, 115, 397–403. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.B.; Chen, W.J.; Ellis, B.; Hoffman, C.; Overton, L.; Rink, M.; Smith, A.; Marshall, C.J.; Wood, E.R. A scintillation proximity assay for the raf/MEK/ERK kinase cascade: High-throughput screening and identification of selective enzyme inhibitors. Anal. Biochem. 1999, 268, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Eisenmesser, E.Z.; Kapust, R.B.; Nawrocki, J.P.; Mazzulla, M.J.; Pannell, L.K.; Waugh, D.S.; Byrd, R.A. Expression, purification, refolding, and characterization of recombinant human interleukin-13: Utilization of intracellular processing. Protein Expr. Purif. 2000, 20, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waugh, D.S. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 2011, 80, 283–293. [Google Scholar] [CrossRef]
- Jenny, R.J.; Mann, K.G.; Lundblad, R.L. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr. Purif. 2003, 31, 1–11. [Google Scholar] [CrossRef]
- Chiang, C.F.; Okou, D.T.; Griffin, T.B.; Verret, C.R.; Williams, M.N.V. Green fluorescent protein rendered susceptible to proteolysis: Positions for protease-sensitive insertions. Arch. Biochem. Biophys. 2001, 394, 229–235. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Tang, L.; Persky, A.M.; Hochhaus, G.; Meibohm, B. Pharmacokinetic aspects of biotechnology products. J. Pharm. Sci. 2004, 93, 2184–2204. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The Impact of PEGylation on Biological Therapies. Biodrugs 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Baumann, A.; Tuerck, D.; Prabhu, S.; Dickmann, L.; Sims, J. Pharmacokinetics, metabolism and distribution of PEGs and PEGylated proteins: Quo vadis? Drug Discov. Today 2014, 19, 1623–1631. [Google Scholar] [CrossRef]
- Da Silva Freitas, D.; Spencer, P.J.; Vassão, R.C.; Abrahão-Neto, J. Biochemical and biopharmaceutical properties of PEGylated uricase. Int. J. Pharm. 2010, 387, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Zaman, R.; Islam, R.A.; Ibnat, N.; Othman, I.; Zaini, A.; Lee, C.Y.; Chowdhury, E.H. Current strategies in extending half-lives of therapeutic proteins. J. Control. Release 2019, 301, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Elsadek, B.; Kratz, F. Impact of albumin on drug delivery—New applications on the horizon. J. Control. Release 2012, 157, 4–28. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Chaudhury, C.; Brooks, C.L.; Carter, D.C.; Robinson, J.M.; Anderson, C.L. Albumin binding to FcRn: Distinct from the FcRn-IgG interaction. Biochemistry 2006, 45, 4983–4990. [Google Scholar] [CrossRef]
- Bern, M.; Sand, K.M.K.; Nilsen, J.; Sandlie, I.; Andersen, J.T. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. J. Control. Release 2015, 211, 144–162. [Google Scholar] [CrossRef]
- Wang, W.; Ou, Y.; Shi, Y. AlbuBNP, a recombinant B-type natriuretic peptide and human serum albumin fusion hormone, as a long-term therapy of congestive heart failure. Pharm. Res. 2004, 21, 2105–2111. [Google Scholar] [CrossRef]
- Melder, R.J.; Osborn, B.L.; Riccobene, T.; Kanakaraj, P.; Wei, P.; Chen, G.; Stolow, D.; Halpern, W.G.; Migone, T.S.; Wang, Q.; et al. Pharmacokinetics and in vitro and in vivo anti-tumor response of an interleukin-2-human serum albumin fusion protein in mice. Cancer Immunol. Immunother. 2005, 54, 535–5475. [Google Scholar] [CrossRef]
- Subramanian, G.M.; Fiscella, M.; Lamousé-Smith, A.; Zeuzem, S.; McHutchison, J.G. Albinterferon α-2b: A genetic fusion protein for the treatment of chronic hepatitis C. Nat. Biotechnol. 2007, 25, 1411–1419. [Google Scholar] [CrossRef]
- Bukrinski, J.T.; Sønderby, P.; Antunes, F.; Andersen, B.; Schmidt, E.G.W.; Peters, G.H.J.; Harris, P. Glucagon-like Peptide 1 Conjugated to Recombinant Human Serum Albumin Variants with Modified Neonatal Fc Receptor Binding Properties. Impact on Molecular Structure and Half-Life. Biochemistry 2017, 56, 4860–4870. [Google Scholar] [CrossRef]
- Kwon, I.; Kirshenbaum, K.; Tirrell, D.A. Breaking the degeneracy of the genetic code. J. Am. Chem. Soc. 2003, 125, 7512–7513. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.E.; Kwon, I. Effects of non-natural amino acid incorporation into the enzyme core region on enzyme structure and function. Int. J. Mol. Sci. 2015, 16, 22735–22753. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Kwon, I. Expansion of bioorthogonal chemistries towards site-specific polymer-protein conjugation. Polym. Chem. 2016, 7, 4584–4598. [Google Scholar] [CrossRef]
- Lim, S.I.; Mizuta, Y.; Takasu, A.; Hahn, Y.S.; Kim, Y.H.; Kwon, I. Site-specific fatty acid-conjugation to prolong protein half-life in vivo. J. Control. Release 2013, 170, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.I.; Hahn, Y.S.; Kwon, I. Site-specific albumination of a therapeutic protein with multi-subunit to prolong activity in vivo. J. Control. Release 2015, 207, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Kim, J.C.; Seong, J.; Tae, G.; Kwon, I. Comparative studies of the serum half-life extension of a protein: Via site-specific conjugation to a species-matched or -mismatched albumin. Biomater. Sci. 2018, 6, 2092–2100. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.I.; Kwon, I. Bioconjugation of therapeutic proteins and enzymes using the expanded set of genetically encoded amino acids. Crit. Rev. Biotechnol. 2016, 5, 803–815. [Google Scholar] [CrossRef]
- Chin, J.W.; Santoro, S.W.; Martin, A.B.; King, D.S.; Wang, L.; Schultz, P.G. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 2002, 124, 9026–9027. [Google Scholar] [CrossRef]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Expand Biological Functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Vilsbøll, T.; Agersø, H.; Krarup, T.; Holst, J.J. Similar elimination rates of glucagon-like peptide-1 in obese type 2 diabetic patients and healthy subjects. J. Clin. Endocrinol. Metab. 2003, 88, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Sharma, D.; Verma, S.; Vaidya, S.; Kalia, K.; Tiwari, V. Recent updates on GLP-1 agonists: Current advancements & challenges. Biomed. Pharmacother. 2018, 108, 952–962. [Google Scholar]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [Green Version]
- Kroeze, W.K.; Sassano, M.F.; Huang, X.P.; Lansu, K.; Mccorvy, J.D.; Giguere, P.M.; Sciaky, N.; Roth, B.L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Madsen, K.; Knudsen, L.B.; Agersoe, H.; Nielsen, P.F.; Thøgersen, H.; Wilken, M.; Johansen, N.L. Structure-activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: Importance of fatty acid length, polarity, and bulkiness. J. Med. Chem. 2007, 50, 6126–6132. [Google Scholar] [CrossRef]
- Deacon, C.F.; Knudsen, L.B.; Madsen, K.; Wiberg, F.C.; Jacobsen, O.; Holst, J.J. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia 1998, 41, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.I.; Cho, J.; Kwon, I. Double clicking for site-specific coupling of multiple enzymes. Chem. Commun. 2015, 51, 13607–13610. [Google Scholar] [CrossRef]
- Manandhar, B.; Ahn, J.M. Glucagon-like peptide-1 (GLP-1) analogs: Recent advances, new possibilities, and therapeutic implications. J. Med. Chem. 2015, 58, 1020–1037. [Google Scholar] [CrossRef]
- Adelhorst, K.; Hedegaard, B.B.; Knudsen, L.B.; Kirk, O. Structure-activity studies of glucagon-like peptide-1. J. Biol. Chem. 1994, 269, 6275–6278. [Google Scholar] [PubMed]
- Watanabe, Y.; Kawai, K.; Ohashi, S.; Yokota, C.; Suzuki, S.; Yamashita, K. Structure-activity relationships of glucagon-like peptide-1(7-36)amide: Insulinotropic activities in perfused rat pancreases, and receptor binding and cyclic AMP production in RINm5F cells. J. Endocrinol. 1994, 140, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Daniel, T.; Buechler, Y.J.; Litzinger, D.C.; Maio, Z.; Putnam, A.M.H.; Kraynov, V.S.; Sim, B.C.; Bussell, S.; Javahishvili, T.; et al. Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl. Acad. Sci. USA 2011, 108, 9060–9065. [Google Scholar] [CrossRef] [Green Version]
- Selis, F.; Schrepfer, R.; Sanna, R.; Scaramuzza, S.; Tonon, G.; Dedoni, S.; Onali, P.; Orsini, G.; Genovese, S. Enzymatic mono-pegylation of glucagon-like peptide 1 towards long lasting treatment of type 2 diabetes. Results Pharma Sci. 2012, 2, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.J.; Park, S.; Kim, D.K.; Cho, E.B.; Hwang, J.I.; Vaudry, H.; Seong, J.Y. Structural and molecular conservation of glucagon-like peptide-1 and its receptor confers selective ligand-receptor interaction. Front. Endocrinol. (Lausanne) 2012, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Zhou, X.E.; Hou, L.; Zhao, L.H.; Liu, B.; Wang, G.; Jiang, Y.; Melcher, K.; Xu, H.E. An intrinsic agonist mechanism for activation of glucagon-like peptide-1 receptor by its extracellular domain. Cell Discov. 2016, 2, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Underwood, C.R.; Garibay, P.; Knudsen, L.B.; Hastrup, S.; Peters, G.H.; Rudolph, R.; Reedtz-Runge, S. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J. Biol. Chem. 2010, 285, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, D. The structure and function of the glucagon-like peptide-1 receptor and its ligands. Br. J. Pharmacol. 2012, 166, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, B.; Feng, D.; Hu, H.; Chu, M.; Qu, Q.; Tarrasch, J.T.; Li, S.; Sun Kobilka, T.; Kobilka, B.K.; et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 2017, 546, 248–253. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bak, M.; Park, J.; Min, K.; Cho, J.; Seong, J.; Hahn, Y.S.; Tae, G.; Kwon, I. Recombinant Peptide Production Platform Coupled with Site-Specific Albumin Conjugation Enables a Convenient Production of Long-Acting Therapeutic Peptide. Pharmaceutics 2020, 12, 364. https://doi.org/10.3390/pharmaceutics12040364
Bak M, Park J, Min K, Cho J, Seong J, Hahn YS, Tae G, Kwon I. Recombinant Peptide Production Platform Coupled with Site-Specific Albumin Conjugation Enables a Convenient Production of Long-Acting Therapeutic Peptide. Pharmaceutics. 2020; 12(4):364. https://doi.org/10.3390/pharmaceutics12040364
Chicago/Turabian StyleBak, Mijeong, Junyong Park, Kiyoon Min, Jinhwan Cho, Jihyoun Seong, Young S. Hahn, Giyoong Tae, and Inchan Kwon. 2020. "Recombinant Peptide Production Platform Coupled with Site-Specific Albumin Conjugation Enables a Convenient Production of Long-Acting Therapeutic Peptide" Pharmaceutics 12, no. 4: 364. https://doi.org/10.3390/pharmaceutics12040364