
Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors
by
 and
and
Ashraf K. El-Damasy
1,2,*,†,
Jong Eun Park
3,†,
Hyun Ji Kim
1,
Jinhyuk Lee
4,5,
Eun-Kyoung Bang
1,
Hoon Kim
3,* and
Gyochang Keum
1,* 1
Center for Brain Technology, Brain Science Institute, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea
2
Department of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
3
Department of Pharmacy, and Research Institute of Life Pharmaceutical Sciences, Sunchon National University, Suncheon 57922, Republic of Korea
4
Disease Target Structure Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Daejeon 34141, Republic of Korea
5
Department of Bioinformatics, KRIBB School of Bioscience, University of Science and Technology (UST), Daejeon 34113, Republic of Korea
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Pharmaceuticals 2023, 16(1), 83; https://doi.org/10.3390/ph16010083
Submission received: 24 November 2022
/
Revised: 31 December 2022
/
Accepted: 3 January 2023
/
Published: 6 January 2023
(This article belongs to the Special Issue Advances in Acetylcholinesterase and Butyrylcholinesterase Inhibitors)
Abstract
:Monoamine oxidase-B (MAO-B), acetylcholinesterase (AChE), and butyrylcholinesterase (BChE) have been considered target enzymes of depression and neurodegenerative diseases, including Alzheimer’s disease (AD). In this study, seventeen N-methyl-piperazine chalcones were synthesized, and their inhibitory activities were evaluated against the target enzymes. Compound 2k (3-trifluoromethyl-4-fluorinated derivative) showed the highest selective inhibition against MAO-B with an IC50 of 0.71 μM and selectivity index (SI) of 56.34, followed by 2n (2-fluoro-5-bromophenyl derivative) (IC50 = 1.11 μM, SI = 16.04). Compounds 2k and 2n were reversible competitive MAO-B inhibitors with Ki values of 0.21 and 0.28 μM, respectively. Moreover, 2k and 2n effectively inhibited AChE with IC50 of 8.10 and 4.32 μM, which underscored their multi-target inhibitory modes. Interestingly, compound 2o elicited remarkable inhibitions over MAO-B, AChE, and BChE with IC50 of 1.19–3.87 μM. A cell-based assay of compounds 2k and 2n against Vero normal cells pointed out their low cytotoxicity. In a docking simulation, 2k showed the lowest energy for MAO-B (−11.6 kcal/mol) with four hydrogen bonds and two π-π interactions. Furthermore, in silico studies were conducted, and disclosed that 2k and 2n are expected to possess favorable pharmacokinetic properties, such as the ability to penetrate the blood–brain barrier (BBB). In view of these findings, compounds 2k and 2n could serve as promising potential candidates for the treatment of neurodegenerative diseases.
1. Introduction
Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the most prevalent neurodegenerative diseases (NDDs) that mainly affect the elderly [1]. AD and PD are characterized by heterogeneous and complex multifaceted pathologies that stem from a combination of diverse genomic, epigenomic, environmental, and metabolic factors [2]. A growing body of evidence disclosed that recovery of brain functional steadiness after a neurologic impairment is unlikely to be accomplished by focusing on a single molecular target [3]. Therefore, the research efforts for the treatment of NDDs are being oriented towards the design of multi-target directed ligands (MTDLs) [4]. However, to strike a balance between the effects of MTDLs acting at their molecular targets represents a major challenge [5].
The oxidative deamination of the biogenic amines in peripheral and central tissues is catalyzed by the flavin adenine dinucleotide (FAD) dependent monoamine oxidases (MAOs)-A and -B [6]. During the catabolism of different monoamine neurotransmitters by MAO-A/B, hydrogen peroxide and reactive oxygen species (ROS) are excessively produced as major by-products, triggering oxidative stress and neuronal cell damage associated with several NDDs [7]. Overexpression of MAO-B levels in the substantia nigra of PD patients, along with the relevance of elevated activity of MAO-B and impairment of cognitive functions in AD patients has been reported [8,9]. In this regard, MAO inhibitors are considered as up-regulating agents for neurotransmitter amines, such as dopamine and noradrenaline with neuroprotective properties [10]. Numerous investigations have reported the beneficial effects of reversible/irreversible MAO-B inhibitors in AD related therapeutic approaches [11,12]. Moreover, acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) inhibitors have shown remarkable roles in the preservation of cholinergic functions and symptomatic improvement in AD [13,14]. In view of MTDLs merits, the contemporary medicinal chemists have focused their efforts, over the past few decades, to develop new chemical entities as multiple-acting MAO-B and AChE/BChE inhibitors [15,16,17,18].
N-Methylpiperazine is a privileged derivative of piperazine moiety, with better lipophilic and steric characters, which renders this motif a proper structural element in maintaining the right balance of pharmacokinetic and pharmacodynamic attributes [19]. The existence of two nitrogens along with the small bulky N-methyl allows N-methylpiperazine to be engaged in various hydrophobic and charge transfer interactions with target enzymes. The favorable drug-like properties of N-methylpiperazine were reflected in several central nervous system (CNS) acting drugs, such as clozapine, loxapine, olanzapine, trifluoperazine, and thiothixene (Figure 1) [20]. Furthermore, N-methylpiperazine was introduced in a number of anticancer kinase inhibitors, to improve water solubility and/or target affinity, such as bosutinib, ponatinib, nintedanib, brigatinib, and gilteritinib [21]. Moreover, recent studies revealed that the replacement of one or both nitrogen atoms in the piperazine ring with different structural motifs could significantly improve MAO-B, and AChE inhibitions [22,23,24]. Based on this perspective, insertion of N-methylpiperazine into the proper scaffolds of certain MAO-B inhibitors would be expected to afford dual MAO-B/AChE inhibitions.
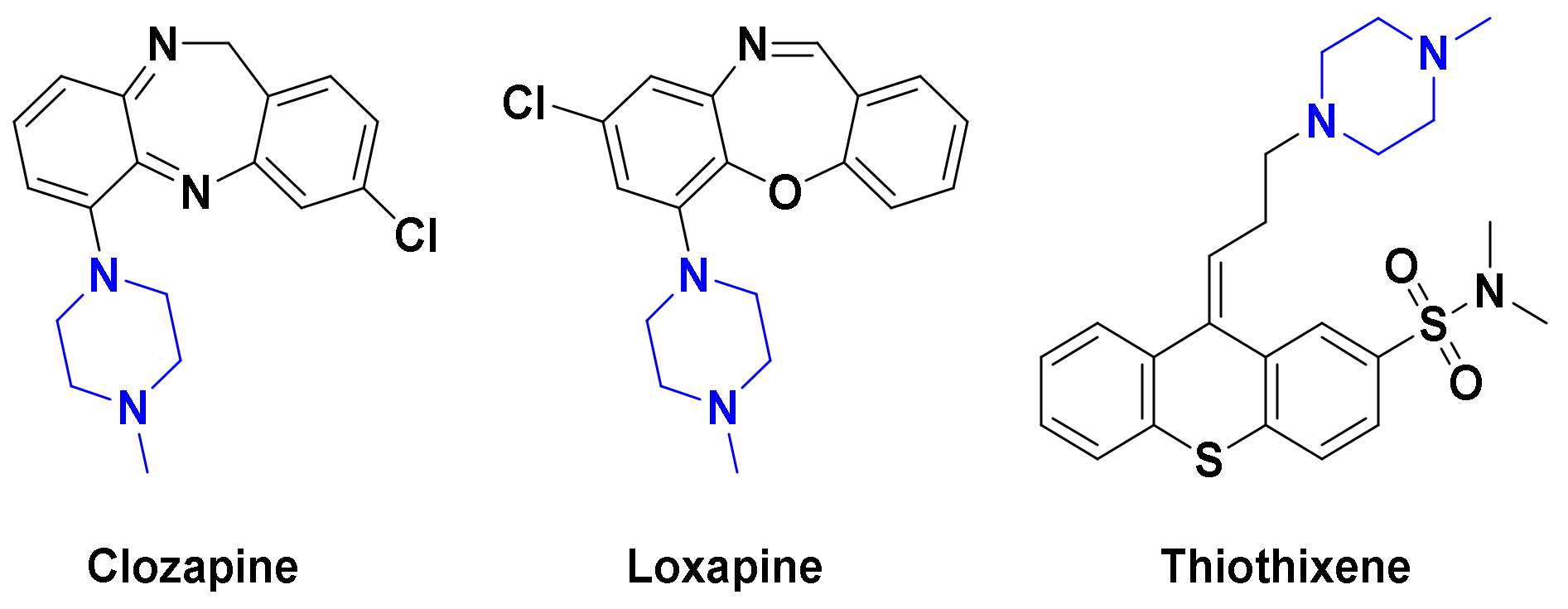
Further, chalcone, 1,3-diphenylprop-2-en-1-one, represents a substantial flexible scaffold for the design of selective MAO-B and/or AChE inhibitors [25], where the existence of propenone (α-β unsaturated ketone) and three rotatable bonds in chalcones can furnish various binding orientations with multiple targets [26]. Recent studies pointed out that conjugation of certain FDA-approved drugs, such as rivastigmine and donepezil, with chalcone scaffold led to generation of multifunctional MAO-B/ChE inhibitors, such as compound I with potent neuroprotective properties for AD [15]. Installing different electron-pulling and pushing groups on the aryl/heteroaryl rings of chalcones can modulate the electrophilic character of the Michael acceptor propanone as exemplified by chalcones II and III [27,28], where the introduction of lipophilic halogens (F, Cl, Br)/electron donation groups (-OMe, -N(Me)2) onto the phenyl B ring of chalcones afford highly selective MAO-B inhibitors such as compound IV [29,30]. On the other hand, the existence of proper alkylamino moieties on the ketone linked ring A (i.e., compound V) was found to be favorable for achieving AChE inhibitory activity [31,32].
As part of our continued endeavors to develop novel chemical entities as potent MAO-B/or AChE inhibitors, we have recently reported compounds PC-10 and PC-11 as piperazine featuring chalcones with promising selective MAO-B inhibitors (Figure 2) [33]. Compounds PC-10 and PC-11 showed IC50 values of 0.65 and 0.71 μM, respectively, against MAO-B, with selectivity index (SI) of 48.3 and 49.2 over MAO-A. Furthermore, kinetics study of PC-10 and PC-11 revealed their competitive mode of MAO-B inhibition with inhibition constant (Ki) of 0.63 and 0.53 μM, respectively. However, these molecules exerted modest activity towards both AChE and BChE with IC50 values of 26.3–28.0 and 36.2–36.4 μM, respectively. Motivated by such findings, as well as the aforementioned considerations highlighting the significance of terminal methyl group (Figure 2), we pursued further structural modifications on compounds PC-10 and PC-11 aiming at design of dual MAO-B/AChE inhibitors with improved potency. Thus, in the current study, seventeen N-methylpiperazine chalcones were designed, synthesized, and biologically tested against MAO-A/B and AChE/BChE. We expected that insertion of the small bulky methyl group on the terminal amine of piperazine would impart AChE inhibitory activity for the target molecules. In addition, diverse mono- and di-substituted phenyl rings, with various lipophilic characters, were investigated to construct a reliable structure activity relationship (SAR) study. To the best of our knowledge, this is the first report harnessing N-methylpiperazine moiety to afford dual MAO-B/AChE chalcone inhibitors.
2. Results and Discussion
2.1. Synthesis of Compounds
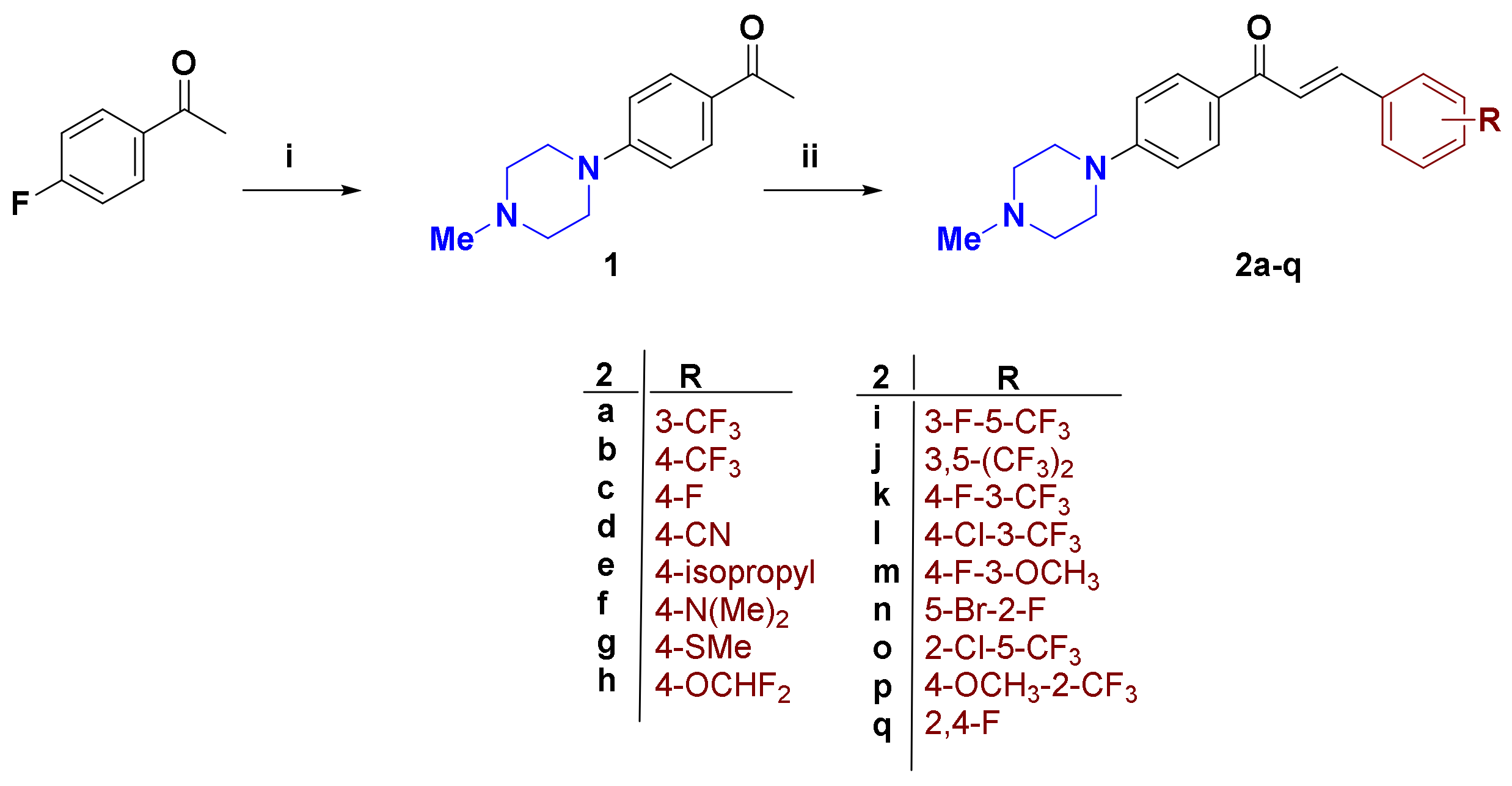
As illustrated in Scheme 1, the synthesis of the target 4-methylpiperazine-containing α,β-unsaturated ketones 2a–q was commenced with preparation of 4-methylpiperazine acetophenone 1 as the key precursor. Heating of 4-fluoroacetophenone with 4-methylpiperazine at 140 °C under neat conditions afforded compound 1 in 84% yield [34]. The Claisen–Schmidt condensation of 1 was with equimolar amounts of various mono/di-substituted benzaldehydes in 40% alcoholic NaOH at room temperature, and furnished the target molecules 2a–q in good yield. The 1H NMR charts of the final compounds showed the 4-methylpiperazine ring protons N-CH2 resonating at 3.42 and 2.57 ppm as triplets, as well as N-CH3 protons as a singlet at 2.36 ppm. The large coupling constant (J = 15.6 Hz) value of Hα and Hβ enone protons confirmed the trans (E) configuration of the 4-methylpiperazine chalcones. The spectra were provided in Supplementary (Figures S1–S3).
2.2. Inhibitory Activities against MAO-A/B, AchE, and BChE
2.2.1. Overview of the Activity
Chalcone is a well-known privileged scaffold as a MAO inhibitor, particularly selective towards MAO-B [35]. From this background, a plethora of structurally diverse chalcone derivatives has been synthesized by our team, and their MAO-A/B inhibition profiles have been reported [33,36,37]. The results revealed that the best members in those chalcone derivatives had IC50 spanning from 0.0021 to 0.65 μM against MAO-B [33,36,37].
In this study, a set of seventeen N-methyl-piperazine chalcone derivatives were synthesized and their inhibitory activities were evaluated against the target enzymes. In the preliminary inhibition assay at 10 μM concentration, all compounds exerted <50% of residual activity for MAO-B, except 2j, whereas all compounds showed >50% of residual activity for MAO-A, except 2p (Table 1). Compound 2k (3-trifluoromethyl-4-fluorinated derivative) elicited the highest selective inhibition against MAO-B with IC50 of 0.71 μM and selectivity index (SI) of 56.34, followed by the 2-fluoro-5-brominated derivative 2n (IC50 = 1.11 μM, SI = 16.04) (Table 2, Figure S4). Furthermore, most of the tested chalcone derivatives displayed <50% of residual activity for AChE at 10 μM, except 2g, 2i, 2j, and 2l, whereas most of compounds showed >50% of residual activity for BChE at 10 μM, except 2c, 2e, and 2n–p. Among them, 2b had the highest AChE inhibition with IC50 of 2.26 μM and SI (ChE) of 5.92, followed by 2d with IC50 of 2.38 μM, and SI (ChE) of 10.38. Concerning BChE, 2o stood out as the most potent member with IC50 of 1.19 μM. Remarkably, all the compounds exerted selective dual MAO-B/AChE inhibition, except 2o, disclosing that N-methyl-piperazine chalcone derivatives could be expected as multi-target inhibitors for AD.
2.2.2. SAR
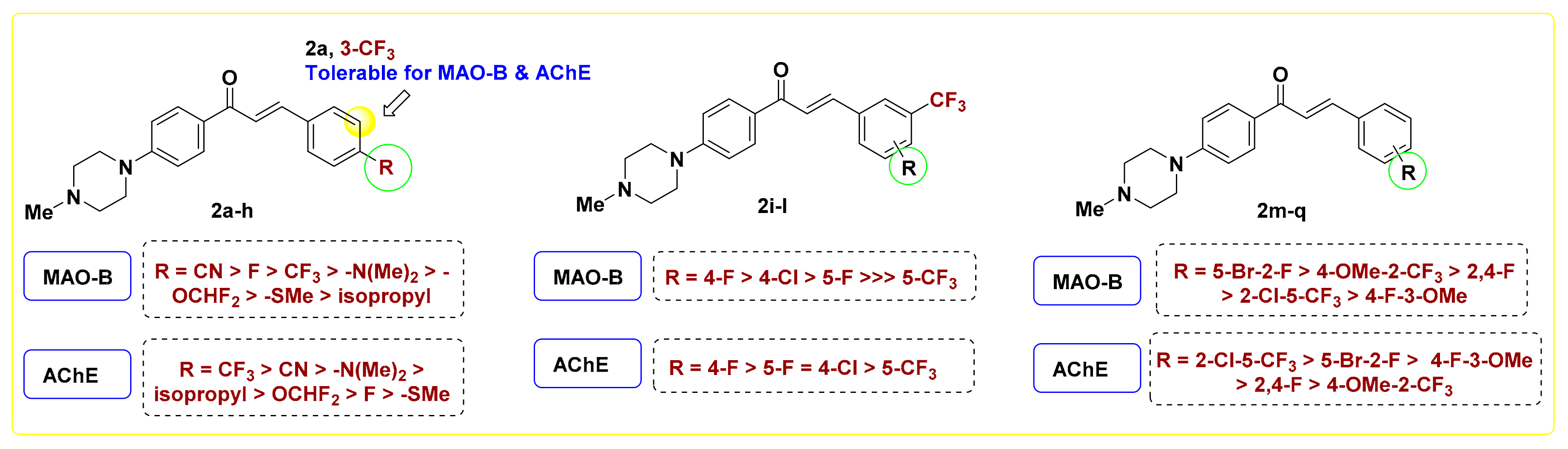
Regarding the monosubstituted chalcones 2a–h, installing an electron withdrawing group (EWG) on the terminal phenyl moiety is favorable for MAO-B inhibition, as observed with 2a–d (IC50 = 2.20–3.10 μM). The two positional isomers 2a and 2b showed comparable activity for MAO-B, however the para-trifluoromethylphenyl derivative 2b elicited 2.5-fold better inhibitory activity against AChE (IC50 = 2.26 μM) than its corresponding congener 2a. Replacing the –CF3 group in 2b with fluorine (2c) retained MAO-B inhibitory potency, while leading to ~4.6-fold reduction in AChE inhibition (2c, IC50 = 10.3 μM), pointing out that the trifluoromethyl moiety is favorable for AChE suppression. Such a finding was further emphasized by compounds 2g and 2h, which are equipotent against MAO-B, while the 4-difluoromethoxy member 2h exerted better potency for AChE (IC50 = 5.82 μM) than its corresponding methylthio derivative 2g (IC50 = 12.41 μM). Replacing the carbon of isopropyl substituted chalcone 2e (IC50 = 8.36 μM) with nitrogen (2f) resulted in ~2.5-fold augmentation in MAO-B inhibition (IC50 = 3.29 μM). Upon comparing the activity of 2b, 2c, and 2f with their corresponding methyl lacking piperazine congeners PC11, PC10, and PC5 [33], respectively. It was found that incorporation of the methyl moiety at piperazine decreased the MAO-B inhibition to certain extent, however it significantly improved the AChE inhibitory potency. For example, the 4-methylpiperazine chalcones 2b and 2f exerted IC50 values of 2.26 μM and 3.03 against AChE, while their relevant desmethyl analogs PC11 and PC5 showed IC50 values of 26.3 μM and >10 μM [33], respectively. Except 2c and 2g, all monosubstituted chalcones elicited IC50 values < 6.0 μM towards AChE, standing as promising dual MAO-B/AChE inhibitors. Among them, the para-cyanophenyl derivative 2b showed the best activity with equipotency towards MAO-B/AChE (IC50 = 2.2 μM), along with highest SI (18.18).
Referring to the disubstituted derivatives 2i–q, it was noticed that the existence of a four-halogen moiety (F/Cl) adjacent to meta-trifluoromethyl is optimal for MAO-B inhibition. For example, compound 2k bearing 3-trifluoromethyl-4-fluorophenyl showed superior potency against MAO-B (IC50 = 0.71 μM) compared to its 3,5-positional isomer 2i (IC50 = 2.33 μM). Upon comparing the 4-halogenated-3-trifluoromerthylphenyl derivatives 2k and 2l, it was found that 2k is relatively more potent than its corresponding 4-chloro congener 2l over MAO-B, AChE, and BChE. Such a finding is in agreement with the recent report of Rullo et al., which underscored the importance of fluorinated motifs in achieving sound MAO-B inhibition [38]. Worth noting is that replacing fluorine in 2i (IC50 = 2.33 μM) with a trifluoromethyl group (2j) led to dramatic drop in MAO-B inhibition (IC50 = 15.70 μM). In addition, the introduction of a 3-methoxy group, in place of the 3-trifluoromethyl motif of 2k, afforded the chalcone 2m with significantly reduced potency for MAO-B (IC50 = 6.76 μM), yet with reasonable inhibition towards AChE and BChE. Upon inspection of the activity of 2,5-disubstituted chalcones 2n and 2o, it was observed that compound 2o, the positional isomer of 2l, displayed a distinct multi-target inhibitory effect over MAO-B, AChE, and particularly BChE (IC50 = 1.19 μM). As an exception to the noticed dual selective MAO-B/AChE inhibition of this set of 4-methylpiperazine, the 2,4-difluoro member 2p elicited moderate MAO-A potency (IC50 = 6.67 μM) with a low SI value of 3.06. In view of these biochemical assay results and as illustrated in Figure 3, it is evident that both the nature and position of the substituent installed on terminal phenyl ring possess critical roles in modulating the inhibitory potency towards MAO-A/B and AChE/BChE.
2.3. Inhibition Pattern Analysis of 2k and 2n
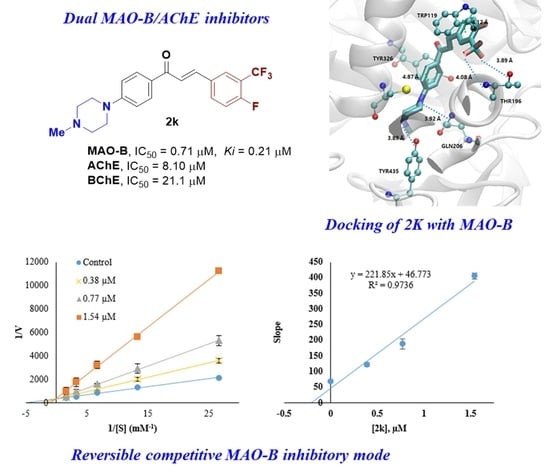
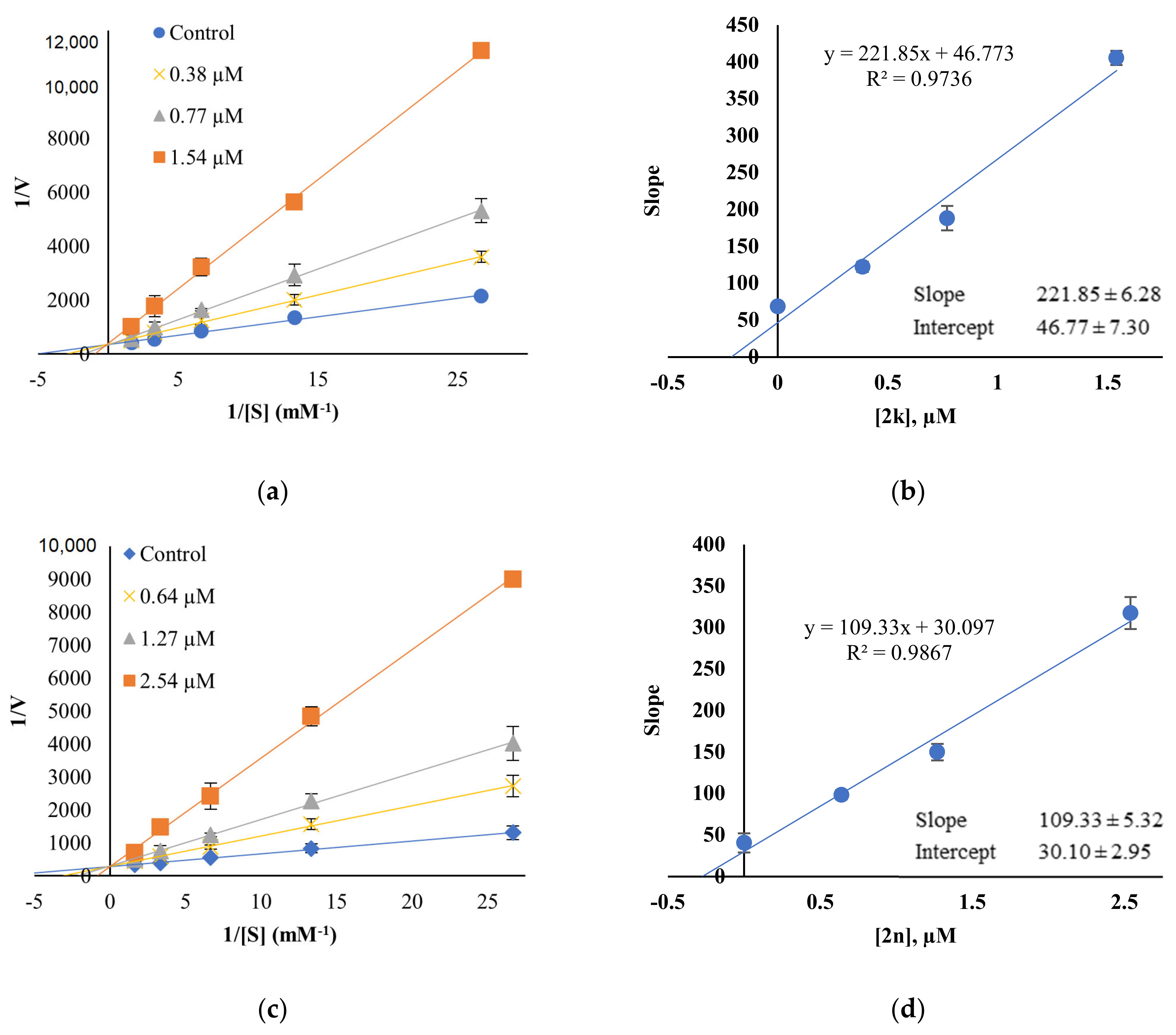
A kinetic study of the most active compounds 2k and 2n against MAO-B was performed to analyze the inhibition patterns using Lineweaver–Burk plots, and their Ki values were determined using their secondary plots. The Lineweaver–Burk plots of 2k and 2n, matched in Y-axis, showed that Ki values were 0.21 ± 0.03 μM and 0.28 ± 0.01 μM, respectively, which points out that both compounds are competitive inhibitors of MAO-B (Figure 4).
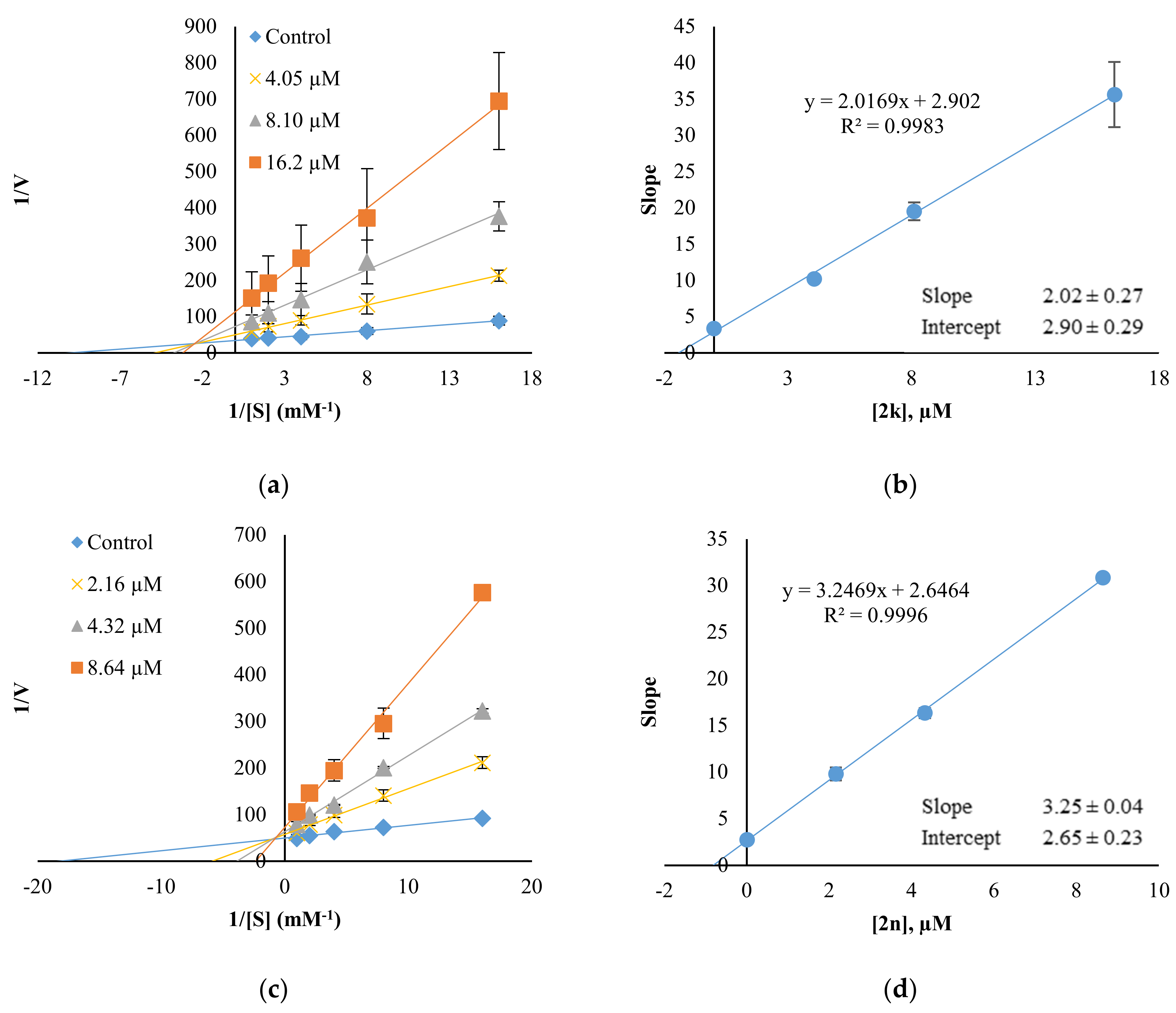
Since 2k and 2n displayed AChE inhibition with IC50 of 8.10 and 4.32 µM, respectively, kinetics for AChE were also performed (Figure 5). 2k and 2n were found to be mixed type inhibitors for AChE, showing contact points in second quadrants with Ki values of 1.44 ± 0.33 and 0.82 ± 0.08 µM, respectively.
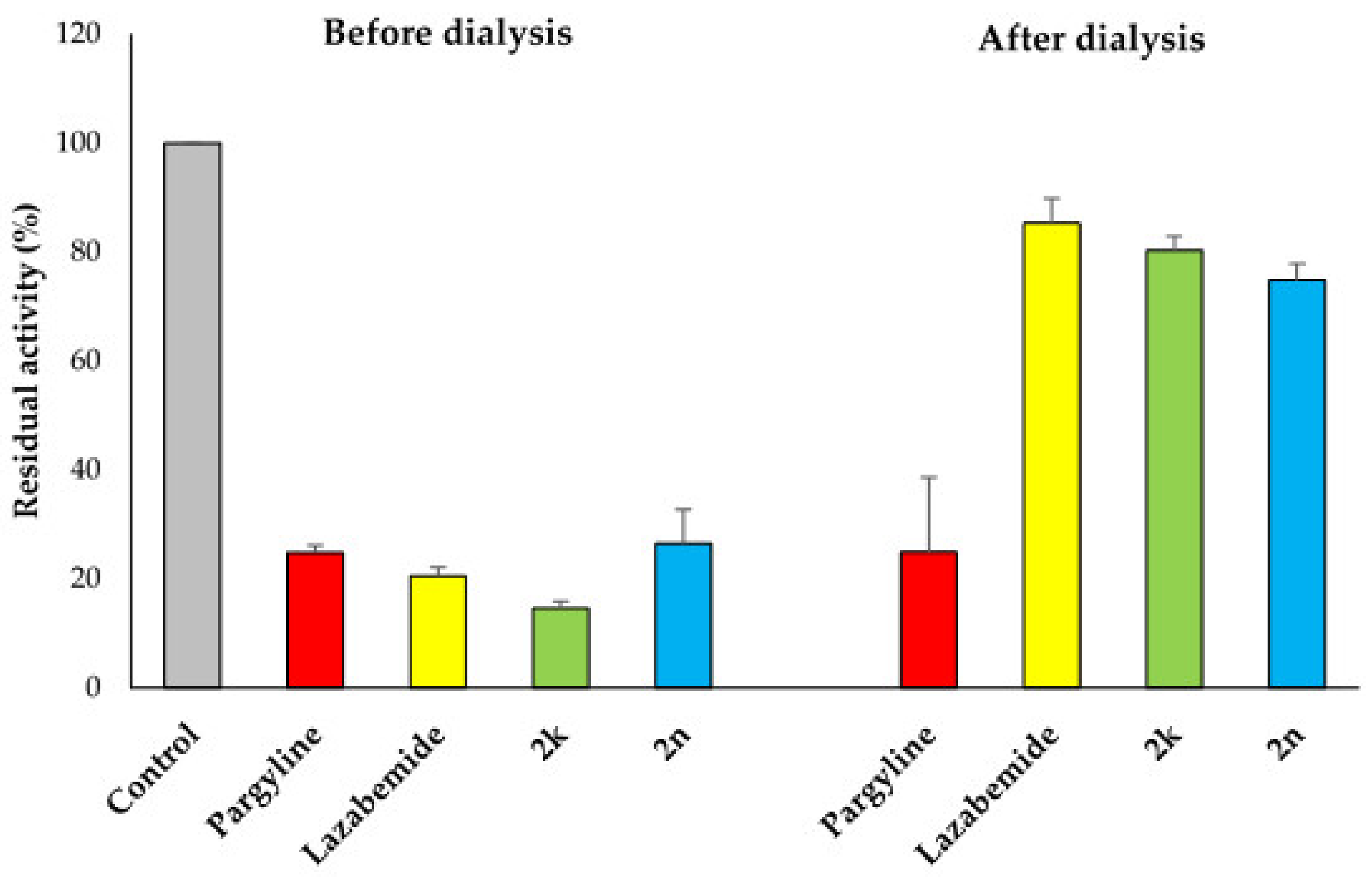
In reversibility tests of 2k and 2n for MAO-B using the dialysis method, their residual activities were efficiently recovered from 14.63% and 26.47% before dialysis, respectively, to 80.26% and 74.83% after dialysis, respectively (Figure 6). In references, pargyline, an irreversible MAO-B inhibitor, displayed no recovery of the residual activity, i.e., 24.81% to 24.88%, whereas lazabemide, a reversible MAO-B inhibitor, showed efficient recovery of the residual activity, i.e., from 20.53% to 98.78%. From these results, it is concluded that 2k and 2n are reversible MAO-B inhibitors.
In a previous study, piperazine chalcone derivatives were also analyzed for their inhibitory activities against MAO-B [33], and p-fluorinated piperazine chalcone derivative PC10 showed the highest reversible competitive inhibition for MAO-B with an IC50 value of 0.65 µM (Ki = 0.50 µM). On the other hand, 2k, the most potent MAO-B inhibitor in this study, also showed reversible competitive inhibition with IC50 of 0.71 µM (Ki = 0.21 µM), followed by 2n (IC50 = 1.11 µM, Ki = 0.28 µM). Interestingly, 2k and 2n elicited effective inhibition against AChE with IC50 values of 8.10 and 4.32 µM, respectively, in contrast to PC10 which showed a moderate inhibition with IC50 of 28.0 µM [33]. 2k and 2n showed mixed type inhibition for AChE with Ki values of 1.44 and 0.82 µM, respectively; however, there were no kinetics data of PC10 against AChE [33]. We previously reported about AChE inhibition of morpholine-based chalcones, which have a similar structure to the piperazine chalcone; however, the lead compounds MO5 and MO9 exerted competitive and non-competitive inhibition, not mixed-type, for AChE with Ki values of 2.52 µM and 7.04 µM, respectively, which are remarkably lower than those of 2k and 2n [39]. These findings suggest that N-methyl piperazine chalcones possess comparable or better Ki values for MAO-B than their corresponding piperazine chalcones, as well as possessing higher affinities with multi-target inhibitions for targeting AD.
2.4. Cytotoxicity Study of 2k and 2n
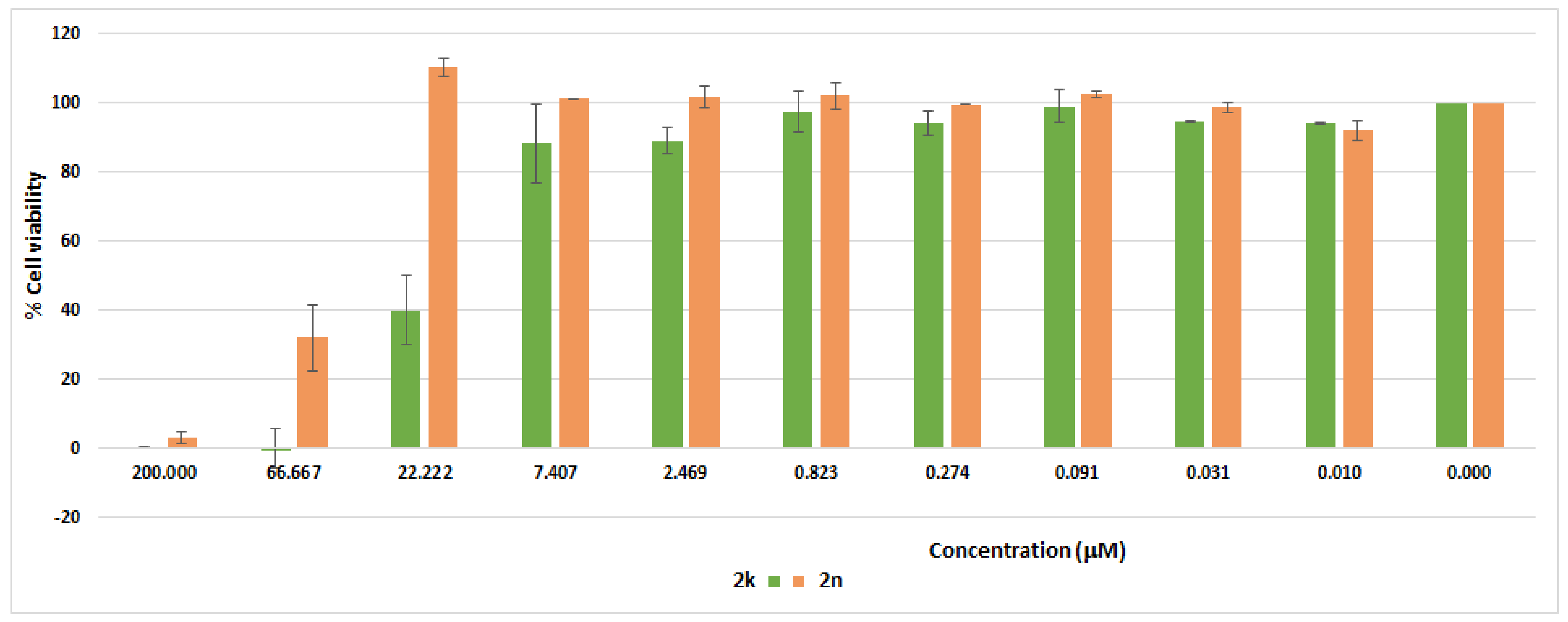
The cytotoxicity profile of 2k and 2n on the cellular viability of VERO normal cells was investigated at ten different concentrations (0.0102–200 µM), using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. As depicted in Figure 7, compounds 2k and 2n were non-toxic to normal VERO cells with IC50 values of 19.93 µM and 62.04 µM, respectively, pointing out their suitability as potential therapeutic candidates.
2.5. Computational Studies
2.5.1. Docking Simulations of 2k and 2n
Docking simulations of the best two members 2k and 2n with MAO-A (pdb code: 2Z5X), MAO-B (pdb code: 4A79), and AChE (pdb code: 6O4W) were performed using AUTODOCK-VINA [40]. The catalytic sites of MAO-A, MAO-B, and AChE are highlighted, as depicted in Figure 8A, 8B, and 8C, respectively. Six combinations of docking simulations were performed (MAO-A-2k and MAOA-2n in Figure 8D, MAO-B-2k and MAO-B-2n in Figure 8E, and AChE-2k and AChE-2n in Figure 8F). The docked compounds were clustered based on their position on the protein. Each top lowest energy conformation in the catalytic region of the three proteins is tabulated in Table 3. Each lowest energy compound near the catalytic region is sketched, with their docking energies in Figure 8D–F. Among the generated conformations, compound 2k has the lowest energy with MAO-B (−11.6 kcal/mol), indicating the tightest binding which is consistent with the experimental IC50 value. That tight binding stems from the existence of several hydrogen bonding interactions (blue dash lines), as shown in Figure 9. The main key residues are THR196, GLN206, and TYR435, and the crucial interacting functional groups in 2k are CF3 and two nitrogen atoms in piperazine. Because the side chain of THR196 can rotate freely and stay in three conformers, Gauche+ (60° of torsional angle in N-CA-CB-OG1), Gauche- (−60°), and trans (180°), the hydrogen bonds of the donor, OG1 in THR196 could enhance its binding affinity toward the –CF3 of 2k. The nitrogen atom in the backbone of THR196 can also engage in hydrogen bonds with the –CF3. Two terminus atoms in the side chain (–NE2 for GLN206 and -OH for TYR435) can share binding interactions toward the two nitrogen atoms in the piperazine of 2k. Although the number of hydrogen bonds in MAO-B-2k is four hydrogen bonds, the MAO-A-2k also has four hydrogen bonds. However, MAO-B-2k possesses lower binding energy and the number of π-π interaction is two, as shown in Figure 8, which is higher than one in MAO-A-2k. Two π-π interactions are found between (1) TYR326 and the central planar ring, and (2) TRP119 and the fluorine aromatic ring. In particular, the first π-π interaction between TYR326 and the central ring of 2k plays an important role in inhibitory selectivity of MAO proteins. The MAO-B has TYR326, and the corresponding residue in MAO-A is ILE335. The structural difference confirms that the inhibitor selectivity of MAO-A and MAO-B is caused by π-π interaction [41]. The docking of compounds 2k and 2n on AChE had higher binding energies than the MAO series. However, AChE showed a higher number of hydrogen bonds and the same number of π-π interactions. Therefore, both compounds 2k and 2n could have tighter binding on AchE than the MAO-A protein. As revealed from the docking simulation results, compound 2k was the most favorable inhibitor in MAO-B in terms of the lowest binding energy and higher number of hydrogen bonds along with π-π interactions, and these results are in good agreement with the IC50 or Ki experiment results. In AChE inhibition, Ki results were not in accordance with the results of the docking data; 2n (Ki = 0.82 uM) bound more tightly on AChE than 2k (Ki = 1.44 uM), but differences in the docking data of AChE were smaller than those of MAO-B. This discrepancy might come from the difference in inhibition modes; AChE inhibition by 2k and 2n was mixed type with a different degree, while MAO-B inhibition was competitive. In addition, docking data were calculated based on a combination of various factors such as hydrogen bonding, electrostatic bonding, van der Waals forces, dissolvent effects, and the flexibilities of compounds [42]. Therefore, mixed inhibition degrees and microelement environments of the binding sites might influence the experimental Ki values, though docking values were not exactly in line with the Ki values.
2.5.2. Pharmacokinetic Prediction of 2k and 2n
As a part of our study, ADME prediction for compounds 2k and 2n was performed. 2k and 2n were predicted to have high gastrointestinal absorption, blood–brain barrier (BBB) permeability, and no substrates of P-glycoprotein (P-gp). 2k inhibited cytochrome P450 (CYP) 2C19, 2D6, and 3A4, whereas 2n inhibited CYP 2C19, 2C9, and 2D6 (Table 4). Furthermore, 2k and 2n were predicted to have no violations for Lipinski rules (Table 5) [44]. Accordingly, 2k and 2n would be expected to possess favorable pharmacokinetic properties for the treatment of neurodegeneration disorders.
3. Materials and Methods
3.1. Chemistry
General: All solvents and reagents were obtained from commercial suppliers and were used directly without further purification. The reaction progress was monitored on a TLC plate (Merck, silica gel 60 F254, Darmstadt, Germany). Melting points were measured using OptiMelt MPA100 melting point apparatus and were uncorrected. FT-IR spectra were scanned using Frontier IR SP8000 MIR DTGS / KBr / Al / Ext beams with Spotlight 200i microscopy (PerkinElmer Inc., MA, USA). 1H and 13C NMR spectra were recorded on a Bruker Advance 400 MHz spectrometer, using deuterated chloroform. Chemical shifts (δ) are given in parts per million (ppm) upfield from tetramethylsilane (TMS) as an internal standard, and s, d, t, and m are presented as singlet, doublet, doublet triplet and multiplet, respectively. Coupling constants (J) are reported in hertz (Hz). High resolution mass spectra (HRMS) were recorded on a Waters Acquity UPLC/Synapt G2 QTOF MS or JMS 700 (Jeol, Tokyo, Japan) mass spectrometer.
3.2. Synthesis
3.2.1. 1-(4-(4-Methylpiperazin-1-yl)phenyl)ethan-1-one (1) [34]
A mixture of 1-(4-fluorophenyl)ethan-1-one (2.5 g, 18 mmol) and 4-methylpiperazine (9 g, 90 mmol) was stirred and heated in a sealed tube at 140 °C for 12 h. The reaction mixture was cooled down to room temperature and poured into ice water. The precipitated solid was filtered under vacuum and dried to afford the title compound as a pure yellow solid, 3.5 g (84.2% yield). 1H and 13C NMR data are consistent with the reported in literature.
3.2.2. General Procedure for Synthesis of Compounds 2a–q
To a solution of compound 1 (250 mg, 1.15 mmol) and the appropriate aromatic aldehyde (1.15 mmol) in 95% ethanol (5 mL), 40% alcoholic sodium hydroxide (1 mL) was added. The reaction mixture was stirred at rt for 0.5–4 h, and the formed precipitate was filtered, washed with cold ethanol, dried and crystallized from ethanol to afford the target compounds, as a yellow amorphous powder, in their pure form.
- 1.
- (E)-1-(4-(4-methylpiperazin-1-yl)phenyl)-3-(3-(trifluoromethyl)phenyl)prop-2-en-1-one (2a)
Yield 89%; mp = 162–163 °C, IR (KBr, cm−1): 2798, 1654, 1608, 1584, 1252, 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 9.0 Hz, 2H), 7.84 (s, 1H), 7.77–7.73 (m, 2H), 7.60–7.56 (m, 2H), 7.49 (t, J = 7.7 Hz, 1H), 6.88 (d, J = 9.0 Hz, 2H), 3.37 (t, J = 5.1 Hz, 4H), 2.52 (t, J = 5.1 Hz, 4H), 2.32 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.31, 154.22, 141.04, 136.17, 131.47, 131.16, 130.79, 129.42, 127.73, 126.33 (d, J = 3 Hz), 124.49 (d, J = 4 Hz), 123.91 (d, J = 271 Hz), 123.68, 113.44, 54.69, 47.13, 46.11; HRMS (ESI-TOF) m/z calcd for C21H22F3N2O [M+H]+: 375.1684, found: 375.1701.
- 2.
- (E)-1-(4-(4-methylpiperazin-1-yl)phenyl)-3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one (2b)
Yield 85%; mp = 190–191 °C, IR (KBr, cm−1): 2800, 1652, 1606, 1586, 1256, 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 15.7 Hz, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.65–7.60 (m, 3H), 6.91 (d, J = 8.9 Hz, 2H), 3.40 (t, J = 5.0 Hz, 4H), 2.55 (t, J = 5.0 Hz, 4H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.36, 154.25, 140.98, 138.79, 131.39 (q, J = 32 Hz), 130.80, 128.34, 127.73, 125.80 (q, J = 3.7 Hz), 124.29, 123.94 (d, J = 271 Hz), 113.45, 54.70, 47.13, 46.13; HRMS (ESI-TOF) m/z calcd for C21H22F3N2O [M+H]+: 375.1684, found: 375.1693.
- 3.
- (E)-3-(4-fluorophenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2c)
Yield 74%; mp = 157–158 °C, IR (KBr, cm−1): 2795, 1648, 1605, 1593, 1413, 1220, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 9.0 Hz, 2H), 7.75 (d, J = 15.6 Hz, 1H), 7.62 (dd, J = 8.6, 5.4 Hz, 2H), 7.48 (d, J = 15.6 Hz, 1H), 7.09 (t, J = 8.6 Hz, 2H), 6.91 (d, J = 9.0 Hz, 2H), 3.40 (t, J = 5.1 Hz, 4H), 2.56 (t, J = 5.1 Hz, 4H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.78, 163.81 (d, J = 249 Hz), 154.12, 141.79, 131.61 (d, J = 3.6 Hz), 130.67, 130.12 (d, J = 8.3 Hz), 128.13, 121.75 (d, J = 2.1 Hz), 116.00 (d, J = 22 Hz), 113.52, 54.75, 47.24, 46.16; HRMS (ESI-TOF) m/z calcd for C20H22FN2O [M+H]+: 325.1716, found: 325.1722.
- 4.
- (E)-4-(3-(4-(4-methylpiperazin-1-yl)phenyl)-3-oxoprop-1-en-1-yl)benzonitrile (2d)
Yield 84%; mp = 218–220 °C, IR (KBr, cm−1): 2801, 2224, 1650, 1603, 1582, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 9.0 Hz, 2H), 7.74 (d, J = 15.6 Hz, 1H), 7.70–7.67 (m, 4H), 7.63 (d, J = 15.6 Hz, 1H), 6.92 (d, J = 9.0 Hz, 2H), 3.42 (t, J = 5.0 Hz, 4H), 2.56 (t, J = 5.0 Hz, 4H), 2.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.06, 154.32, 140.39, 139.76, 132.61, 130.85, 128.55, 127.54, 125.21, 118.54, 113.43, 112.99, 54.71, 47.11, 46.15; HRMS (ESI-TOF) m/z calcd for C21H22N3O [M+H]+: 332.1763, found: 332.1774.
- 5.
- (E)-3-(4-isopropylphenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2e)
Yield 78.3%; mp = 135–136 °C, IR (KBr, cm−1): 2842, 1649, 1601, 1583, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.9 Hz, 2H), 7.78 (d, J = 15.6 Hz, 1H), 7.57 (d, J = 8.2 Hz, 2H), 7.52 (d, J = 15.6 Hz, 1H), 7.27 (d, J = 8.1 Hz, 2H), 6.92 (d, J = 9.0 Hz, 2H), 3.39 (t, J = 5.1 Hz, 4H), 2.94 (quintet, J = 6.9 Hz, 1H), 2.56 (t, J = 5.1 Hz, 4H), 2.35 (s, 3H), 1.27 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 188.22, 154.04, 151.45, 143.25, 133.00, 130.64, 128.43, 127.02, 121.16, 113.57, 54.77, 47.30, 46.16, 34.12, 23.82; HRMS (ESI-TOF) m/z calcd for C23H29N2O [M+H]+: 349.228, found: 349.2291.
- 6.
- (E)-3-(4-(dimethylamino)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2f)
Yield 59.4%; mp = 182–184 °C, IR (KBr, cm−1): 2795, 1640, 1595, 1548, 1433, 1340, 1258, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 15.4 Hz, 1H), 7.53 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 15.4 Hz, 1H), 6.90 (d, J = 8.9 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 3.35 (t, J = 4.7 Hz, 4H), 3.00 (s, 6H), 2.54 (t, J = 4.8 Hz, 4H), 2.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.27, 153.78, 151.77, 144.10, 130.37, 130.14, 129.12, 123.09, 116.85, 113.63, 111.87, 54.79, 47.42, 46.17, 40.16; HRMS (ESI-TOF) m/z calcd for C22H28N3O [M+H]+: 350.2232, found: 350.2239.
- 7.
- (E)-1-(4-(4-methylpiperazin-1-yl)phenyl)-3-(4-(methylthio)phenyl)prop-2-en-1-one (2g)
Yield 81%; mp = 166–167 °C, IR (KBr, cm−1): 2842, 1645, 1605, 1585, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.7 Hz, 2H), 7.74 (d, J = 15.6 Hz, 1H), 7.54 (d, J = 7.8 Hz, 2H), 7.52 (d, J = 14.9 Hz, 1H), 7.23 (d, J = 8.2 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 3.38 (t, J = 4.7 Hz, 4H), 2.54 (t, J = 4.7 Hz, 4H), 2.49 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.88, 154.04, 142.54, 141.65, 131.88, 130.62, 128.67, 128.30, 126.03, 121.00, 113.52, 54.75, 47.25, 46.17, 15.22; HRMS (ESI-TOF) m/z calcd for C21H25N2OS [M+H]+: 353.1687, found: 353.1694.
- 8.
- (E)-3-(4-(difluoromethoxy)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2h)
Yield 92%; mp = 151–152 °C, IR (KBr, cm−1): 2798, 1650, 1605, 1605, 1589, 1224, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.8 Hz, 2H), 7.74 (d, J = 15.6 Hz, 1H), 7.63 (d, J = 8.6 Hz, 2H), 7.50 (d, J = 15.6 Hz, 1H), 7.14 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 8.9 Hz, 2H), 6.56 (t, J = 73.5 Hz, 1H), 3.39 (t, J = 4.9 Hz, 4H), 2.55 (t, J = 4.9 Hz, 4H), 2.35 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 187.68, 154.14, 152.34, 141.59, 132.56, 130.68, 129.81, 128.03, 122.10, 119.59, 115.65 (t, J = 259 Hz), 113.49, 54.73, 47.20, 46.14; HRMS (ESI-TOF) m/z calcd for C21H23F2N2O2 [M+H]+: 373.1727, found: 373.1732.
- 9.
- (E)-3-(3-fluoro-5-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2i)
Yield 79%; mp = 167–168 °C, IR (KBr, cm−1): 2800, 1654, 1608, 1250, 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 9.0 Hz, 2H), 7.72 (d, J = 15.6 Hz, 1H), 7.66 (s, 1H), 7.59 (d, J = 15.6 Hz, 1H), 7.49 (d, J = 9.1 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 6.92 (d, J = 9.0 Hz, 2H), 3.42 (t, J = 5.1 Hz, 4H), 2.57 (t, J = 5.1 Hz, 4H), 2.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 186.95, 162.75 (d, J = 247 Hz), 154.34. 139.70, 138.82 (d, J = 8.0 Hz), 130.87, 127.50, 124.93, 120.62, 117.99, 117.77, 113.90, 113.69, 113.45, 54.73, 47.14, 46.15; HRMS (ESI-TOF) m/z calcd for C21H21F4N2O [M+H]+: 393.159, found: 393.1592.
- 10.
- (E/Z)-3-(3,5-bis(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2j)
Yield 78% (E/Z, 4:1); mp = 160–162 °C, IR (KBr, cm−1): 2801, 1655, 1583, 1253, 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 12 Hz, 4H), 7.87–7.81 (m, 1H) 7.79 (d, J = 15.6 Hz, 1H), 7.66 (d, J = 15.6 Hz, 1H), 6.93 (d, J = 9.0 Hz, 2H), 3.43 (t, J = 5.0 Hz, 4H), 2.57 (t, J = 5.0 Hz, 4H), 2.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 186.72, 154.39, 139.18, 137.57, 132.39 (d, J = 33 Hz), 130.93, 127.77, 127.35, 125.49, 123.12 (d, J = 271 Hz), 122.98, 113.44, 54.71, 47.10, 46.14; HRMS (ESI-TOF) m/z calcd for C22H21F6N2O [M+H]+: 443.1558, found: 443.1561.
- 11.
- (E)-3-(4-fluoro-3-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2k)
Yield 82.3%; mp = 133–135 °C, IR (KBr, cm−1): 2805, 1652, 1605, 1466, 1225, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.8 Hz, 2H), 7.86 (d, J = 4.4 Hz, 1H), 7.80–7.77 (m, 1H), 7.73 (d, J = 15.6 Hz, 1H), 7.53 (d, J = 15.6 Hz, 1H), 7.23 (d, J = 9.8 Hz, 1H), 6.92 (d, J = 8.8 Hz, 2H), 3.41 (t, J = 5.0 Hz, 4H), 2.56 (t, J = 5.0 Hz, 4H), 2.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.15, 154.27, 141.44, 139.97, 133.60, 133.52, 131.99, 130.79, 130.66, 127.67, 126.61, 123.45, 121.04, 117.72, 117.62 (d, J = 21 Hz), 113.50 (d, J = 5.0 Hz), 54.73, 47.16, 46.15; HRMS (ESI-TOF) m/z calcd for C21H21F4N2O [M+H]+: 393.1590, found: 393.1595.
- 12.
- (E)-3-(4-chloro-3-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2l)
Yield 77.4%; mp = 139–140 °C, IR (KBr, cm−1): 2807, 1650, 1605, 1583, 1365, 1253, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.9 Hz, 2H), 7.92 (d, J = 1.1 Hz, 1H), 7.72 (d, J = 15.6 Hz, 1H), 7.69 (s, 1H), 7.58 (d, J = 15.6 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 6.92 (d, J = 9.0 Hz, 2H), 3.41 (t, J = 5.0 Hz, 4H), 2.56 (t, J = 5.0 Hz, 4H), 2.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.08, 154.29, 139.89, 134.40, 133.40, 132.09, 132.05, 130.82, 129.03 (d, J = 31 Hz), 127.61, 126.82 (d, J = 5.2 Hz), 124.05, 122.61 (d, J = 272 Hz), 113.45, 54.73, 47.15, 46.15; HRMS (ESI-TOF) m/z calcd for C21H21ClF3N2O [M+H]+: 409.1294, found: 409.1303.
- 13.
- (E)-3-(4-fluoro-3-methoxyphenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2m)
Yield 84%; mp = 153–154 °C, IR (KBr, cm−1): 2794, 1646, 1602, 1578, 1235, 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 15.6 Hz, 1H), 7.44 (d, J = 15.6 Hz, 1H), 7.20–7.16 (m, 2H), 7.10–7.05 (m, 1H), 6.90 (d, J = 8.8 Hz, 2H), 3.93 (s, 3H), 3.38 (t, J = 4.9 Hz, 4H), 2.54 (t, J = 4.8 Hz, 4H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.89, 154.13, 153.63 (d, J = 250 Hz), 147.97 (d, J = 11 Hz), 142.17, 132.04 (d, J = 3.8 Hz), 130.67, 128.12, 121.92, 121.49 (d, J = 7.0 Hz), 116.46 (d, J = 19 Hz), 113.51, 112.92, 56.34, 54.74, 47.24, 46.13; HRMS (ESI-TOF) m/z calcd for C21H24FN2O2 [M+H]+: 355.1822, found: 355.1826.
- 14.
- (E)-3-(5-bromo-2-fluorophenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2n)
Yield 64%; mp = 133–135 °C, IR (KBr, cm−1): 2803, 1651, 1590, 1303, 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 9.0 Hz, 2H), 7.78 (d, J = 15.7 Hz, 1H), 7.74 (d, J = 2.4 Hz, 1H), 7.63 (d, J = 15.8 Hz, 1H), 7.43 (ddd, J = 8.7, 4.5, 2.5 Hz, 1H), 7.00 (dd, J = 10, 8.8 Hz, 1H), 6.90 (d, J = 9.0 Hz, 2H), 3.40 (t, J = 5.1 Hz, 4H), 2.55 (t, J = 5.1 Hz, 4H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.26, 160.50 (d, J = 252 Hz), 154.22, 133.90, 133.79, 131.75 (d, J = 3.0 Hz), 130.84, 127.68, 125.63 (d, J = 6.2 Hz), 125.49 (d, J = 13 Hz), 117.99 (d, J = 24 Hz), 116.98 (d, J = 3.0 Hz), 113.44, 54.71, 47.13, 46.15; HRMS (ESI-TOF) m/z calcd for C20H21BrFN2O [M+H]+: 403.0821, found: 403.0836.
- 15.
- (E)-3-(2-chloro-5-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2o)
Yield 85.4%; mp = 128–129 °C, IR (KBr, cm−1): 2800, 1651, 1591, 1255, 1H NMR (400 MHz, CDCl3) δ 8.11 (dd, J = 15.7, 1.8 Hz, 1H), 8.00 (d, J = 7.3 Hz, 2H), 7.98 (d, J = 11.5 Hz, 1H), 7.58–7.54 (m, 3H), 6.92 (dd, J = 9.0, 2.2 Hz, 2H), 3.41 (s, 4H), 2.55 (s, 4H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.05, 154.29, 138.64, 137.17, 134.63, 130.93, 130.82, 129.60 (d, J = 33 Hz), 127.47, 126.90, 126.30, 124.52 (d, J = 3.6 Hz), 123.57 (d, J = 270 Hz), 113.43, 54.70, 47.09, 46.13; HRMS (ESI-TOF) m/z calcd for C21H21ClF3N2O [M+H]+: 409.1294, found: 409.1295.
- 16.
- (E)-3-(4-methoxy-2-(trifluoromethyl)phenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2p)
Yield 91%; mp = 133–134 °C, IR (KBr, cm−1): 2801, 1650, 1592, 1320, 1H NMR (400 MHz, CDCl3) δ 8.06 (dd, J = 15.4, 2.0 Hz, 1H), 7.97 (d, J = 9.0 Hz, 2H), 7.79 (d, J = 8.7 Hz, 1H), 7.39 (d, J = 15.4 Hz, 1H), 7.20 (d, J = 2.6 Hz, 1H), 7.07 (dd, J = 8.7, 2.5 Hz, 1H), 6.89 (d, J = 9.0 Hz, 2H), 3.86 (s, 3H), 3.38 (t, J = 5.1 Hz, 4H), 2.54 (t, J = 5.0 Hz, 4H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.69, 160.37, 154.11, 138.03, 130.75, 130.48 (d, J = 30 Hz), 129.48, 127.88, 126.40, 124.26, 123.77 (d, J = 273 Hz), 117.23, 113.46, 112.04 (q, J = 5.8 Hz), 55.65, 54.72, 47.18, 46.13; HRMS (ESI-TOF) m/z calcd for C22H24F3N2O2 [M+H]+: 405.1790, found: 405.1800.
- 17.
- (E)-3-(2,4-difluorophenyl)-1-(4-(4-methylpiperazin-1-yl)phenyl)prop-2-en-1-one (2q)
Yield 85%; mp = 138–141 °C, IR (KBr, cm−1): 2802, 1651, 1588, 1427, 1292, 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.9 Hz, 2H), 7.81 (d, J = 15.8 Hz, 1H), 7.65–7.52 (m, 2H), 6.94–6.62 (m, 4H), 3.40 (t, J = 5.0 Hz, 4H), 2.55 (t, J = 5.0 Hz, 4H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.71, 154.09 (d, J = 15 Hz), 136.07, 134.76, 130.83 (d, J = 4.8 Hz), 130.75, 129.27 (d, J = 269 Hz), 124.22 (d, J = 7.1 Hz), 119.90, 113.54, 113.49, 112.10, 111.85, 104.92, 104.66, 104.40, 64.09, 54.73, 47.29, 47.19, 46.15; HRMS (ESI-TOF) m/z calcd for C20H21F2N2O [M+H]+: 343.1622, found: 343.1622.
3.3. Monoamine Oxidase (MAO) and Cholinesterase (ChE) Biological Studies
Materials: Monoamine oxidase A and B (MAO-A and MAO-B), acetyl and butyrylcholinesterase (AChE and BChE), kynuramine, benzylamine, acetylthiocholine iodide (ATCI), S-butyrylthiocholine iodide (BTCI), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), toloxatone, clorgyline, lazabemide, pargyline, donepezil, and dimethyl sulfoxide (DMSO) were obtained from Sigma Aldrich (St. Louis, MO, USA).
3.3.1. Enzyme Inhibition Assays
MAO assays were performed as described previously [45], i.e., ~0.09 U/mL of MAO-A and MAO-B were reacted with 0.06 mM kynuramine and 0.3 mM benzylamine, respectively, in 50 mM sodium phosphate (pH 7.2). The absorbances of reaction mixtures of MAO-A and MAO-B were observed at 316 nm and 250 nm, respectively, for 30 min under kinetic mode of the spectrophotometer (OPTIZEN, K-Lab, Dajeon, Republic of Korea). For inhibition assay, the compounds were dissolved in DMSO and added into the reaction mixture. The final concentration of DMSO in the reaction mixture was less than 1% to avoid solvent disturbance.
For measuring cholinesterase (ChE) activity, the Ellman method [46] was employed with a slight modification. The AChE and BchE with ~0.02 U/mL were reacted with 0.5 mM ATCI and BTCI, respectively, followed by addition of 0.5 mM DTNB. The absorbance of the reaction mixture was continuously observed at 412 nm for 15 min under the kinetic mode of the spectrophotometer. In the inhibition assay, the enzyme and the compound were preincubated in reaction buffer for 15 min, followed by adding substrates and DTNB.
3.3.2. Kinetics and Reversibility Analysis
To analyze the inhibitory patterns of compounds 2k and 2n, which showed the most potent MAO-B inhibition with the lowest IC50, kinetics and reversibility tests were conducted [47]. In kinetics for MAO-B, 0.038, 0.075, 0.15, 0.3, and 0.6 mM of benzylamine were used to construct Lineweaver–Burk plots, and the compounds were added with concentrations of ~1/2× IC50, IC50, and 2× IC50 for a secondary plot to obtain Km and Ki values. In the kinetics for AChE, 0.0625, 0.125, 0.25, 0.5, 1.0 mM of ATCI were used in the presence of DTNB, and the compounds were added to be ~1/2× IC50, IC50, and 2× IC50 concentrations for the secondary plot to obtain Km and Ki values.
Reversibility of the compounds for MAO-B was measured using the dialysis method. The compounds were treated with ~1.5× IC50 for 2k and ~2× IC50 for 2n, respectively, with the reaction mixture without substrate. The reaction mixture was placed in a dialysis tube (DiaEasyTM Dialyzer (800 µL) MWCO 6–8 kDa, Biovision, CA, USA) and dialyzed for 6 h. After that, the 0.3 mM of benzylamine was added into the reaction mixture for measuring enzyme activity. The relative activity to the control (without inhibitors) before and after dialysis was calculated, respectively. The lazabemide and pargyline were used as standard reversible and irreversible MAO-B inhibitors, respectively.
3.4. Cell Cytotoxicity Study
3.4.1. Materials
The reference compound staurosporine was purchased from Sigma-Aldrich (Saint Louis, MO, USA). Compounds 2k and 2n were dissolved in DMSO as 50 mM stock. CellTiter 96® Non-Radioactive Cell Proliferation Assay (MTT) reagent was purchased from Promega (Cat# G4000). The normal Vero cell line was purchased from ATCC. Normal Vero cells were cultured with EMEM medium plus 10% FBS. In total, 100 µg/mL penicillin and 100 µg/mL streptomycin were added to the culture media. Cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
3.4.2. Method
Compounds 2k and 2n and staurosporine were diluted with DMSO in 10-dose and 3-fold serial dilution starting at 50 mM (test compounds) and 10 mM (staurosporine). The test compounds (100 nL) and staurosporine (25 nL) were delivered from the source plate to the wells of 384-well cell culture plates by Echo 550 Liquid Handler. Culture medium (25 µL) containing 2000 of normal Vero cells was added to the 384-well cell culture plates, and the plates were incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air for 72 h. MTT Dye Solution (8 µL) was added to each well, and the plates were incubated at 37 °C in a CO2 incubator for 6 h. After incubation, the formazan was produced in the cells. Solubilization/Stop Solution (30 µL) was added to each well and the plates were incubated for another 1 h at 37 °C in a CO2 incubator to dissolve the formazan crystals. The absorbance of each sample was measured at 590 nm using Envision 2104 Multilabel Reader (PerkinElmer, Santa Clara, CA, USA), and the cell viability was determined based on the quantification of the color intensity in each culture well (duplicate assay mode). The IC50 curves were plotted, and the IC50 values were calculated using the GraphPad Prism 4 program based on a sigmoidal dose–response equation.
3.5. Molecular Docking
Docking simulation of three enzymes, MAO-A, MAO-B, and AChE with compounds 2k and 2n were performed using AUTODOCK-VINA [40]. The protein structures of MAO-A, MAO-B, and AChE are PDB (Protein Data Bank) ID of 2Z5X, 4A79, and 6O4W, respectively. MAO-B and AChE have a homogenous complex with two chains, A and B. We used two programs; the first program predicts a binding site, and the second program, Pck, is a pocket search program, not a binding site. Because the pocket and the binding site are not identical, we cannot find another binding site working under allosteric inhibition when the docking simulation runs only in the binding site. The first program is for understanding the enzyme, and the second one is for general docking procedure. For the docking simulation, the first chain, A, was chosen. The binding sites of two proteins were determined using a binding pocket program, DPSP (dockable pocket site prediction: as an in-house program). It searches and collects all of the ligands existing on similar sequence structures, and obtains pockets available on the MAO and AChE proteins. Compounds 2k and 2n were sketched, energy minimized, and converted to the general structure format, i.e., PDB, using the Marvin program (ChemAxon; http://www.chemaxon.com, accessed on 1 July 2022). For the docking simulation, the pockets in these proteins were searched utilizing Pck pocket detection program (http://schwarz.benjamin.free.fr/Work/Pck/home.htm, accessed on 1 July 2022), where it detects the amino acids forming pockets and measures their volumes. Twelve, thirteen, and fourteen pockets over 100 Å3 of volume were found in MAO-A, MAO-B, and AChE, respectively. These pockets consist of 168, 158, and 101 amino acids for MAO-A, MAO-B, and AChE. Compounds 2k and 2n were put on Carbon α atom positions in these pocket residues, and the docking simulations were run ten times with different random seeds, i.e., a total of 1680, 1580, and 1010 simulations were carried out for MAO-A, MAO-B, and AChE, respectively. The box size with a length of 15 Å was used to prevent the compounds from drifting from the center of the pocket residue. With these docking poses, the clustering based on the center of mass was performed to group these docking conformations using CHARMM (Chemistry at HARvard Macromolecular Mechanics) [48]. The compounds were ranked by the lowest energy of the group, the largest number of the group, and the lowest energy conformation in the group. The molecular structures were drawn by VMD (visual molecular dynamics) visualizer program (https://www.ks.uiuc.edu/Research/vmd/, accessed on 1 July 2022). Hydrogen bond and π-π interaction analysis between each compound and proteins were calculated based on a geometric analysis. First, the acceptor and donor atoms of hydrogen bonds were assigned and then their Cartesian coordinates were obtained. Second, all of the pairs between acceptor and donor atoms were arranged to measure geometric distance and angle. The used hydrogen bonding criteria is the distance below 4.5 Å between the acceptor and donor atoms. The π-π interaction forms between two close parallel aromatic rings. First, the aromatic rings were sought, and then the atoms forming the aromatic rings were found. The centroid of these atoms was calculated and the Cartesian coordinate was obtained. The distance between the two centroids was measured, in the case that both aromatic rings are close and parallel.
3.6. Pharmacokinetic Prediction of the 2k and 2n Using the SwissADME Web Tool
The pharmacokinetic and physicochemical properties such as gastrointestinal (GI) absorption, BBB permeability, P-glycoprotein (P-gp) substrate, cytochrome P450 (CYP), and Lipinski parameters [43] of 2k and 2n were predicted using the web tool of SwissADME (http://www.swissadme.ch, accessed on 1 July 2022) [49].
4. Conclusions
In this report, we describe the synthesis of new N-methyl-piperazine chalcones, and their MAOs and AChE inhibition profiles. Most of the tested compounds exerted potent MAO-B inhibition with considerable SI along with moderate AChE inhibition. The fluorinated substituted derivatives 2k and 2n inhibited MAO-B with Ki values of sub-micromolar range. The kinetics study of 2k and 2n disclosed their competitive reversible MAO-B inhibitory pattern. A cell-based assay of 2k and 2n against normal Vero cells revealed their minimized cytotoxicity. A docking simulation predicted that 2k showed the lowest energy for MAO-B (−11.6 kcal/mol) with four hydrogen bonds and two π-π interactions. In silico ADME/Tox prediction studies of 2k and 2n pointed out their favorable drug-like properties. Overall, compounds 2k and 2n could serve as promising MAO-B/AChE inhibitors for the treatment of NDD.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph16010083/s1, this section includes; Figure S1, 1H NMR and 13C NMR spectra; Figure S2, HRMS charts; Figure S3, IR spectra; Figure S4, IC50 curves of 2k and 2n against MAO-B.
Author Contributions
Conceptualization, A.K.E.-D., H.K. and G.K.; synthesis and characterization of target compounds, A.K.E.-D. and H.J.K.; biochemical enzyme assays, J.E.P.; docking simulation, J.L.; validation and data curation, J.E.P., E.-K.B. and H.K.; writing—original draft preparation, A.K.E.-D. and J.L.; writing—review and editing, H.K. and G.K.; supervision, H.K. and G.K.; project administration, G.K.; funding acquisition, A.K.E.-D., E.-K.B., H.K. and G.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Research Council of Science & Technology (NST) grant by the Korean government (MSIT) (No. CAP-20-01-KRIBB), the National Research Foundation of Korea (NRF) grant funded by the Korean government (NRF-2022R1A2B5B01002536), the Korea Research Fellowship Program grant through NRF funded by the Ministry of Science and ICT (2019H1D3A1A0107088214, A.K. El-Damasy), and the Institutional Program grant by the Korea Institute of Science and Technology (2E31512).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J.A. Novel Approaches for the Treatment of Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.F.; Xu, S.T.; Zhu, Z.Y.; Xu, J.Y. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Soacha, D.A.; Scheiner, M.; Decker, M. Multi-target-directed-ligands acting as enzyme inhibitors and receptor ligands. Eur. J. Med. Chem. 2019, 180, 690–706. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.D.; Dias, K.S.T.; Gontijo, V.S.; Ortiz, C.J.C.; Viegas, C. Multi-Target Directed Drugs as a Modern Approach for Drug Design Towards Alzheimer’s Disease: An Update. Curr. Med. Chem. 2018, 25, 3491–3525. [Google Scholar] [CrossRef]
- Vianello, R.; Repič, M.; Mavri, J. How are biogenic amines metabolized by monoamine oxidases? Eur. J. Org. Chem. 2012, 2012, 7057–7065. [Google Scholar] [CrossRef]
- Jenner, P.; Olanow, C.W. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 1996, 47, 161S–170S. [Google Scholar] [CrossRef]
- Robottom, B.J. Efficacy, safety, and patient preference of monoamine oxidase B inhibitors in the treatment of Parkinson’s disease. Patient Prefer. Adher. 2011, 5, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carradori, S.; Secci, D.; Bolasco, A.; Chimenti, P.; D’Ascenzio, M. Patent-related survey on new monoamine oxidase inhibitors and their therapeutic potential. Expert Opin. Ther. Pat. 2012, 22, 759–801. [Google Scholar] [CrossRef]
- Rehuman, N.A.; Mathew, B.; Jat, R.K.; Nicolotti, O.; Kim, H. A Comprehensive Review of Monoamine Oxidase-A Inhibitors in their Syntheses and Poteneies. Comb. Chem. High Throughput Scr. 2020, 23, 898–914. [Google Scholar] [CrossRef]
- Park, J.-H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Lu, C.J.; Sun, Y.; Mao, F.; Luo, Z.H.; Su, T.; Jiang, H.L.; Shan, W.J.; Li, X.S. Multitarget-Directed Benzylideneindanone Derivatives: Anti-beta-Amyloid (A beta) Aggregation, Antioxidant, Metal Chelation, and Monoamine Oxidase B (MAO-B) Inhibition Properties against Alzheimer’s Disease. J. Med. Chem. 2012, 55, 8483–8492. [Google Scholar] [CrossRef] [PubMed]
- Viayna, E.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Cisternas, P.; Oliva, C.A.; Sánchez-López, E.; Ettcheto, M.; Bartolini, M.; De Simone, A.; Ricchini, M. Discovery of a potent dual inhibitor of acetylcholinesterase and butyrylcholinesterase with antioxidant activity that alleviates Alzheimer-like pathology in old APP/PS1 mice. J. Med. Chem. 2020, 64, 812–839. [Google Scholar] [CrossRef] [PubMed]
- Tasso, B.; Catto, M.; Nicolotti, O.; Novelli, F.; Tonelli, M.; Giangreco, I.; Pisani, L.; Sparatore, A.; Boido, V.; Carotti, A. Quinolizidinyl derivatives of bi-and tricyclic systems as potent inhibitors of acetyl-and butyrylcholinesterase with potential in Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 2170–2184. [Google Scholar] [CrossRef]
- Sang, Z.P.; Bai, P.; Ban, Y.J.; Wang, K.R.; Wu, A.G.; Mi, J.; Hu, J.Q.; Xu, R.; Zhu, G.F.; Wang, J.T.; et al. Novel donepezil-chalcone-rivastigmine hybrids as potential multifunctional anti-Alzheimer’s agents: Design, synthesis, In Vitro biological evaluation, In Vivo and In Silico studies. Bioorg. Chem. 2022, 127, 106007. [Google Scholar] [CrossRef]
- Sang, Z.P.; Song, Q.; Cao, Z.C.; Deng, Y.; Tan, Z.H.; Zhang, L. Design, synthesis and evaluation of novel dimethylamino chalcone-O-alkylamines derivatives as potential multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2021, 216, 113310. [Google Scholar] [CrossRef]
- Reis, J.; Cagide, F.; Valencia, M.E.; Teixeira, J.; Bagetta, D.; Perez, C.; Uriarte, E.; Oliveira, P.J.; Ortuso, F.; Alcaro, S.; et al. Multi-target-directed ligands for Alzheimer’s disease: Discovery of chromone-based monoamine oxidase/cholinesterase inhibitors. Eur. J. Med. Chem. 2018, 158, 781–800. [Google Scholar] [CrossRef]
- Yamali, C.; Engin, F.S.; Bilginer, S.; Tugrak, M.; Ozgun, D.O.; Ozli, G.; Levent, S.; Saglik, B.N.; Ozkay, Y.; Gul, H.I. Phenothiazine-based chalcones as potential dual-target inhibitors toward cholinesterases (AChE, BuChE) and monoamine oxidases (MAO-A, MAO-B). J. Heterocycl. Chem. 2021, 58, 161–171. [Google Scholar] [CrossRef]
- Rathi, A.K.; Syed, R.; Shin, H.S.; Patel, R.V. Piperazine derivatives for therapeutic use: A patent review (2010-present). Expert Opin. Ther. Pat. 2016, 26, 777–797. [Google Scholar] [CrossRef]
- Brito, A.F.; Moreira, L.K.S.; Menegatti, R.; Costa, E.A. Piperazine derivatives with central pharmacological activity used as therapeutic tools. Fund Clin. Pharmacol. 2019, 33, 13–24. [Google Scholar] [CrossRef]
- Ayala-Aguilera, C.C.; Valero, T.; Lorente-Macias, A.; Baillache, D.J.; Croke, S.; Unciti-Broceta, A. Small Molecule Kinase Inhibitor Drugs (1995–2021): Medical Indication, Pharmacology, and Synthesis. J. Med. Chem. 2022, 65, 1047–1131. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Mantha, A.K.; Kumar, V. Synthesis, biological evaluation and molecular modeling studies of phenyl-/benzhydrylpiperazine derivatives as potential MAO inhibitors. Bioorg. Chem. 2018, 77, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Jevtic, I.I.; Lai, T.H.; Penjisevic, J.Z.; Dukic-Stefanovic, S.; Andric, D.B.; Brust, P.; Kostic-Rajacic, S.V.; Teodoro, R. Newly Synthesized Fluorinated Cinnamylpiperazines Possessing Low In Vitro MAO-B Binding. Molecules 2020, 25, 4941. [Google Scholar] [CrossRef] [PubMed]
- Modh, R.P.; Kumar, S.P.; Jasrai, Y.T.; Chikhalia, K.H. Design, Synthesis, Biological Evaluation, and Molecular Modeling of Coumarin-Piperazine Derivatives as Acetylcholinesterase Inhibitors. Arch. Pharm. 2013, 346, 793–804. [Google Scholar] [CrossRef]
- Guglielmi, P.; Mathew, B.; Secci, D.; Carradori, S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J Med. Chem. 2020, 205, 112650. [Google Scholar] [CrossRef]
- Zhuang, C.L.; Zhang, W.; Sheng, C.Q.; Zhang, W.N.; Xing, C.G.; Miao, Z.Y. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yanez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. Chalcones: A Valid Scaffold for Monoamine Oxidases Inhibitors. J. Med. Chem. 2009, 52, 2818–2824. [Google Scholar] [CrossRef]
- Mathew, B.; Ucar, G.; Mathew, G.E.; Mathew, S.; Purapurath, P.K.; Moolayil, F.; Mohan, S.; Gupta, S.V. Monoamine Oxidase Inhibitory Activity: Methyl- versus Chlorochalcone Derivatives. Chemmedchem 2016, 11, 2649–2655. [Google Scholar] [CrossRef]
- Hammuda, A.; Shalaby, R.; Rovida, S.; Edmondson, D.E.; Binda, C.; Khalil, A. Design and synthesis of novel chalcones as potent selective monoamine oxidase-B inhibitors. Eur. J. Med. Chem. 2016, 114, 162–169. [Google Scholar] [CrossRef]
- Lakshminarayanan, B.; Baek, S.C.; Lee, J.P.; Kannappan, N.; Mangiatordi, G.F.; Nicolotti, O.; Subburaju, T.; Kim, H.; Mathew, B. Ethoxylated Head of Chalcones as a New Class of Multi-Targeted MAO Inhibitors. Chemistryselect 2019, 4, 6614–6619. [Google Scholar] [CrossRef]
- Xiao, G.Y.; Li, Y.; Qiang, X.M.; Xu, R.; Zheng, Y.X.Z.; Cao, Z.C.; Luo, L.; Yang, X.; Sang, Z.P.; Su, F.; et al. Design, synthesis and biological evaluation of 4’-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2017, 25, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.Q.; Qiang, X.M.; Song, Q.; Cao, Z.C.; Ye, C.Y.; He, Y.X.; Deng, Y.; Zhang, L. Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorg. Chem. 2020, 94, 103477. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Oh, J.M.; Baty, R.S.; Batiha, G.E.S.; Parambi, D.T.; Gambacorta, N.; Nicolotti, O.; Kim, H. Piperazine-substituted chalcones: A new class of MAO-B, AChE, and BACE-1 inhibitors for the treatment of neurological disorders. Environ. Sci. Pollut. Res. 2021, 28, 38855–38866. [Google Scholar] [CrossRef] [PubMed]
- Heiser, U.; Niestroj, A.; Zeitlmann, L. N-Pyridinyl Carboxamides as Cyclin-Dependent Kinase Inhibitors Useful in the Treatment of Diseases. 2011, WO2011110612.
- Rangarajan, T.; Mathew, B. Recent Updates on Pyrazoline Derivatives as Promising Candidates for Neuropsychiatric and Neurodegenerative Disorders. Curr. Trends Med. Chem. 2021, 21, 2695–2714. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Baek, S.C.; Lee, J.P.; Tondo, A.R.; Nicolotti, O.; Kim, H.; Mathew, B. Design, synthesis and biological evaluation of oxygenated chalcones as potent and selective MAO-B inhibitors. Bioorg. Chem. 2019, 93, 103335. [Google Scholar]
- Jeong, G.S.; Kaipakasseri, S.; Lee, S.R.; Marraiki, N.; Batiha, G.E.S.; Dev, S.; Palakkathondi, A.; Kavully, F.S.; Gambacorta, N.; Nicolotti, O.; et al. Selected 1, 3-benzodioxine-containing chalcones as multipotent oxidase and acetylcholinesterase inhibitors. ChemMedChem 2020, 15, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Rullo, M.; Cipolloni, M.; Catto, M.; Colliva, C.; Miniero, D.V.; Latronico, T.; De Candia, M.; Benicchi, T.; Linusson, A.; Giacchè, N. Probing Fluorinated Motifs onto Dual AChE-MAO B Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Early-ADME Studies. J. Med. Chem. 2022, 65, 3962–3977. [Google Scholar] [CrossRef]
- Sasidharan, R.; Eom, B.H.; Heo, J.H.; Park, J.E.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Manju, S.L.; Mathew, B.; et al. Morpholine-based chalcones as dual-acting monoamine oxidase-B and acetylcholinesterase inhibitors: Synthesis and biochemical investigations. J. Enzym. Inhib. Med. Chem. 2021, 36, 188–197. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Son, S.Y.; Ma, A.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-angstrom resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Soto, C.S.; Honig, B. Evaluating conformational free energies: The colony energy and its application to the problem of loop prediction. Proc. Natl. Acad. Sci. USA 2002, 99, 7432–7437. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Ryu, H.W.; Kang, M.-G.; Park, D.; Oh, S.-R.; Kim, H. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett. 2016, 26, 4714–4719. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Oh, J.M.; Jang, H.-J.; Kim, W.J.; Kang, M.-G.; Baek, S.C.; Lee, J.P.; Park, D.; Oh, S.-R.; Kim, H. Calycosin and 8-O-methylretusin isolated from Maackia amurensis as potent and selective reversible inhibitors of human monoamine oxidase-B. Int. J. Biol. Macromol. 2020, 151, 441–448. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar]
Figure 1.
Representative examples of CNS acting FDA-approved drugs possessing N-methylpiperazine.

Figure 2.
Representative examples of reported chalcones as selective MAO-B inhibitors or dual MAO-B/AChE or BChE inhibitors, and chemical structure of the target compounds 2k–2q.
Figure 2.
Representative examples of reported chalcones as selective MAO-B inhibitors or dual MAO-B/AChE or BChE inhibitors, and chemical structure of the target compounds 2k–2q.

Scheme 1.
Reagents and reaction conditions: (i) 4-Methylpiperazine, neat, 140 °C, 12 h, 84%; (ii) Benzaldehyde derivative, 40% ethanolic sodium hydroxide, 95% ethanol, rt, 0.5–4 h, 59–92%.
Scheme 1.
Reagents and reaction conditions: (i) 4-Methylpiperazine, neat, 140 °C, 12 h, 84%; (ii) Benzaldehyde derivative, 40% ethanolic sodium hydroxide, 95% ethanol, rt, 0.5–4 h, 59–92%.

Figure 3.
SAR analysis of the target compounds 2a–q for MAO-B/AChE inhibition.

Figure 4.
Lineweaver–Burk plots (a,c) and their secondary plots (b,d) of 2k and 2n for MAO-B, respectively. Five concentrations of benzylamine used were 0.0375, 0.075, 0.15, 0.3 and 0.6 mM, respectively, and three concentrations of inhibitor were ~1/2×, 1×, and 2 × IC50: 0.38, 0.77, and 1.54 µM for 2k; 0.64, 1.27, and 2.54 µM for 2n. The Km value for MAO-B was 0.20 mM.
Figure 4.
Lineweaver–Burk plots (a,c) and their secondary plots (b,d) of 2k and 2n for MAO-B, respectively. Five concentrations of benzylamine used were 0.0375, 0.075, 0.15, 0.3 and 0.6 mM, respectively, and three concentrations of inhibitor were ~1/2×, 1×, and 2 × IC50: 0.38, 0.77, and 1.54 µM for 2k; 0.64, 1.27, and 2.54 µM for 2n. The Km value for MAO-B was 0.20 mM.

Figure 5.
Lineweaver–Burk plots (a,c) and their secondary plots (b,d) of 2k and 2n for AChE. Five concentrations of ATCI were used; 0.0625, 0.125, 0.25, 0.5 and 1.0 mM, respectively, and three concentrations of inhibitor were ~1/2×, 1×, and 2 × IC50: 4.05, 8.10, and 16.20 µM for 2k; 2.16, 4.32, and 8.64 µM for 2n. The Km value for AChE was 0.075 mM.
Figure 5.
Lineweaver–Burk plots (a,c) and their secondary plots (b,d) of 2k and 2n for AChE. Five concentrations of ATCI were used; 0.0625, 0.125, 0.25, 0.5 and 1.0 mM, respectively, and three concentrations of inhibitor were ~1/2×, 1×, and 2 × IC50: 4.05, 8.10, and 16.20 µM for 2k; 2.16, 4.32, and 8.64 µM for 2n. The Km value for AChE was 0.075 mM.

Figure 6.
Reversibility tests of 2k and 2n for MAO-B using dialysis method. MAO-B was treated with ~2 × IC50 of pargyline (0.28 µM), lazabemide (0.22 µM), 2k (1.16 µM), and 2n (2.54 µM) before dialysis for 6 h. The residual activities were determined before and after dialysis to confirm the reversibility pattern using enzyme assay described in Materials and Methods.
Figure 6.
Reversibility tests of 2k and 2n for MAO-B using dialysis method. MAO-B was treated with ~2 × IC50 of pargyline (0.28 µM), lazabemide (0.22 µM), 2k (1.16 µM), and 2n (2.54 µM) before dialysis for 6 h. The residual activities were determined before and after dialysis to confirm the reversibility pattern using enzyme assay described in Materials and Methods.

Figure 7.
Cytotoxicity profile of compounds 2k and 2n over normal Vero cells. The cells were incubated with various concentrations of 2k and 2n for 24 h (0.01, 0.03, 0.09, 0.27, 0.82, 2.47, 7.41, 22.2, 66.7, and 200 µM) and MTT assays were conducted. Data are expressed as the mean ± SD of duplicate assay.
Figure 7.
Cytotoxicity profile of compounds 2k and 2n over normal Vero cells. The cells were incubated with various concentrations of 2k and 2n for 24 h (0.01, 0.03, 0.09, 0.27, 0.82, 2.47, 7.41, 22.2, 66.7, and 200 µM) and MTT assays were conducted. Data are expressed as the mean ± SD of duplicate assay.

Figure 8.
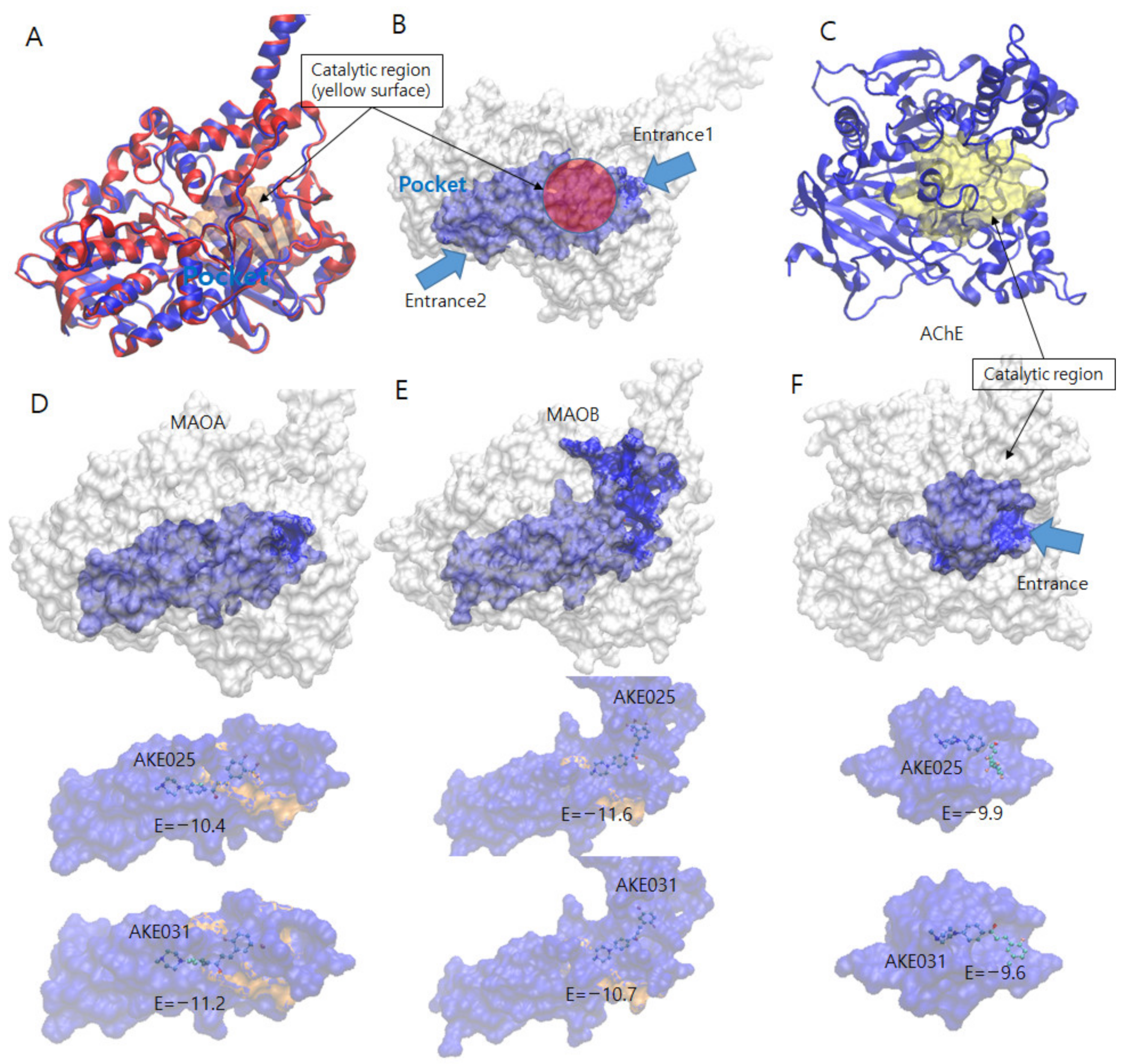
Molecular docking of 2k and 2n on MAO-A, MAO-B, and AChE. (A) Superimposed MAO family structure. Both structures are drawn by Cartoon model shaded by blue (MAO-A) and red color (MAO-B). The catalytic regions are drawn by transparent yellow surface model in the center. (B) Pocket and catalytic region of MAOs. The protein model is drawn by white surface model. MAO family structure has large pocket (blue surface model) in the center and connected to the catalytic region (red circle) in the center. The pocket has two entrances toward the catalytic regions (two blue arrows). (C) AChE protein structure drawn by blue cartoon model and yellow transparent surface model (catalytic region). The compound entrances toward the catalytic region are indicated by blue arrows. Docking simulation results of compounds 2k and 2n into MAO-A (D), MAO-B (E), and AChE (F). The MAOs structures are sketched by white sphere model and the binding pockets that exist on the center are shaded by blue color. The below panels show the binding pocket shade by transparent blue spheres for the existing compound to show. The binding catalytic regions are shown by transparent yellow colors. The compounds are bound in the pocket and near the catalytic region.
Figure 8.
Molecular docking of 2k and 2n on MAO-A, MAO-B, and AChE. (A) Superimposed MAO family structure. Both structures are drawn by Cartoon model shaded by blue (MAO-A) and red color (MAO-B). The catalytic regions are drawn by transparent yellow surface model in the center. (B) Pocket and catalytic region of MAOs. The protein model is drawn by white surface model. MAO family structure has large pocket (blue surface model) in the center and connected to the catalytic region (red circle) in the center. The pocket has two entrances toward the catalytic regions (two blue arrows). (C) AChE protein structure drawn by blue cartoon model and yellow transparent surface model (catalytic region). The compound entrances toward the catalytic region are indicated by blue arrows. Docking simulation results of compounds 2k and 2n into MAO-A (D), MAO-B (E), and AChE (F). The MAOs structures are sketched by white sphere model and the binding pockets that exist on the center are shaded by blue color. The below panels show the binding pocket shade by transparent blue spheres for the existing compound to show. The binding catalytic regions are shown by transparent yellow colors. The compounds are bound in the pocket and near the catalytic region.

Figure 9.
The putative 3D binding mode of 2k with MAO-B. The π-π binding interactions are indicated by green thick dash lines with the distance between two centroids of the rings, and the hydrogen bonds are depicted by blue thick dash lines with the distance between donor and acceptor atoms. The detailed analyses including donor and acceptor atoms and the distance are tabulated on right of the Figure.
Figure 9.
The putative 3D binding mode of 2k with MAO-B. The π-π binding interactions are indicated by green thick dash lines with the distance between two centroids of the rings, and the hydrogen bonds are depicted by blue thick dash lines with the distance between donor and acceptor atoms. The detailed analyses including donor and acceptor atoms and the distance are tabulated on right of the Figure.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Preliminary assay of compounds 2a–q against MAO-A, MAO-B, AChE, and BChE at 10 µM.
| ID | Residual Activities at 10 μM (%) | |||
|---|---|---|---|---|
| MAO-A | MAO-B | AChE | BChE | |
| 2a | 117.97 ± 7.73 | 12.78 ± 0.79 | 34.94 ± 5.19 | 58.65 ± 6.80 |
| 2b | 88.19 ± 6.87 | 21.02 ± 4.02 | 18.99 ± 0.55 | 58.50 ± 1.38 |
| 2c | 103.47 ± 8.84 | 22.73 ± 1.61 | 49.42 ± 4.66 | 49.67 ± 3.70 |
| 2d | 72.35 ± 2.50 | 16.67 ± 4.71 | 24.26 ± 0.95 | 71.53 ± 5.25 |
| 2e | 60.56 ± 3.98 | 44.09 ± 3.04 | 17.75 ± 1.85 | 49.32 ± 1.28 |
| 2f | 83.58 ± 4.22 | 21.33 ± 3.77 | 26.37 ± 1.85 | 76.51 ± 3.35 |
| 2g | 97.18 ± 1.99 | 40.00 ± 1.67 | 50.39 ± 4.43 | 74.66 ± 4.48 |
| 2h | 55.22 ± 4.12 | 17.33 ± 3.77 | 37.99 ± 0.92 | 73.92 ± 3.35 |
| 2i | 114.29 ± 6.06 | 20.35 ± 2.47 | 51.49 ± 4.92 | 63.72 ± 6.24 |
| 2j | 97.14 ± 4.04 | 67.78 ± 5.36 | 57.12 ± 5.39 | 86.03 ± 1.04 |
| 2k | 96.43 ± 3.03 | −8.72 ± 0.82 | 46.85 ± 2.58 | 61.76 ± 5.54 |
| 2l | 85.88 ± 1.66 | 0.00 ± 1.79 | 68.50 ± 1.32 | 73.02 ± 1.75 |
| 2m | 78.82 ± 1.66 | 39.87 ± 6.26 | 27.61 ± 1.90 | 66.83 ± 2.10 |
| 2n | 55.63 ± 1.00 | 6.45 ± 3.04 | 30.55 ± 1.11 | 43.21 ± 2.88 |
| 2o | 92.54 ± 4.23 | 22.87 ± 2.26 | 37.60 ± 0.00 | 6.46 ± 3.05 |
| 2p | 44.53 ± 3.31 | 15.00 ± 3.93 | 37.84 ± 7.64 | 28.85 ± 4.08 |
| 2q | 72.66 ± 3.31 | 13.11 ± 2.06 | 33.78 ± 0.27 | 55.29 ± 0.68 |
Results are expressed as the means ± SD of at least two experiments.
Table 2.
Inhibitory activities (IC50, μM) and selectivity indices of compounds 2a–q against MAO-A, MAO-B, AChE, and BChE.
Table 2.
Inhibitory activities (IC50, μM) and selectivity indices of compounds 2a–q against MAO-A, MAO-B, AChE, and BChE.
| Compound No. | IC50 (μM) | SI (MAO) | SI (ChE) | |||
|---|---|---|---|---|---|---|
| MAO-A | MAO-B | AChE | BChE | |||
| 2a | >40 | 2.58 ± 0.19 | 5.70 ± 0.22 | 13.89 ± 2.22 | 15.50 | 2.44 |
| 2b | >40 | 3.10 ± 0.04 | 2.26 ± 0.29 | 13.37 ± 0.27 | 12.90 | 5.92 |
| 2c | >40 | 2.98 ± 0.18 | 10.30 ± 1.03 | 18.41 ± 3.17 | 13.42 | 1.79 |
| 2d | >40 | 2.20 ± 0.06 | 2.38 ± 0.13 | 24.71 ± 2.00 | 18.18 | 10.38 |
| 2e | 16.21 ± 1.74 | 8.36 ± 0.39 | 3.75 ± 0.82 | 14.36 ± 0.40 | 1.94 | 3.83 |
| 2f | >40 | 3.29 ± 0.08 | 3.03 ± 0.31 | >40 | 12.16 | >13.20 |
| 2g | >40 | 6.74 ± 0.03 | 12.41 ± 1.96 | >40 | 5.93 | >3.22 |
| 2h | >40 | 6.14 ± 0.43 | 5.82 ± 0.51 | >40 | 6.51 | >6.87 |
| 2i | >40 | 2.33 ± 0.14 | 13.93 ± 0.08 | 25.30 ± 2.96 | 17.17 | 1.82 |
| 2j | >40 | 15.70 ± 1.17 | 15.36 ± 2.43 | >40 | 2.55 | >2.60 |
| 2k | >40 | 0.71 ± 0.03 | 8.10 ± 0.69 | 21.09 ± 0.23 | 56.34 | 2.60 |
| 2l | >40 | 1.41 ± 0.04 | 14.07 ± 0.54 | 26.09 ± 1.28 | 28.37 | 1.85 |
| 2m | >40 | 6.76 ± 0.82 | 4.45 ± 0.24 | 18.61 ± 1.35 | 5.92 | 4.18 |
| 2n | 17.80 ± 0.28 | 1.11 ± 0.06 | 4.32 ± 0.39 | 9.44 ± 0.05 | 16.04 | 2.19 |
| 2o | >40 | 3.20 ± 0.32 | 3.87 ± 0.18 | 1.19 ± 0.06 | 12.50 | 0.31 |
| 2p | 6.67 ± 0.63 | 2.18 ± 0.01 | 7.17 ± 0.60 | 7.58 ± 0.25 | 3.06 | 1.06 |
| 2q | 16.52 ± 1.75 | 3.03 ± 0.13 | 6.37 ± 0.17 | 10.89 ± 0.90 | 5.45 | 1.71 |
| Toloxatone | 1.08 ± 0.03 | - | - | - | - | - |
| Lazabemide | 0.11 ± 0.02 | - | - | - | - | |
| Clorgyline | 0.007 ± 0.001 | - | - | - | - | - |
| Pargyline | - | 0.14 ± 0.01 | - | - | - | - |
| Tacrine | - | - | 0.27 ± 0.02 | 0.060 ± 0.002 | - | - |
| Donepezil | - | - | 0.010 ± 0.002 | 0.180 ± 0.004 | - | - |
| PC5 * | 34.1 ± 2.51 | 2.31 ± 0.46 | - | - | 14.8 | |
| PC10 * | 31.4 ± 3.50 | 0.65 ± 0.023 | 28.0 ± 2.43 | 36.4 ± 2.36 | ||
| PC11 * | 34.9 ± 4.10 | 0.71 ± 0.0035 | 26.3 ± 1.29 | 36.2 ± 3.65 | ||
Results are expressed as the means ± SD of at least two experiments. SI values: MAO, MAO-B selectivity over MAO-A; ChE, AChE selectivity over BChE, based on their respective IC50 values. *, from ref [33].
Table 3.
Detailed docking results of compounds 2k and 2n on MAO-A, MAO-B, and AChE a.
| Enzyme | Cpds | No. of Cpds | Ave. Energy | Lowest Energy (Rank) | Free Energy | No. of HB | No. of π-π/Aro. |
|---|---|---|---|---|---|---|---|
| MAO-A | 2k | 32 | −5.97 ± 2.56 | −10.40 (2) | −12.48 | 4 | 1/5 |
| 2n | 50 | −5.88 ± 2.42 | −11.20 (2) | −13.55 | 2 | 1/4 | |
| MAO-B | 2k | 10 | −5.47 ± 3.70 | −11.60 (1) | −12.98 | 4 | 2/7 |
| 2n | 16 | −5.33 ± 3.31 | −10.70 (1) | −12.36 | 2 | 2/7 | |
| AChE | 2k | 41 | −7.81 ± 1.15 | −9.90 (1) | −12.13 | 7 | 1/9 |
| 2n | 39 | −7.74 ± 1.25 | −9.60 (1) | −11.80 | 4 | 1/9 |
a Each first lowest energy compound in the catalytic region is chosen. The result is tabulated in terms of number of compounds existing on the pocket (No. of Cpds), average energy on the pocket, the number of docking compounds on the same pocket, lowest energy with cluster rank with Parentheses, free energy, number of Hydrogen bonding, and number of π-π interaction/aromatic rings. Energy unit is kcal/mol. The free energy is the colony energy [43] calculated by E–kT log(N), where E is the lowest energy, N is the number of compounds in the catalytic region. The k is Boltzmann constant and T is room temperature, 300 K. The free energy considers both energetic and entropic contribution.
Table 4.
Predicted pharmacokinetic properties of 2k and 2n.
| Compound No. | GI Absorption | BBB Permeant | P-gp Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor | Log Kp (Skin Permeation) |
|---|---|---|---|---|---|---|---|---|---|
| 2k | High | Yes | No | No | Yes | No | Yes | Yes | −5.42 cm/s |
| 2n | High | Yes | No | No | Yes | Yes | Yes | No | −5.62 cm/s |
GI: gastrointestinal; BBB: blood–brain barrier; P-gp: P-glycoprotein; CYP: Cytochrome P.
Table 5.
Physicochemical properties of 2k and 2n.
| Compound No. | Mw | cLog Po/w | HBD | HBA | TPSA (Å2) | RB | Lipinski Violations * |
|---|---|---|---|---|---|---|---|
| 2k | 392.40 | 4.48 | 0 | 6 | 23.55 | 5 | 0 |
| 2n | 403.30 | 4.03 | 0 | 3 | 23.55 | 4 | 0 |
Mw: molecular weight; cLog P: consensus Log Po/w; HBD: H-bond donors; HBA: H-bond acceptors; TPSA: topological polar surface area; RB: rotatable bonds. * Lipinski violation is indicated by counting the number of violations in the Lipinski’s rule, i.e., MW < 500, cLog Po/w < 5.00, HBD < 5, HBA < 10, TPSA < 140 Å2, and RB < 10.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
El-Damasy, A.K.; Park, J.E.; Kim, H.J.; Lee, J.; Bang, E.-K.; Kim, H.; Keum, G. Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors. Pharmaceuticals 2023, 16, 83. https://doi.org/10.3390/ph16010083
AMA Style
El-Damasy AK, Park JE, Kim HJ, Lee J, Bang E-K, Kim H, Keum G. Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors. Pharmaceuticals. 2023; 16(1):83. https://doi.org/10.3390/ph16010083
Chicago/Turabian StyleEl-Damasy, Ashraf K., Jong Eun Park, Hyun Ji Kim, Jinhyuk Lee, Eun-Kyoung Bang, Hoon Kim, and Gyochang Keum. 2023. "Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors" Pharmaceuticals 16, no. 1: 83. https://doi.org/10.3390/ph16010083
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.