Using Supercritical Fluid Technology as a Green Alternative During the Preparation of Drug Delivery Systems

Abstract
:1. Introduction
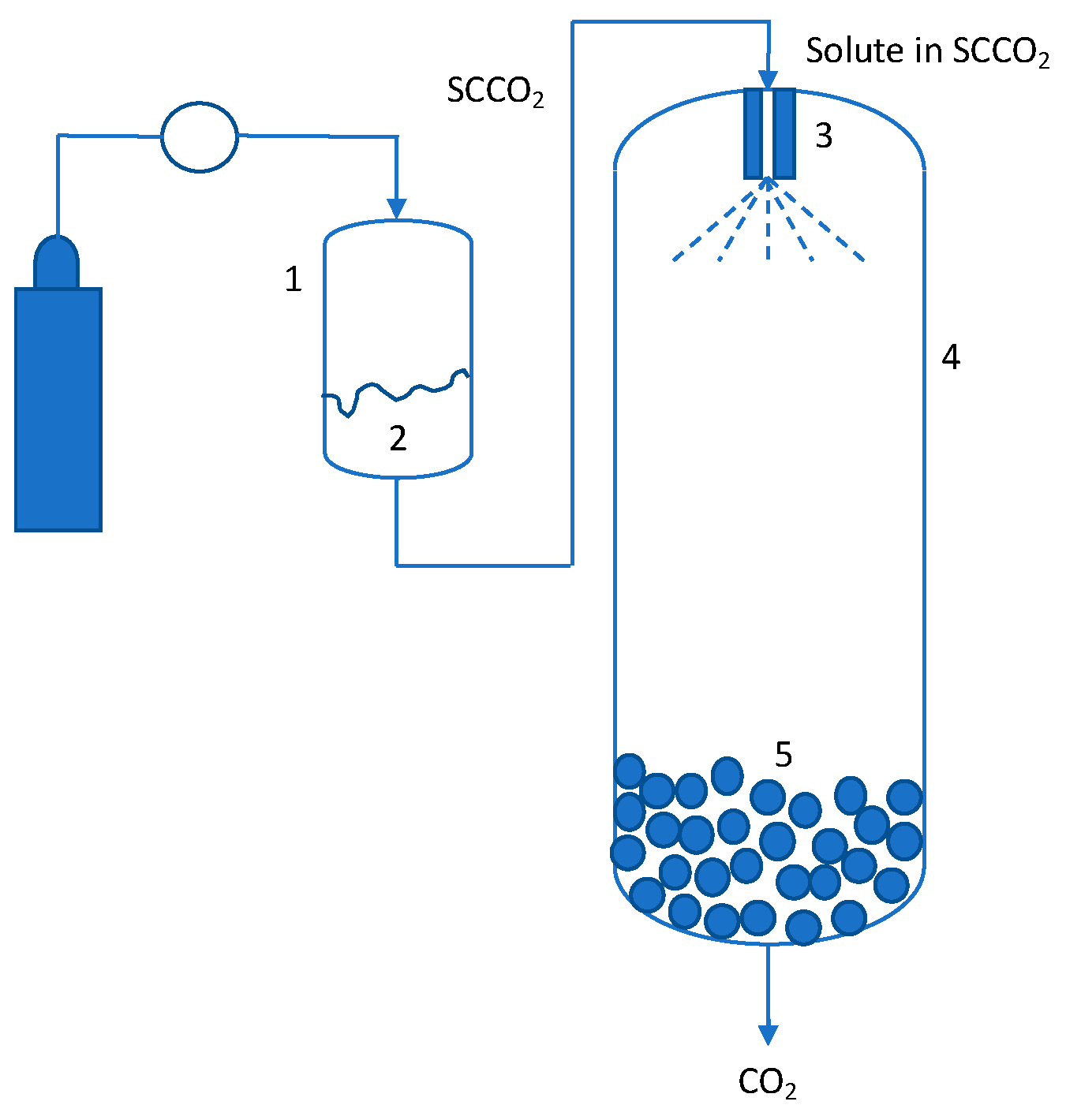
2. SCF-Based Manufacturing Technologies
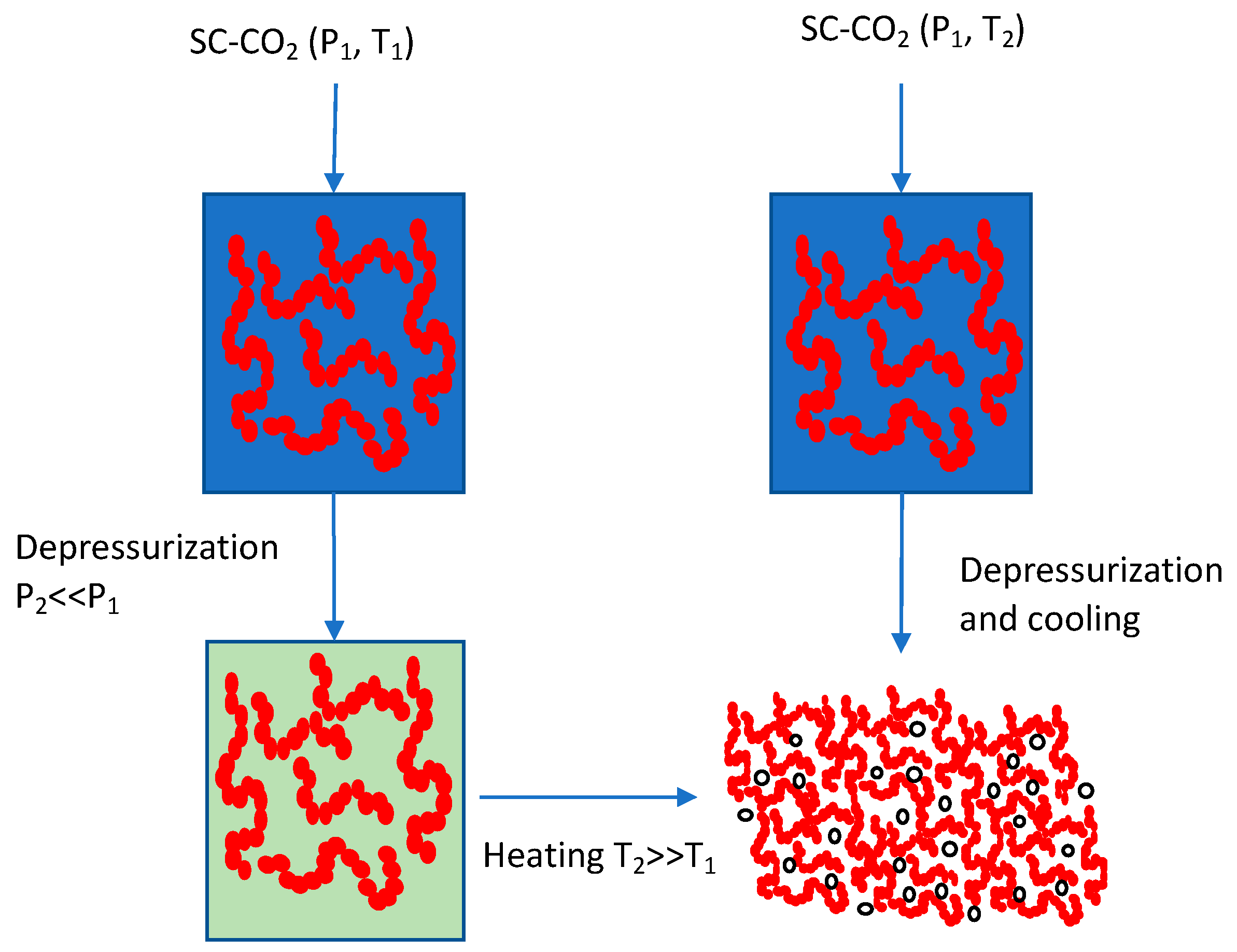
2.1. Rapid Expansion of Supercritical Solutions (RESS) and Related Processes
2.2. Gas Antisolvent (GAS)/Supercritical Antisolvent (SAS) and Related Processes
3. Discussion
3.1. Active Pharmaceutical Ingredient (API) Particle Size, Shape, and Polymorphic Form
3.1.1. Form Control and Polymorphism
3.1.2. Particle Engineering and Micronization
3.1.3. Section Summary
- SCF technology can be harnessed to produce novel polymorphic forms or metastable solid forms that are difficult to obtain using conventional crystallization techniques.
- SCF technology can be used to tailor morphology and control the polymorphic form of crystalline API in a reproducible manner.
- These methods produce free flowing micronized powders with reduced adhesion and cohesion that are more amenable to being aerosolized, making them very attractive for pulmonary delivery applications.
- SCF-based micronization is particularly useful for thermally labile materials such as recombinant proteins and antibodies.
- From a commercial standpoint, application of SCF-based micronization is more suitable for high value drug delivery applications rather than traditional oral dosage solid forms.
3.2. Polymeric Nano- and Micro-Particles
Section Summary
- SCF-based processes, including RESS, SAS, and PGSS, have been reported to permit production of drug-loaded polymeric nano- and micro-particles with distinct advantages over traditional techniques such as solvent emulsification-based methods.
- PGSS-based processes remain the most commonly used and have been reported for encapsulation of small molecules, peptides, and proteins in polymer particles via organic solvent-free processing.
- The high prevalence of these methods, as described in the literature, does not correlate with their limited use in commercial processes. To overcome this gap, significant investment and investigation are required.
3.3. Polymeric Membranes
Section Summary
- Application of SCF has been demonstrated in making polymer membranes for a diverse range of materials such as nylon 6, polystyrene, and cellulose acetate. Production of porous membrane scaffolds without collapse has been demonstrated.
- SCF-based methods provide greater control of pore size and structure when compared to traditional methods using organic solvents.
- The use of SCFs can also lead to efficient downstream processing, as no additional post-treatment may be required to remove organic solvents.
3.4. Aerogels
3.4.1. Preparation of Aerogels
3.4.2. Utility of Aerogels
Inorganic Aerogels for Drug Delivery
Organic Aerogels for Drug Delivery and Other Applications
Specialized Aerogels
3.4.3. Section Summary
- Aerogels are a unique class of ultra-light porous materials used as drug delivery systems, owing to their high porosity, increased surface area, and low density that helps to achieve high drug loadings, influence the in vivo release kinetics of drugs, and improve bioavailability of poorly soluble compounds and the physical stability of the loaded active.
- Aerogels are produced via a two-step method of sol–gel polymerization, followed by a drying step involving the removal of the pore solvent to produce a rigid gel. Supercritical fluid drying is the most effective drying method to produce mechanically stable aerogels that do no collapse and retain their porosity and texture.
- Both organic and inorganic aerogels are extensively used for drug delivery, the latter being preferred due to biocompatibility and biodegradability. Inorganic aerogels are mainly silica-based, whereas bio-based aerogels are made in conjunction with organic polymers such as chitin, cellulose, etc. Specialized aerogels such as functionalized, composite, and layered aerogels are also being used to render additional physico-chemical attributes to modulate drug delivery further.
3.5. Microporous Foams
Section Summary
- Supercritical foaming is one of the many diverse applications of SCF technology that is used in producing three-dimensional drug-releasing microporous polymeric foams, which have a wide variety of biomedical applications.
- Nucleation and bubble growth are key to foaming and are affected by several processing conditions, as well as physico-chemical and viscoelastic properties of the polymer melt and SCF. CO2 is the SCF of choice for producing polymeric foams.
- Batch, extrusion, and foam injection molding are used for manufacturing of microporous foams.
- Polymeric foams produced by SCF technology are used for drug delivery of both hydrophilic and hydrophobic drugs that can be encapsulated in these foams.
3.6. Solid Lipid Nanoparticles
Section Summary
- Drug-loaded SLN preparation by SFEE, PGSS, RESS, and GAS processes has been described with a wide variety of drugs, lipids, and processing conditions.
- A key area for current and future development remains NLC production via SCF-based processes, which allow for higher drug loading capacity and improved drug release characteristics.
3.7. Preparation of Liposomes
Section Summary
- SCF CO2 has been successfully employed to demonstrate the production of liposomes. The recent demonstration of continuous production of liposomes using the SuperLip process is encouraging.
- In order for widespread use of SCF CO2-based processes to occur, meaningful control experiments have to be done comparing SCF CO2-based with microfluidics-based techniques, which are now increasingly being used by researchers in the field of liposomes.
4. Conclusions and Perspectives
- In the context of oral delivery, SCF-based methods have been touted as a way to reproducibly control particle size and morphology, and have been compared with conventional methods such as jet and bead milling. While SCF is indeed better than conventional technologies, more studies are needed to establish their superiority over emerging bottom-up methods such as microfluidics and flash nanoprecipitation. Secondly, from a practical perspective, there is a very high bar for introduction of new technologies in the pharmaceutical industry due to regulatory considerations. In this scenario, the use of SCF-based technology to reduce particle size and improve flow for oral delivery provides only incremental rather than transformative benefit to make the case for a shift in manufacturing technologies. However, for specialized applications such as pulmonary delivery, SCF may clearly provide the advantage over conventional milling technologies in terms of improved aerosolization, ability to combine multiple drugs, and potential ease of formulation due to reduced surface energy of the particles. Thus, choice of application areas should be carefully considered to enable commercial translation of SCF-based technologies.
- Detailed modeling efforts have been undertaken to understand phase equilibria under pressure for SCFs. However, the predictability of these models has not been good. This is especially a problem when multiple phases are involved in the SCF-operation, such as co-solvents and polymeric carriers. In these cases, extensive experimental data are needed to establish the process parameters for SCF-based methods. This has led to the belief that SCF-based technologies suffer from scale-up issues and are not reproducible and robust. Thus, there is a need for more fundamental understanding of thermodynamics and kinetics of SCF-based processes.
- Finally, SCF-based methods may have significant advantages in the manufacturing of novel drug delivery systems such as solid lipid nanoparticles and liposomes. However, very few studies have been reported in this area and more work needs to be done to understand the potential of SCFs. SCF-based methods should be compared and contrasted against appropriate controls, such as microfluidics and flash nanoprecipitation.
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Topics Covered in Review | Reference |
|---|---|
| A general overview of use of supercritical fluid (SCF) techniques for size control and encapsulation. | [50] |
| An excellent overview of the use of supercritical CO2-based techniques for particle size control. | [81] |
| Review of experimental solubility data of solids in SCFs. | [210] |
| A specific review on supercritical CO2-assisted extrusion process for foaming of biopolymers. | [161] |
| Review focused primarily on the use of SCF methodologies in novel drug delivery systems. | [82] |
| A specific review of SCF-based techniques for making drug delivery systems for cancer therapy. | [211] |
| Descriptions of SCF-related manufacturing processes as related to co-crystal production. | [212] |
| Overview of the use of SCF to process biopolymers for tissue engineering applications. | [213] |
| Overview of SCF-assisted impregnation process for the production of drug eluting implants. | [214] |
| Detailed description of SCF-based manufacturing processes with primary focus of particle size control. | [215] |
References
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Gref, R.; Minamitake, Y.; Peracchia, M.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.D.; Kiran, E.J. Formation of polymer particles with supercritical fluids: A review. Supercrit. Fluids 2005, 34, 287–308. [Google Scholar] [CrossRef]
- Vemavarapu, C.; Mollan, M.J.; Lodaya, M.; Needham, T.E. Design and process aspects of laboratory scale SCF particle formation systems. Int. J. Pharm. 2005, 292, 1. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.F.; Eckert, C.A. Phase Equilibria for Supercritical Fluid Process Design. Am. Inst. Chem. Eng. J. 1989, 35, 1409–1427. [Google Scholar] [CrossRef]
- Davies, O.R.; Lewis, A.L.; Whitaker, M.J.; Tai, H.; Shakesheff, K.M.; Howdle, S.M. Applications of supercritical CO2 in the fabrication of polymer systems for drug delivery and tissue engineering. Adv. Drug Deliv. Rev. 2008, 60, 373–387. [Google Scholar] [CrossRef]
- Pasquali, I.; Bettini, R. Are pharmaceutics really going supercritical? Int. J. Pharm. 2008, 364, 176–187. [Google Scholar] [CrossRef]
- Sinko, P.J. Martin’s Physical Pharmacy and Pharmaceutical Sciences; Physical Chemical and Biopharmaceutical Principles in the Pharmaceutical Sciences, 5th ed.; Lippincott Williams and Wilkins: Baltimore, MD, USA, 2011. [Google Scholar]
- Kalani, M.; Yunus, R. Application of supercritical antisolvent method in drug encapsulation: A review. Int. J. Nanomed. 2011, 6, 1429. [Google Scholar] [CrossRef]
- Wu, K.; Li, J. Precipitation of a biodegradable polymer using compressed carbon dioxide as antisolvent. J. Supercrit. Fluids 2008, 46, 211. [Google Scholar] [CrossRef]
- Montes, A.; Gordillo, M.D.; Pereyra, C.; Martinez de la Ossa, E.J. Particles Formation Using Supercritical Fluids. In Mass Transfer—Advanced Aspects; Nakajima, H., Ed.; InTech: London, UK, 2011; pp. 461–480. [Google Scholar]
- Yasuji, T.; Takeuchi, H.; Kawashima, Y. Particle design of poorly water-soluble drug substances using supercritical fluid technologies. Adv. Drug Deliv. Rev. 2008, 60, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Parhi, R.; Suresh, P. Supercritical Fluid Technology: A Review. J. Adv. Pharm. Sci. Technol. 2013, 1, 13. [Google Scholar] [CrossRef]
- Kompella, U.; Koushik, K. Preparation of drug delivery systems using supercritical fluid technology. Crit. Rev. Ther. Drug Carrier Syst. 2001, 18, 173–199. [Google Scholar] [PubMed]
- Djerafi, R.; Masmoudi, Y.; Crampon, C.; Meniai, A.; Badens, E. Supercritical anti-solvent precipitation of ethyl cellulose. J. Supercrit. Fluids 2015, 105, 92. [Google Scholar] [CrossRef]
- Castor, T.P. Phospholipids nanosomes. Curr. Drug Deliv. 2005, 2, 329–340. [Google Scholar] [CrossRef]
- Matson, D.W.; Fulton, J.L.; Petersen, R.C.; Smith, R.D. Rapid expansion of supercritical fluid solutions: Solute formation of powders, thin films, and fibers. Ind. Eng. Chem. Res. 1987, 26, 2298–2306. [Google Scholar] [CrossRef]
- Matson, D.W.; Petersen, R.C.; Smith, R.D. The preparation of polycarbosilane powders and fibers during rapid expansion of supercritical fluid solutions. Mater. Lett. 1986, 4, 429–432. [Google Scholar] [CrossRef]
- Petersen, R.C.; Matson, D.W.; Smith, R.D. Rapid precipitation of low vapor pressure solids from supercritical fluid solutions: The formation of thin films and powders. J. Am. Chem. Soc. 1986, 108, 2100–2102. [Google Scholar] [CrossRef]
- Tom, J.W.; Debenedetti, P.G. Particle formation with supercritical fluids—A review. J. Aerosol Sci. 1991, 22, 555–584. [Google Scholar] [CrossRef]
- Bagheri, H.; Ali Mansoori, G.; Hashemipour, H. A novel approach to predict drugs solubility in supercritical solvents for ress process using various cubic eos-mixing rule. J. Mol. Liq. 2018, 261, 174–188. [Google Scholar] [CrossRef]
- Debenedetti, P.G.; Tom, J.W.; Kwauk, X.; Yeo, S.D. Rapid expansion of supercritical solutions (ress): Fundamentals and applications. Fluid Phase Equilibria 1993, 82, 311–321. [Google Scholar] [CrossRef]
- Helfgen, B.; Türk, M.; Schaber, K. Hydrodynamic and aerosol modelling of the rapid expansion of supercritical solutions (ress-process). J. Supercrit. Fluids 2003, 26, 225–242. [Google Scholar] [CrossRef]
- Türk, M. Formation of small organic particles by ress: Experimental and theoretical investigations. J. Supercrit. Fluids 1999, 15, 79–89. [Google Scholar] [CrossRef]
- Meziani, M.J.; Sun, Y.-P. Protein-conjugated nanoparticles from rapid expansion of supercritical fluid solution into aqueous solution. J. Am. Chem. Soc. 2003, 125, 8015–8018. [Google Scholar] [CrossRef]
- Pathak, P.; Meziani, M.J.; Desai, T.; Sun, Y.-P. Nanosizing drug particles in supercritical fluid processing. J. Am. Chem. Soc. 2004, 126, 10842–10843. [Google Scholar] [CrossRef]
- Sun, Y.-P.; Rollins, H.W. Preparation of polymer-protected semiconductor nanoparticles through the rapid expansion of supercritical fluid solution. Chem. Phys. Lett. 1998, 288, 585–588. [Google Scholar] [CrossRef]
- Sun, Y.-P.; Rollins, H.W.; Guduru, R. Preparations of nickel, cobalt, and iron nanoparticles through the rapid expansion of supercritical fluid solutions (ress) and chemical reduction. Chem. Mater. 1999, 11, 7–9. [Google Scholar] [CrossRef]
- Thakur, R.; Gupta, R.B. Rapid expansion of supercritical solution with solid cosolvent (ress−sc) process: Formation of griseofulvin nanoparticles. Ind. Eng. Chem. Res. 2005, 44, 7380–7387. [Google Scholar] [CrossRef]
- Thakur, R.; Gupta, R.B. Formation of phenytoin nanoparticles using rapid expansion of supercritical solution with solid cosolvent (ress-sc) process. Int. J. Pharm. 2006, 308, 190–199. [Google Scholar] [CrossRef]
- Thakur, R.; Gupta, R.B. Rapid expansion of supercritical solution with solid cosolvent (ress-sc) process: Formation of 2-aminobenzoic acid nanoparticle. J. Supercrit. Fluids 2006, 37, 307–315. [Google Scholar] [CrossRef]
- Padrela, L.; Rodrigues, M.A.; Tiago, J.; Velaga, S.P.; Matos, H.A.; de Azevedo, E.G. Insight into the mechanisms of cocrystallization of pharmaceuticals in supercritical solvents. Cryst. Growth Des. 2015, 15, 3175–3181. [Google Scholar] [CrossRef]
- Padrela, L.; Rodrigues, M.A.; Velaga, S.P.; Matos, H.A.; de Azevedo, E.G. Formation of indomethacin–saccharin cocrystals using supercritical fluid technology. Eur. J. Pharm. Sci. 2009, 38, 9–17. [Google Scholar] [CrossRef]
- Gallagher, P.M.; Coffey, M.P.; Krukonis, V.J.; Klasutis, N. Gas antisolvent recrystallization: New process to recrystallize compounds insoluble in supercritical fluids. In Supercritical Fluid Science and Technology; American Chemical Society: Washington, DC, USA, 1989; Volume 406, pp. 334–354. [Google Scholar]
- Moneghini, M.; Kikic, I.; Voinovich, D.; Perissutti, B.; Alessi, P.; Cortesi, A.; Princivalle, F.; Solinas, D. Study of the solid state of carbamazepine after processing with gas anti-solvent technique. Eur. J. Pharm. Biopharm. 2003, 56, 281–289. [Google Scholar] [CrossRef]
- Moneghini, M.; Kikic, I.; Voinovich, D.; Perissutti, B.; Filipović-Grčić, J. Processing of carbamazepine–peg 4000 solid dispersions with supercritical carbon dioxide: Preparation, characterisation, and in vitro dissolution. Int. J. Pharm. 2001, 222, 129–138. [Google Scholar] [CrossRef]
- Moribe, K.; Tozuka, Y.; Yamamoto, K. Supercritical carbon dioxide processing of active pharmaceutical ingredients for polymorphic control and for complex formation. Adv. Drug Deliv. Rev. 2008, 60, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Ober, C.A.; Gupta, R.B. Formation of itraconazole—Succinic acid cocrystals by gas antisolvent cocrystallization. AAPS Pharm. Sci. Tech. 2012, 13, 1396–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, P.; Shekunov, B.Y.; Yim, D.; Cipolla, D.; Boyd, B.; Farr, S. Production of solid lipid nanoparticle suspensions using supercritical fluid extraction of emulsions (sfee) for pulmonary delivery using the aerx system. Adv. Drug Deliv. Rev. 2007, 59, 444–453. [Google Scholar] [CrossRef]
- Shekunov, B.Y.; Chattopadhyay, P.; Seitzinger, J.; Huff, R. Nanoparticles of poorly water-soluble drugs prepared by supercritical fluid extraction of emulsions. Pharm. Res. 2006, 23, 196–204. [Google Scholar] [CrossRef]
- Werling, J.O.; Debenedetti, P.G. Numerical modeling of mass transfer in the supercritical antisolvent process: Miscible conditions. J. Supercrit. Fluids 2000, 18, 11–24. [Google Scholar] [CrossRef]
- Yeo, S.-D.; Lim, G.-B.; Debendetti, P.G.; Bernstein, H. Formation of microparticulate protein powder using a supercritical fluid antisolvent. Biotechnol. Bioeng. 1993, 41, 341–346. [Google Scholar] [CrossRef]
- Chattopadhyay, P.; Gupta, R.B. Production of griseofulvin nanoparticles using supercritical CO2 antisolvent with enhanced mass transfer. Int. J. Pharm. 2001, 228, 19–31. [Google Scholar] [CrossRef]
- Chattopadhyay, P.; Gupta, R.B. Protein nanoparticles formation by supercritical antisolvent with enhanced mass transfer. AIChE J. 2002, 48, 235–244. [Google Scholar] [CrossRef]
- Bleich, J.; Müller, B.W. Production of drug loaded microparticles by the use of supercritical gases with the aerosol solvent extraction system (ases) process. J. Microencapsul. 1996, 13, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Bleich, J.; Müller, B.W.; Waßmus, W. Aerosol solvent extraction system—A new microparticle production technique. Int. J. Pharm. 1993, 97, 111–117. [Google Scholar] [CrossRef]
- Reverchon, E. Supercritical-assisted atomization to produce micro- and/or nanoparticles of controlled size and distribution. Ind. Eng. Chem. Res. 2002, 41, 2405–2411. [Google Scholar] [CrossRef]
- Reverchon, E.; Caputo, G.; De Marco, I. Role of phase behavior and atomization in the supercritical antisolvent precipitation. Ind. Eng. Chem. Res. 2003, 42, 6406–6414. [Google Scholar] [CrossRef]
- Soh, S.H.; Lee, L.Y. Microencapsulation and Nanoencapsulation Using Supercritical Fluid (SCF) techniques. Pharmaceutics 2019, 11, 21. [Google Scholar] [CrossRef] [Green Version]
- Moribe, K.; Tsutsumi, S.-I.; Morishita, S.; Shinozaki, H.; Tozuka, Y.; Oguchi, T.; Yamamoto, K. Micronization of Phenylbutazone by Rapid Expansion of Supercritical CO2 Solution. Chem. Pharm. Bull. 2005, 53, 1025–1028. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki, H.; Oguchi, T.; Suzuki, S.; Aoki, K.; Sako, T.; Morishita, S.; Tozuka, Y.; Moribe, K.; Yamamoto, K. Micronization and Polymorphic Conversion of Tolbutamide and Barbital by Rapid Expansion of Supercritical Solutions. Drug Dev. Ind. Pharm. 2006, 32, 877–891. [Google Scholar] [CrossRef]
- Bettini, R.; Bonassi, L.; Castoro, V.; Rossi, A.; Zema, L.; Gazzaniga, A.; Giordano, F. Solubility and Conversion of Carbamazepine Polymorphs in Supercritical Carbon Dioxide. Eur. J. Pharm. Sci. 2001, 13, 281–286. [Google Scholar] [CrossRef]
- Tozuka, Y.; Kawada, D.; Oguchi, T.; Yamamoto, K. Supercritical Carbon Dioxide Treatment as a Method for Polymorph Preparation of Deoxycholic Acid. Int. J. Pharm. 2003, 263, 45–50. [Google Scholar] [CrossRef]
- Bouchard, A.; Jovanović, N.; Hofland, G.W.; Crommelin, D.J.; Jiskoot, W.; Witkamp, G.-J. Ways of Manipulating the Polymorphism of Glycine During Supercritical Fluid Crystallisation. J. Supercrit. Fluids 2008, 44, 422–432. [Google Scholar] [CrossRef]
- Benali, M.; Boumghar, Y. Supercritical Fluid-Assisted Drying. In Handbook of Industrial Drying, 4th ed.; Mujumdar, A.S., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 1261–1269. [Google Scholar]
- Velaga, S.P.; Berger, R.; Carlfors, J. Supercritical Fluids Crystallization of Budesonide and Flunisolide. Pharm. Res. 2002, 19, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Kordikowski, A.; Shekunov, T.; York, P. Polymorph Control of Sulfathiazole in Supercritical CO2. Pharm. Res. 2001, 18, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Kordikowski, A.; York, P.; Latham, D. Resolution of Ephedrine in Supercritical CO2: A Novel Technique for The Separation of Chiral Drugs. J. Pharm. Sci. 1999, 88, 786–791. [Google Scholar] [CrossRef]
- Oakes, R.S.; Clifford, A.A.; Bartle, K.D.; Pett, M.T.; Rayner, C.M. Sulfur Oxidation in Supercritical Carbon Dioxide: Dramatic Pressure Dependent Enhancement of Diastereoselectivity for Sulfoxidation of Cysteine Derivatives. Chem. Commun. 1999, 3, 247–248. [Google Scholar] [CrossRef]
- Rehman, M.; Shekunov, B.Y.; York, P.; Lechuga-Ballesteros, D.; Miller, D.P.; Tan, T.; Colthorpe, P. Optimisation of Powders for Pulmonary Delivery Using Supercritical Fluid Technology. Eur. J. Pharm. Sci. 2004, 22, 1–17. [Google Scholar] [CrossRef]
- Steckel, H.; Müller, B.W. Metered-dose Inhaler Formulation of Fluticasone-17-propionate Micronized with Supercritical Carbon Dioxide Using the Alternative Propellant HFA-227. Int. J. Pharm. 1998, 173, 25–33. [Google Scholar] [CrossRef]
- Okamoto, H.; Danjo, K. Application of Supercritical Fluid to Preparation of Powders of High-molecular Weight Drugs for Inhalation. Adv. Drug Deliv. Rev. 2008, 60, 433–446. [Google Scholar] [CrossRef]
- Shoyele, S.A.; Cawthorne, S. Particle Engineering Techniques for Inhaled Biopharmaceuticals. Adv. Drug Deliv. Rev. 2006, 58, 1009–1029. [Google Scholar] [CrossRef]
- Türk, M.; Hils, P.; Helfgen, B.; Schaber, K.; Martin, H.-J.; Wahl, M.A. Micronization of Pharmaceutical Substances by the Rapid Expansion of Supercritical Solutions (RESS): A Promising Method to Improve Bioavailability of Poorly Soluble Pharmaceutical Agents. J. Supercrit. Fluids 2002, 22, 75–84. [Google Scholar] [CrossRef]
- Perrut, M.; Jung, J.; Leboeuf, F. Enhancement of Dissolution Rate of Poorly-soluble Active Ingredients by Supercritical Fluid Processes: Part I: Micronization of Neat Particles. Int. J. Pharm. 2005, 288, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Reverchon, E.; De Marco, I. Supercritical Antisolvent Micronization of Cefonicid: Thermodynamic Interpretation of Results. J. Supercrit. Fluids 2004, 31, 207–215. [Google Scholar] [CrossRef]
- Reverchon, E.; De Marco, I.; Della Porta, G. Rifampicin Microparticles Production by Supercritical Antisolvent Precipitation. Int. J. Pharm. 2002, 243, 83–91. [Google Scholar] [CrossRef]
- Reverchon, E.; Della Porta, G. Micronization of Antibiotics by Supercritical Assisted Atomization. J. Supercrit. Fluids 2003, 26, 243–252. [Google Scholar] [CrossRef]
- Reverchon, E.; Della Porta, G. Terbutaline Microparticles Suitable for Aerosol Delivery Produced by Supercritical Assisted Atomization. Int. J. Pharm. 2003, 258, 1–9. [Google Scholar] [CrossRef]
- Reverchon, E.; Spada, A. Erythromycin Micro-particles Produced by Supercritical Fluid Aatomization. Powder Tech. 2004, 141, 100–108. [Google Scholar] [CrossRef]
- Yeo, S.-D.; Lee, J.-C. Crystallization of Sulfamethizole Using the Supercritical and Liquid Antisolvent Processes. J. Supercrit. Fluids 2004, 30, 315–323. [Google Scholar] [CrossRef]
- Kröber, H.; Teipel, U. Materials Processing with Supercritical Antisolvent Precipitation: Process Parameters and Morphology of Tartaric Acid. J. Supercrit. Fluids 2002, 22, 229–235. [Google Scholar] [CrossRef]
- York, P. Strategies for Particle Design Using Supercritical Fluid Technologies. Pharm. Sci. Technol. Today 1999, 2, 430–440. [Google Scholar] [CrossRef]
- Bristow, S.; Shekunov, T.; Shekunov, B.Y.; York, P. Analysis of the Supersaturation and Precipitation Process with Supercritical CO2. J. Supercrit. Fluids 2001, 21, 257–271. [Google Scholar] [CrossRef]
- Hanna, M.; York, P. Method and Apparatus for the Formation of Particles. UK Patent 5,851,453, 22 December 1998. [Google Scholar]
- Palakodaty, S.; York, P. Phase Behavioral Effects on Particle Formation Processes Using Supercritical Fluids. Pharm. Res. 1999, 16, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Muhrer, G.; Mazzotti, M. Precipitation of Lysozyme Nanoparticles from Dimethyl Sulfoxide Using Carbon Dioxide as Antisolvent. Biotechnol. Prog. 2003, 19, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Peiriço, N.; Matos, H.; De Azevedo, E.G.; Lobato, M.; Almeida, A. Microcomposites Theophylline/hydrogenated Palm oil from a PGSS Process for Controlled Drug Delivery Systems. J. Supercrit. Fluids 2004, 29, 175–184. [Google Scholar] [CrossRef]
- Velaga, S.P.; Ghaderi, R.; Carlfors, J. Preparation and Characterization of Hydrocortisone Particles Using a Supercritical Fluids Extraction Process. Int. J. Pharm. 2002, 231, 155–166. [Google Scholar] [CrossRef]
- Padrela, L.; Rodrigues, M.A.; Duarte, A.; Dias, A.M.A.; Braga, M.E.M.; de Sousa, H.C. Supercritical carbon dioxide-based technologies for the production of drug nanoparticles/nanocrystals—A comprehensive review. Adv. Drug Deliv. Rev. 2018, 131, 22–78. [Google Scholar] [CrossRef]
- Kankala, R.K.; Zhang, Y.S.; Wang, S.-B.; Lee, C.-H.; Chen, A.-Z. Supercritical Fluid Technology: An Emphasis on Drug Delivery and Related Biomedical Applications. Adv. Healthc. Mater. 2017, 6, 1700433. [Google Scholar] [CrossRef] [Green Version]
- Dalvi, S.V.; Azad, M.A.; Dave, R. Precipitation and stabilization of ultrafine particles of Fenofibrate in aqueous suspensions by RESOLV. Powder Technol. 2013, 236, 75–84. [Google Scholar] [CrossRef]
- Sane, A.; Limtrakul, J. Formation of retinyl palmitate-loaded poly(l-lactide) nanoparticles using rapid expansion of supercritical solutions into liquid solvents (RESOLV). J. Supercrit. Fluids 2009, 51, 230–237. [Google Scholar] [CrossRef]
- Campardelli, R.; Baldino, L.; Reverchon, E. Supercritical fluids applications in nanomedicine. J. Supercrit. Fluids 2015, 101, 193–214. [Google Scholar] [CrossRef]
- Kalani, M.; Yunus, R.; Abdullah, N. Optimizing supercritical antisolvent process parameters to minimize the particle size of paracetamol nanoencapsulated in L-polylactide. Int. J. Nanomed. 2011, 6, 1101–1105. [Google Scholar] [CrossRef] [Green Version]
- Kalani, M. Effect of supercritical fluid density on nanoencapsulated drug particle size using the supercritical antisolvent method. Int. J. Nanomed. 2012, 7, 2165–2172. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-S.; Kim, J.-S.; Park, H.J.; Cho, W.K.; Cha, K.-H.; Hwang, S.-J. Enhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int. J. Nanomed. 2011, 6, 2997–3009. [Google Scholar]
- Weidner, E.; Steiner, R.; Knez, Z. Powder generation from polyethyleneglycols with compressible fluids. Process Technol. Proc. 1996, 12, 223–228. [Google Scholar]
- Jordan, F.; Naylor, A.; Kelly, C.A.; Howdle, S.M.; Lewis, A.; Illum, L. Sustained release hGH microsphere formulation produced by a novel supercritical fluid technology: In vivo studies. J. Controll. Release 2010, 141, 153–160. [Google Scholar] [CrossRef]
- Perinelli, D.R.; Cespi, M.; Bonacucina, G.; Naylor, A.; Whitaker, M.; Lam, J.K.W.; Howdle, S.M.; Casettari, L.; Palmieri, G.F. PEGylated Biodegradable Polyesters for PGSS Microparticles Formulation: Processability, Physical and Release Properties. Curr. Drug Deliv. 2016, 13, 673–681. [Google Scholar] [CrossRef]
- Baldino, L.; Cardea, S.; Reverchon, E. A supercritical CO2 assisted electrohydrodynamic process used to produce microparticles and microfibers of a model polymer. J. CO2 Util. 2019, 33, 532–540. [Google Scholar]
- Sarrade, S.; Guizard, C.; Rios, G. New Applications of Supercritical Fluids and Supercritical Fluids Processes in Separation. Sep. Purif. Technol. 2003, 32, 57–63. [Google Scholar] [CrossRef]
- Kho, Y.W.; Kalika, D.S.; Knutson, B.L. Precipitation of Nylon 6 Membranes Using Compressed Carbon Dioxide. Polymer 2001, 42, 6119–6127. [Google Scholar] [CrossRef]
- Reverchon, E.; Cardea, S. Formation of Cellulose Acetate Membranes Using a Supercritical Fluid Assisted Process. J. Membr. Sci. 2004, 240, 187–195. [Google Scholar] [CrossRef]
- Matsuyama, H.; Yano, H.; Maki, T.; Teramoto, M.; Mishima, K.; Matsuyama, K. Formation of Porous Flat Membrane by Phase Separation with Supercritical CO2. J. Membr. Sci. 2001, 194, 157–163. [Google Scholar] [CrossRef]
- Matsuyama, H.; Yamamoto, A.; Yano, H.; Maki, T.; Teramoto, M.; Mishima, K.; Matsuyama, K. Effect of Organic Solvents on Membrane Formation by Phase Separation with Supercritical CO2. J. Membr. Sci. 2002, 204, 81–87. [Google Scholar] [CrossRef]
- Reverchon, E.; Cardea, S. PVDF− HFP Membrane Formation by Supercritical CO2 Processing: Elucidation of Formation Mechanisms. Ind. Eng. Chem. Res. 2006, 45, 8939–8945. [Google Scholar] [CrossRef]
- Şahin, İ.; Özbakır, Y.; Inönü, Z.; Ulker, Z.; Erkey, C. Kinetics of supercritical drying of gels. Gels 2018, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Aerogels. Uses for Supercritical Fluids. Available online: http://www.appliedseparations.com/aerogels.html (accessed on 15 September 2019).
- García-González, C.; Alnaief, M.; Smirnova, I. Polysaccharide-based aerogels—Promising biodegradable carriers for drug delivery systems. Carbohydr. Polym. 2011, 86, 1425–1438. [Google Scholar]
- Tan, C.; Fung, B.M.; Newman, J.K.; Vu, C. Organic aerogels with very high impact strength. Adv. Mater. 2001, 13, 644–646. [Google Scholar] [CrossRef]
- Placin, F.; Desvergne, J.-P.; Cansell, F. Organic low molecular weight aerogel formed in supercritical fluids. J. Mater. Chem. 2000, 10, 2147–2149. [Google Scholar] [CrossRef]
- Raman, S.; Gurikov, P.; Smirnova, I. Hybrid alginate based aerogels by carbon dioxide induced gelation: Novel technique for multiple applications. J. Supercrit. Fluids 2015, 106, 23–33. [Google Scholar] [CrossRef]
- Robitzer, M.; David, L.; Rochas, C.; Di Renzo, F.; Quignard, F. Nanostructure of calcium alginate aerogels obtained from multistep solvent exchange route. Langmuir 2008, 24, 12547–12552. [Google Scholar] [CrossRef]
- Cardea, S.; Pisanti, P.; Reverchon, E. Generation of chitosan nanoporous structures for tissue engineering applications using a supercritical fluid assisted process. J. Supercrit. Fluids 2010, 54, 290–295. [Google Scholar] [CrossRef]
- Subrahmanyam, R.; Gurikov, P.; Dieringer, P.; Sun, M.; Smirnova, I. On the road to biopolymer aerogels—Dealing with the solvent. Gels 2015, 1, 291–313. [Google Scholar] [CrossRef] [Green Version]
- Caputo, G.; Scognamiglio, M.; De Marco, I. Nimesulide adsorbed on silica aerogel using supercritical carbon dioxide. Chem. Eng. Res. Des. 2012, 90, 1082–1089. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, B.; Quan, G.; Li, F.; Wu, Q.; Dian, L.; Dong, Y.; Li, G.; Wu, C. Increasing the oral bioavailability of poorly water-soluble carbamazepine using immediate-release pellets supported on SBA-15 mesoporous silica. Int. J. Nanomed. 2012, 7, 5807–5818. [Google Scholar]
- Bouledjouidja, A.; Masmoudi, Y.; Van Speybroeck, M.; Schueller, L.; Badens, E. Impregnation of Fenofibrate on mesoporous silica using supercritical carbon dioxide. Int. J. Pharm. 2016, 499, 1–9. [Google Scholar] [CrossRef]
- Smirnova, I.; Mamic, J.; Arlt, W. Adsorption of Drugs on Silica Aerogels. Langmuir 2003, 19, 8521–8525. [Google Scholar] [CrossRef]
- Belhadj-Ahmed, F.; Badens, E.; Llewellyn, P.; Denoyel, R.; Charbit, G. Impregnation of vitamin E acetate on silica mesoporous phases using supercritical carbon dioxide. J. Supercrit. Fluids 2009, 51, 278–286. [Google Scholar] [CrossRef]
- Smirnova, I.; Suttiruengwong, S.; Seiler, M.; Arlt, W. Dissolution Rate Enhancement by Adsorption of Poorly Soluble Drugs on Hydrophilic Silica Aerogels. Pharm. Dev. Technol. 2005, 9, 443–452. [Google Scholar] [CrossRef]
- Matsuyama, K.; Hayashi, N.; Yokomizo, M.; Kato, T.; Ohara, K.; Okuyama, T. Supercritical carbon dioxide-assisted drug loading and release from biocompatible porous metal—Organic frameworks. J. Mater. Chem. B 2014, 2, 7551–7558. [Google Scholar] [CrossRef]
- Smirnova, I. Aerogels: Current Status and Challenges for The Future. J. Supercrit. Fluids 2015, 106, 1. [Google Scholar] [CrossRef]
- Martins, M.; Barros, A.A.; Quraishi, S.; Gurikov, P.; Raman, S.; Smirnova, I.; Duarte, A.R.C.; Reis, R.L. Preparation of Macroporous Alginate-based Aerogels for Biomedical Applications. J. Supercrit. Fluids 2015, 106, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Tkalec, G.; Knez, Ž.; Novak, Z. Fast Production of High-methoxyl Pectin Aerogels for Enhancing the Bioavailability of Low-soluble Drugs. J. Supercrit. Fluids 2015, 106, 16–22. [Google Scholar] [CrossRef]
- Giri, T.K.; Thakur, A.; Alexander, A.; Badwaik, H.; Tripathi, D.K. Modified Chitosan Hydrogels as Drug Delivery and Tissue Engineering Systems: Present Status and Applications. Acta Pharm. Sin. B 2012, 2, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Hile, D.D.; Amirpour, M.L.; Akgerman, A.; Pishko, M.V. Active growth factor delivery from poly(d,l-lactide-co-glycolide) foams prepared in supercritical CO2. J. Control. Release 2000, 66, 177–185. [Google Scholar] [CrossRef]
- Cabezas, L.I.; Gracia, I.; García, M.T.; de Lucas, A.; Rodríguez, J.F. Production of biodegradable porous scaffolds impregnated with 5-fluorouracil in supercritical CO2. J. Supercrit. Fluids 2013, 80, 1–8. [Google Scholar] [CrossRef]
- Cabezas, L.I.; Fernández, V.; Mazarro, R.; Gracia, I.; de Lucas, A.; Rodríguez, J.F. Production of biodegradable porous scaffolds impregnated with indomethacin in supercritical CO2. J. Supercrit. Fluids 2012, 63, 155–160. [Google Scholar] [CrossRef]
- Yoganathan, R.; Mammucari, R.; Foster, N.R. Impregnation of ibuprofen into polycaprolactone using supercritical carbon dioxide. J. Phys. Conf. Ser. 2010, 215, 012087. [Google Scholar] [CrossRef]
- Betz, M.; García-González, C.; Subrahmanyam, R.; Smirnova, I.; Kulozik, U. Preparation of Novel Whey Protein-based Aerogels as Drug Carriers for Life Science Applications. J. Supercrit. Fluids 2012, 72, 111–119. [Google Scholar] [CrossRef]
- Marin, M.A.; Mallepally, R.R.; McHugh, M.A. Silk fibroin aerogels for drug delivery applications. J. Supercrit. Fluids 2014, 91, 84–89. [Google Scholar] [CrossRef]
- Mehling, T.; Smirnova, I.; Guenther, U.; Neubert, R.H.H. Polysaccharide-based aerogels as drug carriers. J. Non Cryst. Solids 2009, 355, 2472–2479. [Google Scholar] [CrossRef]
- García-González, C.A.; Jin, M.; Gerth, J.; Alvarez-Lorenzo, C.; Smirnova, I. Polysaccharide-based Aerogel Microspheres for Oral Drug Delivery. Carbohydr. Polym. 2015, 117, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Ulker, Z.; Erkey, C. An Emerging Platform for Drug Delivery: Aerogel Based Systems. J. Control. Release 2014, 177, 51–63. [Google Scholar] [CrossRef]
- Rao, A.V.; Kalesh, R.R. Comparative studies of the physical and hydrophobic properties of TEOS based silica aerogels using different co-precursors. Sci. Technol. Adv. Mater. 2003, 4, 509. [Google Scholar] [CrossRef]
- Rao, A.V.; Kulkarni, M.M.; Amalnerkar, D.; Seth, T. Superhydrophobic silica aerogels based on methyltrimethoxysilane precursor. J. Non Cryst. Solids 2003, 330, 187–195. [Google Scholar]
- Lee, K.-H.; Kim, S.-Y.; Yoo, K.-P. Low-density, hydrophobic aerogels. J. Non Cryst. Solids 1995, 186, 18–22. [Google Scholar] [CrossRef]
- Ilhan, F.; Fabrizio, E.F.; McCorkle, L.; Scheiman, D.A.; Dass, A.; Palczer, A. Hydrophobic monolithic aerogels by nanocasting polystyrene on amine-modified silica. J. Mater. Chem. 2006, 16, 3046–3054. [Google Scholar] [CrossRef]
- Katti, A.; Shimpi, N.; Roy, S.; Lu, H.; Fabrizio, E.F.; Dass, A. Chemical, physical, and mechanical characterization of isocyanate cross-linked amine-modified silica aerogels. Chem. Mater. 2006, 18, 285–296. [Google Scholar] [CrossRef]
- Wen, J.; Wilkes, G.L. Organic/inorganic hybrid network materials by the sol− gel approach. Chem. Mater. 1996, 8, 1667–1681. [Google Scholar] [CrossRef]
- Zhang, G.; Dass, A.; Rawashdeh, A.-M.M.; Thomas, J.; Counsil, J.A.; Sotiriou-Leventis, C. Isocyanate-crosslinked silica aerogel monoliths: Preparation and characterization. J. Non-Cryst. Solids 2004, 350, 152–164. [Google Scholar] [CrossRef]
- Leventis, N.; Sadekar, A.; Chandrasekaran, N.; Sotiriou-Leventis, C. Click synthesis of monolithic silicon carbide aerogels from polyacrylonitrile-coated 3D silica networks. Chem. Mater. 2010, 22, 2790–2803. [Google Scholar] [CrossRef]
- Meador, M.A.B.; Fabrizio, E.F.; Ilhan, F.; Dass, A.; Zhang, G.; Vassilaras, P. Cross-linking amine-modified silica aerogels with epoxies: Mechanically strong lightweight porous materials. Chem. Mater. 2005, 17, 1085–1098. [Google Scholar] [CrossRef] [Green Version]
- Novak, B.M.; Auerbach, D.; Verrier, C. Low-density, mutually interpenetrating organic-inorganic composite materials via supercritical drying techniques. Chem. Mater. 1994, 6, 282–286. [Google Scholar] [CrossRef]
- Shaikh, R.P.; Pillay, V.; Choonara, Y.E.; du Toit, L.C.; Ndesendo, V.M.; Bawa, P. A review of multi-responsive membranous systems for rate-modulated drug delivery. Aaps Pharmscitech 2010, 11, 441–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SinnáAw, M. A multi-drug delivery system with sequential release using titania nanotube arrays. Chem. Commun. 2012, 48, 3348–3350. [Google Scholar]
- Sundararaj, S.C.; Thomas, M.V.; Peyyala, R.; Dziubla, T.D.; Puleo, D.A. Design of a multiple drug delivery system directed at periodontitis. Biomaterials 2013, 34, 8835–8842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo-de Santiago, G.; Portales-Cabrera, C.G.; Portillo-Lara, R.; Araiz-Hernández, D.; Del Barone, M.C.; García-López, E.; Rojas-de Gante, C.; De Santiago, M.D.L.A.; Segoviano-Ramírez, J.C.; García-Lara, S. Supercritical CO2 Foaming of Thermoplastic Materials Derived From Maize: Proof-of-concept Use in Mammalian Cell Culture Applications. PLoS ONE 2015, 10, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, E.; Kiran, E. Foaming of polymers with supercritical fluids and perspectives on the current knowledge gaps and challenges. J. Supercrit. Fluids 2018, 134, 157–166. [Google Scholar] [CrossRef]
- Tayton, E.; Purcell, M.; Aarvold, A.; Smith, J.; Kalra, S.; Briscoe, A.; Shakesheff, K.; Howdell, S.M.; Dunlop, D.G.; Oreffo, R.O.C. Supercritical CO2 Fluid-foaming of Polymers to Increase Porosity: A Method to Improve the Mechanical and Biocompatibility Characteristics for Use as a Potential Alternative to Allografts in Impaction Bone Grafting? Acta Biomater. 2012, 8, 1918–1927. [Google Scholar] [CrossRef]
- Lee, L.Y.; Ranganath, S.H.; Fu, Y.; Zheng, J.L.; Lee, H.S.; Wang, C.-H.; Smith, K.A. Paclitaxel Release from Micro-porous PLGA Disks. Chem. Eng. Sci. 2009, 64, 4341–4349. [Google Scholar] [CrossRef]
- Nie, H.; Lee, L.Y.; Tong, H.; Wang, C.-H. PLGA/Chitosan Composites from a Combination of Spray Drying and Supercritical Fluid Foaming Techniques: New Carriers for DNA Delivery. J. Control. Release 2008, 129, 207–214. [Google Scholar] [CrossRef]
- Ong, Y.X.J.; Lee, L.Y.; Davoodi, P.; Wang, C.H. Production of Drug-releasing Biodegradable Microporous Scaffold Using a Two-step Micro-encapsulation/Supercritical Foaming Process. J. Supercrit. Fluids 2018, 133, 263–269. [Google Scholar] [CrossRef]
- Matos, R.L.; Lu, T.; McConville, C.; Leeke, G.; Ingram, A. Analysis of Curcumin Precipitation and Coating on Lactose by the Integrated Supercritical Antisolvent-fluidized Bed Process. J. Supercrit. Fluids 2018, 141, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.K.; Beckman, E.J. Nucleation and growth in microcellular materials: Supercritical CO2 as foaming agent. AIChE J. 1995, 41, 357–367. [Google Scholar] [CrossRef]
- Sarver, J.A.; Sumey, J.L.; Williams, M.L.; Bishop, J.P.; Dean, D.M.; Kiran, E. Foaming of poly (ethylene-co-vinyl acetate) and poly (ethylene-co-vinyl acetate-co-carbon monoxide) and their blends with carbon dioxide. J. Appl. Polym. Sci. 2018, 135, 45841. [Google Scholar] [CrossRef]
- Wang, K.; Wang, S.; Wu, F.; Pang, Y.; Zhai, W.; Zheng, W. Supercritical CO2 in controlling phase morphology of polypropylene/polystyrene blends and the corresponding mechanical properties and foamability. Polym. Bull. 2016, 73, 941–957. [Google Scholar] [CrossRef]
- Elkovitch, M.D.; Tomasko, D.L.; Lee, L.J. Supercritical carbon dioxide assisted blending of polystyrene and poly (methyl methyacrylate). Polym. Eng. Sci. 1999, 39, 2075–2084. [Google Scholar] [CrossRef]
- Taki, K.; Waratani, Y.; Ohshima, M. Preparation of Nanowells on a PS-b-PMMA Copolymer Thin Film by CO2 Treatment. Macromol. Mater. Eng. 2008, 293, 589–597. [Google Scholar] [CrossRef]
- Kiran, E. Supercritical Fluids and Polymers—The Year in Review–2014. J. Supercrit. Fluids 2016, 110, 126–153. [Google Scholar] [CrossRef]
- Salerno, A.; Oliviero, M.; Di Maio, E.; Iannace, S. Thermoplastic Foams From Zein and Gelatin. Int. Polym. Process 2007, 22, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Salerno, A.; Di Maio, E.; Iannace, S.; Netti, P. Tailoring the Pore Structure of PCL Scaffolds for Tissue Engineering Prepared via Gas Foaming of Multi-phase Blends. J. Porous Mater. 2012, 19, 181–188. [Google Scholar] [CrossRef]
- White, L.J.; Hutter, V.; Tai, H.; Howdle, S.M.; Shakesheff, K.M. The Effect of Processing Variables on Morphological and Mechanical Properties of Supercritical CO2 Foamed Scaffolds for Tissue Engineering. Acta Biomater. 2012, 8, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Salerno, A.; Di Maio, E.; Iannace, S.; Netti, P. Tuning the Microstructure and Biodegradation of Three-phase Scaffolds for Bone Regeneration Made of PCL, Zein, and HA. J. Cell. Plast. 2011, 47, 245–260. [Google Scholar] [CrossRef]
- Tomasko, D.L.; Burley, A.; Feng, L.; Yeh, S.-K.; Miyazono, K.; Nirmal-Kumar, S.; Kusaka, I.; Koelling, K. Development of CO2 for Polymer Foam Applications. J. Supercrit. Fluids 2009, 47, 493–499. [Google Scholar] [CrossRef]
- Curia, S.; De Focatiis, D.S.; Howdle, S.M. High-pressure Rheological Analysis of CO2-induced Melting Point Depression and Viscosity Reduction of Poly (ε-caprolactone). Polymer 2015, 69, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Kiran, E. Foaming Strategies for Bioabsorbable Polymers in Supercritical Fluid Mixtures. Part, I. Miscibility and Foaming of Poly (l-lactic acid) in Carbon Dioxide + Acetone Binary Fluid Mixtures. J. Supercrit. Fluids 2010, 54, 296–307. [Google Scholar] [CrossRef]
- Chauvet, M.; Sauceau, M.; Fages, J. Extrusion assisted by supercritical CO2: A review on its application to biopolymers. J. Supercrit. Fluids 2017, 120, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.; Sun, G.; Gao, F.; Liu, T.; Esseghir, M.; Zhao, L. Effect of phase compatibility on the foaming behavior of LDPE/HDPE and LDPE/PP blends with subcritical CO2 as the blowing agent. J. Supercrit. Fluids 2017, 120, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Bunjes, H. Lipid nanoparticles for the delivery of poorly water-soluble drugs. J. Pharm. Pharmacol. 2010, 62, 1637–1645. [Google Scholar] [CrossRef]
- Almeida, A.J.; Souto, E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv. Drug Deliv. Rev. 2007, 59, 478–490. [Google Scholar] [CrossRef]
- Alessi, P.; Cortesi, A.; Kikic, I.; Vecchione, F. Plasticization of polymers with supercritical carbon dioxide: Experimental determination of glass-transition temperatures. J. Appl. Polym. Sci. 2003, 88, 2189–2193. [Google Scholar] [CrossRef]
- Spilimbergo, S.; Luca, G.; Elvassore, N.; Bertucco, A. Effect of high-pressure gases on phase behaviour of solid lipids. J. Supercrit. Fluids 2006, 38, 289–294. [Google Scholar] [CrossRef]
- Wang, X.; Chen, H.; Guo, Y.; Su, Y.; Wang, H.; Li, J. Preparation of ibuprofen/lipid composite microparticles by supercritical fluid technique. Front. Chem. Eng. China 2008, 2, 361–367. [Google Scholar] [CrossRef]
- Pestieau, A.; Krier, F.; Lebrun, P.; Brouwers, A.; Streel, B.; Evrard, B. Optimization of a PGSS (particles from gas saturated solutions) process for a fenofibrate lipid-based solid dispersion formulation. Int. J. Pharm. 2015, 485, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Pedro, A.S.; Villa, S.D.; Caliceti, P.; Melo, S.A.B.V.d.; Albuquerque, E.C.; Bertucco, A.; Salmaso, S. Curcumin-loaded solid lipid particles by PGSS technology. J. Supercrit. Fluids 2016, 107, 534–541. [Google Scholar] [CrossRef]
- Salmaso, S.; Bersani, S.; Elvassore, N.; Bertucco, A.; Caliceti, P. Biopharmaceutical characterisation of insulin and recombinant human growth hormone loaded lipid submicron particles produced by supercritical gas micro-atomisation. Int. J. Pharm. 2009, 379, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, S.; Elvassore, N.; Bertucco, A.; Caliceti, P. Production of Solid Lipid Submicron Particles for Protein Delivery Using a Novel Supercritical Gas Assisted Melting Atomization Process. J. Pharm. Sci. 2009, 98, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Akbari, Z.; Amanlou, M.; Karimi-Sabet, J.; Golestani, A.; Shariaty Niassar, M. Production of Ibuprofen-Loaded Solid Lipid Nanoparticles Using Rapid Expansion of Supercritical Solution. J. Nano Res. 2015, 31, 15–29. [Google Scholar] [CrossRef]
- Akbari, Z.; Amanlou, M.; Karimi-Sabet, J.; Golestani, A.; Niasar, M.S. Characterization of Carbamazepine-Loaded Solid Lipid Nanoparticles Prepared by Rapid Expansion of Supercritical Solution. Trop. J. Pharm. Res. 2015, 13, 1955. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Ng, W.K.; Kanaujia, P.; Kim, S.; Tan, R.B.H. Formulation design, preparation and physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic drug: Effects of process variables. Colloids Surf. B Biointerfaces 2011, 88, 483–489. [Google Scholar] [CrossRef]
- Westesen, K.; Bunjes, H.; Koch, M.H.J. Physicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J. Control. Release 1997, 48, 223–236. [Google Scholar] [CrossRef]
- Gonçalves, V.S.S.; Matias, A.A.; Rodríguez-Rojo, S.; Nogueira, I.D.; Duarte, C.M.M. Supercritical fluid precipitation of ketoprofen in novel structured lipid carriers for enhanced mucosal delivery—A comparison with solid lipid particles. Int. J. Pharm. 2015, 495, 302–311. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Liu, Q.; Boyd, B.J. Liposomes in biosensors. Analyst 2013, 138, 391–409. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.W.; Hill, M.W.; Bangham, E. Large volume liposomes by an ether evaporation method. Biochem. Biophys. Acta 1976, 443, 629–634. [Google Scholar] [CrossRef]
- Batzri, S.; Korn, E.D. Interaction of phospholipid vesicles with cells endocytes.and fusion as alternate mechanisms for the uptake of lipid-soluble and watersoluble molecules. J. Cell Biol. 1975, 66, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Batzri, S.; Korn, E.D. Single bilayer liposomes prepared without sonication. Biochim. Biophys. Acta 1973, 298, 1015–1019. [Google Scholar] [CrossRef]
- Deamer, D.; Bangham, A. Large volume liposomes by an ether vaporization method. Biochim. Biophys. Acta 1976, 443, 629–634. [Google Scholar] [CrossRef]
- Karn, P.R.; Vanic, Z.; Pepic, I.; Škalko-Basnet, N. Mucoadhesive liposomal delivery systems: The choice of coating material. Drug Dev. Ind. Pharm. 2011, 37, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Szoka, F.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [Green Version]
- Shew, R.L.; Deamer, D.W. A novel method for encapsulation of macromolecules in liposomes. Biochim. Biophys. Acta 1985, 816, 1–8. [Google Scholar] [CrossRef]
- Barenholz, Y.; Amselem, S.; Lichtenberg, D. A new method for the preparation of phospholipid vesicles (liposomes)-French Press. FEBS Lett. 1979, 99, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Kagawa, Y.; Racker, E. Partial resolution of enzymes catalyzing oxidative phosphorylation. J. Biol. Chem. 1971, 246, 5477–5487. [Google Scholar]
- Otake, K.; Imura, T.; Sakai, H.; Abe, M. Development of a new preparation method of liposomes using supercritical carbon dioxide. Langmuir 2001, 17, 3898–3901. [Google Scholar] [CrossRef]
- Castor, T.P. Methods and Apparatus for Making Liposomes Using Critical, Supercritical or Near Critical Fluids. U.S. Patent 5,554,382, 26 May 1994. [Google Scholar]
- Zhong, J.; Dai, L.C. Liposomal preparation by supercritical fluids technology. Afr. J. Biotechnol. 2011, 10, 16406–16413. [Google Scholar] [CrossRef]
- Otake, K.; Shimomura, T.; Goto, T.; Imura, T.; Furuya, T.; Yoda, S. Preparation of liposomes using an improved supercritical reverse phase evaporation method. Langmuir 2006, 22, 2543–2550. [Google Scholar] [CrossRef] [PubMed]
- Santo, I.E.; Campardelli, R.; Albuquerque, E.C.; de Melo, S.V.; Della Porta, G.; Reverchon, E. Liposomes preparation using a supercritical fluid assisted continuous process. Chem. Eng. J. 2014, 249, 153–159. [Google Scholar] [CrossRef]
- Trucillo, P.; Campardelli, R.; Reverchon, E. Supercritical CO2 assisted liposomes formation: Optimization of the lipidic layer for an efficient hydrophilic drug loading. J. CO2 Util. 2017, 18, 181–188. [Google Scholar] [CrossRef]
- Zhao, L.; Temelli, F.; Curtis, J.M.; Chen, L. Preparation of liposomes using supercritical carbon dioxide technology: Effects of phospholipids and sterols. Food Res. Int. 2015, 77, 63–72. [Google Scholar] [CrossRef]
- Fei, X.; Heyang, J.; Yaping, Z.; Xinqiu, G. Supercritical Antisolvent-based Technology for Preparation of Vitamin D3 Proliposome and Its Characteristics. Chin. J. Chem. Eng. 2011, 19, 1039–1046. [Google Scholar]
- Hu, D.; Lin, C.; Liu, L.; Li, S.; Zhao, Y. Preparation, characterization, and in vitro release investigation of lutein/zein nanoparticles via solution enhanced dispersion by supercritical fluids. J. Food Eng. 2012, 109, 545–552. [Google Scholar] [CrossRef]
- Elvira, C.; Fanovich, A.; Fernández, M.; Fraile, J.; San Román, J.; Domingo, C. Evaluation of drug delivery characteristics of microspheres of PMMA-PCL-cholesterol obtained by supercritical-CO2 impregnation and by dissolution-evaporation techniques. J. Control. Release 2004, 99, 231–240. [Google Scholar] [CrossRef]
- Badens, E.; Magnan, C.; Charbit, G. Microparticles of soy lecithin formed by supercritical processes. Biotechnol. Bioeng. 2001, 72, 194–204. [Google Scholar] [CrossRef]
- Maqbool, F.; Moyle, P.; Tan, M.; Thurecht, K.J.; Falconer, J. Preparation of albendazole-loaded liposomes by supercritical carbon dioxide processing. Artif. Cells Nanomed. Biotechnol. 2018, 46, S1186–S1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, S.; Patel, D.; Surti, N.; Misra, A. Preparation of PEGylated liposomes of docetaxel using supercritical fluid technology. J. Supercrit. Fluids 2010, 54, 110–119. [Google Scholar] [CrossRef]
- Campardelli, R.; Trucillo, P.; Reverchon, E. A Supercritical Fluid-Based Process for the Production of Fluorescein-Loaded Liposomes. Ind. Eng. Chem. Res. 2016, 55, 5359–5365. [Google Scholar] [CrossRef]
- Trucillo, P.; Campardelli, R.; Reverchon, E. Production of liposomes loaded with antioxidants using a supercritical CO2 assisted process. Powder Technol. 2018, 323, 155–162. [Google Scholar] [CrossRef]
- Zhao, L.; Temelli, F.; Curtis, J.M.; Chen, L. Encapsulation of lutein in liposomes using supercritical carbon dioxide. Food Res. Int. 2017, 100, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Temelli, F.; Chen, L. Encapsulation of anthocyanin in liposomes using supercritical carbon dioxide: Effects of anthocyanin and sterol concentrations. J. Funct. Foods 2017, 34, 159–167. [Google Scholar] [CrossRef]
- Frederiksen, L.; Anton, K.; Hoogevest, V.A.N.; Keller, P.; Leuenberger, H.R.H. Preparation of liposomes encapsulating water-soluble compounds using supercritical carbon dioxide. J. Pharm. Sci. 1997, 86, 921–928. [Google Scholar] [CrossRef]
- Otake, K.; Shimomura, T.; Goto, T.; Imura, T.; Furuya, T.; Yoda, S.; Takebayashi, Y.; Sakai, H.; Abe, M. One-step preparation of chitosan-coated cationic liposomes by an improved supercritical reverse-phase evaporation method. Langmuir 2006, 22, 4054–4059. [Google Scholar] [CrossRef]
- Karn, P.R.; Cho, W.; Hwang, S.J. Liposomal drug products and recent advances in the synthesis of supercritical fluid-mediated liposomes. Nanomedicine 2013, 8, 1529–1548. [Google Scholar] [CrossRef]
- Shashidhar, G.M.; Pravin, G.V.; Manohar, B. Nano-engineering of liposomes using a supercritical CO2 mediated gas anti-solvent method. RSC Adv. 2016, 6, 57739–57750. [Google Scholar] [CrossRef]
- Lesoin, L.; Crampon, C.; Boutin, O.; Badens, E. Preparation of liposomes using the supercritical anti-solvent (SAS) process and comparison with a conventional method. J. Supercrit. Fluids 2011, 57, 162–174. [Google Scholar] [CrossRef]
- Knez, Z.; Cör, D.; Knez Hrnčič, M. Solubility of Solids in Sub- and Supercritical Fluids: A Review 2010–2017. J. Chem. Eng. Data 2018, 63, 860–884. [Google Scholar] [CrossRef]
- Tabernero, A.; González-Garcinuño, Á.; Galán, M.A.; del Valle, E.M.M. Survey of supercritical fluid techniques for producing drug delivery systems for a potential use in cancer therapy. Rev. Chem. Eng. 2016, 32, 507–532. [Google Scholar] [CrossRef]
- Pando, C.; Cabanas, A.; Cuadra, I.A. Preparation of pharmaceutical co-crystals through sustainable processes using supercritical carbon dioxide: A review. RSC Adv. 2016, 6, 71134–71150. [Google Scholar] [CrossRef]
- Salerno, A.; Pascual, C.D. Bio-based polymers, supercritical fluids and tissue engineering. Process Biochem. 2015, 50, 826–838. [Google Scholar] [CrossRef]
- Champeau, M.; Thomassin, J.M.; Tassaing, T.; Jérôme, C. Drug loading of polymer implants by supercritical CO2 assisted impregnation: A review. J. Control. Release 2015, 209, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, N. Production of micro and nano particles of pharmaceutical by supercritical carbon dioxide. J. Supercrit. Fluids 2015, 100, 129–141. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakravarty, P.; Famili, A.; Nagapudi, K.; Al-Sayah, M.A. Using Supercritical Fluid Technology as a Green Alternative During the Preparation of Drug Delivery Systems. Pharmaceutics 2019, 11, 629. https://doi.org/10.3390/pharmaceutics11120629
Chakravarty P, Famili A, Nagapudi K, Al-Sayah MA. Using Supercritical Fluid Technology as a Green Alternative During the Preparation of Drug Delivery Systems. Pharmaceutics. 2019; 11(12):629. https://doi.org/10.3390/pharmaceutics11120629
Chicago/Turabian StyleChakravarty, Paroma, Amin Famili, Karthik Nagapudi, and Mohammad A. Al-Sayah. 2019. "Using Supercritical Fluid Technology as a Green Alternative During the Preparation of Drug Delivery Systems" Pharmaceutics 11, no. 12: 629. https://doi.org/10.3390/pharmaceutics11120629