
Identification of New L-Heptanoylphosphatidyl Inositol Pentakisphosphate Derivatives Targeting the Interaction with HIV-1 Gag by Molecular Modelling Studies

 ,
,  ,
,  , , and
, , and
Abstract
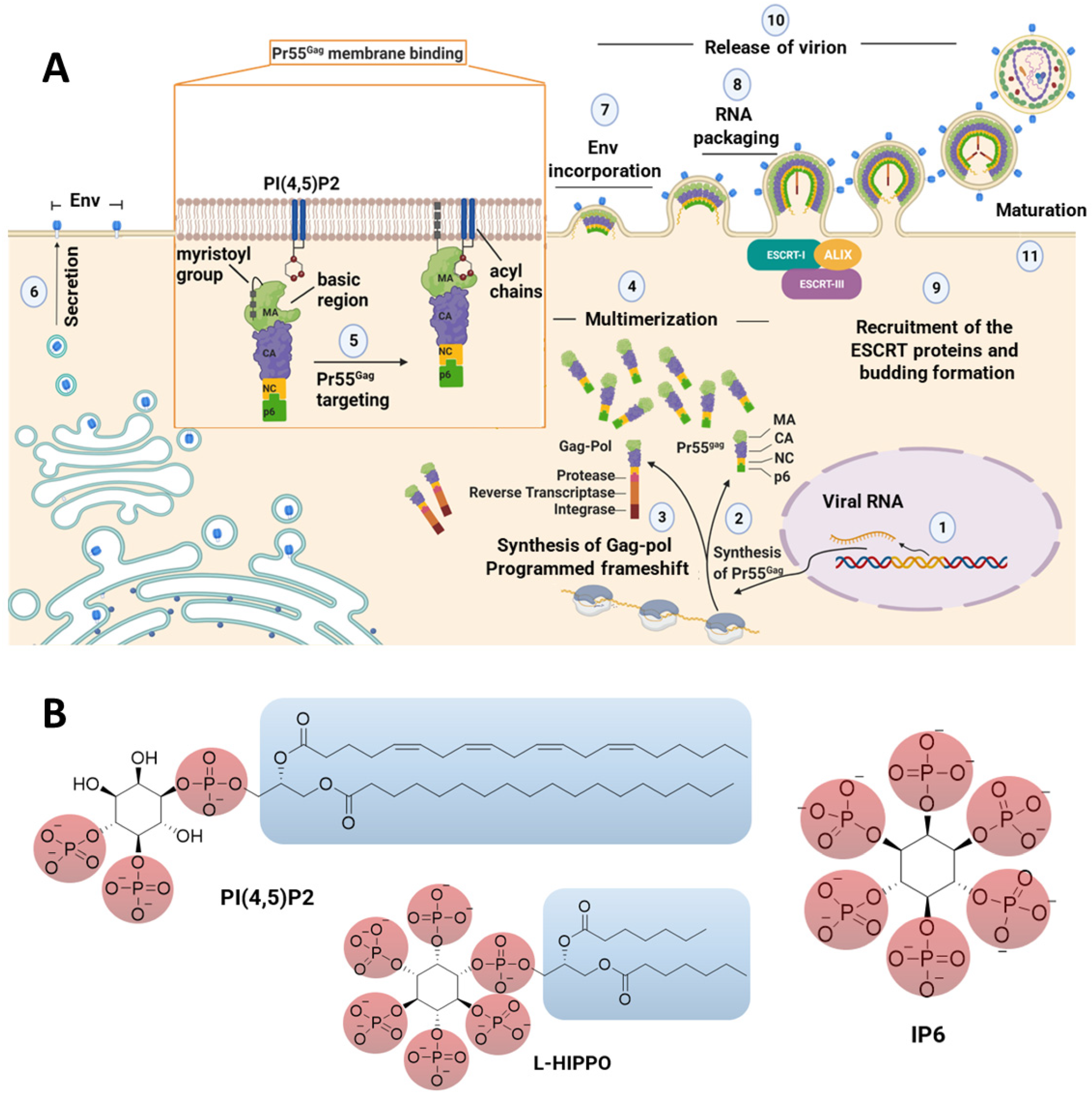
:1. Introduction
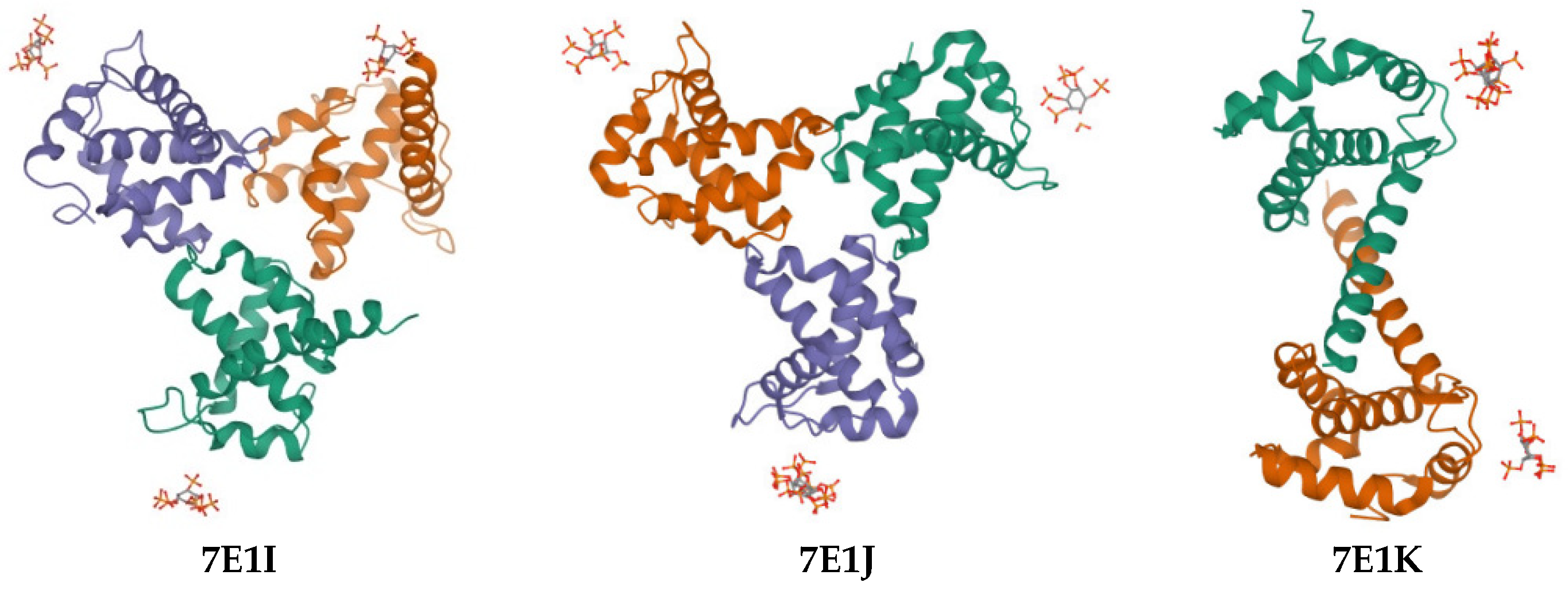
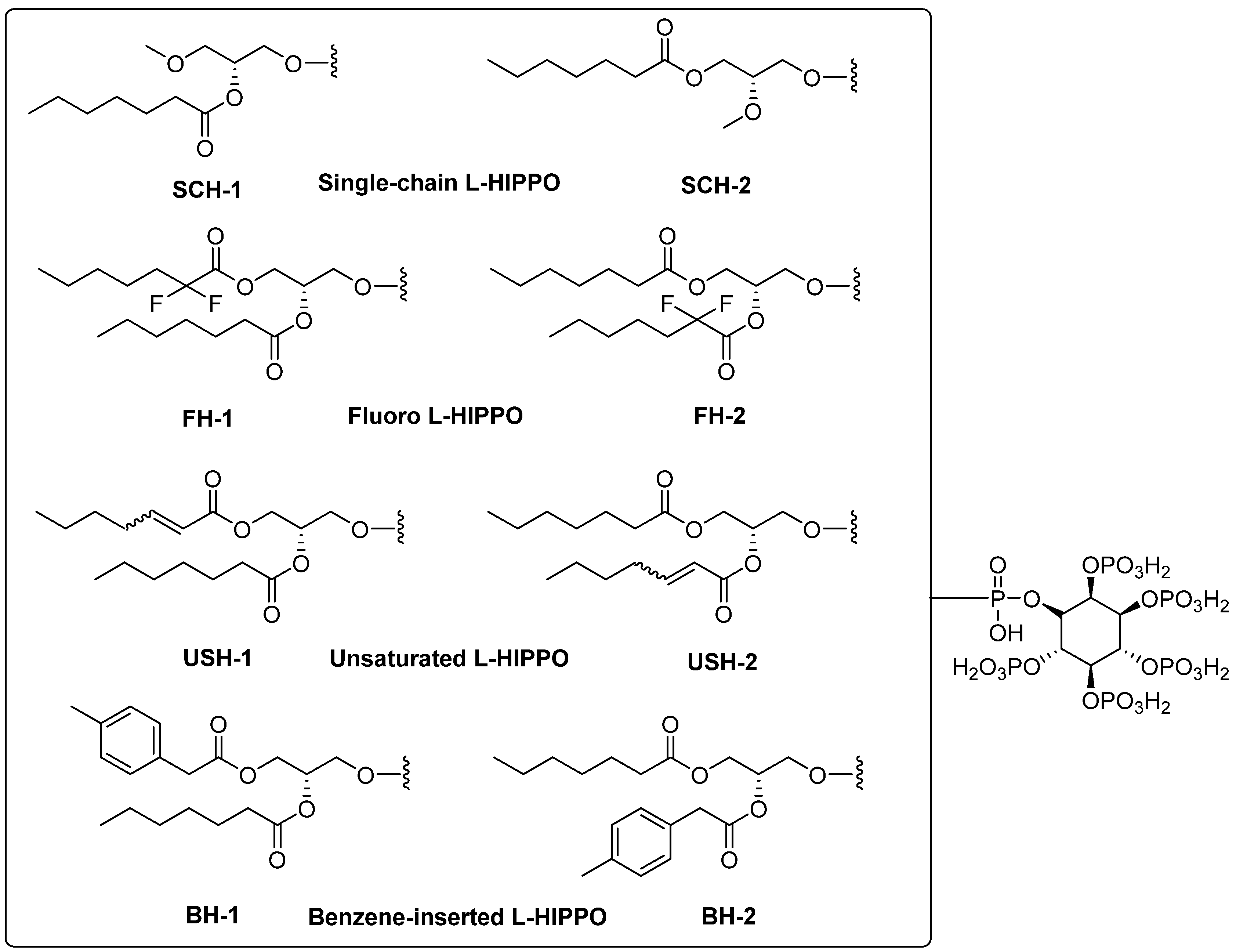
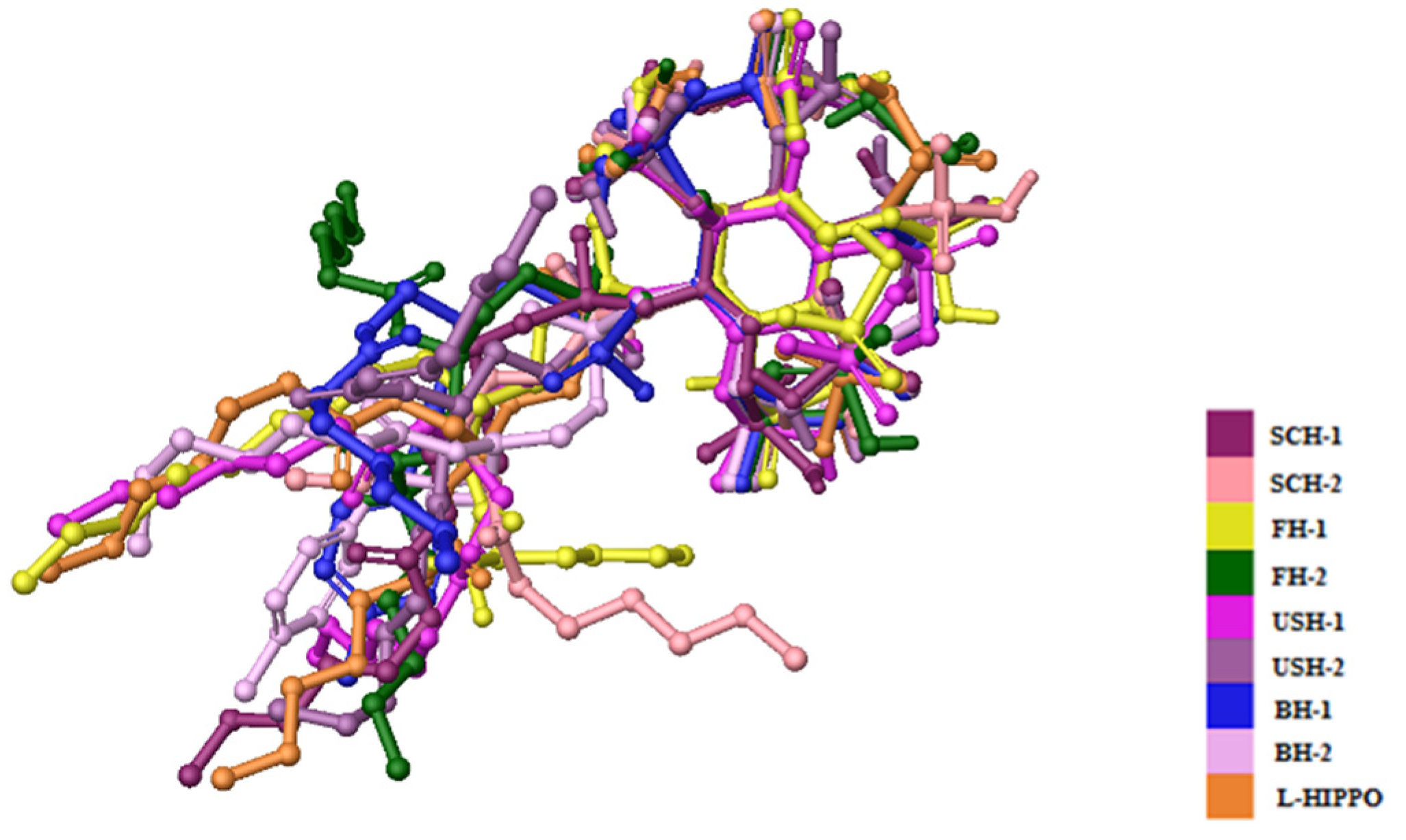
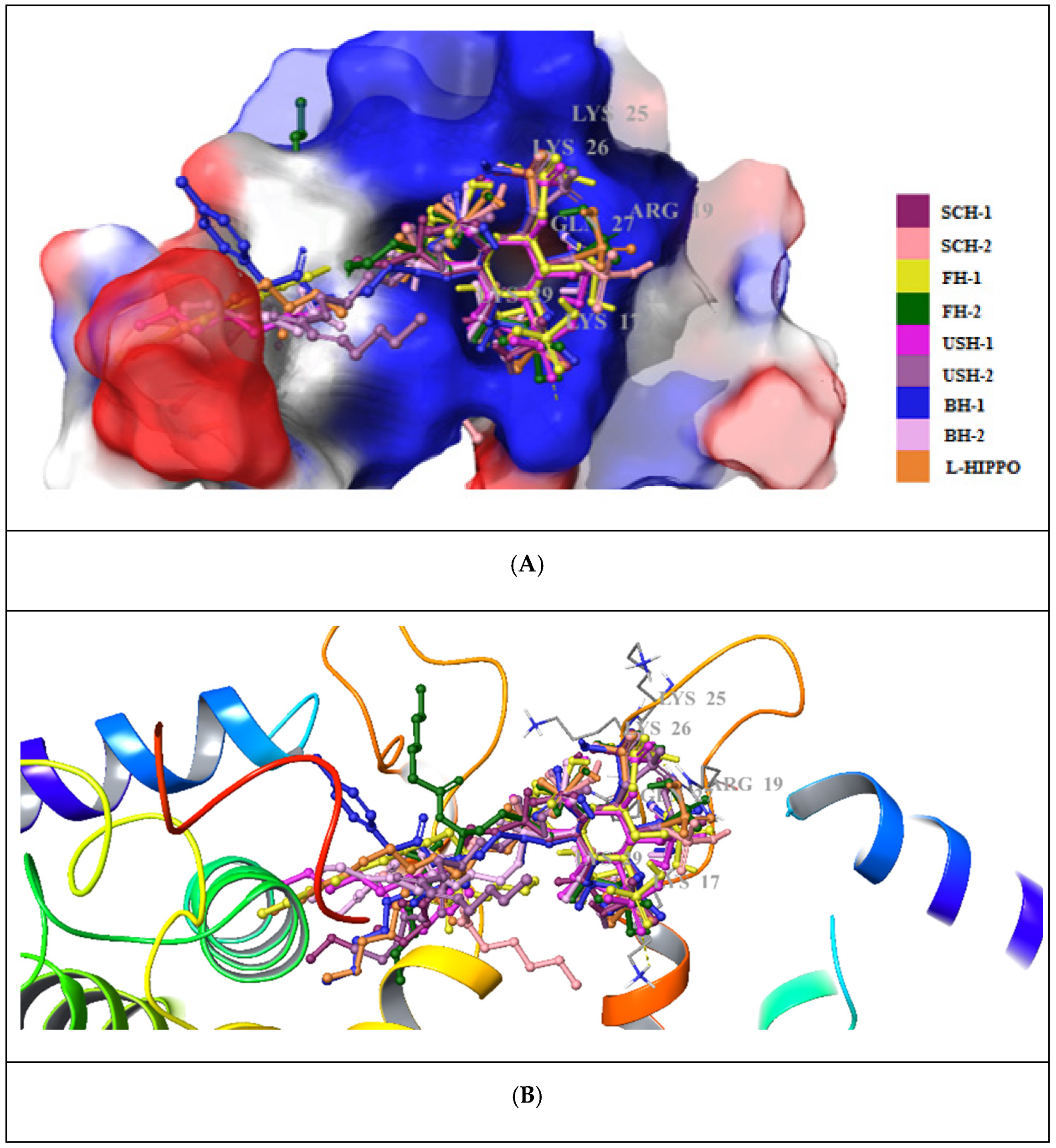
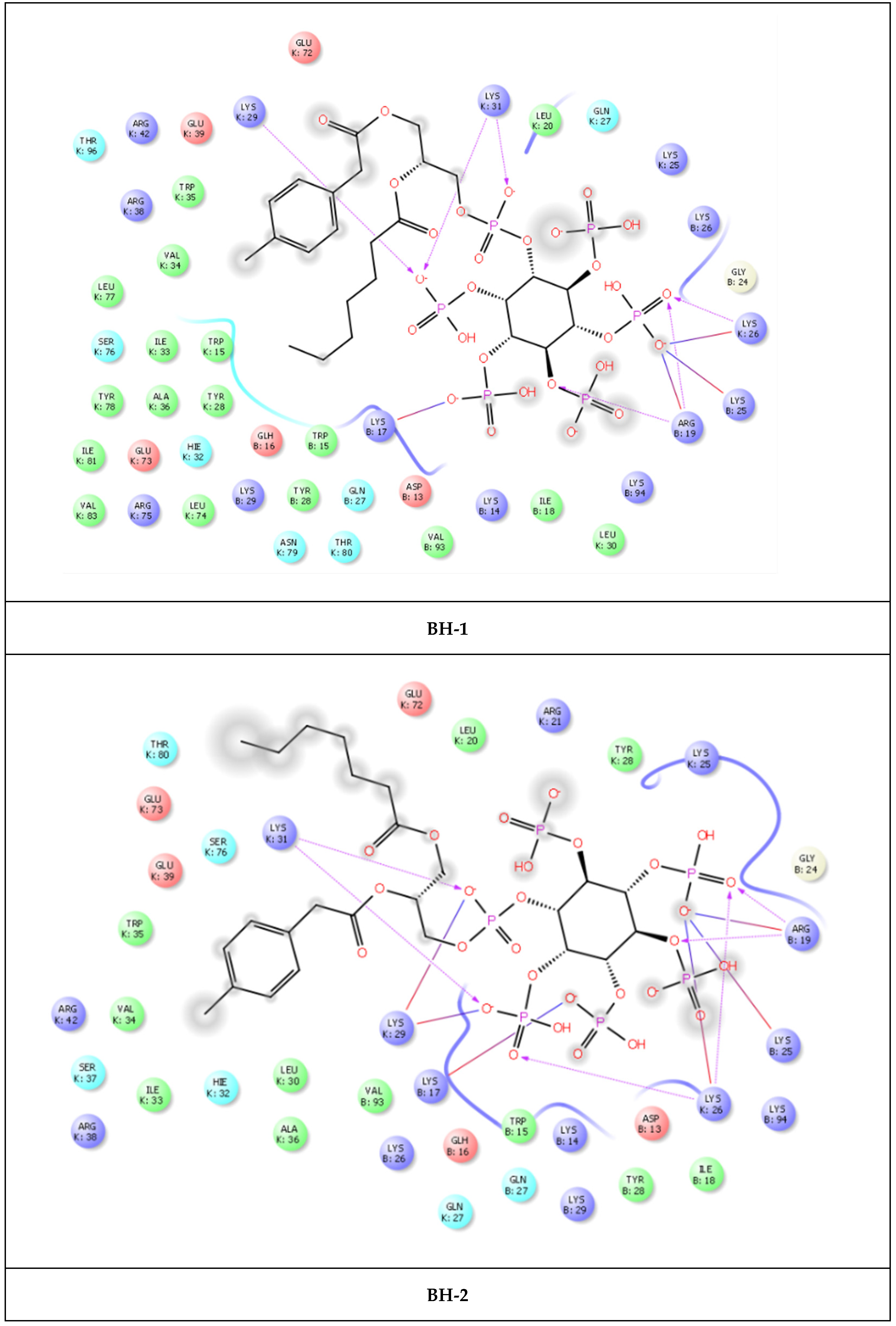
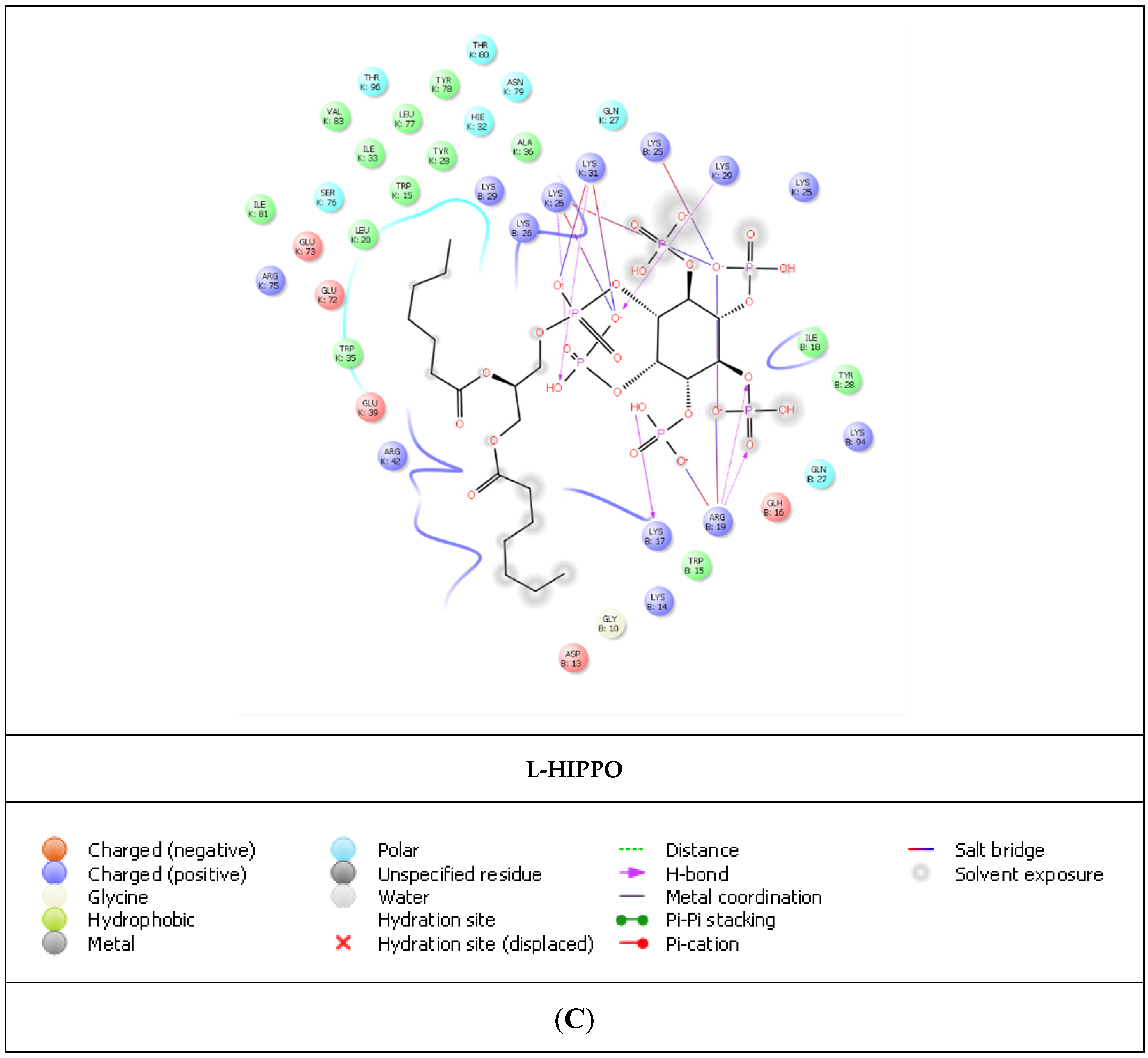
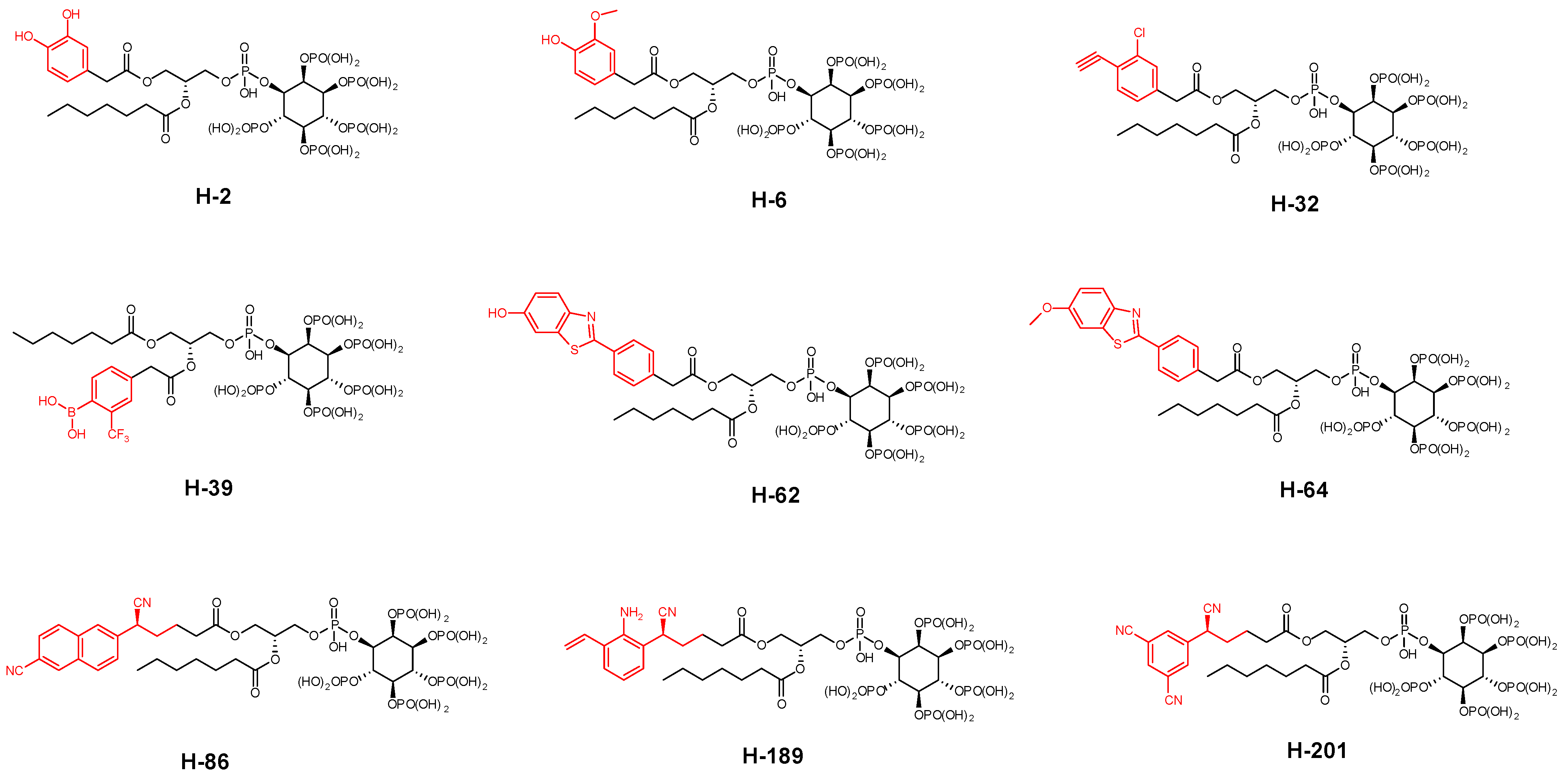
2. Results
- Removal of a heptanoyl group to reduce hydrophobicity.
- Introduction of a fluoro group to increase hydrophobicity.
- Insertion of a double bond into the heptanoyl group in order to alter the π electron.
- Insertion of a benzene ring into the heptanoyl group in order to alter the π electron.
3. Discussion
4. Materials and Methods
4.1. In Silico Docking Assessment and ADME Prediction
4.1.1. Ligand Library Creation
4.1.2. Protein Preparation
4.1.3. Docking Grid Generation
4.1.4. Ligand Preparation
4.1.5. Docking Experiments
4.1.6. In Silico ADME Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adamson, C.S.; Salzwedel, K.; Freed, E.O. Virus maturation as a new HIV-1 therapeutic target. Expert Opin. Ther. Targets 2009, 13, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Elinder, M. Towards a New Generation of Anti-HIV Drugs: Interaction Kinetic Analysis of Enzyme Inhibitors Using SPR-biosensors. Ph.D. Thesis, Acta Universitatis Upsaliensis, Uppsala, Sweden, 9 June 2011. [Google Scholar]
- Hamard-Peron, E.; Muriaux, D. Retroviral matrix and lipids, the intimate interaction. Retrovirology 2011, 8, 15. [Google Scholar] [CrossRef]
- Ghanam, R.H.; Samal, A.B.; Fernandez, T.F.; Saad, J.S. Role of the HIV-1 Matrix Protein in Gag Intracellular Trafficking and Targeting to the Plasma Membrane for Virus Assembly. Front. Microbiol. 2012, 3, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedbury, P.R.; Freed, E.O. The role of matrix in HIV-1 envelope glycoprotein incorporation. Trends Microbiol. 2014, 22, 372–378. [Google Scholar] [CrossRef] [Green Version]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarlata, S.; Carter, C. Role of HIV-1 Gag domains in viral assembly. Biochim. Biophys. Acta 2003, 1614, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Chen, B. HIV Capsid Assembly, Mechanism, and Structure. Biochemistry 2016, 55, 2539–2552. [Google Scholar] [CrossRef]
- Briggs, J.A.; Riches, J.D.; Glass, B.; Bartonova, V.; Zanetti, G.; Kräusslich, H.G. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. USA 2009, 106, 11090–11095. [Google Scholar] [CrossRef] [Green Version]
- Alfadhli, A.; Staubus, A.O.; Tedbury, P.R.; Novikova, M.; Freed, E.O.; Barklis, E. Analysis of HIV-1 Matrix-Envelope Cytoplasmic Tail Interactions. J. Virol. 2019, 93, e01079-19. [Google Scholar] [CrossRef] [PubMed]
- Bush, D.L.; Vogt, V.M. In Vitro Assembly of Retroviruses. Annu. Rev. Virol. 2014, 1, 561–580. [Google Scholar] [CrossRef]
- Lingappa, J.R.; Reed, J.C.; Tanaka, M.; Chutiraka, K.; Robinson, B.A. How HIV-1 Gag assembles in cells: Putting together pieces of the puzzle. Virus Res. 2014, 193, 89–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Sundquist, W.I.; Kräusslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.; Curtis, J.E.; Ratcliff, W.; Clark, P.K.; Crist, R.M.; Lebowitz, J.; Krueger, S.; Rein, A. Conformation of the HIV-1 Gag protein in solution. J. Mol. Biol. 2007, 365, 812–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.A.; Zhao, Z.; Clark, P.K.; Tarasov, S.; Alexandratos, J.N.; Campbell, S.J.; Kvaratskhelia, M.; Lebowitz, J.; Rein, A. Interactions between HIV-1 Gag molecules in solution: An inositol phosphate-mediated switch. J. Mol. Biol. 2007, 365, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Loeliger, E.; Luncsford, P.; Kinde, I.; Beckett, D.; Summers, M.F. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. USA 2004, 101, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resh, M.D. A myristoyl switch regulates membrane binding of HIV-1 Gag. Proc. Natl. Acad. Sci. USA 2004, 101, 417–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.P.; Datta, S.A.; Rein, A.; Rouzina, I.; Musier-Forsyth, K. Matrix domain modulates HIV-1 Gag’s nucleic acid chaperone activity via inositol phosphate binding. J. Virol. 2011, 85, 1594–1603. [Google Scholar] [CrossRef] [Green Version]
- Dick, R.A.; Vogt, V.M. Membrane interaction of retroviral Gag proteins. Front. Microbiol. 2014, 5, 187. [Google Scholar] [CrossRef] [Green Version]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar] [CrossRef] [PubMed]
- Deshmuk, L.; Ghirlando, R.; Clore, G.M. Conformation and dynamics of the Gag polyprotein of the human immunodeficiency virus 1 studied by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef] [Green Version]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Chukkapalli, V.; Hogue, I.B.; Boyko, V.; Hu, W.S.; Ono, A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J. Virol. 2008, 82, 2405–2417. [Google Scholar] [CrossRef] [Green Version]
- Alfadhli, A.; Still, A.; Barklis, E. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J. Virol. 2009, 83, 12196–12203. [Google Scholar] [CrossRef] [Green Version]
- Dalton, A.K.; Ako-Adjei, D.; Murray, P.S.; Murray, D.; Vogt, V.M. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J. Virol. 2007, 81, 6434–6445. [Google Scholar] [CrossRef] [Green Version]
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Charlier, L.; Louet, M.; Chaloin, L.; Fuchs, P.; Martinez, J.; Muriaux, D.; Favard, C.; Floquet, N. Coarse-grained simulations of the HIV-1 matrix protein anchoring: Revisiting its assembly on membrane domains. Biophys. J. 2014, 106, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Anraku, K.; Fukuda, R.; Takamune, N.; Misumi, S.; Okamoto, Y.; Otsuka, M.; Fujita, M. Highly sensitive analysis of the interaction between HIV-1 Gag and phosphoinositide derivatives based on surface plasmon resonance. Biochemistry 2010, 49, 5109–5116. [Google Scholar] [CrossRef]
- Campbell, S.; Fisher, R.J.; Towler, E.M.; Fox, S.; Issaq, H.J.; Wolfe, T.; Phillips, L.R.; Rein, A. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl. Acad. Sci. USA 2001, 98, 10875–10879. [Google Scholar] [CrossRef]
- Dick, R.A.; Zadrozny, K.K.; Xu, C.; Schur, F.K.M.; Lyddon, T.D.; Ricana, C.L.; Wagner, J.M.; Perilla, J.R.; Ganser-Pornillos, B.K.; Johnson, M.C.; et al. Inositol phosphates are assembly co-factors for HIV-1. Nature 2018, 560, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; Márquez, C.L.; McEwan, W.A.; Dickson, C.F.; Jacques, D.A.; Anandapadamanaban, M.; Bichel, K.; Towers, G.J.; Saiardi, A.; Böcking, T.; et al. IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. eLife 2018, 7, e35335. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; Faysal, K.M.R.; Kleinpeter, A.; Wilson, M.S.C.; Vaysburd, M.; Fletcher, A.J.; Novikova, M.; Böcking, T.; Freed, E.O.; Saiardi, A.; et al. Cellular IP6 Levels Limit HIV Production while Viruses that Cannot Efficiently Package IP6 Are Attenuated for Infection and Replication. Cell Rep. 2019, 29, 3983–3996.e4. [Google Scholar] [CrossRef] [Green Version]
- Jennings, J.; Shi, J.; Varadarajan, J.; Jamieson, P.J.; Aiken, C. The Host Cell Metabolite Inositol Hexakisphosphate Promotes Efficient Endogenous HIV-1 Reverse Transcription by Stabilizing the Viral Capsid. mBio 2020, 11, e02820-20. [Google Scholar] [CrossRef]
- Ricaña, C.L.; Dick, R.A. Inositol Phosphates and Retroviral Assembly: A Cellular Perspective. Viruses 2021, 13, 2516. [Google Scholar] [CrossRef]
- Sowd, G.A.; Aiken, C. Inositol phosphates promote HIV-1 assembly and maturation to facilitate viral spread in human CD4+ T cells. PLoS Pathog. 2021, 17, e1009190. [Google Scholar]
- Tateishi, H.; Anraku, K.; Koga, R.; Okamoto, Y.; Fujita, M.; Otsuka, M. Design and synthesis of lipid-coupled inositol 1,2,3,4,5,6-hexakisphosphate derivatives exhibiting high-affinity binding for the HIV-1 MA domain. Org. Biomol. Chem. 2014, 12, 5006. [Google Scholar] [CrossRef]
- Lewin, S.R.; Rasmussen, T.A. Kick and kill for HIV latency. Lancet 2020, 395, 844–846. [Google Scholar] [CrossRef]
- Cillo, A.R.; Mellors, J.W. Which therapeutic strategy will achieve a cure for HIV-1? Curr. Opin. Virol. 2016, 18, 14–19. [Google Scholar] [CrossRef]
- Pace, M.; Frater, J. Curing HIV by ‘kick and kill’: From theory to practice? Expert Rev. Anti Infect. Ther. 2019, 17, 383–386. [Google Scholar] [CrossRef]
- Kim, J.T.; Zhang, T.H.; Carmona, C.; Lee, B.; Seet, C.S.; Kostelny, M.; Shah, N.; Chen, H.; Farrell, K.; Soliman, M.S.A.; et al. Latency reversal plus natural killer cells diminish HIV reservoir in vivo. Nat. Commun. 2022, 13, 121. [Google Scholar] [CrossRef]
- Tateishi, H.; Monde, K.; Anraku, K.; Koga, R.; Hayashi, Y.; Ciftci, H.I.; DeMirci, H.; Higashi, T.; Motoyama, K.; Arima, H.; et al. A clue to unprecedented strategy to HIV eradication: “Lock-in and apoptosis”. Sci. Rep. 2017, 7, 8957. [Google Scholar] [CrossRef] [Green Version]
- Ciftci, H.; Tateishi, H.; Koiwai, K.; Koga, R.; Anraku, K.; Monde, K.; Dağ, Ç.; Destan, E.; Yuksel, B.; Ayan, E.; et al. Structural insight into host plasma membrane association and assembly of HIV-1 matrix protein. Sci. Rep. 2021, 11, 15819. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Sierra, R.G.; Yoon, C.H.; Su, Z.; Tateishi, H.; Koga, R.; Kotaro, K.; Yumoto, F.; Senda, T.; Liang, M.; et al. Serial Femtosecond X-Ray Diffraction of HIV-1 Gag MA-IP6 Microcrystals at Ambient Temperature. Int. J. Mol. Sci. 2019, 20, 1675. [Google Scholar] [CrossRef] [Green Version]
- Van Den Driessche, G.; Fourches, D. Adverse drug reactions triggered by the common HLA-B*57:01 variant: A molecular docking study. J. Cheminform. 2017, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Close-range electrostatic interactions in proteins. Chembiochem 2002, 3, 604–617. [Google Scholar] [CrossRef]
- Debiec, K.T.; Gronenborn, A.M.; Chong, L.T. Evaluating the strength of salt bridges: A comparison of current biomolecular force fields. J. Phys. Chem. B 2014, 118, 6561–6569. [Google Scholar] [CrossRef]
- Nittinger, E.; Inhester, T.; Bietz, S.; Meyder, A.; Schomburg, K.T.; Lange, G.; Klein, R.; Rarey, M. Large-Scale Analysis of Hydrogen Bond Interaction Patterns in Protein-Ligand Interfaces. Med. Chem. 2017, 60, 4245–4257. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [Green Version]
- Amani, P.; Habibpour, R.; Karami, L.; Hofmann, A. Docking Screens of Noncovalent Interaction Motifs of the Human Subtype-D2 Receptor-75 Schizophrenia Antipsychotic Complexes with Physicochemical Appraisal of Antipsychotics. ACS Chem. Neurosci. 2021, 12, 2218–2232. [Google Scholar] [CrossRef] [PubMed]
- Beak, M.; Park, S.; Kim, J.H.; Eom, H.J.; Lee, H.Y.; Kim, Y.H.; Lee, J.; Nam, S. Second-Generation JK-206 Targets the Oncogenic Signal Mediator RHOA in Gastric Cancer. Cancers 2022, 14, 1604. [Google Scholar] [CrossRef] [PubMed]
- Jayaswal, A.; Mishra, H.; Mishra, A.; Shah, K. Examining pharmacodynamic and pharmacokinetic properties of eleven analogues of saquinavir for HIV protease inhibition. Arch. Virol. 2019, 164, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Puhl, A.C.; Garzino Demo, A.; Makarov, V.A.; Ekins, S. New targets for HIV drug discovery. Drug Discov. Today 2019, 24, 1139–1147. [Google Scholar] [CrossRef]
- Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The Paradox of HIV Blood-Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci. 2020, 43, 695–708. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
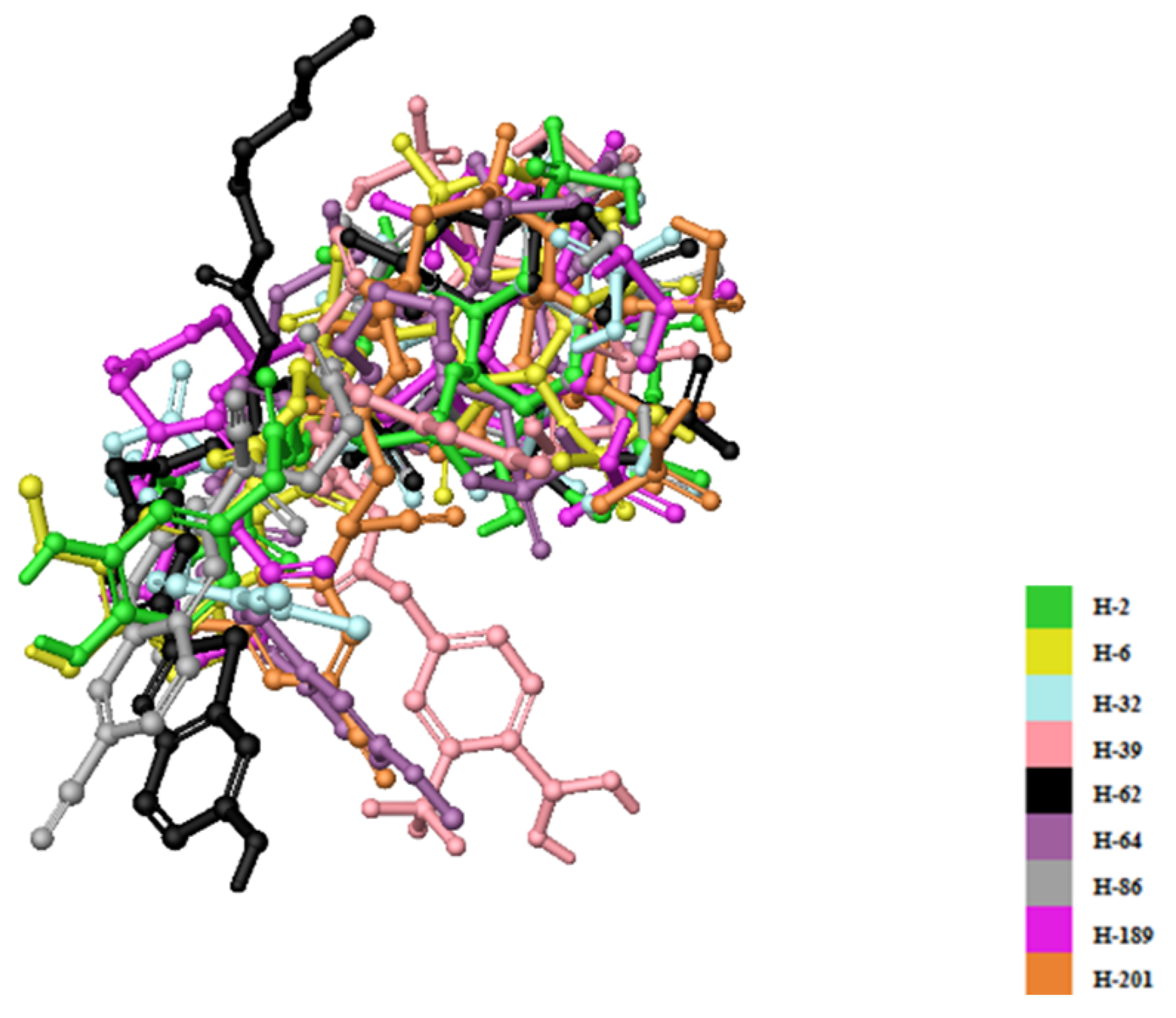{kind=link}
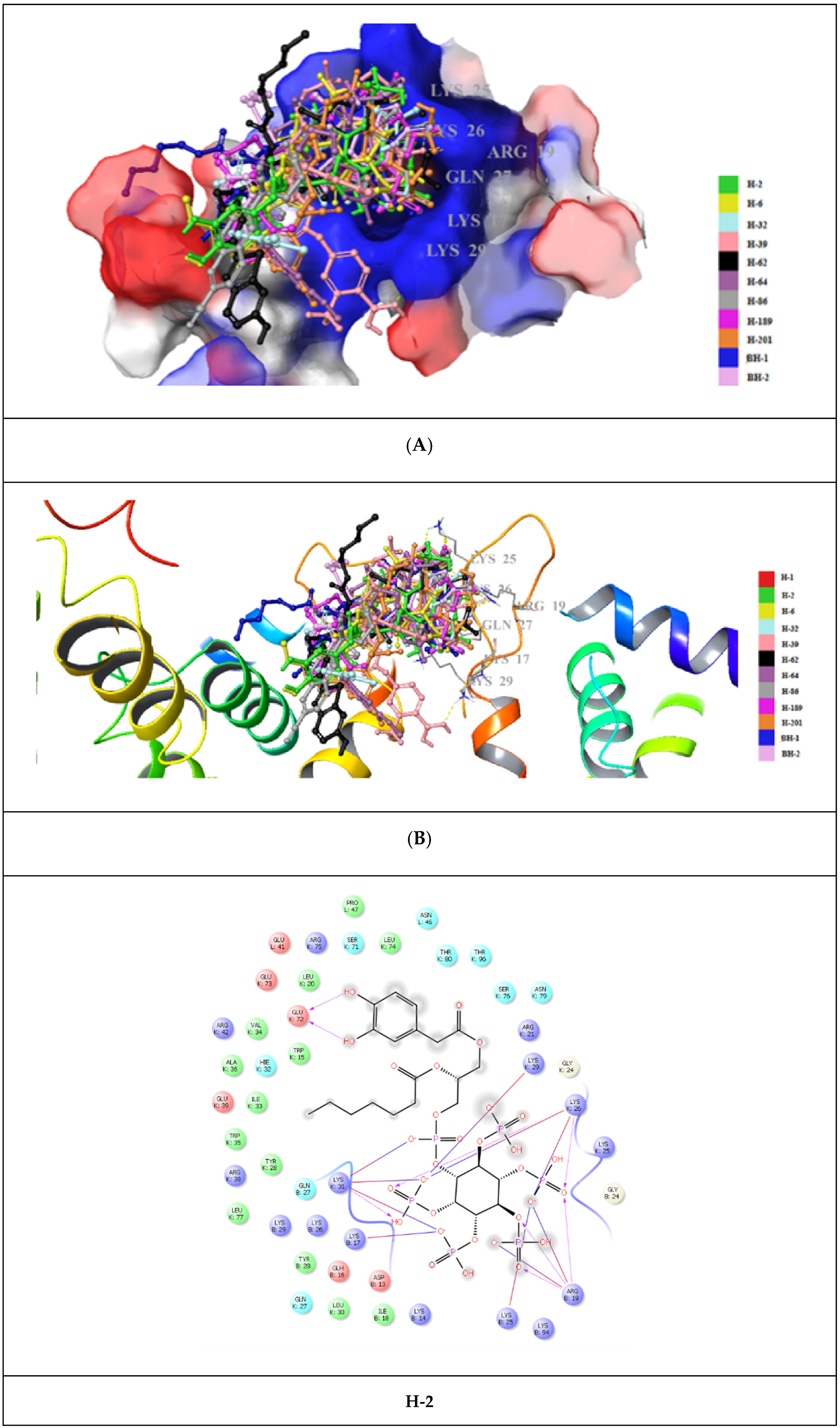{kind=link}
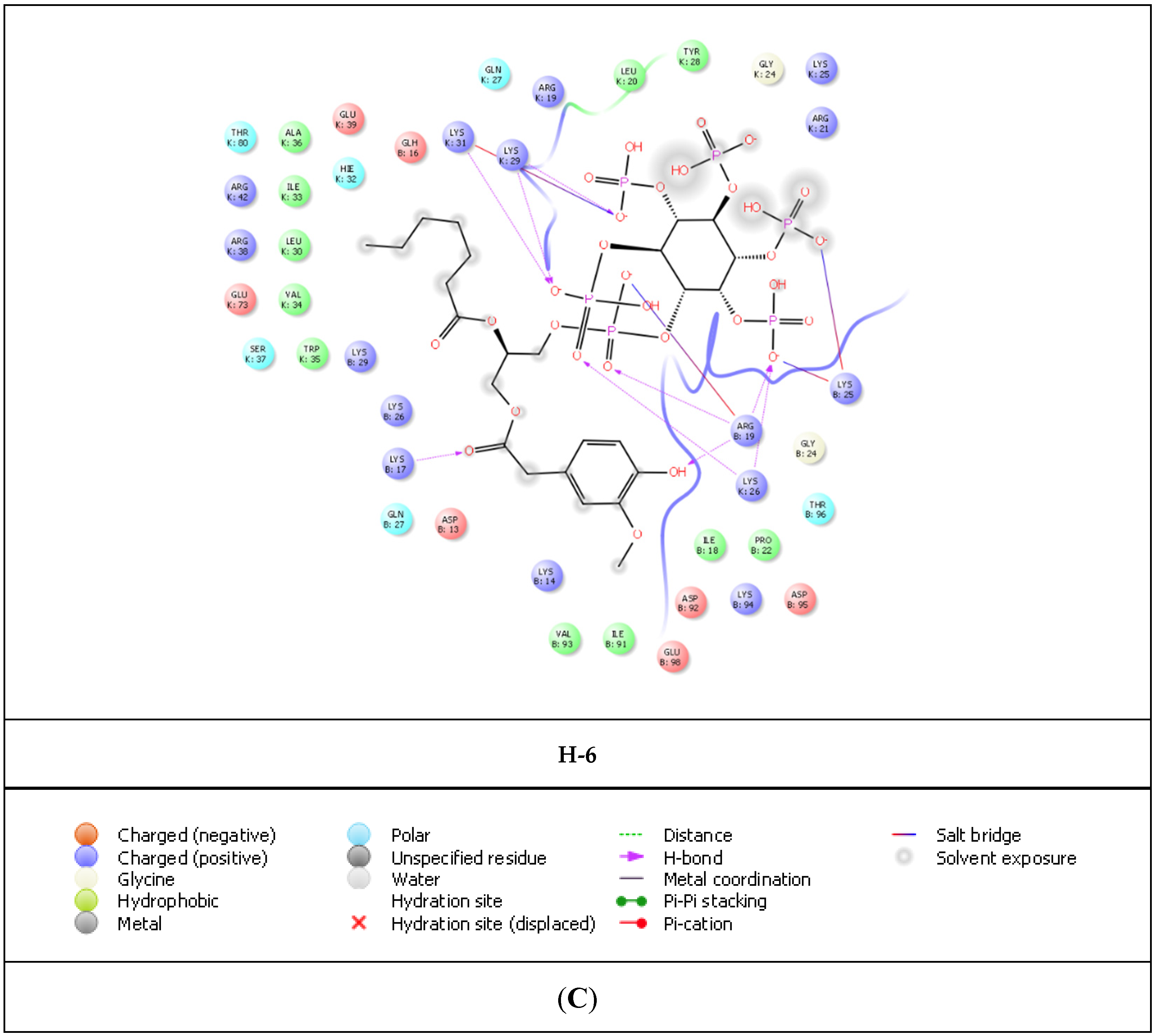{kind=link}
| Compound | PDB IDs | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 7E1I | 7E1J | 7E1K | |||||||
| Docking Score | Glide Gscore | Glide Emodel | Docking Score | Glide Gscore | Glide Emodel | Docking Score | Glide Gscore | Glide Emodel | |
| SCH-1 | −6.018 | −6.032 | −188.439 | −4.634 | −4.649 | −51.858 | −5.809 | −5.823 | −94.442 |
| SCH-2 | −6.153 | −6.167 | −166.921 | −3.943 | −3.957 | −46.579 | −6.029 | −6.043 | −105.004 |
| FH-1 | −6.268 | −6.282 | −158.902 | −4.407 | −4.407 | −53.872 | −5.627 | −4.483 | −88.529 |
| FH-2 | −5.303 | −5.303 | −162.556 | −5.777 | −5.777 | −74.193 | −5.699 | −5.712 | −86.413 |
| USH-1 | −6.259 | −6.273 | −141.432 | −3.770 | −3.784 | −32.005 | −6.221 | −6.235 | −110.130 |
| USH-2 | −6.246 | −6.259 | −133.188 | −3.660 | −3.660 | −35.692 | −6.106 | −6.119 | −101.126 |
| BH-1 | −8.123 | −8.137 | −158.580 | −6.253 | −6.953 | −40.835 | −7.484 | −7.497 | −101.439 |
| BH-2 | −8.074 | −8.088 | −153.840 | −6.070 | −6.070 | −41.406 | −7.221 | −7.235 | −102.794 |
| IP6 | −6.914 | −6.210 | −162.260 | −4.677 | −4.692 | −60.450 | −5.916 | −5.931 | −109.656 |
| L-HIPPO | −7.259 | −7.273 | −141.432 | −5.770 | −5.784 | −32.005 | −7.107 | −7.121 | −110.130 |
| Compound | Residue | Distance | |
|---|---|---|---|
| H-Bond | Salt-Bridge Formation | ||
| L-HIPPO | Arg19 | 1.83 and 1.61 | 4.31 |
| Lys25 | 4.20 | ||
| Lys26 | 2.07 | 4.67 | |
| Lys29 | 1.96 | ||
| Lys31 | 2.23 | 3.65 | |
| BH-1 | Arg19 | 1.57 and 1.93 | 4.20 |
| Lys25 | 2.76 | ||
| Lys26 | 2.16 | ||
| Lys29 | 2.41 | ||
| Lys31 | 2.40 and 2.53 | ||
| BH-2 | Arg19 | 2.02 | |
| Lys25 | 2.88 | ||
| Lys26 | 2.54 | 4.97 | |
| Lys29 | 2.62 | 4.72 | |
| Lys31 | 1.94 and 1.77 | ||
| Compound | PDB IDs | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 7E1I | 7E1J | 7E1K | |||||||
| Docking Score | Glide Gscore | Glide Emodel | Docking Score | Glide Gscore | Glide Emodel | Docking Score | Glide Gscore | Glide Emodel | |
| H-2 | −11.587 | −11.564 | −190.864 | −9.236 | −9.175 | −40.375 | −10.587 | −10.432 | −120.138 |
| H-6 | −11.615 | −11.629 | −192.714 | −9.675 | −9.689 | −84.165 | −10.777 | −10.790 | −96.017 |
| H-32 | −11.023 | −11.037 | −192.043 | −9.424 | −9.438 | −38.360 | −10.140 | −10.153 | −107.200 |
| H-39 | −11.341 | −11.355 | −185.992 | −9.432 | −9.466 | −44.267 | −10.213 | −10.227 | −94.803 |
| H-62 | −11.275 | −11.290 | −191.833 | −9.289 | −9.303 | −54.908 | −10.314 | −10.328 | −162.156 |
| H-64 | −11.110 | −11.124 | −174.378 | −9.213 | −9.227 | −51.333 | −10.715 | −10.729 | −126.444 |
| H-86 | −11.298 | −11.312 | −177.756 | −9.033 | −9.047 | −77.899 | −10.249 | −10.262 | −101.827 |
| H-189 | −11.498 | −11.452 | −180.730 | −9.625 | −9.639 | −82.109 | −10.373 | −10.387 | −106.364 |
| H-201 | −11.403 | −11.417 | −186.951 | −9.020 | −9.034 | −38.228 | −10.348 | −10.962 | −117.939 |
| Compound | CIQPlogS* | QPlogBB* | CNS* | QPlogPo/w* | Rule of Five* | Rule of Three* |
|---|---|---|---|---|---|---|
| H-2 | −4.571 | −7.647 | −2 | −0.425 | 3 | 1 |
| H-6 | −4.910 | −9.260 | −2 | −0.496 | 3 | 1 |
| H-32 | −5.752 | −8.159 | −2 | 1.176 | 3 | 1 |
| H-39 | −6.432 | −10.160 | −2 | 1.016 | 3 | 1 |
| H-62 | −6.072 | −8.966 | −2 | 1.112 | 3 | 1 |
| H-64 | −5.639 | −9.209 | −2 | 0.229 | 3 | 1 |
| H-86 | −6.173 | −10.299 | −2 | 0.223 | 3 | 1 |
| H-189 | −4.966 | −8.993 | −2 | −0.100 | 3 | 1 |
| H-201 | −5.885 | −9.326 | −2 | −1.441 | 3 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciftci, H.; Sever, B.; Ayan, E.; Can, M.; DeMirci, H.; Otsuka, M.; TuYuN, A.F.; Tateishi, H.; Fujita, M. Identification of New L-Heptanoylphosphatidyl Inositol Pentakisphosphate Derivatives Targeting the Interaction with HIV-1 Gag by Molecular Modelling Studies. Pharmaceuticals 2022, 15, 1255. https://doi.org/10.3390/ph15101255
Ciftci H, Sever B, Ayan E, Can M, DeMirci H, Otsuka M, TuYuN AF, Tateishi H, Fujita M. Identification of New L-Heptanoylphosphatidyl Inositol Pentakisphosphate Derivatives Targeting the Interaction with HIV-1 Gag by Molecular Modelling Studies. Pharmaceuticals. 2022; 15(10):1255. https://doi.org/10.3390/ph15101255
Chicago/Turabian StyleCiftci, Halilibrahim, Belgin Sever, Esra Ayan, Mustafa Can, Hasan DeMirci, Masami Otsuka, Amaç Fatih TuYuN, Hiroshi Tateishi, and Mikako Fujita. 2022. "Identification of New L-Heptanoylphosphatidyl Inositol Pentakisphosphate Derivatives Targeting the Interaction with HIV-1 Gag by Molecular Modelling Studies" Pharmaceuticals 15, no. 10: 1255. https://doi.org/10.3390/ph15101255