
Synthesis, Characterization, and Biological Evaluation of New Derivatives Targeting MbtI as Antitubercular Agents

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
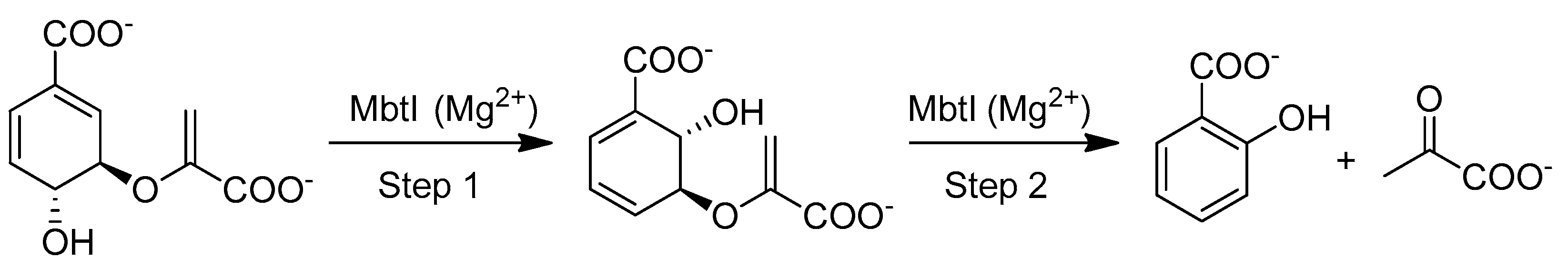
2. Results
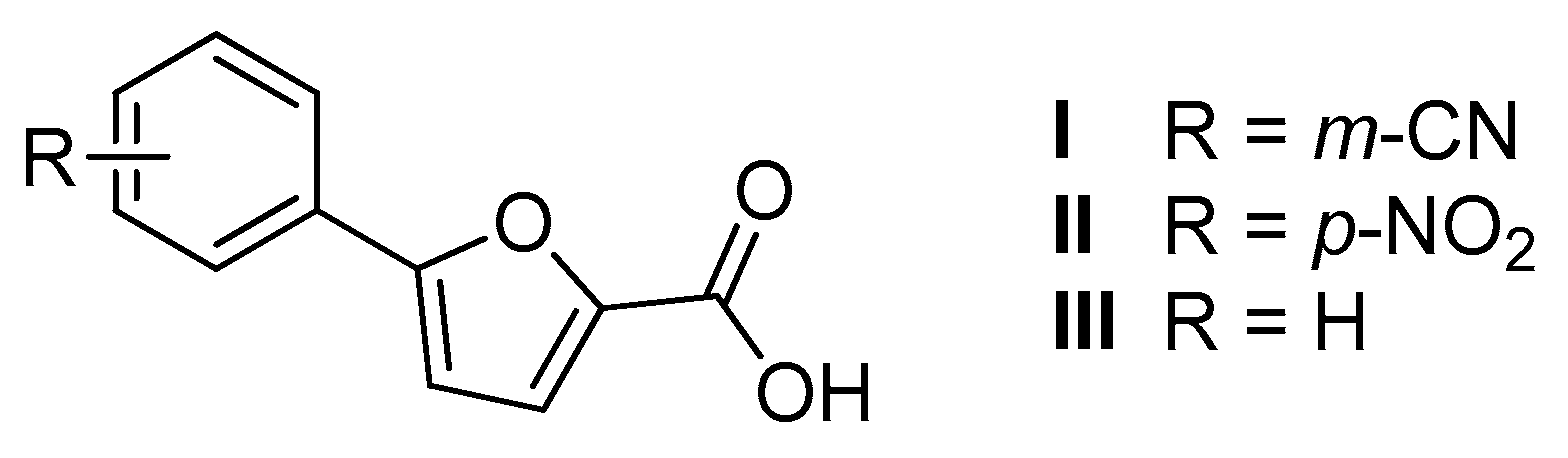
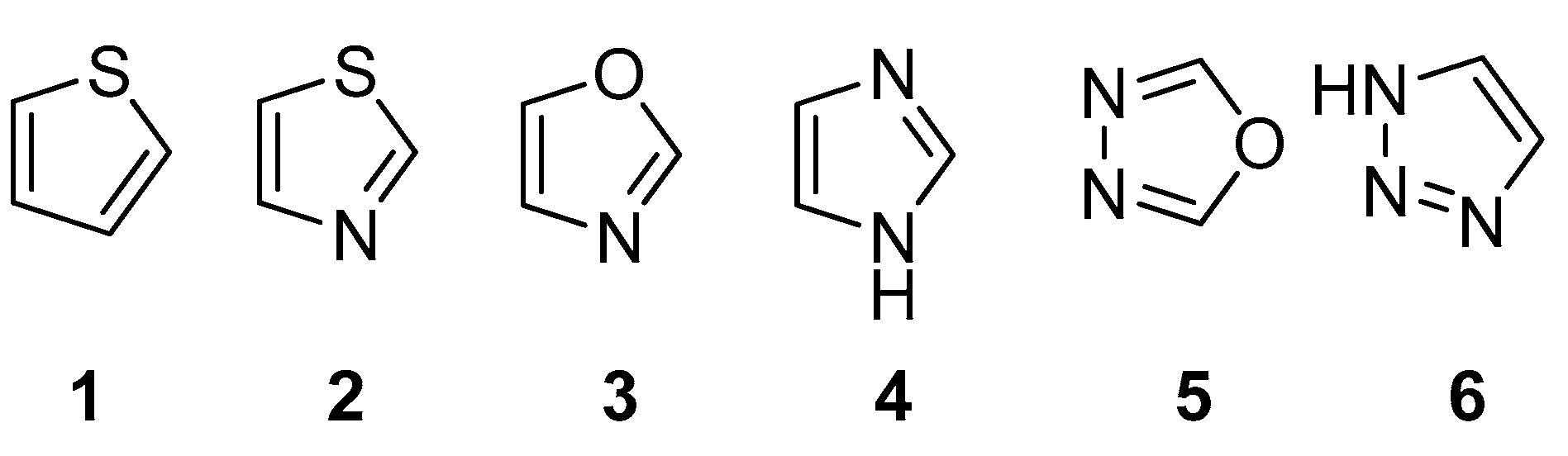
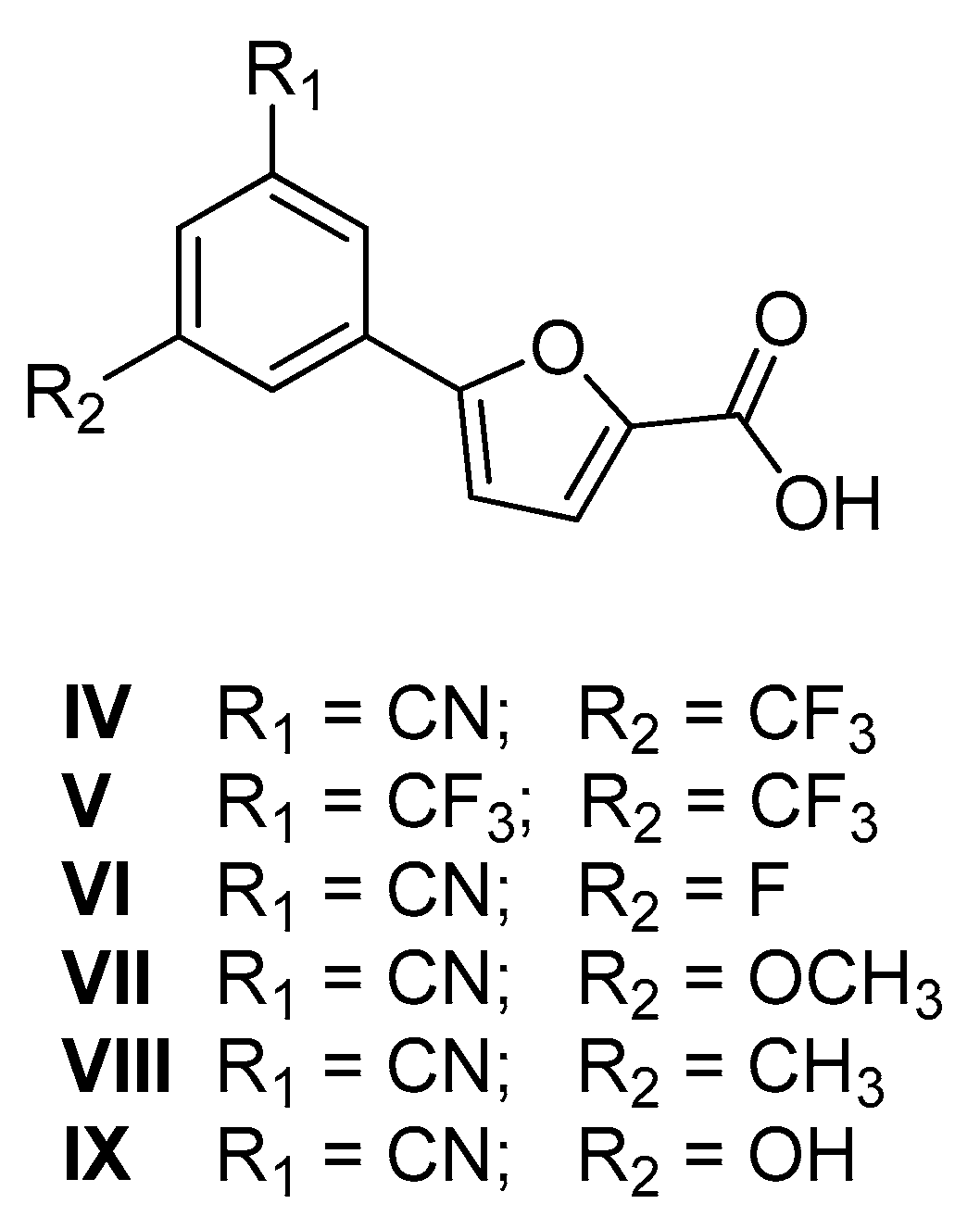
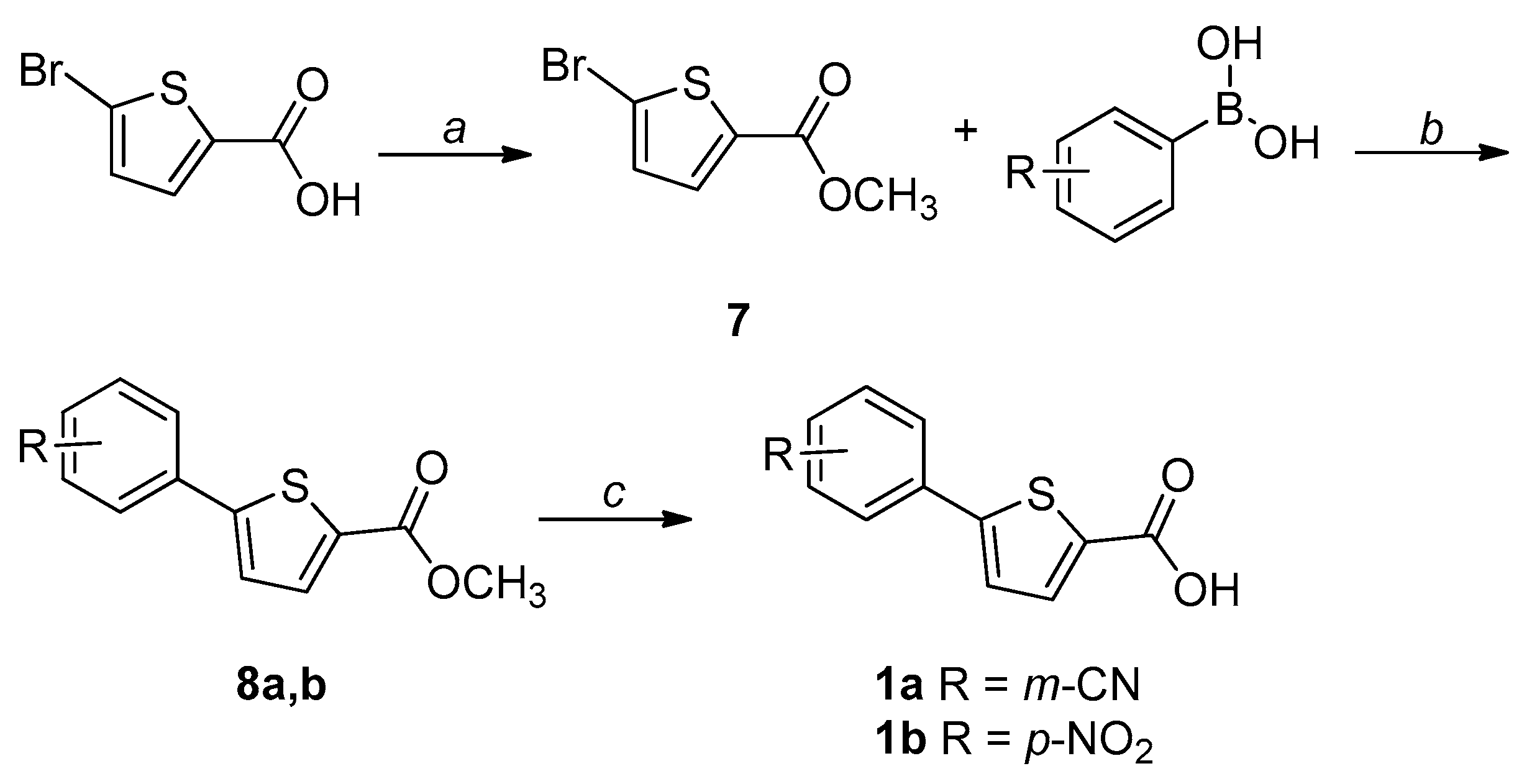
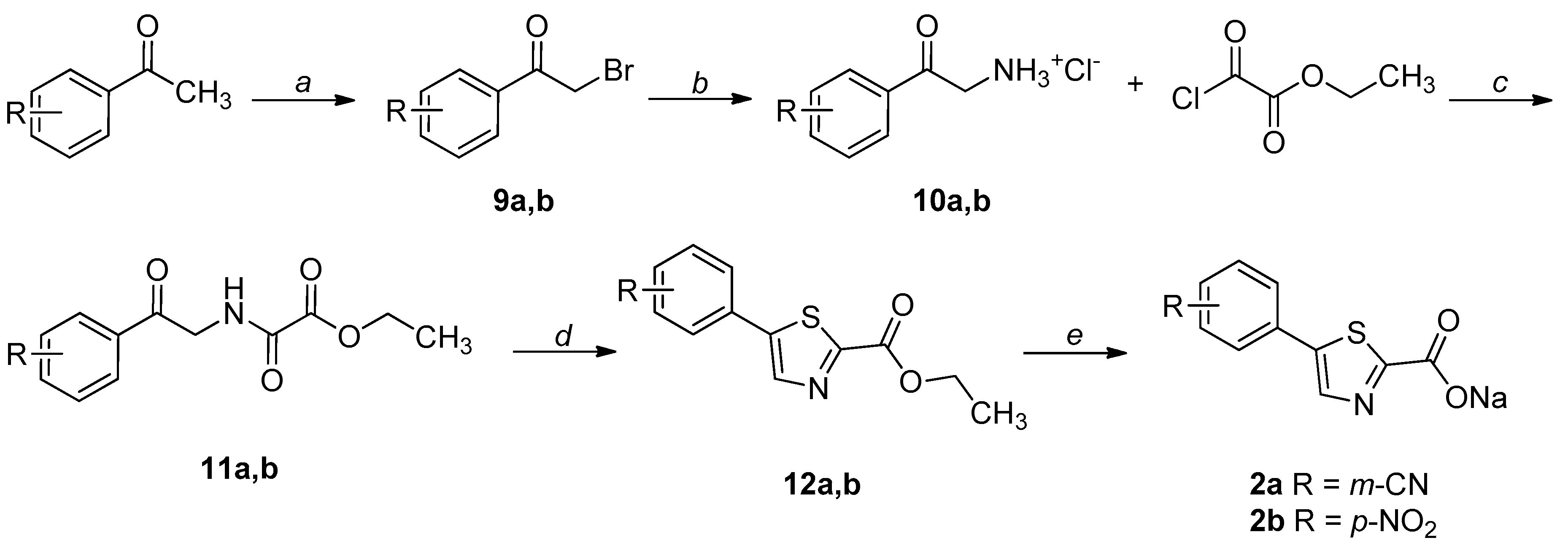
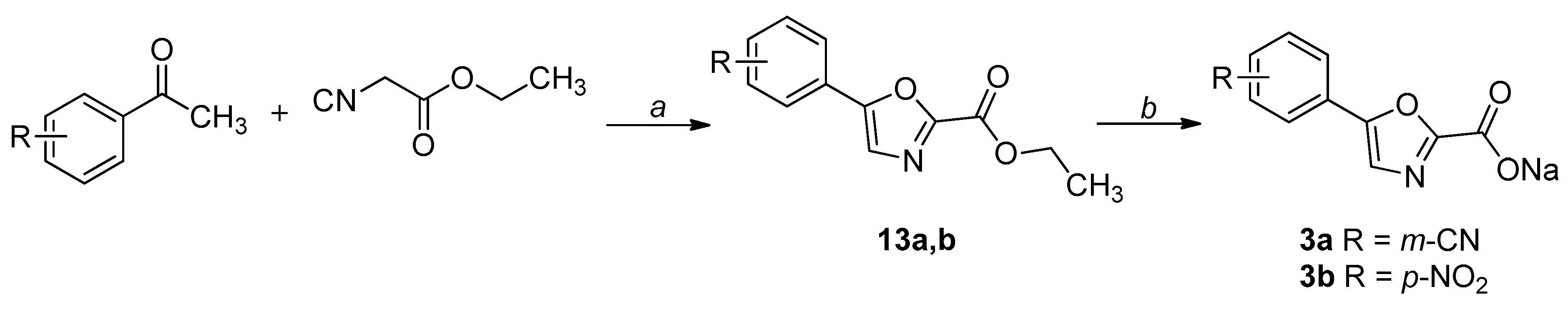
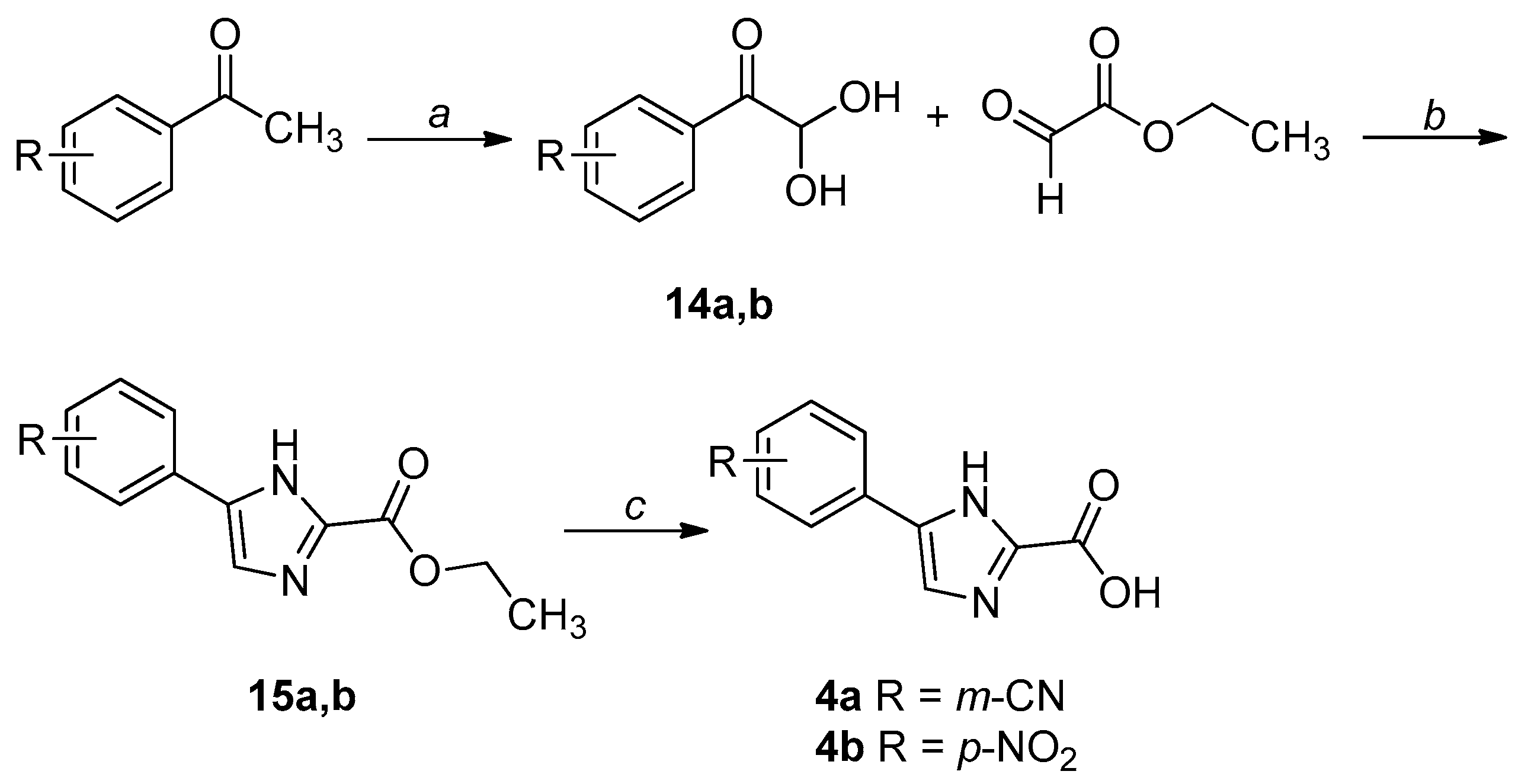
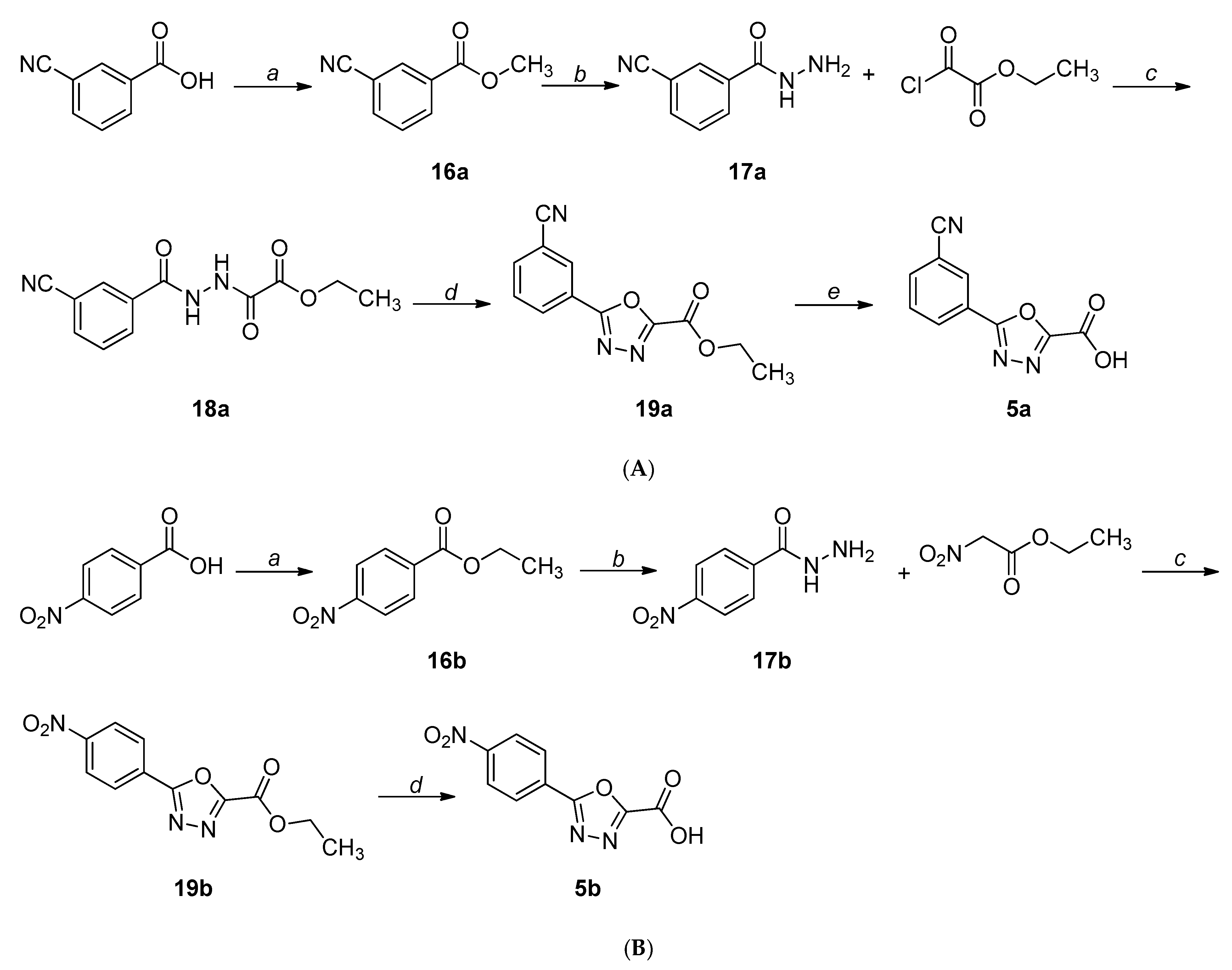
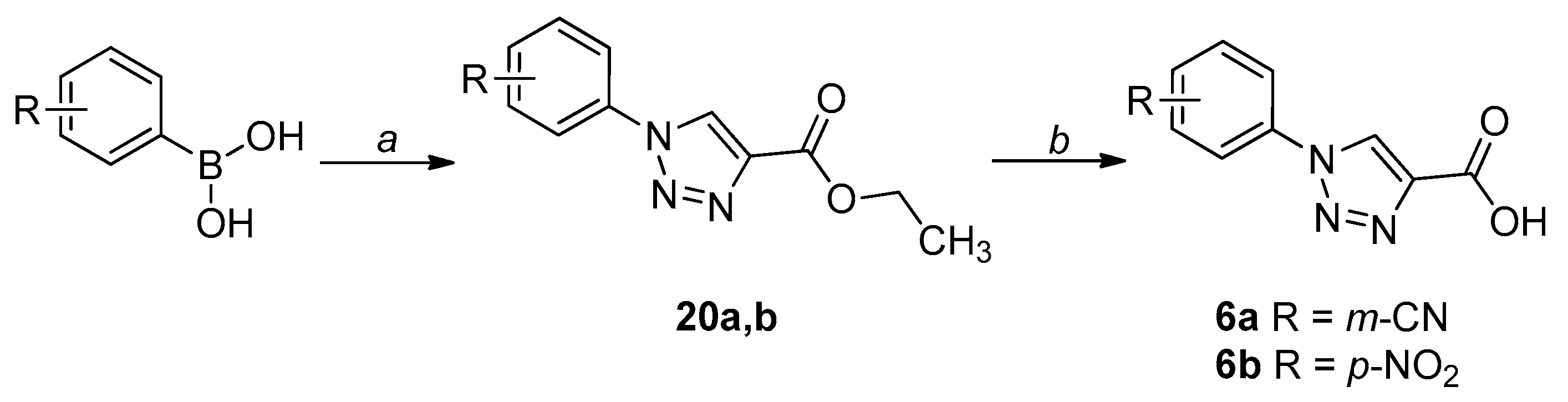
2.1. Chemistry
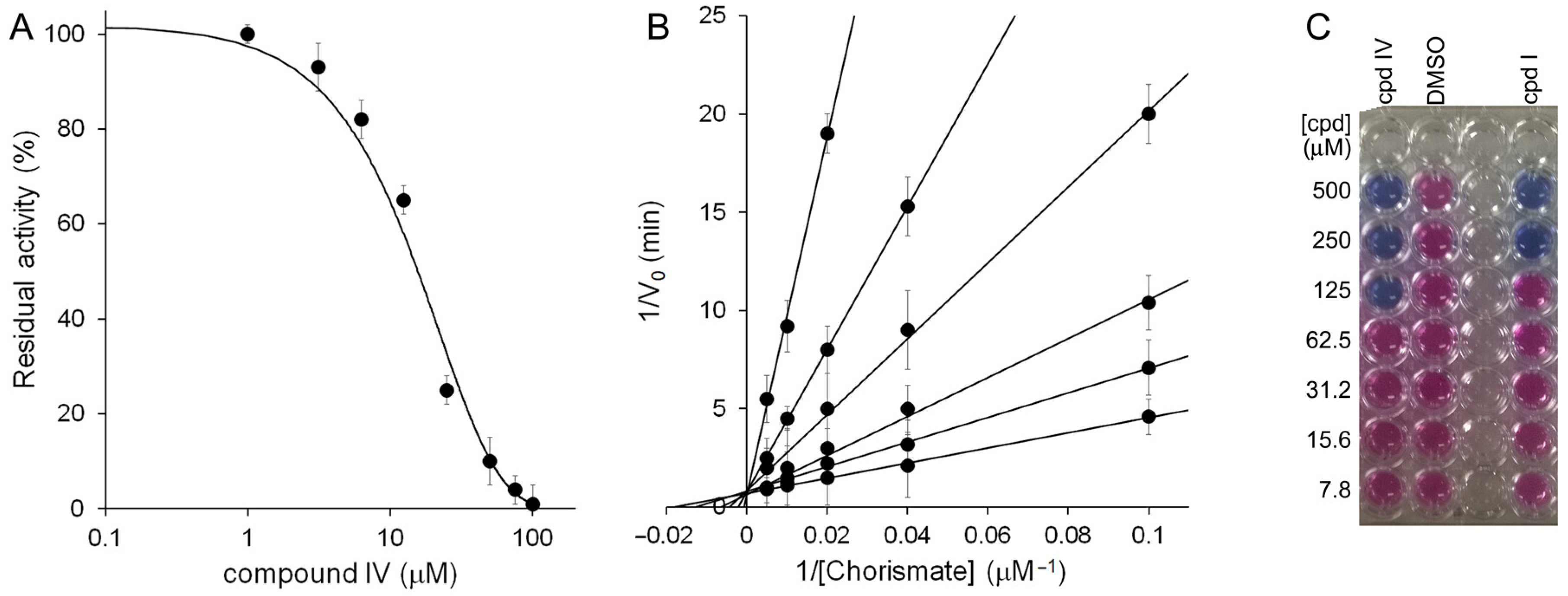
2.2. Biological Studies
3. Discussion
4. Material and Methods
4.1. Chemistry
4.2. Biological Activities
4.2.1. MbtI Enzymatic Assays
4.2.2. MIC Determination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001313-1.
- Xu, K.; Liang, Z.C.; Ding, X.; Hu, H.; Liu, S.; Nurmik, M.; Bi, S.; Hu, F.; Ji, Z.; Ren, J.; et al. Nanomaterials in the Prevention, Diagnosis, and Treatment of Mycobacterium Tuberculosis Infections. Adv. Healthc. Mater. 2018, 7, 1700509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truzzi, E.; Meneghetti, F.; Mori, M.; Costantino, L.; Iannuccelli, V.; Maretti, E.; Domenici, F.; Castellano, C.; Rogers, S.; Capocefalo, A.; et al. Drugs/Lamellae Interface Influences the Inner Structure of Double-Loaded Liposomes for Inhaled Anti-TB Therapy: An In-Depth Small-Angle Neutron Scattering Investigation. J. Colloid Interface Sci. 2019, 541, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brozek, J.L.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin. Infect. Dis. 2016, 63, e147–e195. [Google Scholar] [CrossRef]
- Falzon, D.; Jaramillo, E.; Schünemann, H.J.; Arentz, M.; Bauer, M.; Bayona, J.; Blanc, L.; Caminero, J.A.; Daley, C.L.; Duncombe, C.; et al. WHO Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis: 2011 Update. Eur. Respir. J. 2011, 38, 516–528. [Google Scholar] [CrossRef]
- Fanzani, L.; Porta, F.; Meneghetti, F.; Villa, S.; Gelain, A.; Lucarelli, A.P.; Parisini, E. Mycobacterium tuberculosis Low Molecular Weight Phosphatases (MPtpA and MPtpB): From Biological Insight to Inhibitors. Curr. Med. Chem. 2015, 22, 3110–3132. [Google Scholar] [CrossRef] [PubMed]
- Stelitano, G.; Sammartino, J.C.; Chiarelli, L.R. Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis. Molecules 2020, 25, 1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Sammartino, J.C.; Costantino, L.; Gelain, A.; Meneghetti, F.; Villa, S.; Chiarelli, L.R. An Overview on the Potential Antimycobacterial Agents Targeting Serine/Threonine Protein Kinases from Mycobacterium tuberculosis. Curr. Top. Med. Chem. 2019, 19, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, F.; Villa, S.; Gelain, A.; Barlocco, D.; Chiarelli, L.R.; Pasca, M.R.; Costantino, L. Iron Acquisition Pathways as Targets for Antitubercular Drugs. Curr. Med. Chem. 2016, 23, 4009–4026. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Sieminski, P.J.; Owens, C.P.; Goulding, C.W. Iron Acquisition in Mycobacterium tuberculosis. Chem. Rev. 2019, 119, 1193–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-K.; Liu, F.; Fiers, W.D.; Sun, W.-M.; Guo, J.; Liu, Z.; Aldrich, C.C. Synthesis of Transition-State Inhibitors of Chorismate Utilizing Enzymes from Bromobenzene cis -1,2-Dihydrodiol. J. Org. Chem. 2017, 82, 3432–3440. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Stelitano, G.; Gelain, A.; Pini, E.; Chiarelli, L.R.; Sammartino, J.C.; Poli, G.; Tuccinardi, T.; Beretta, G.; Porta, A.; et al. Shedding X-ray Light on the Role of Magnesium in the Activity of M. tuberculosis Salicylate Synthase (MbtI) for Drug Design. J. Med. Chem. 2020, 63, 7066–7080. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Beretta, G.; Gelain, A.; Pini, E.; Sammartino, J.C.; Stelitano, G.; Barlocco, D.; Costantino, L.; Lapillo, M.; et al. New Insight into Structure-Activity of Furan-based Salicylate Synthase (MbtI) Inhibitors as Potential Antitubercular Agents. J. Enzym. Inhib. Med. Chem. 2019, 34, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and Development of Novel Salicylate Synthase (MbtI) Furanic Inhibitors as Antitubercular Agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Pini, E.; Poli, G.; Tuccinardi, T.; Chiarelli, L.; Mori, M.; Gelain, A.; Costantino, L.; Villa, S.; Meneghetti, F.; Barlocco, D. New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies. Molecules 2018, 23, 1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinsberg, O. Über β-Naphtolsulfid und Iso-β-naphtolsulfid. J. Prakt. Chem. 1916, 93, 277–301. [Google Scholar] [CrossRef]
- Chauhan, P.M.S.; Sunduru, N.; Sharma, M. Recent Advances in the Design and Synthesis of Heterocycles as Anti-Tubercular Agents. Future Med. Chem. 2010, 2, 1469–1500. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kumar, S.K.; Maurya, V.K.; Mehta, B.K.; Ahmad, H.; Dwivedi, A.K.; Chaturvedi, V.; Thakur, T.S.; Sinha, S. S-Enantiomer of the Antitubercular Compound S006-830 Complements Activity of Frontline TB Drugs and Targets Biogenesis of Mycobacterium tuberculosis Cell Envelope. ACS Omega 2017, 2, 8453–8465. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.K.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259.e25. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.; Kumar, P.; Parashar, V.; Vilchèze, C.; Veyron-Churlet, R.; Freundlich, J.S.; Barnes, S.W.; Walker, J.R.; Szymonifka, M.J.; Marchiano, E.; et al. Antituberculosis Thiophenes Define a Requirement for Pks13 in Mycolic Acid Biosynthesis. Nat. Chem. Biol. 2013, 9, 499–506. [Google Scholar] [CrossRef]
- Nayak, S.; Gaonkar, S.L. A Review on Recent Synthetic Strategies and Pharmacological Importance of 1,3-Thiazole Derivatives. Mini Rev. Med. Chem. 2018, 19, 215–238. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Azzali, E.; Couto, I.; Costantino, G.; Pieroni, M.; Viveiros, M. Adjuvant Therapies Against Tuberculosis: Discovery of a 2-Aminothiazole Targeting Mycobacterium tuberculosis Energetics. Future Microbiol. 2018, 13, 1383–1402. [Google Scholar] [CrossRef]
- Azzali, E.; Machado, D.; Kaushik, A.; Vacondio, F.; Flisi, S.; Cabassi, C.S.; Lamichhane, G.; Viveiros, M.; Costantino, G.; Pieroni, M. Substituted N-Phenyl-5-(2-(phenylamino)thiazol-4-yl)isoxazole-3-carboxamides Are Valuable Antitubercular Candidates that Evade Innate Efflux Machinery. J. Med. Chem. 2017, 60, 7108–7122. [Google Scholar] [CrossRef]
- Tyagi, S.; Nuermberger, E.; Yoshimatsu, T.; Williams, K.; Rosenthal, I.; Lounis, N.; Bishai, W.; Grosset, J. Bactericidal Activity of the Nitroimidazopyran PA-824 in a Murine Model of Tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 2289–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieroni, M.; Wan, B.; Zuliani, V.; Franzblau, S.G.; Costantino, G.; Rivara, M. Discovery of Antitubercular 2,4-diphenyl-1H-imidazoles from Chemical Library Repositioning and Rational Design. Eur. J. Med. Chem. 2015, 100, 44–49. [Google Scholar] [CrossRef] [PubMed]
- De, S.S.; Khambete, M.P.; Degani, M.S. Oxadiazole Scaffolds in Anti-Tuberculosis Drug Discovery. Bioorg. Med. Chem. Lett. 2019, 29, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; He, Y.; Zhang, X.; Xu, J.; Luo, Y.; Wang, Y.; Franzblau, S.G.; Yang, Z.; Chan, R.J.; Liu, Y.; et al. Targeting Mycobacterium Protein Tyrosine Phosphatase B for Antituberculosis Agents. Proc. Natl. Acad. Sci. USA 2010, 107, 4573–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, K.S.; Jadhav, R.D.; Kandre, S.; Guha, T.; Reddy, M.M.K.; Brahma, M.K.; Deshmukh, N.J.; Dixit, A.; Doshi, L.; Srinivasan, S.; et al. Evaluation of Thiazole Containing Biaryl Analogs as Diacylglycerol Acyltransferase 1 (DGAT1) Inhibitors. Eur. J. Med. Chem. 2013, 65, 337–347. [Google Scholar] [CrossRef]
- Wu, X.; Geng, X.; Zhao, P.; Zhang, J.; Wu, Y.D.; Wu, A.X. I2-Promoted Formal [3+2] Cycloaddition of α-Methylenyl Isocyanides with Methyl Ketones: A Route to 2,5-Disubstituted Oxazoles. Chem. Commun. 2017, 53, 3438–3441. [Google Scholar] [CrossRef]
- Curreli, F.; Kwon, Y.D.; Belov, D.S.; Ramesh, R.R.; Kurkin, A.V.; Altieri, A.; Kwong, P.D.; Debnath, A.K. Synthesis, Antiviral Potency, in Vitro ADMET, and X-ray Structure of Potent CD4 Mimics as Entry Inhibitors That Target the Phe43 Cavity of HIV-1 gp120. J. Med. Chem. 2017, 60, 3124–3153. [Google Scholar] [CrossRef]
- Leung, D.; Du, W.; Hardouin, C.; Cheng, H.; Hwang, I.; Cravatt, B.F.; Boger, D.L. Discovery of an Exceptionally Potent and Selective Class of Fatty Acid Amide Hydrolase Inhibitors Enlisting Proteome-Wide Selectivity Screening: Concurrent Optimization of Enzyme Inhibitor Potency and Selectivity. Bioorg. Med. Chem. Lett. 2005, 15, 1423–1428. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Khamraev, V.; Aksenov, N.A.; Kirilov, N.K.; Domenyuk, D.A.; Zelensky, V.A.; Rubin, M. Electrophilic Activation of Nitroalkanes in Efficient Synthesis of 1,3,4-Oxadiazoles. RSC Adv. 2019, 9, 6636–6642. [Google Scholar] [CrossRef] [Green Version]
- Devine, S.M.; Mulcair, M.D.; Debono, C.O.; Leung, E.W.W.; Nissink, J.W.M.; Lim, S.S.; Chandrashekaran, I.R.; Vazirani, M.; Mohanty, B.; Simpson, J.S.; et al. Promiscuous 2-Aminothiazoles (PrATs): A Frequent Hitting Scaffold. J. Med. Chem. 2015, 58, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox Filtering Computations for Chemical Biology and Early Stages Drug Discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free WebTool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, T.M.; Bouloc, N.; Buxton, R.S.; Chugh, J.; Lougheed, K.E.A.; Osborne, S.A.; Saxty, B.; Smerdon, S.J.; Taylor, D.L.; Whalley, D. Substituted Aminopyrimidine Protein Kinase B (PknB) Inhibitors Show Activity Against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2012, 22, 3349–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyam, M.; Shilkar, D.; Verma, H.; Dev, A.; Sinha, B.N.; Brucoli, F.; Bhakta, S.; Jayaprakash, V. The Mycobactin Biosynthesis Pathway: A Prospective Therapeutic Target in the Battle against Tuberculosis. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Vasan, M.; Neres, J.; Williams, J.; Wilson, D.J.; Teitelbaum, A.M.; Remmel, R.P.; Aldrich, C.C. Inhibitors of the Salicylate Synthase (MbtI) from Mycobacterium tuberculosis Discovered by High-Throughput Screening. ChemMedChem 2010, 5, 2079–2087. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Series A R = m-CNPh | Series B R = p-NO2Ph | |||
|---|---|---|---|---|---|
| %RA * | IC50 ** (µM) | %RA * | IC50 ** (µM) | ||
 | I | II | |||
| 3.1 ± 1.0 | 6.3 ± 0.9 | 18.2 ± 5.1 | 7.6 ± 1.6 | ||
 | 1a | 1b | |||
| 23.7 ± 5.0 | 35.5 ± 1.9 | 22.5 ±10.8 | 18.6 ± 1.7 | ||
 | 2a | 2b | |||
| 24.8 ± 1.8 | 41.9 ± 7.3 | 32.8 ± 2.3 | - | ||
 | 3a | 3b | |||
| 38.0 ± 7.6 | - | 23.0 ± 7.8 | 21.1 ± 2.7 | ||
 | 4a | 4b | |||
| 21.2 ± 1.0 | 39.3 ± 3.0 | 18.2 ± 4.9 | 27.5 ± 2.3 | ||
 | 5a | 5b | |||
| 75.0 ± 9.8 | - | 59.0 ± 4.3 | - | ||
 | 6a | 6b | |||
| 67.9 ± 6.6 | - | 32.7 ± 7.8 | - | ||
| Entry | % RA * | IC50 ** (µM) |
|---|---|---|
| IV | 0.7 ± 2.7 | 15.5 ± 3.1 |
| V | 8.9 ± 1.6 | 18.8 ± 6.8 |
| VI | 8.7 ± 1.4 | 17.3 ± 3.1 |
| VII | 10.5 ± 3.9 | 14.5 ± 2.1 |
| VIII | 16.7 ± 3.8 | 29.1 ± 1.8 |
| IX | 15.3 ± 2.6 | 33.5 ± 3.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, M.; Stelitano, G.; Chiarelli, L.R.; Cazzaniga, G.; Gelain, A.; Barlocco, D.; Pini, E.; Meneghetti, F.; Villa, S. Synthesis, Characterization, and Biological Evaluation of New Derivatives Targeting MbtI as Antitubercular Agents. Pharmaceuticals 2021, 14, 155. https://doi.org/10.3390/ph14020155
Mori M, Stelitano G, Chiarelli LR, Cazzaniga G, Gelain A, Barlocco D, Pini E, Meneghetti F, Villa S. Synthesis, Characterization, and Biological Evaluation of New Derivatives Targeting MbtI as Antitubercular Agents. Pharmaceuticals. 2021; 14(2):155. https://doi.org/10.3390/ph14020155
Chicago/Turabian StyleMori, Matteo, Giovanni Stelitano, Laurent R. Chiarelli, Giulia Cazzaniga, Arianna Gelain, Daniela Barlocco, Elena Pini, Fiorella Meneghetti, and Stefania Villa. 2021. "Synthesis, Characterization, and Biological Evaluation of New Derivatives Targeting MbtI as Antitubercular Agents" Pharmaceuticals 14, no. 2: 155. https://doi.org/10.3390/ph14020155