Modulation of Host Lipid Pathways by Pathogenic Intracellular Bacteria

Vector Borne Disease Laboratories, Department of Pathobiological Sciences, LSU School of Veterinary Medicine, Baton Rouge, LA 70803, USA
*
Author to whom correspondence should be addressed.
Pathogens 2020, 9(8), 614; https://doi.org/10.3390/pathogens9080614
Submission received: 27 June 2020
/
Revised: 17 July 2020
/
Accepted: 25 July 2020
/
Published: 28 July 2020
(This article belongs to the Special Issue Virulence Mechanisms of Rickettsiae)
{kind=link}
{kind=link}
Abstract
:Lipids are a broad group of molecules required for cell maintenance and homeostasis. Various intracellular pathogens have developed mechanisms of modulating and sequestering host lipid processes for a large array of functions for both bacterial and host cell survival. Among the host cell lipid functions that intracellular bacteria exploit for infection are the modulation of host plasma membrane microdomains (lipid rafts) required for efficient bacterial entry; the recruitment of specific lipids for membrane integrity of intracellular vacuoles; and the utilization of host lipid droplets for the regulation of immune responses and for energy production through fatty acid β-oxidation and oxidative phosphorylation. The majority of published studies on the utilization of these host lipid pathways during infection have focused on intracellular bacterial pathogens that reside within a vacuole during infection and, thus, have vastly different requirements for host lipid metabolites when compared to those intracellular pathogens that are released into the host cytosol upon infection. Here we summarize the mechanisms by which intracellular bacteria sequester host lipid species and compare the modulation of host lipid pathways and metabolites during host cell infection by intracellular pathogens residing in either a vacuole or within the cytosol of infected mammalian cells. This review will also highlight common and unique host pathways necessary for intracellular bacterial growth that could potentially be targeted for therapeutic intervention.
1. Introduction
Lipids are broadly described as hydrophobic or amphipathic molecules that are involved in various biological functions including mediating immune signaling, regulating vesicle trafficking, providing energy storage and production, and structural membrane composition [1,2,3,4,5,6,7,8,9,10]. Lipids are a large, diverse group of molecules that provide various specific functions required by the host cell to maintain homeostasis. These lipids can be divided into various classes that are highly organized and provide very specialized functions [5]. Furthermore, because of the essentiality and multifunctional property of host lipids for cell maintenance, intracellular pathogens have adapted mechanisms to employ a number of these lipids throughout infection for both host cell regulation, and pathogen replication and sustainability [11].
Intracellular pathogens modify host lipid composition by regulating host cell uptake of exogenous lipids from the environment and modulating the expression of enzymes involved in the host de novo lipid biosynthetic pathways [12,13,14,15,16]. This alteration in host lipid composition can be beneficial to the pathogen by providing a number of different functions including: (i) Employing storage and/or free lipids for energy production. Fatty acids within host lipid storage compartments, known as lipid droplets (LDs), and free fatty acids can be catabolized by various methods, such as lipolysis, lipophagy, and selective breakdown via phospholipases. These processes differ in that lipolysis releases lipids through hydrolysis, lipophagy utilizes host autophagy machinery to breakdown lipids within lipid droplets, and phospholipases selectively catabolize specific species of lipids some of which are associated with LDs. The lipids released can then be used in the β-fatty acid oxidation (FAO) pathway, which enzymatically breaks down fatty acids to acetyl-CoA for use in oxidative phosphorylation (OXPHOS) for energy production [17]. Pathogens are also able to release and sequester metabolites within these catabolic pathways by use of bacterial phospholipases for self-generation of energy. The regulation of host lipid metabolism to promote shifts in energy production pathways has also been implemented in modulating host cell persistence, pathogen sustainability, and anti-inflammatory mechanisms. (ii) Pathogen membrane production. Some intracellular pathogens have been shown to have unique lipids within their membrane structures that are unlike common bacterial lipids and that are unable to be made by the pathogen machinery encoded in the genome. Thus, is it likely that the lipids are acquired from the host and are incorporated into the pathogen membrane. (iii) Invasion of host signaling pathways and the host immune system. Lipid organization within host membranes play an important role in the regulation of signaling pathways by creating “hot spots,” known as lipid rafts, for enabling the association of receptors with external and internal stimuli. These functional, highly organized membrane regions are utilized by various intracellular bacterial pathogens for entry into the host cell [18]. There are also fatty acid molecules released from LDs, such as arachidonic acids, that are precursors of host immune modulators, such as prostaglandins and leukotrienes, and have a key role in the presentation of disease during pathogen infection [19,20,21].
Human pathogenic intracellular bacteria have developed mechanisms that initiate the employment of host lipids to cause disease during mammalian infection. Among the functions that require host lipids are bacterial entry into the host cell, formation of an intracellular compartment, metabolic requirements for bacterial survival and replication, and disease initiation and progression. However, because intracellular bacterial pathogens differ in the mechanisms used to colonize cells, it is likely that host lipid manipulation during infection varies between different bacterial species [22]. Specifically, intracellular bacteria can be separated into those that persist in a vacuolar compartment inside the host cell and those that survive and replicate within the host cell cytosol. Together, the differences among the intracellular bacteria indicate a requirement of host cell lipids that varies among and within vacuolar and cytosolic groups. This review addresses current knowledge on the alteration and use of host lipids during human pathogenic intracellular bacterial infection. The research discussed highlights differences in vacuolar and cytosolic bacteria’s use of host lipids for pathogen membrane structure, host LD alterations during infection, and lipid utilization for energy production.
2. Intracellular Bacteria That Replicate Inside a Vacuole during Infection
As briefly mentioned previously, vacuolar bacterial pathogens are defined as species that remain within the vacuole formed upon entry into the host cell, though not all vacuoles are created equally [22,23]. The pathogen-containing vacuoles (PCVs) are very specific to each pathogen with differences in the suitable intravacuolar acidity for pathogen growth. The vacuole markers and composition are indicative of the stages of vacuole transport pathways that the PCV is derived from or halted in and the presence of intravacuolar nutrients and associated host cell organelles. Many vacuolar pathogens, including Coxiella burnetii, Chlamydia spp., Mycobacterium spp., Anaplasma phagocytophilum, Ehrlichia chaffeensis, Brucella abortus, Legionella pneumophila, and Salmonella spp., have been shown to manipulate host lipids during intracellular infection for an array of defined and speculated functions involved in bacterial and vacuolar membrane production, foam cell presentation and immune modulator production, and energy production.
2.1. Host Lipids Provide Structural Integrity to Pathogen-Associated Membranes
Bacterial membranes are limited in their composition of lipid species when compared to their eukaryotic counterparts. The predominant lipids present in standard Gram-negative and Gram-positive bacterial membranes are cardiolipin, phosphatidylglycerol (PG), phosphatidylethanolamine (PE), phosphatidic acid (PA), and phosphatidylserine (PS), with machinery lacking for biosynthesis of cholesterol and, in some intracellular bacteria, phosphatidylcholine (PC). In general, bacterial membrane lipids are unsaturated and can exhibit distinct branching patterns among the various bacteria [24,25]. Many vacuolar intracellular bacteria recruit and incorporate specific host lipids as structural components for both the host-derived PCVs and bacterial membrane. Although, intracellular bacteria do not encode the enzymes necessary for production of cholesterol and other lipids commonly found within eukaryotic cells, they have adapted mechanisms for modification of sequestered host lipids for incorporation into structural membranes during infection.
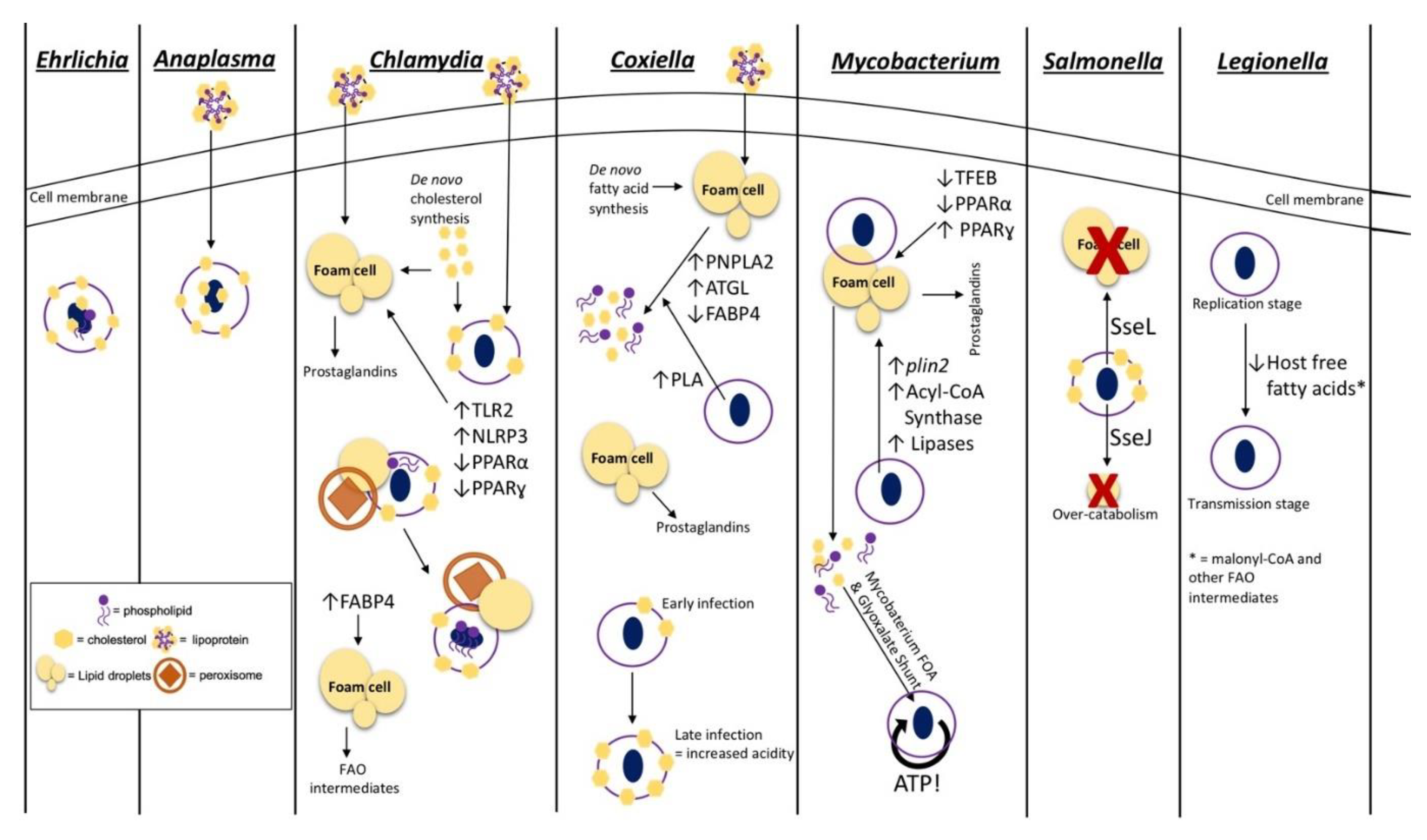
Chlamydia spp. are vacuolar pathogens that typically target epithelial cells in areas of species-specific tropism and can actively replicate in vacuoles within recruited macrophages during infection [26]. A characteristic of the Chlamydia-PCV is that the membrane contains Golgi apparatus-derived membrane markers indicative of a non-acidic, nutrient-rich vacuole unlikely to interact with the lysosome during late stages of the endocytic pathway [27,28]. Association of the PCV with the host Golgi apparatus, which acts as a nutrient-rich recycling and trafficking organelle, also provides nutrients to the bacteria. During human and mouse fibroblast infection, the Chlamydia-containing vacuole contains cholesterol-rich microdomains that comprise an abundance of PCV membrane-associated bacterial proteins (Figure 1). These bacterial PCV proteins are important for interacting with the host proteins, such as Src-family kinases, to modulate vacuole trafficking via the host microtubule network. This trafficking is required to facilitate the PCV and Golgi-derived endosome fusion for nutrient sharing and genetic exchange required for the development of disease [29,30,31]. In addition to the incorporation of host cholesterol into the PCV, Chlamydia spp., such as C. trachomatis and C. pneumoniae, also actively recruit host cholesterol and free fatty acids through initiation of exogenous lipoprotein recruitment and de novo lipid synthesis for fatty acid overaccumulation within host cells [27,32] (Figure 1). Interestingly, during C. trachomatis infection of Hep-2 epithelial cells and mouse and human fibroblasts, host compartments, such as lipid droplets (LDs) and host peroxisomes, along with free fatty acids, colocalize at the PCV [33,34]. The association of these host cellular components at the PCV allows for efficient peroxisome-driven lipolysis of associated fatty acids, specifically PC and lysophosphatidylcholine (lyso-PC), that are unable to be made by C. trachomatis. It is important to note that although peroxisome-driven lipolysis is a known source of lipids for the bacteria, lipid droplet-associated lipases are also likely involved in providing the required lipids for C. pneumoniae growth [33]. Thus, these events allow C. trachomatis to scavenge and modify the catabolized lipids for incorporation in the bacterial membrane [33,35,36,37] (Figure 1). Both host and chlamydial proteins play crucial roles in the sequestering and modification of host PC for incorporation into the bacterial membrane. Specifically, the proteins involved in PC modification are the human proteins, human acyl-CoA:lyso-PC acyltransferase 1, CLA1 lipid transporter, and human acyl-CoA carrier 6, along with the pathogen protein, chlamydial lyso-PC acyltransferase. The combination of these proteins ultimately re-acetylates host PC for bacterial use. These bacterial and host modifications provide PC species with an increased branch number, making these sequestered lipids similar to typical bacteria-made membrane lipids suitable for the production of bacterial progeny [24,38,39,40,41]. C. trachomatis also encodes phosphatidylserine decarboxylase and acyl-ACP synthase enzymes supporting the notion that these bacteria have the ability to modify and utilize host lipids during infection [42,43]. Although C. trachomatis is able to modify host PCs, this bacteria is also able to produce other phospholipid species utilizing the bacterial type II fatty acid synthesis (FASII) pathway, which when combined with host lipid modification of scavenged lipids, allows for efficient reproduction during intracellular infection [44].
Anaplasma phagocytophilum and Ehrlichia chaffeensis are closely related bacterial species that halt the phagosome–lysosome fusion step of the endocytic pathway and reside within a non-acidic, early and late endosome, respectively, during intracellular infection [45,46]. During mammalian infection, A. phagocytophilum targets neutrophils, and E. chaffeensis predominantly infects monocytes [46]. Similar to C. trachomatis, A. phagocytophilum and E. chaffeensis, require cholesterol for persistence but do not genetically encode for anabolic cholesterol pathways; thus, cholesterol present within bacterial compartments is captured from the host [47,48]. Similar to C. trachomatis PCVs, A. phagocytophilum PCVs contain cholesterol-rich domains to enable microtubule trafficking for nutrient-rich endosome fusion, though the specific protein:protein interactions involved have yet to be elucidated [49]. However, unlike Chlamydia spp., A. phagocytophilum infection of HL-60 promyeloblasts does not require host de novo cholesterol synthesis and, instead, actively stimulates uptake of lipoproteins by the host cell and recruitment to the PCV through regulation of the Niemann–Pick disease type C1 (NPC1)-mediated intracellular cholesterol trafficking [50,51,52]. The mechanism for sequestering cholesterol from the host cell during E. chaffeensis infection is not well defined; however, host cholesterol is incorporated into the bacterial membranes of both A. phagocytophilum and E. chaffeensis (Figure 1). Both A. phagocytophilum and E. chaffeensis lack the lipid-A protective layer and traditional lipopolysaccharide structures seen in most Gram-negative bacterial outer membranes. Therefore, it is likely that the addition of host cholesterol within the bacterial cell membrane is important in providing rigidity and protection that these bacteria otherwise lack during intracellular infection [50,53]. Recently, E. chaffeensis was also shown to encode a partial glycerophospholipid pathway indicating a potential requirement of host glycerophospholipids and intermediates for the production of various membrane lipids. This requirement for host membrane intermediates is supported by the finding that E. chaffeensis replication in monocytic THP-1 cells is diminished when host de novo fatty acid synthesis and subsequent phospholipid production is inhibited [49]. The process of sequestering host lipids for E. chaffeensis membrane construction is thought to be initiated by a type IV secretion system (T4SS) bacterial effector protein(s), as lipid acquisition is perturbed when bacterial protein synthesis and T4SS secretion is inhibited [49].
Conversely, C. burnetii promotes phagolysosome maturation, during macrophage infection, to form an acidic parasitophorous vacuole that contains an abundance of host cholesterol and other sterols in the PCV membrane [54]. Though C. burnetii infection does not require cholesterol specifically for successful infection, the regulation of the overall sterol composition within the parasitophorous vacuole is necessary for the regulation of infection stages and growth of the PCV [55]. More recent studies indicate that the sterol composition in this PCV is important for regulation of the v-ATPase pump required to maintain optimum acidity within the Coxiella-containing vacuole. Specifically, this study showed a correlation of an increase in host cholesterol within the PCV membrane and increases in PCV acidity during later stages of mouse embryonic fibroblast C. burnetii infection [56,57] (Figure 1). Inhibition of bacterial protein synthesis followed by a decrease in sterol recruitment suggests that host sterol composition within the PCV is actively regulated by Coxiella effector proteins, which likely modulate endosomal trafficking and cholesterol acquisition [58,59]. C. burnetii also contains an active sterol reductase, which catalyzes the last step in the cholesterol biosynthesis pathway, implying a possible use of host sterol intermediates for bacterial completion of cholesterol production [60]. Unlike other intracellular pathogens such as Chlamydia spp., E. chaffeensis, and A. phagocytophilum, the use of host cholesterol or lipids for C. burnetii membrane production is undefined. The presence of complete bacterial fatty acid and phospholipid biosynthesis pathways within the C. burnetii genome suggests that host phospholipid incorporation may not be required for replication. Nevertheless, as described for C. trachomatis, host lipid incorporation in bacterial membranes may be coupled with endogenous bacterial membrane production pathways for improved intracellular replication [60].
Several studies support the requirement of host lipids for the structural integrity of obligate intracellular pathogens discussed above; however, the requirement of host lipids for de novo membrane synthesis in facultative intracellular bacterial species is not well defined. Genetically, facultative intracellular pathogens contain a higher number of complete biosynthesis pathways overall compared to obligate intracellular bacteria, and the ability to survive and replicate extracellularly indicates a lower reliance on the host for required nutrients [61,62]. However, facultative intracellular Salmonella spp. infect macrophages and reside within undeveloped phagosomes that contains host cholesterol thought to be acquired during entry, which plays a role in early vacuolar trafficking [63]. Similar to Chlamydia, the cholesterol within the Salmonella-PCV is actively recruited to the membrane and is an important regulator of host:pathogen protein interactions by providing structured “hot spots,” such as lipid rafts, on the PCV membrane that facilitate Salmonella protein presentation and association with host proteins in the cytosol [64]. This active recruitment is achieved through the type III secretion system (T3SS) effector protein, SseJ, which has acyltransferase activity [65,66,67,68] (Figure 1). A mutation in sseJ during mouse infection attenuates virulence and causes a decrease in PCV production, supporting the requirement and use of SseJ as an acyltransferase used to modify membrane lipids for incorporation into the PCV [68].
2.2. Host Lipid-Droplet Modulation
Lipid droplets (LDs) are a host cellular compartments used to store predominantly neutral, esterified cholesterol and triacylglycerols, surrounded by a phospholipids monolayer with lipid-associated proteins, which can serve various functions including energy production and immune response modulation [69,70]. Some pathogens, such as Chlamydia pneumoniae, Chlamydia trachomatis, Coxiella burnetii, and M. tuberculosis produce foam cells during macrophage infection, characterized by an overaccumulation of LDs within the host cell [71]. This lipid accumulation in macrophages and the subsequent alteration of metabolism and immune signaling correlate with changes in macrophage polarization, which have been shown to be important during macrophage infection by various intracellular pathogens [72,73].
C. pneumoniae and C. trachomatis induce a “foamy” cell phenotype during macrophage infection by employing a bacterial cholesterol esterification enzyme, CT149, to form cholesterol esters that are subsequently stored in LDs [42]. During macrophage infection, C. pneumoniae also actively downregulates host transcription factors, peroxisome proliferator-activated receptors (PPAR) α and ɣ. These two proteins are both known to be involved in the regulation of LD breakdown and cholesterol efflux, providing a likely mechanism for the formation of the foam cell phenotype during infection [74,75,76,77]. Immune regulation also occurs through the stimulation of the host inflammasome, NLRP3, and toll-like receptor (TLR) 2 to subsequently increase LD formation in infected macrophages. However, the function(s) or consequences of host lipid overaccumulation into LDs during infection by C. pneumoniae is not yet clearly elucidated [78,79]. Conversely, C. trachomatis employs the modulation of fatty acids within host LDs for immune response regulation by recruiting host triacylglycerol lipases to the LDs associated with the bacterial PCVs. These host lipases breakdown the stored triacylglycerols within the LDs into immune signaling intermediates, such as arachidonic acid, to be converted into immune mediators, such as prostaglandins, for enhancement of tissue damage and overall disease during mammalian infection [40,80]. Stimulation of lipases for prostaglandin production can be induced by pathogen infection of a single cell and by host cell–to–host cell signaling caused by the infection. Therefore, infected and uninfected bystander cells within a mammalian host are likely involved in overall prostaglandin production during infection. As described previously, lipids are also scavenged from PCV-associated LDs for bacterial membrane production [24,38,39,40] (Figure 1). It is important to note that although LDs are a major source of lipids for C. trachomatis during intracellular growth, these host structures are not required during mouse embryonic fibroblast infection. Interestingly, during inhibition of LD formation in mouse embryonic fibroblasts, C. trachomatis is able to efficiently grow when free fatty acids are supplemented. This suggests that the bacteria are able to recruit free fatty acids and other host lipids not related to LDs to the PCV as a way of compensating for a lack of lipids within LDs [81].
Similarly, C. burnetii’s formation of foam cells is actively modulated by bacterial effector proteins and is dependent on an increase in fatty acid recruitment to the endoplasmic reticulum for packaging into host LDs [82]. During C. burnetii infection of macrophages, the expression of host proteins involved in LD breakdown, including patatin-like phospholipase domain containing protein 2 (PNPLA2) or adipose triglyceride lipase (ATGL), acyl-CoA:cholesterol transferase, and fatty acid binding protein (FABP4), is modulated suggesting a requirement of host lipids within LDs, such as cholesterol esters and triacylglycerols, during infection [82,83,84,85]. The regulation of ATGL and FABP4 indicate a release of free fatty acids and cholesterol from host LDs for C. burnetii nutrient acquisition and membrane production as described previously [82]. C. burnetii also encodes a bacterial phospholipase A enzyme that is involved in accumulating free fatty acids, possibly for membrane and energy production or immune regulation during infection [86]. Similar to C. trachomatis, C. burnetii infection also utilizes prostaglandins derived from host LD intermediates to promote intracellular growth and disease presentation [87,88,89] (Figure 1). Interestingly, the inhibition of acyl-CoA anabolism and de novo lipid synthesis involved in production of host LDs enhances C. burnetii growth during macrophage infection. Conversely, pharmacological inhibition of LD catabolism is detrimental to C. burnetii infection. Together, these results suggest that lipids derived from host LDs and likely exogenous-derived lipids are essential for modulation of the host immune response and nutrient acquisition during C. burnetii macrophage infection [82].
Little is known about the mechanisms employed to modulate host LDs during A. phagocytophilum and E. chaffeensis infection of the respective target cells. During infection of promyelocytic cells, A. phagocytophilum increases expression of perilipin 1 (PLIN1), the major host protein important for regulation of LD formation and lipolysis, suggesting a possible role of host LDs as a source of cholesterol and fatty acids. However, the exact function of host LDs for A. phagocytophilum infection is still not yet elucidated [90,91,92,93,94]. Interestingly, E. chaffeensis has not been shown to have an association with host LDs or LD-related host molecules. Similar to A. phagocytophilum, the requirement of host cholesterol for successful infection implies a possible use of host LDs for cholesterol acquisition during infection.
Facultative intracellular pathogens from the genus Mycobacterium provide classic examples of producing a foam cell phenotype required for persistence during alveolar macrophage and overall mammalian infection. Many bacterial and host mechanisms are employed to produce and maintain the foam cell state during mycobacterial infection with various Mycobacterium spp. For example, M. tuberculosis and M. leprae infection increases transcription of bacterial genes involved in LD accumulation, plin2 (coding for perilipin 2) and acyl-CoA synthase, while M. tuberculosis also modulates host transcription factors PPARα and ɣ, similar to C. pneumoniae [95,96,97,98,99,100,101]. Unlike C. pneumoniae, M. tuberculosis’s growth during infection is increased by an upregulation in PPARɣ and subsequent triacylglycerol production and accumulation of host LDs. M. tuberculosis growth is also downregulated when LD catabolism is increased by transcription factor EB (TFEB)-initiation and subsequent PPARα expression. Together, these data suggest an importance of LD overaccumulation during macrophage infection [96,97]. Similar to that of C. pneumoniae and C. burnetii, M. leprae infection also increases prostaglandin production, which then stimulates other immune modulatory molecules, such as IL-10 and TGFβ required for bacterial survival and persistence [100,102]. Interestingly, M. tuberculosis is capable of producing triacylglycerol-rich bacterial LD storages during the dormancy stage of infection [98,99,103,104,105]; however, M. tuberculosis still associates with host LDs suggesting utilization of host storages for nutrient acquisition to increase efficiency and persistence during long-term mammalian infection [106]. M. tuberculosis and M. leprae also encode phospholipase C and other bacterial lipases used to release free fatty acids from storage lipids as carbon sources for increased infection efficiency [107,108] (Figure 1). Similar to C. trachomatis, M. tuberculosis is also able to survive in the absence of host LDs suggesting a bacterial mechanism for sequestering free fatty acids from other host sources [109].
Host LD accumulation may also be indicative of the virulence potential of various Mycobacterium strains. For instance, it is speculated that the formation of LDs is detrimental to less pathogenic strains of M. tuberculosis during infection likely due to the increased availability of signaling precursors able to be released from host LDs to produce a bactericidal response [110]. Conversely, infection with the highly pathogenic M. tuberculosis strain, H37Rv, supports that increased host LD formation is beneficial for bacterial growth and persistence, in that, the signaling molecules from LDs assist in producing the anti-inflammatory response required for the production of necrotic tissue and granuloma formation during severe mammalian infection [111].
Comparably, Brucella abortus, a facultative, intracellular bacteria that resides within the endoplasmic reticulum-associated vacuoles, increases host PPARɣ expression during infection. However, as observed for M. tuberculosis, this increase potentially modulates host triacylglycerol production and subsequent accumulation in LDs [112]. Another facultative intracellular pathogen that is thought to associate with host LDs is Salmonella typhimurium. Unlike, the pathogens described above, studies have suggested that the association of S. typhimurium with host LDs is based on the defined functionality of T3SS effector proteins, such as SseJ and SseL. As mentioned previously, SseJ not only has acyltransferase activity but also possesses cholesterol esterase activity similar to the C. trachomatis enzyme used to increase host LD production described above [65,66,67,68]. Infection of host cells with a C. trachomatis sseJ mutant results in a decrease in PCV production and in a decrease of LD accumulation. Together, these data support the use of SseJ as a cholesterol esterase for accumulation of stored cholesterol in host LDs during infection [67,68]. On the other hand, SseL contains deubiquitnase activity and sseL mutants stimulate an overaccumulation of host LDs (Figure 1). This strongly suggests that SseL and SseJ play a role in the maintenance of lipid homeostasis during infection, for direct bacterial use, and likely play roles in the modulation of the host response as described for other pathogens mentioned [113,114].
2.3. Fatty Acid β-Oxidation for Energy Production
The ability of bacterial pathogens to generate energy is required for survival and replication during infection of a mammalian host. There are a well-known and characterized range of biosynthesis pathways, both host and bacterial, that have the potential to be employed for energy production. A wide range of bacteria utilize glucose, or a sugar carbon source derivative, as a quick energy source. Glucose is most efficiently used through glycolysis followed by cellular respiration or other aerobic processes [115]. However, glucose and other sugars are not the only carbon sources available and utilized for energy production. Lipids as a carbon source provide more energy per molecule and can be processed through fatty acid β-oxidation (FAO), a pathway found in both microbes and eukaryotes, to produce acetyl-CoA for use in cellular respiration [115,116]. Generally, extracellular bacteria depend less on host processes and encode for the energy pathways necessary for efficient survival, growth, and disease progression. In contrast, intracellular bacteria rely more heavily on nutrients provided by host biosynthesis processes for efficient infection and growth.
As mentioned previously, facultative intracellular bacteria typically have functional pathways that allow these organisms to survive and replicate in both extracellular and intracellular environments, whereas obligate intracellular pathogens rely heavily on host metabolites during infection [61,62]. For example, M. tuberculosis employs multiple mechanisms to sequester host lipids as a carbon source. This facultative intracellular bacteria encodes phospholipases, such as phospholipase C, to breakdown host lipids before employment of bacterial mammalian cell entry (Mce) protein transporter complexes and homologs of flavin adenine dinucleotide (Fad) transporter protein to import cholesterol and fatty acids into the bacterial cell [117,118,119]. Cholesterol and fatty acids are the primary carbon source used for M. tuberculosis’s production of energy, making host lipid production important for sustainable and efficient infection [119,120,121,122]. Interestingly, M. tuberculosis also encodes for 200 distinct enzymes used for lipid metabolism that are involved in shuttling and processing degraded fatty acids during in vivo infection [107,123,124,125]. There are two main pathways utilized by M. tuberculosis for bacterial lipid catabolism, i.e., FAO and the glycoxylate shunt, with the latter being the dominant pathway. Interestingly, the M. tuberculosis genome has a large amount of redundancy regarding predicted enzymes within the FAO pathway to predominantly catabolize lipids into acetyl-CoA, which is then shuttled to the tricarboxylic acid cycle (TCA) indicating a potential significance of this secondary pathway during infection [126,127] (Figure 1). The utilization of various lipids as an energy source and the redundancy of lipid metabolic pathways allow M. tuberculosis to have enhanced plasticity and adaptability and to persist during long-term mammalian infection [128]. It is important to note that because of the plasticity of M. tuberculosis metabolism, cholesterol is not required for active M. tuberculosis infection. Nevertheless, the use of cholesterol as a major carbon source is necessary for increased persistence during chronic infections [129]. Although M. tuberculosis is able to self-process exogenous cholesterol and lipids as a carbon source through bacterial FAO, a recent study indicates a requirement of active host FAO during infection. Host FAO is important for regulation of antibacterial responses such as reactive oxygen species production and phagosome acidification. M. tuberculosis, therefore, is thought to stimulate FAO during infection to promote downregulation of these bactericidal responses [130].
Interestingly, the obligate intracellular bacteria, Chlamydia spp. lack a large array of metabolic and lipid biosynthesis pathway thereby relying on the host for pathway intermediates and scavenged lipid nutrients [131]. As described previously, C. pneumoniae replication requires the host lipid transport protein, FABP4, which regulates the release of free fatty acids from the host LDs, typically for host FAO and subsequent ATP production. These data suggest that C. pneumoniae may utilize host lipids as an intracellular energy source [132] (Figure 1). In addition, a facultative intracellular bacterium, Legionella pneumophila, infects alveolar macrophages and replicates within an ER-derived PCV and requires host FAO during infection [133,134,135]. The host FAO regulation has not been shown to be used as a method of energy production for the bacteria but instead is a mechanism for modulating bacterial growth states during the transition from a replicative stage upon host cell entry. This shift in bacterial stages is regulated by nutrient acquisition in the form of ER-derived vacuole fusion with the PCV [136]. Later in the replication stage, modulation of sphingolipid metabolism and autophagy initiates auto-phagolysosome fusion with the PCV providing lysosomal characteristics such as encoded lipolytic enzymes giving potential for breakdown and utilization of other nutrients such as lipids [133,137,138]. Once the PCV is depleted of the nutrients required for active replication, L. pneumophila shifts to a “transmission phenotype,” characterized by bacterial flagella activation and a downregulation in bacterial metabolism. During the life stages of intracellular infection, L. pneumophila monitors depletion of fatty acid levels, including host malonyl-CoA and intermediates of FAO-derived acyl-chain breakdown, to initiate the stringent response triggered by ppGpp synthetases and SPoT, which regulates the phenotypic shifts required during the L. pneumophila lifecycle [134,135,139,140,141,142] (Figure 1).
3. Intracellular Bacteria That Replicate within the Cytosol
In contrast to the pathogenic bacteria described above, there is a smaller group of intracellular bacteria that does not actively modulate trafficking of an intracellular vesicular compartment but rather employs mechanisms to escape a membranous vacuole after entry into the host cell to survive and reproduce within the host cytosol. To escape the vacuole, some cytosolic pathogens, including Listeria monocytogenes and various pathogenic Rickettsia species (spp.), employ bacterial phospholipases and toxins to breakdown the vacuole membrane for exit into the cytoplasm [143,144,145,146,147,148,149,150,151]. Similar to vacuolar pathogens, there is large genomic diversity within the group of cytosolic intracellular bacteria comprised of both facultative and obligate intracellular bacteria [61,62].
3.1. Host Lipids Provide Structural Integrity to Bacterial Membranes
The phospholipid and lipid membrane spp. that compose bacterial membranes are similar for most bacterial species as described previously. A distinct difference when discussing membranes associated with vacuolar pathogens and cytosolic pathogens is the presence of a PCV during vacuolar bacterial infection. As discussed previously, various host lipids are recruited to the PCV membrane to provide a variety of functions for the vacuolar bacteria. Cytosolic bacteria, however, do not remain within a vacuole; thus, the incorporation of host lipids into pathogen-associated membranes during replication is limited to the bacterial membrane. Most studies discussing the incorporation of host lipids into the cytosolic bacterial membranes focused on elucidating bacterial membrane compositions and speculated that various lipid classes originated from the infected host cell.
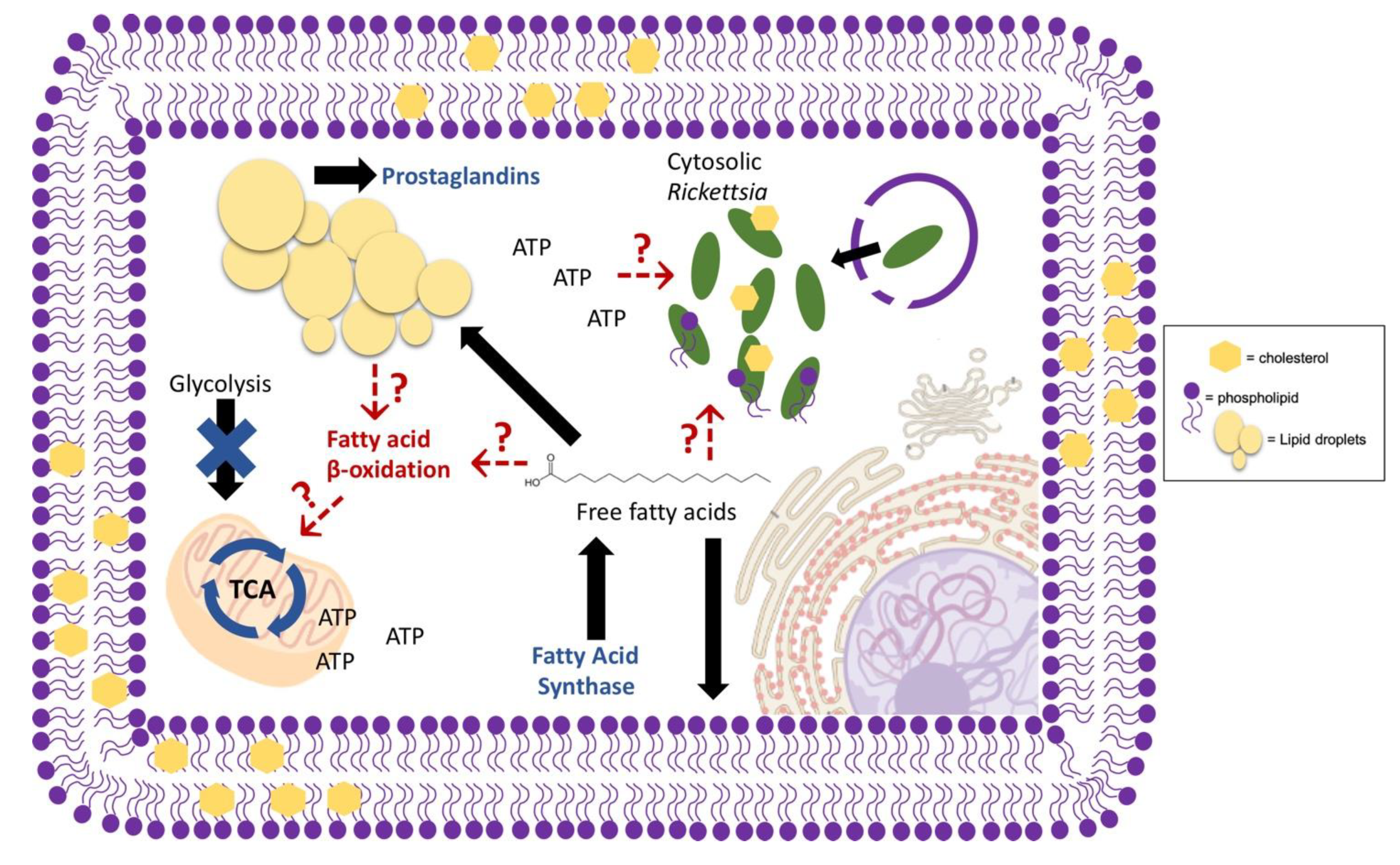
Genomic analysis of various Rickettsia spp. and another related bacterial species, Orientia tsutsugamushi, indicates gaps within the biosynthetic pathways necessary to construct the bacterial cell wall and membrane structure. This suggests a requirement of host lipid metabolites for membrane production during intracellular replication and growth [152,153,154]. A more recent genomic study denoted multiple lipid anabolic pathways that are missing or incomplete, within various rickettsial genomes, implying a large reliance on the host to provide the enzymes and intermediates necessary for full functional lipid biosynthesis pathways. Among the lipid metabolic pathways lacking or incomplete in the described Rickettsia spp. are pathways involved in lipopolysaccharide production and fatty acid and PC synthesis, supporting the implication that there is a heavy rickettsial reliance on host enzymes and metabolic intermediates for lipid products required for adequate building of the bacterial membrane [155]. Interestingly, previous experiments investigating the phospholipid composition of Rickettsia prowazekii in chicken embryo yolk sacs indicated the presence of PC and cardiolipins, later found to compose 5.1% and 2.1% of the membrane lipid composition, respectively [156] (Figure 2). Gram-negative bacterial membranes are predominantly composed of saturated and branched fatty acid chains, with limited sterol structures [24,25]. PC is not a normal component of the canonical Gram-negative inner or outer membrane, suggesting that the PC is likely sequestered from the host [157]. Studies have also shown that Rickettsia rickettsii membranes contain a moderate amount of unsaturated lipids and that R. prowazekii membranes contain cholesterol, in further support that membrane components may be actively acquired from the infected host cell [156,158]. Recently, a proteomic study defining host alterations during Rickettsia conorii infection of macrophages showed that inhibition of host fatty acid synthase (FASN), an enzyme required for lipid biosynthesis, is detrimental for bacterial replication and survival. A major role in the host cell and possible requirement of FASN for R. conorii growth is the production of phospholipids for membrane production, though the function of host FASN-produced lipids is not defined [159] (Figure 2).
3.2. Fatty Acid β-Oxidation for Energy Production
As described for intracellular pathogens that reside inside a vacuole, energy metabolism is an essential factor involved in the maintenance and sustainability of active infection. Due to the genomic decay and increased reliance on host processes and the importance of energy for efficient intracellular pathogen infection, much research has been done to elucidate the host energy pathways required during infection. Although there is an increased reliance on the host metabolism during intracellular infection, some pathogens still contain energy pathways necessary for self-sustainable energy production. One of the major energy-producing pathways is FAO, which catabolizes lipids, either acquired from exogenous lipids, de novo lipids, or lipid storages, for acetyl-CoA generation and use in host or bacterial OXPHOS. It is important to note that when comparing energy metabolism between vacuolar and cytosolic intracellular pathogens, the nutrients within the cellular compartments are vastly different. For example, vacuolar bacteria have adapted to utilize a diverse range of metabolites that are recruited and associated with PCVs during infection. In contrast, cytosolic bacteria do not employ this strategy and are limited to using available metabolites within the cytosol for energy production.
A study of host metabolites present during L. monocytogenes infection of Drosophila melanogaster, for example, showed an overall downregulation in energy metabolites, such as triglycerides, glycogen storages, and other intermediates required for the major energy pathways, FAO and glycolysis. This downregulated shift in metabolism is speculated to be an antibacterial response to prevent bacterial use of energy stores or to be a bacterial mechanism employed to block antibacterial responses, such as use of major energy metabolites for regulation of reactive oxygen species used by the host cell as an antibacterial mechanism, as seen with M. tuberculosis [130,160].
Similar to Mycobacterium spp. variation in virulent potential, Rickettsia spp. also vary in their capacity to cause disease in mammals. A recent study demonstrated that a recognized human pathogen, R. conorii, is able to invade into and efficiently replicate within both non-phagocytic and phagocytic mammalian cells. In contrast, Rickettsia montanensis, a species that is not recognized as pathogenic to mammals, can proliferate in epithelial cells but is rapidly killed intracellularly in macrophage-like THP-1 cells. This study suggested that growth within professional phagocytic cells may be a key determinant to distinguish pathogenic Rickettsia spp. from non-pathogenic species [161]. In addition, a proteomic study revealed the differential host protein changes that occur when macrophage-like THP-1 cells are infected with either R. conorii or R. montanensis. In the R. conorii infected cells, host glycolysis and other sugar metabolic processes were downregulated while proteins involved in OXPHOS were upregulated. In contrast, infection with R. montanensis did not result in these changes. The observed overall increase in OXPHOS and decrease in glycolysis during R. conorii infection of macrophages, strongly suggested that non-sugar carbon sources, such as lipids, are likely important for energy production. This proteomic analysis also indicated a shift in host lipid metabolic pathways and a requirement of the anabolic FASN, discussed previously, for de novo production of host lipids (Figure 2). These metabolic shifts are also not observed during R. montanensis infection of macrophages. Taken together, these results suggest that the ability to stimulate specific host metabolic pathways within macrophages and possibly other target cells likely contributes to the virulence potential of R. conorii and related pathogenic spotted fever group Rickettsia species [159]. Similar to R. conorii, the closely related cytosolic bacteria O. tsutsugamushi also shifts the host cell metabolic response to decrease host glycolysis during in vivo infection of a mouse model, suggesting a likely use of lipids as carbon sources. However, the O. tsutsugamushi preferred carbon source for energy production during infection is still unclear [162]. Although the use of the newly synthesized lipid species is yet to be defined, it is possible that these fatty acids could be used as a carbon source for ATP production during rickettsial macrophage infection [159]. In support of this hypothesis, a previous study of another human pathogenic species, Rickettsia typhi, identified a gene, tlc1, present in rickettsial genomes that encodes active transporter that is sufficient for ATP transport [163]. Similarly, the O. tsutsugamushi genome also contains genes predicted to encode redundant ATP transport systems and lacks certain enzymes necessary for ATP production [164]. Together, these data strongly suggest that this class of obligate, intracellular bacteria acquire host-derived ATP to meet metabolic requirements.
Another pathogen that manipulates the host during infection for regulation of host energy production is the facultative intracellular bacteria, Francisella tularensis. Similar to Rickettsia, studies have suggested that F. tularensis initiates utilization and acquisition of alternative carbon sources, such as glutamate and fatty acids, during infection. These sequestered metabolites are used for mitochondrial respiration and host ATP production during early stages of infection. Although the host metabolic profile is known, the exact mechanism and advantage to the host and/or bacteria has yet to be defined [165].
3.3. Host Lipid-Droplet Modulation
Unlike some of the vacuolar pathogens, the foam cell phenotype is not defined among cytosolic pathogens. O. tsutsugamushi stimulates an increase in triacylglycerol-rich LDs in murine fibroblasts suggesting that these LDs may serve as a nutrient source for energy production [166]. Conversely, infection of Drosophila melanogaster with L. monocytogenes, results in a decrease in triacylglycerols and subsequent LD structures during infection in this model system, suggesting lipids within host LDs are likely being utilized during infection for functions such as host energy production discussed previously [160]. In contrast, rickettsial species have not been associated with the utilization of host LDs for energy production; however, these bacteria do encode phospholipases within the genome. Phospholipases are enzymes that have been linked with other bacterial species described previously for generation of LD-derived lipid immune regulators. Specifically, the phospholipase A2 enzymes have been shown to be present and functional in R. typhi and R. prowazekii suggesting the ability to actively release fatty acids from host LDs for functions yet to be elucidated [150,151,167,168,169]. Among the R. prowazekii and R. typhi phospholipase enzymes are RP534 and RT0522, respectively, that are proteins homologous to Pseudomonas aeruginosa, ExoU, which hydrolyzes fatty acids from LD for the production of prostaglandins. Some studies have demonstrated that Rickettsia spp. stimulate an increase in prostaglandin production during in vivo and in vitro infection models indicating that these mechanisms discussed could be used for the production of lipid immune modulators derived from host LDs as described for C. pneumoniae, C. burnetii, and M. leprae [170,171,172] (Figure 2). Similarly, L. monocytogenes and Shigella dysenteriae also increase prostaglandin levels during in vivo infection of a mouse model for immune suppression, suggesting LD release of lipid modulators, however, the mechanism has yet to be defined [173,174]. In addition, F. tularensis, a facultative intracellular bacteria that resides within the late phagosome prior to release into the cytosol, modulates host TLR2 signaling and the transcription factor PPARα, which as described for C. pneumoniae can have implications on LD production during macrophage infection [175,176,177,178]. Moreover, similar to other cytosolic bacterial infections, F. tularensis macrophage infection also increases host prostaglandin production via host phospholipases [178,179,180]; although, bacterial factors directly involved in prostaglandin release and other LD functions during F. tularensis infection are still unknown.
4. Conclusions
Both obligate and facultative intracellular bacteria have developed several strategies to regulate and manipulate host processes for efficient intracellular growth. Among these is the modulation of host lipid metabolism that has been demonstrated not only to be required for the maintenance of host cell homeostasis but also to be implemented in various intracellular infections for increased persistence and disease progression. The key lipid pathways employed by intracellular bacterial pathogens for proliferation and sustainable infection include, but are not limited to, membrane fatty acid biosynthesis, LD regulation, and lipid catabolism through FAO for energy production. The current knowledge on host lipid metabolism utilization by various species of intracellular bacteria is fairly limited, in that, much of the previous research addresses the recruitment and sequestering of lipids to the PCV for various functions during infection with bacteria that reside within a vacuole. As demonstrated here, there are large experimental gaps, specifically in facultative and cytosolic intracellular bacteria, where host lipids have the potential to play specific roles in infection; however, the functions and mechanisms involved in this use of host lipid metabolic processes are not well defined compared to some of their vacuolar counterparts. Therefore, it is necessary to further investigate the importance of host lipids for intracellular bacterial infection to better understand the host:pathogen relationship required to provide potential targets for increases in therapeutics against these human disease causing agents.
Author Contributions
Conceptualization, J.J.M.; writing—original draft preparation, P.E.A. and J.J.M.; writing—review and editing, P.E.A. and J.J.M.; funding acquisition, J.J.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by an award (AI072606) from the National Institutes of Health, National Institute of Allergy and Infectious Diseases to J.J.M. This work was also funded in part by a Graduate Student stipend provided to P.E.A. by the Department of Pathobiological Sciences at the LSU School of Veterinary Medicine.
Acknowledgments
The authors would like to thank current members of the Martinez Laboratory at the LSU School of Veterinary Medicine, Department of Pathobiological Sciences for helpful suggestions regarding this review.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Bell, R.M. Protein kinase C activation by diacylglycerol second messengers. Cell 1986, 45, 631–632. [Google Scholar] [CrossRef]
- Benveniste, J. Platelet-activating factor, a new mediator of anaphylaxis and immune complex deposition from rabbit and human basophils. Nature 1974, 249, 581–582. [Google Scholar] [CrossRef]
- Berridge, M.; Irvine, R. Inositol phosphates and cell signalling. Nature 1989, 341, 197–205. [Google Scholar] [CrossRef]
- Hannun, Y.; Bell, R. Functions of sphingolipids and sphingolipid breakdown products in cellular regulation. Science 1989, 243, 500–507. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef] [Green Version]
- Haucke, V.; Di Paolo, G. Lipids and lipid modifications in the regulation of membrane traffic. Curr. Opin. Cell Biol. 2007, 19, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Moonlennar, W.; Kruijer, W.; Tilly, B.; Verlaan, L.; Bierman, A.; de Laat, S. Growth factor-like action of phosphatidic acid. Nature 1986, 323, 171–173. [Google Scholar] [CrossRef]
- Nishizuka, Y. Studies and perspectives of protein kinase C. Science 1986, 233, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Vaz, W.L. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 269–295. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, V.; Aitken, J.; Cleves, A.; Dowhan, W. An essential role for a phospholipid transfer protein in yeast Golgi function. Nature 1990, 347, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer-Janssen, Y.P.; van Galen, J.; Batenburg, J.J.; Helms, J.B. Lipids in host-pathogen interactions: Pathogens exploit the complexity of the host cell lipidome. Prog. Lipid Res. 2010, 49, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, A.; Gill, R.K.; Hodges, K.; Ramaswamy, K.; Hecht, G.; Dudeja, P.K. Enteropathogenic Escherichia coli inhibits butyrate uptake in Caco-2 cells by altering the apical membrane MCT1 level. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G30–G35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agaisse, H.; Burrack, L.; Philips, J.; Rubin, E.; Perrimon, N.; Higgins, D. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science 2005, 309, 1248–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapadia, S.; Chisari, F. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philips, J.; Rubin, E.; Perrimon, N. Drosophila RNAi Screen Reveals CD36 Family Member Required for Mycobacterial Infection. Science 2005, 309, 1251–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, A.; Pezacki, J.; Wodicka, L.; Brideau, A.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.; Schultz, P.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Toledo, A.; Benach, J.L. Hijacking and Use of Host Lipids by Intracellular Pathogens. Microbiol. Spectr. 2015, 3, 637–666. [Google Scholar] [CrossRef] [Green Version]
- Roingeard, P.; Melo, R.C. Lipid droplet hijacking by intracellular pathogens. Cell. Microbiol. 2017, 19, e12688. [Google Scholar] [CrossRef]
- Bozza, P.T.; Bakker-Abreu, I.; Navarro-Xavier, R.A.; Bandeira-Melo, C. Lipid body function in eicosanoid synthesis: An update. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 205–213. [Google Scholar] [CrossRef]
- Pereira-Dutra, F.S.; Teixeira, L.; de Souza Costa, M.F.; Bozza, P.T. Fat, fight, and beyond: The multiple roles of lipid droplets in infections and inflammation. J. Leukoc. Biol. 2019, 106, 563–580. [Google Scholar] [CrossRef]
- Kumar, Y.; Valdivia, R.H. Leading a sheltered life: Intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 2009, 5, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, E.D.; Smith, J.A.; Ficht, T.A.; Samuel, J.E.; de Figueiredo, P. Space: A Final Frontier for Vacuolar Pathogens. Traffic 2016, 17, 461–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohlenkamp, C.; Deiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. [Google Scholar] [CrossRef] [Green Version]
- Renner, L.; DB, W. Cardiolipin microdomains localize to negatively curved regions of Escherchia coli membranes. Proc. Natl. Acad. Sci. USA 2011, 108, 6264–6269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ooij, C.; Apodaca, G.; Engel, J. Characterization of the Chlamydia trachomatis Vacuole and Its Interaction with the Host Endocytic Pathway in HeLa Cells. Infect. Immun. 1997, 65, 758–766. [Google Scholar] [CrossRef] [Green Version]
- Carabeo, R.A.; Mead, D.J.; Hackstadt, T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc. Natl. Acad. Sci. USA 2003, 100, 6771–6776. [Google Scholar] [CrossRef] [Green Version]
- Hackstadt, T.; Rockey, D.D.; Heinzen, R.A.; Scidmore, M.A. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 1996, 15, 964–977. [Google Scholar] [CrossRef]
- Mital, J.; Lutter, E.I.; Barger, A.C.; Dooley, C.A.; Hackstadt, T. Chlamydia trachomatis inclusion membrane protein CT850 interacts with the dynein light chain DYNLT1 (Tctex1). Biochem. Biophys. Res. Commun. 2015, 462, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Mital, J.; Miller, N.J.; Fischer, E.R.; Hackstadt, T. Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cell. Microbiol. 2010, 12, 1235–1249. [Google Scholar] [CrossRef] [Green Version]
- Richards, T.S.; Knowlton, A.E.; Grieshaber, S.S. Chlamydia trachomatis homotypic inclusion fusion is promoted by host microtubule trafficking. BMC Microbiol. 2013, 13, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilk, S.D.; Cockrell, D.C.; Luterbach, C.; Hansen, B.; Knodler, L.A.; Ibarra, J.A.; Steele-Mortimer, O.; Heinzen, R.A. Bacterial colonization of host cells in the absence of cholesterol. PLoS Pathog. 2013, 9, e1003107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, Y.; Cocchiaro, J.; Valdivia, R.H. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr. Biol. 2006, 16, 1646–1651. [Google Scholar] [CrossRef] [Green Version]
- Saka, H.A.; Thompson, J.W.; Chen, Y.S.; Dubois, L.G.; Haas, J.T.; Moseley, A.; Valdivia, R.H. Chlamydia trachomatis Infection Leads to Defined Alterations to the Lipid Droplet Proteome in Epithelial Cells. PLoS ONE 2015, 10, e0124630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Cherian, P.T.; Frank, M.W.; Rock, C.O. Chlamydia trachomatis Relies on Autonomous Phospholipid Synthesis for Membrane Biogenesis. J. Biol. Chem. 2015, 290, 18874–18888. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Dodson, V.J.; Frank, M.W.; Rock, C.O. Chlamydia trachomatis Scavenges Host Fatty Acids for Phospholipid Synthesis via an Acyl-Acyl Carrier Protein Synthetase. J. Biol. Chem. 2015, 290, 22163–22173. [Google Scholar] [CrossRef] [Green Version]
- Boncompain, G.; Muller, C.; Meas-Yedid, V.; Schmitt-Kopplin, P.; Lazarow, P.B.; Subtil, A. The intracellular bacteria Chlamydia hijack peroxisomes and utilize their enzymatic capacity to produce bacteria-specific phospholipids. PLoS ONE 2014, 9, e86196. [Google Scholar] [CrossRef] [Green Version]
- Soupene, E.; Rothschild, J.; Kuypers, F.A.; Dean, D. Eukaryotic protein recruitment into the Chlamydia inclusion: Implications for survival and growth. PLoS ONE 2012, 7, e36843. [Google Scholar] [CrossRef]
- Soupene, E.; Wang, D.; Kuypers, F.A. Remodeling of host phosphatidylcholine by Chlamydia acyltransferase is regulated by acyl-CoA binding protein ACBD6 associated with lipid droplets. Microbiologyopen 2015, 4, 235–251. [Google Scholar] [CrossRef]
- Cocchiaro, J.L.; Kumar, Y.; Fischer, E.R.; Hackstadt, T.; Valdivia, R.H. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. USA 2008, 105, 9379–9384. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.V.; Abdelrahman, Y.M.; Peters, J.; Naher, N.; Belland, R.J. Chlamydia trachomatis utilizes the mammalian CLA1 lipid transporter to acquire host phosphatidylcholine essential for growth. Cell. Microbiol. 2016, 18, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.; Onguri, V.; Nishimoto, S.K.; Marion, T.N.; Byrne, G.I. The Chlamydia trachomatis CT149 protein exhibits esterase activity in vitro and catalyzes cholesteryl ester hydrolysis when expressed in HeLa cells. Microbes Infect. 2012, 14, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Soupene, E.; Kuypers, F.A. Phosphatidylserine decarboxylase CT699, lysophospholipid acyltransferase CT775, and acyl-ACP synthase CT776 provide membrane lipid diversity to Chlamydia trachomatis. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Abdelrahman, Y.M.; Robertson, R.M.; Cox, J.V.; Belland, R.J.; White, S.W.; Rock, C.O. Type II fatty acid synthesis is essential for the replication of Chlamydia trachomatis. J. Biol. Chem. 2014, 289, 22365–22376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truchan, H.K.; Cockburn, C.L.; Hebert, K.S.; Magunda, F.; Noh, S.M.; Carlyon, J.A. The Pathogen-Occupied Vacuoles of Anaplasma phagocytophilum and Anaplasma marginale Interact with the Endoplasmic Reticulum. Front. Cell. Infect. Microbiol. 2016, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikihisa, Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: Subversive manipulators of host cells. Nat. Rev. Microbiol. 2010, 8, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Mulye, M.; Clemente, T.M.; Justis, A.V.; Gilk, S.D. Manipulation of Host Cholesterol by Obligate Intracellular Bacteria. Front. Cell. Infect. Microbiol. 2017, 7, 165. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Rikihisa, Y. Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid A biosynthesis and incorporate cholesterol for their survival. Infect. Immun. 2003, 71, 5324–5331. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Grandinetti, G.; Hartnell, L.M.; Bliss, D.; Subramaniam, S.; Rikihisa, Y. Host membrane lipids are trafficked to membranes of intravacuolar bacterium Ehrlichia chaffeensis. Proc. Natl. Acad. Sci. USA 2020, 117, 8032–8043. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Q.; Lin, M.; Rikihisa, Y. Cholesterol-dependent anaplasma phagocytophilum exploits the low-density lipoprotein uptake pathway. PLoS Pathog. 2009, 5, e1000329. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Q.; Rikihisa, Y. Subversion of NPC1 pathway of cholesterol transport by Anaplasma phagocytophilum. Cell. Microbiol. 2012, 14, 560–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Q.; Lin, M.; Huang, W.; Rikihisa, Y. Infection by Anaplasma phagocytophilum Requires Recruitment of Low-Density Lipoprotein Cholesterol by Flotillins. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Rikihisa, Y. Obligatory intracellular parasitism by Ehrlichia chaffeensis and Anaplasma phagocytophilum involves caveolae and glycosylphosphatidylinositol-anchored proteins. Cell. Microbiol. 2003, 5, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, E.; Colombo, M.I.; Heinzen, R.A. The Coxiella burnetii parasiophorous vacuole. Adv. Exp. Med. Biol. 2012, 984, 141–169. [Google Scholar]
- Howe, D.; Heinzen, R.A. Coxiella burnetii inhabits a cholesterol-rich vacuole and influences cellular cholesterol metabolism. Cell. Microbiol. 2006, 8, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Mulye, M.; Samanta, D.; Winfree, S.; Heinzen, R.A.; Gilk, S.D. Elevated Cholesterol in the Coxiella burnetii Intracellular Niche Is Bacteriolytic. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Dragan, A.L.; Kurten, R.C.; Voth, D.E. Characterizing Early Stages of Human Alveolar Infection by the Q Fever Agent, Coxiella burnetii. Infect. Immun. 2019, 87, e00028-19. [Google Scholar] [CrossRef] [Green Version]
- Newton, H.J.; McDonough, J.A.; Roy, C.R. Effector protein translocation by the Coxiella burnetii Dot/Icm type IV secretion system requires endocytic maturation of the pathogen-occupied vacuole. PLoS ONE 2013, 8, e54566. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Clemente, T.M.; Schuler, B.E.; Gilk, S.D. Coxiella burnetii Type 4B Secretion System-dependent manipulation of endolysosomal maturation is required for bacterial growth. PLoS Pathog. 2019, 15, e1007855. [Google Scholar] [CrossRef] [Green Version]
- Gilk, S.D.; Beare, P.A.; Heinzen, R.A. Coxiella burnetii expresses a functional Delta24 sterol reductase. J. Bacteriol. 2010, 192, 6154–6159. [Google Scholar] [CrossRef] [Green Version]
- Merhej, V.; Royer-Carenzi, M.; Pontarotti, P.; Raoult, D. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 2009, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casadevall, A. Evolution of intracellular pathogens. Annu. Rev. Microbiol. 2008, 62, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holden, D.W. Trafficking of the Salmonella Vacuole in Macrophages. Traffic 2002, 3, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Catron, D.M.; Sylvester, M.D.; Lange, Y.; Kadekoppala, M.; Jones, B.D.; Monack, D.M.; Falkow, S.; Haldar, K. The Salmonella-containing vacuole is a major site of intracellular cholesterol accumulation and recruits the GPI-anchored protein CD55. Cell. Microbiol. 2002, 4, 315–328. [Google Scholar] [CrossRef] [PubMed]
- MacIntyre, S.; Buckley, J.T. Presence of glycerophospholipid: Cholesterol acyltransferase and phospholipase in culture supernatant of Aeromonas hydrophila. J. Bacteriol. 1978, 135, 402–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, J.T. Substrate specificity of bacterial glycerophospholipid:cholesterol acyltransferase. Biochemistry 1982, 21, 6699–6703. [Google Scholar] [CrossRef]
- Nawabi, P.; Catron, D.M.; Haldar, K. Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol. Microbiol. 2008, 68, 173–185. [Google Scholar] [CrossRef]
- Ruiz-Albert, J.; Yu, X.J.; Beuzon, C.R.; Blakey, A.N.; Galyov, E.E.; Holden, D.W. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol. Microbiol. 2002, 44, 645–661. [Google Scholar] [CrossRef] [Green Version]
- Libbing, C.L.; McDevitt, A.R.; Azcueta, R.P.; Ahila, A.; Mulye, M. Lipid Droplets: A Significant but Understudied Contributor of Host(-)Bacterial Interactions. Cells 2019, 8, 354. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Ley, K.; Miller, Y.I.; Hedrick, C.C. Monocyte and macrophage dynamics during atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1506–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Becker, L.; Pritchard, D.K.; Gharib, S.A.; Wijsman, E.; Bammler, T.K.; Beyer, R.P.; Vaisar, T.; Oram, J.F.; Heinecke, J.W. Cholesterol Accumulation Regulates the Expression of Macrophage Proteins Implicated in Proteolysis and Complement. Activation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2910–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashkin, P.; Dragulev, B.; Ley, K. Macrophage differentiation to foam cells. Curr. Pharm. Des. 2005, 11, 3061–3072. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.; He, P.; Cheng, B.; Liu, W.; Wang, Y.F.; Wan, J.J. Chlamydia pneumoniae induces macrophage-derived foam cell formation via PPAR alpha and PPAR gamma-dependent pathway. Cell Biol. Int. 2009, 33, 301–308. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Mei, C.; Cheng, B.; Liu, W.; Wang, Y.; Wan, J. Chlamydia pneumoniae induces macrophage-derived foam cell formation by up-regulating acyl-coenzyme A: Cholesterol acyltransferase 1. Microbes Infect. 2009, 11, 157–163. [Google Scholar] [CrossRef]
- Cheng, B.; Wu, X.; Sun, S.; Wu, Q.; Mei, C.; Xu, Q.; Wu, J.; He, P. MAPK-PPARalpha/gamma signal transduction pathways are involved in Chlamydia pneumoniae induced macrophage-derived foam cell formation. Microb. Pathog. 2014, 69–70, 1–8. [Google Scholar] [CrossRef]
- Barbier, O.; Torra, I.P.; Duguay, Y.; Blanquart, C.; Fruchart, J.C.; Glineur, C.; Staels, B. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Itoh, R.; Murakami, I.; Chou, B.; Ishii, K.; Soejima, T.; Suzuki, T.; Hiromatsu, K. Chlamydia pneumoniae harness host NLRP3 inflammasome-mediated caspase-1 activation for optimal intracellular growth in murine macrophages. Biochem. Biophys. Res. Commun. 2014, 452, 689–694. [Google Scholar] [CrossRef]
- Cao, F.; Castrillo, A.; Tontonoz, P.; Re, F.; Byrne, G.I. Chlamydia pneumoniae—Induced macrophage foam cell formation is mediated by Toll-like receptor 2. Infect. Immun. 2007, 75, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, E.Y.; Lad, S.P.; Mikolon, D.P.; Iacobelli-Martinez, M.; Li, E. Activation of lipid metabolism contributes to interleukin-8 production during Chlamydia trachomatis infection of cervical epithelial cells. Infect. Immun. 2005, 73, 4017–4024. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Recuero-Checa, M.A.; Fan, F.Y.; Dean, D. Chlamydia trachomatis regulates growth and development in response to host cell fatty acid availability in the absence of lipid droplets. Cell. Microbiol. 2018, 20, e12801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulye, M.; Zapata, B.; Gilk, S.D. Altering lipid droplet homeostasis affects Coxiella burnetii intracellular growth. PLoS ONE 2018, 13, e0192215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Q.; Robertson, S.J.; Howe, D.; Barrows, L.F.; Heinzen, R.A. Comparative DNA microarray analysis of host cell transcriptional responses to infection by Coxiella burnetii or Chlamydia trachomatis. Ann. N. Y. Acad. Sci. 2003, 990, 701–713. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.A.; Newton, H.J.; Klum, S.; Swiss, R.; Agaisse, H.; Roy, C.R. Host pathways important for Coxiella burnetii infection revealed by genome-wide RNA interference screening. mBio 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, S.; Ayoubi, P.; Shaw, E.I. Coxiella burnetii Nine Mile II proteins modulate gene expression of monocytic host cells during infection. BMC Microbiol. 2010, 10, 244. [Google Scholar] [CrossRef] [Green Version]
- Stead, C.M.; Cockrell, D.C.; Beare, P.A.; Miller, H.E.; Heinzen, R.A. A Coxiella burnetii phospholipase A homolog pldA is required for optimal growth in macrophages and developmental form lipid remodeling. BMC Microbiol. 2018, 18, 1–9. [Google Scholar] [CrossRef]
- Koster, F.T.; Williams, J.C.; Goodwin, J.S. Cellular immunity in Q fever: Modulation of responsiveness by a suppressor T cell-monocyte circuit. J. Immunol. 1985, 135, 1067–1072. [Google Scholar]
- Shannon, J.G.; Heinzen, R.A. Adaptive immunity to the obligate intracellular pathogen Coxiella burnetii. Immunol. Res. 2009, 43, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Izzo, A.A.; Marmion, B.P. Variation in interferon-gamma responses to Coxiella burnetii antigens with lymphocytes from vaccinated or naturally infected subjects. Clin. Exp. Immunol. 1993, 94, 507–515. [Google Scholar] [CrossRef]
- Manzano-Roman, R.; Almazán, C.; Naranjo, V.; Blouin, E.F.; Kocan, K.M.; De La Fuente, J. Expression of perilipin in human promyelocytic cells in response to Anaplasma phagocytophilum infection results in modified lipid metabolism. Med Microbiol. 2008, 57, 159–163. [Google Scholar] [CrossRef]
- De la Fuente, J.; Ayoubi, P.; Blouin, E.F.; Almazán, C.; Naranjo, V.; Kocan, K.; Maurin, M. Gene expression profiling of human promyelocytic cells in response to infection with Anaplasma phagocytophilum. Cell. Microbiol. 2005, 7, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Tansey, J.; Sztalryd, C.; Hlavin, E.; Kimmel, A.; Londos, C. The central role of perilipin a in lipid metabolism and adipocyte lipolysis. IUBMB Life 2004, 56, 379–385. [Google Scholar] [CrossRef]
- Moore, H.P.; Silver, R.B.; Mottillo, E.P.; Bernlohr, D.A.; Granneman, J.G. Perilipin targets a novel pool of lipid droplets for lipolytic attack by hormone-sensitive lipase. J. Biol. Chem. 2005, 280, 43109–43120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, H.; Souza, S.C.; Zhang, H.H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Salamon, H.; Bruiners, N.; Lakehal, K.; Shi, L.; Ravi, J.; Yamaguchi, K.D.; Gennaro, M.L. Cutting edge: Vitamin D regulates lipid metabolism in Mycobacterium tuberculosis infection. J. Immunol. 2014, 193, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.; Sirakova, T.; Kolattukudy, P. An acyl-CoA synthetase in Mycobacterium tuberculosis involved in triacylglycerol accumulation during dormancy. PLoS ONE 2014, 9, e114877. [Google Scholar] [CrossRef]
- Daniel, J.; Kapoor, N.; Sirakova, T.; Sinha, R.; Kolattukudy, P. The perilipin-like PPE15 protein in Mycobacterium tuberculosis is required for triacylglycerol accumulation under dormancy-inducing conditions. Mol. Microbiol. 2016, 101, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Mattos, K.A.; D’Avila, H.; Rodrigues, L.S.; Oliveira, V.G.; Sarno, E.N.; Atella, G.C.; Pereira, G.M.; Bozza, P.T.; Pessolani, M.C. Lipid droplet formation in leprosy: Toll-like receptor-regulated organelles involved in eicosanoid formation and Mycobacterium leprae pathogenesis. J. Leukoc. Biol. 2010, 87, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Tanigawa, K.; Suzuki, K.; Nakamura, K.; Akama, T.; Kawashima, A.; Wu, H.; Hayashi, M.; Takahashi, S.; Ikuyama, S.; Ito, T.; et al. Expression of adipose diffrentiation-related protein (ADRP) and perilipin in macrophages infected with Mycobacterium leprae. FEMS Microbiol. Lett. 2008, 289, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Avila, H.; Melo, R.C.; Parreira, G.G.; Werneck-Barroso, E.; Castro-Faria-Neto, H.C.; Bozza, P.T. Mycobacterium bovis bacillus. Calmette-Guerin induces TLR2-mediated formation of lipid bodies: Intracellular domains for eicosanoid synthesis in vivo. J. Immunol. 2006, 176, 3087–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, J.; Maamar, H.; Deb, C.; Sirakova, T.D.; Kolattukudy, P.E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011, 7, e1002093. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.M.; Adams, K.L.; Zilisch, J.E.; Bretl, D.J.; Sato, H.; Anderson, D.M.; Zahrt, T.C. Rv2744c Is a PspA Ortholog That Regulates Lipid Droplet Homeostasis and Nonreplicating Persistence in Mycobacterium Tuberculosis. J. Bacteriol. 2016, 198, 1645–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elamin, A.A.; Stehr, M.; Spallek, R.; Rohde, M.; Singh, M. The Mycobacterium tuberculosis Ag85A is a novel diacylglycerol acyltransferase involved in lipid body formation. Mol. Microbiol. 2011, 81, 1577–1592. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffé, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Cruz, D.; Watson, A.D.; Miller, C.S.; Montoya, D.; Ochoa, M.T.; Sieling, P.A.; Gutierrez, M.A.; Navab, M.; Reddy, S.T.; Witztum, J.L.; et al. Host-derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J. Clin. Investig. 2008, 118, 2917–2928. [Google Scholar] [CrossRef] [Green Version]
- Knight, M.; Braverman, J.; Asfaha, K.; Gronert, K.; Stanley, S. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-gamma HIF-1alpha signaling and supports host defense. PLoS Pathog. 2018, 14, e1006874. [Google Scholar] [CrossRef]
- Dunn, P.L.; North, R.J. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect. Immun. 1995, 63, 3428–3437. [Google Scholar] [CrossRef] [Green Version]
- Jaisinghani, N.; Dawa, S.; Singh, K.; Nandy, A.; Menon, D.; Bhandari, P.D.; Khare, G.; Tyagi, A.; Gandotra, S. Necrosis Driven Triglyceride Synthesis Primes Macrophages for Inflammation During Mycobacterium tuberculosis Infection. Front. Immunol. 2018, 9, 1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, M.N.; Winter, M.G.; Spees, A.M.; den Hartigh, A.B.; Nguyen, K.; Roux, C.M.; Silva, T.M.; Atluri, V.L.; Kerrinnes, T.; Keestra, A.M.; et al. PPARgamma-mediated increase in glucose availability sustains chronic Brucella abortus infection in alternatively activated macrophages. Cell Host Microbe 2013, 14, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Arena, E.T.; Auweter, S.D.; Antunes, L.C.; Vogl, A.W.; Han, J.; Guttman, J.A.; Croxen, M.A.; Menendez, A.; Covey, S.D.; Borchers, C.H.; et al. The deubiquitinase activity of the Salmonella pathogenicity island 2 effector, SseL, prevents accumulation of cellular lipid droplets. Infect. Immun. 2011, 79, 4392–4400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunes, L.C.; Andersen, S.K.; Menendez, A.; Arena, E.T.; Han, J.; Ferreira, R.B.; Borchers, C.H.; Finlay, B.B. Metabolomics reveals phospholipids as important nutrient sources during Salmonella growth in bile in vitro and in vivo. J. Bacteriol. 2011, 193, 4719–4725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passalacqua, K.D.; Charbonneau, M.E.; O’Riordan, M.X.D. Bacterial Metabolism Shapes the Host-Pathogen Interface. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Oxidation of Glucose and Fatty Acids to CO2. In Molecular Cell Biology, 4th ed.; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Raynaud, C.; Guilhot, C.; Rauzier, J.; Bordat, Y.; Pelicic, V.; Manganelli, R.; Smith, I.; Gicquel, B.; Jackson, M. Phospholipases C are involved in the virulence of Mycobacterium tuberculosis. Mol. Microbiol. 2002, 45, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.; Stadthagen, G.; Gicquel, B. Long-Chain multiple methyl-branched fatty acid-containing lipids of Mycobacterium tuberculosis: Biosynthesis, transport, regulation and biological activities. Tuberculosis 2007, 87, 78–86. [Google Scholar] [CrossRef]
- Nazarova, E.V.; Nintague, C.R.; La, T.; Wilburn, K.M.; Sukumar, N.; Lee, W.; Caldwell, S.; Russell, D.G.; VanderVen, B.C. Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. ELife 2017, 6, e26969. [Google Scholar] [CrossRef]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 4376–4380. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.; Wang, C.; Besra, G.S. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2005, 18, 81–101. [Google Scholar] [CrossRef] [Green Version]
- Bloch, H.; Segal, W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J. Bacteriol. 1956, 72, 132. [Google Scholar] [PubMed]
- Lui, K.; Yu, J.; Russell, D.G. Pcka-deficient Mycobacterium bovis BCG shows attenuated virulence in mice and in macrophage. Microbiology 2003, 149, 1829–1835. [Google Scholar]
- Marrero, J.; Rhee, K.Y.; Schnappinger, D.; Pethe, K.; Ehrt, S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc. Natl. Acad. Sci. USA 2010, 107, 9819–9824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinney, J.D.; zu Bentrup, K.H.; Munoz-Elias, E.J.; Miczak, A.; Chen, B.; Chan, W.T.; Swenson, D.; Sacchettini, J.C.; Jacobs, W.R., Jr.; Russell, D.G. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000, 406, 735–738. [Google Scholar] [CrossRef]
- Williams, K.J.; Boshoff, H.I.; Krishnan, N.; Gonzales, J.; Schnappinger, D.; Robertson, B.D. The Mycobacterium tuberculosis beta-oxidation genes echA and fabB3 are dispensible for growth in vitro and in vivo. Tuberculosis 2011, 91, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, J.E.; Clar-Curtiss, J.E. Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences. Proc. Natl. Acad. Sci. USA 1999, 96, 11554–11559. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, L.P.; Fischer, S.M.; Marrero, J.; Nathan, C.; Ehrt, S.; Rhee, K.Y. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 2010, 17, 1122–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989–12994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, P.; He, L.; Zimmerman, M.; Yang, G.; Koster, S.; Ouimet, M.; Wang, H.; Moore, K.J.; Dartois, V.; Schilling, J.D.; et al. Inhibition of Fatty Acid Oxidation Promotes Macrophage Control. of Mycobacterium tuberculosis. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R.; Aravind, L.; Mitchell, W.; Olinger, L.; Tatusov, R.L.; Zhao, Q.; et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Walenna, N.F.; Kurihara, Y.; Chou, B.; Ishii, K.; Soejima, T.; Itoh, R.; Shimizu, A.; Ichinohe, T.; Hiromatsu, K. Chlamydia pneumoniae exploits adipocyte lipid chaperone FABP4 to facilitate fat mobilization and intracellular growth in murine adipocytes. Biochem. Biophys. Res. Commun. 2018, 495, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Sturgill-Koszycki, S.; Swanson, M.S. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J. Exp. Med. 2000, 192, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Trigui, H.; Dudyk, P.; Oh, J.; Hong, J.I.; Faucher, S.P. A regulatory feedback loop between RpoS and SpoT supports the survival of Legionella pneumophila in water. Appl. Environ. Microbiol. 2015, 81, 918–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalebroux, Z.D.; Edwards, R.L.; Swanson, M.S. SpoT governs Legionella pneumophila differentiation in host macrophages. Mol. Microbiol. 2009, 71, 640–658. [Google Scholar] [CrossRef]
- Robinson, C.G.; Roy, C.R. Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell. Microbiol. 2006, 8, 793–805. [Google Scholar] [CrossRef]
- Choy, A.; Dancourt, J.; Mugo, B.; O’Connor, T.J.; Isberg, R.R.; Melia, T.J.; Roy, C.R. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 2012, 338, 1072–1076. [Google Scholar] [CrossRef] [Green Version]
- Rolando, M.; Escoll, P.; Nora, T.; Botti, J.; Boitez, V.; Bedia, C.; Daniels, C.; Abraham, G.; Stogios, P.J.; Skarina, T.; et al. Legionella pneumophila S1P-lyase targets host sphingolipid metabolism and restrains autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 1901–1906. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.L.; Dalebroux, Z.D.; Swanson, M.S. Legionella pneumophilacouples fatty acid flux to microbial differentiation and virulence. Mol. Microbiol. 2009, 71, 1190–1204. [Google Scholar] [CrossRef]
- Hammer, B.K.; Swanson, M.S. Co-ordination of Legionella pneumophila virulence with entry into stationary phase by ppGpp. Mol. Microbiol. 1999, 33, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Dalebroux, Z.D.; Svensson, S.L.; Gaynor, E.C.; Swanson, M.S. ppGpp conjures bacterial virulence. Microbiol. Mol. Biol. Rev. 2010, 74, 171–199. [Google Scholar] [CrossRef] [Green Version]
- Zusman, T.; Gal-Mor, O.; Segal, G. Characterization of a Legionella pneumophila relA insertion mutant and toles of RelA and RpoS in virulence gene expression. J. Bacteriol. 2002, 184, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Weiner, A.; Mellouk, N.; Lopez-Montero, N.; Chang, Y.Y.; Souque, C.; Schmitt, C.; Enninga, J. Macropinosomes are key players in early Shigella invasion and vacuolar escape in epithelial cells. PLoS Pathog. 2016, 12, e1005602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellouk, N.; Weiner, A.; Aulner, N.; Schmitt, C.; Elbaum, M.; Shorte, S.L.; Danckaert, A.; Enninga, J. Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 2014, 16, 517–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, N.; Zhu, Y.; Lu, Q.; Hu, L.; Zheng, Y.; Shao, F. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 2012, 150, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Michel, E.; Reich, K.A.; Favier, R.; Berche, P.; Cossart, P. Attenuated mutants of the intracellular bacterium Listeria monocytogenes obtained by single amino acid subsitutions in listeriolysin O. Mol. Microbiol. 1990, 4, 2167–2178. [Google Scholar] [CrossRef]
- Bielecki, J.; Youngman, P.; Connelly, P.; Portnoy, D.A. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature 1990, 345, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Cossart, P.; Vincente, M.F.; Menguad, J.; Baquero, F.; Peres-Diaz, J.C.; Berche, P. Listeriolysin O is essential for virulence of Listeria monocytogenes:direct evidence obtained by gene complementation. Infect. Immun. 1989, 57, 3629–3636. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, T.; Popov, V.L.; Yu, X.J.; Walker, D.H.; Bouyer, D.H. Expression of the Rickettsia prowazekii pld or tlyC gene in Salmonella enterica serovar Typhimurium mediates phagosomal escape. Infect. Immun. 2005, 73, 6668–6673. [Google Scholar] [CrossRef] [Green Version]
- Driskell, L.O.; Yu, X.J.; Zhang, L.; Liu, Y.; Popov, V.L.; Walker, D.H.; Tucker, A.M.; Wood, D.O. Directed mutagenesis of the Rickettsia prowazekii pld gene encoding phospholipase D. Infect. Immun. 2009, 77, 3244–3248. [Google Scholar] [CrossRef] [Green Version]
- Winkler, H.; Firth, W.; Ballard, J.; Hegarty, B. Role of Phospholipase-Associated Penetration Mechanism in Cell Injury by Rickettsia rickettsii. Infect. Immun. 1983, 40, 840–842. [Google Scholar]
- Chu, H.; Lee, J.H.; Han, S.H.; Kim, S.Y.; Cho, N.H.; Kim, I.S.; Choi, M.S. Exploitation of the endocytic pathway by Orientia tsutsugamushi in nonprofessional phagocytes. Infect. Immun. 2006, 74, 4246–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, T.K.; Cronan, J.E., Jr. Activation of long chain fatty acids with acyl carrier protein: Demonstration of a new enzyme, acyl acyl carrier protein synthetase, in Escherichia coli. Proc. Natl. Acad. Sci. USA 1976, 73, 4374–4378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, K.I.; Tamura, A.; O’hashi, N.; Urakami, H.; Kaya, S.; Fukushi, K. Deficiency of peptidoglycan and lipopolysaccharide components in Rickettsia tsutsugamushi. Infect. Immun. 1987, 55, 2290–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, T.P.; Verhoeve, V.I.; Guillotte, M.L.; Lehman, S.S.; Rennoll, S.A.; Beier-Sexton, M.; Rahman, M.S.; Azad, A.F.; Gillespie, J.J. Wholly Rickettsia! Reconstructed Metabolic Profile of the Quintessential Bacterial Parasite of Eukaryotic Cells. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Zezerov, E.; Loginov, V.; Berezneva, A. Polypeptide and phospholipid composition of the Rickettsia prowazekii membrane and its immunogenic properties. Zhurnal Mikrobiol. Epidemiol. Immunobiol. 1985, 6, 6–13. [Google Scholar]
- Winkler, H.; Miller, E. Phospholipid Composition of Rickettsia prowazeki Grown in Chicken Embryo Yolk Sacs. J. Bacteriol. 1978, 136, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Tzianabos, T.; Moss, C.; McDade, J. Fatty Acid Composition of Rickettsiae. J. Clin. Microbiol. 1981, 13, 603–605. [Google Scholar] [CrossRef] [Green Version]
- Curto, P.; Santa, C.; Allen, P.; Manadas, B.; Simoes, I.; Martinez, J.J. A Pathogen and a Non-pathogen Spotted Fever Group Rickettsia Trigger Differential Proteome Signatures in Macrophages. Front. Cell. Infect. Microbiol. 2019, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Chambers, M.C.; Song, K.H.; Schneider, D.S. Listeria monocytogenes infection causes metabolic shifts in Drosophila melanogaster. PLoS ONE 2012, 7, e50679. [Google Scholar] [CrossRef] [Green Version]
- Curto, P.; Simoes, I.; Riley, S.P.; Martinez, J.J. Differences in Intracellular Fate of Two Spotted Fever Group Rickettsia in Macrophage-Like Cells. Front. Cell. Infect. Microbiol. 2016, 6, 80. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Jung, Y.; Gill, B.; Kim, C.; Hwang, K.J.; Ju, Y.R.; Lee, H.J.; Chu, H.; Hwang, G.S. Metabolic responses to Orientia tsutsugamushi infection in a mouse model. PLoS Negl. Trop. Dis. 2015, 9, e3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audia, J.P.; Winkler, H.H. Study of the five Rickettsia prowazekii proteins annotated as ATP/ADP translocases (Tlc): Only Tlc1 transports ATP/ADP, while Tlc4 and Tlc5 transport other ribonucleotides. J. Bacteriol. 2006, 188, 6261–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, C.K.; Yang, J.S.; Kim, S.; Choi, M.S.; Kim, I.S.; Cho, N.H. Genome-based construction of the metabolic pathways of Orientia tsutsugamushi and comparative analysis within the Rickettsiales order. Comp. Funct. Genom. 2008, 2008, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessop, F.; Schwarz, B.; Heitmann, E.; Buntyn, R.; Wehrly, T.; Bosio, C. Temporal Manipulation of Mitochondrial Function by Virulent Francisella tularensis To Limit Inflammation and Control Cell Death. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, M.; Fukasawa, M.; Satoh, M.; Hanada, K.; Saijo, M.; Uchiyama, T.; Ando, S. The intracellular pathogen Orientia tsutsugamushi responsible for scrub typhus induces lipid droplet formation in mouse fibroblasts. Microbes Infect. 2014, 16, 962–966. [Google Scholar] [CrossRef]
- Rahman, M.S.; Ammerman, N.C.; Sears, K.T.; Ceraul, S.M.; Azad, A.F. Functional characterization of a phospholipase A(2) homolog from Rickettsia typhi. J. Bacteriol. 2010, 192, 3294–3303. [Google Scholar] [CrossRef] [Green Version]
- Winkler, H.; Miller, E. Phospholipase A and the interaction of Rickettsia prowazekii and mouse fibroblasts (L-929 cells). Infect. Immun. 1982, 38, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.H.; Feng, H.M.; Popov, V.L. Rickettsial Phospholipase A2 as a Pathogenic Mechanism in a Model of Cell Injury by Typhus and Spotted Fever Group Rickettsiae. Am. J. Trop. Med. Hygeine 2001, 65, 936–942. [Google Scholar] [CrossRef]
- Hurley, B.P.; Pirzai, W.; Mumy, K.L.; Gronert, K.; McCormick, B.A. Selective eicosanoid-generating capacity of cytoplasmic phospholipase A2 in Pseudomonas aeruginosa-infected epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L286–L294. [Google Scholar] [CrossRef] [Green Version]
- Walker, T.S.; Brown, J.S.; Hoover, C.S.; Morgan, D.A. Endothelial prostaglandin secretion: Effects of typhus rickettsiae. J. Infect. Dis. 1990, 162, 1136. [Google Scholar] [CrossRef]
- Rydkina, E.; Sahni, A.; Baggs, R.B.; Silverman, D.J.; Sahni, S.K. Infection of human endothelial cells with spotted Fever group rickettsiae stimulates cyclooxygenase 2 expression and release of vasoactive prostaglandins. Infect. Immun. 2006, 74, 5067–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuhas, Y.; Weizman, A.; Vanichkin, A.; Ashkenazi, S. Involvement of prostaglandins in an animal model of Shigella-related seizures. J. Neuroimmunol. 2005, 168, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.C.; Richard, G.; Burghoffer, B.; Dagute, G.L. Suppression of Cellular Immunity to Listeria monocytogenes by Activated Macrophages- Mediation by Prostaglandins. Infect. Immun. 1985, 49, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golovliov, I.; Baranov, V.; Krocova, Z.; Kovarova, H.; Sjöstedt, A. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 2003, 71, 5940–5950. [Google Scholar] [CrossRef] [Green Version]
- Santic, M.; Molmeret, M.; Abu Kwaik, Y. Modulation of biogenesis of the Francisella tularensis subsp. novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN gamma. Cell. Microbiol. 2007, 9, 2314. [Google Scholar] [CrossRef]
- Clemens, D.L.; Lee, B.Y.; Horwitz, M.A. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 2004, 72, 3204–3217. [Google Scholar] [CrossRef] [Green Version]
- Crane, D.D.; Ireland, R.; Alinger, J.B.; Small, P.; Bosio, C.M. Lipids derived from virulent Francisella tularensis broadly inhibit pulmonary inflammation via toll-like receptor 2 and peroxisome proliferator-activated receptor alpha. Clin. Vaccine Immunol. 2013, 20, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Navratil, A.R.; Brummett, A.M.; Bryan, J.D.; Woolard, M.D. Francisella tularensis LVS induction of prostaglandin biosynthesis by infected macrophages requires specific host phospholipases and lipid phosphatases. Infect. Immun. 2014, 82, 3299–3311. [Google Scholar] [CrossRef] [Green Version]
- Woolard, M.D.; Wilson, J.E.; Hensley, L.L.; Jania, L.A.; Kawula, T.H.; Drake, J.R.; Frelinger, J.A. Francisella tularensis-Infected Macrophages Release Prostaglandin E2 that Blocks T Cell Proliferation and Promotes a Th2-Like Response. J. Immunol. 2007, 178, 2065–2074. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Mechanisms for acquisition and utilization of host lipids by intracellular pathogens. Intracellular pathogens that reside within a vacuole utilize host lipids for various processes during infection. This model depicts the host lipid pathways and methods exploited by various vacuolar bacterial species to enhance intracellular and, ultimately, mammalian infection. The approaches employed by these pathogenic bacteria include (i) lipid acquisition into pathogen-associated membranes as seen in Ehrlichia, Anaplasma, Chlamydia, and Coxiella (from left to right); (ii) pathogen- and host-stimulated lipid droplet modulation required during Chlamydia, Coxiella, Mycobacterium, and Salmonella infection (from left to right); (iii) immune modulation and lipid inflammatory mediator production from lipid droplets seen during Chlamydia, Coxiella, and Mycobacterium infection (from left to right); (iv) acquirement of host fatty acid β-oxidation (FAO) metabolites for energy production by Mycobacterium and modulation of host FAO regulators by Chlamydia; (v) regulation of bacterial stage transition of Legionella initiated by host intracellular lipid composition. The legend on the bottom left-hand side of the model shows a representative image of the structures used in the panels describing each pathogens’ utilization of host lipids. Toll-like receptor 2 (TLR2); NOD-like receptor protein 3 (NLRP3); peroxisome proliferation activation receptor α (PPARα); peroxisome proliferation activation receptor ɣ (PPARɣ); adipose triglyceride lipase (ATGL); fatty acid binding protein (FABP4); patatin-like phospholipase domain containing protein 2 (PNPLA2); transcription factor EB (TFEB); perilipin 2 (plin2).
Figure 1.
Mechanisms for acquisition and utilization of host lipids by intracellular pathogens. Intracellular pathogens that reside within a vacuole utilize host lipids for various processes during infection. This model depicts the host lipid pathways and methods exploited by various vacuolar bacterial species to enhance intracellular and, ultimately, mammalian infection. The approaches employed by these pathogenic bacteria include (i) lipid acquisition into pathogen-associated membranes as seen in Ehrlichia, Anaplasma, Chlamydia, and Coxiella (from left to right); (ii) pathogen- and host-stimulated lipid droplet modulation required during Chlamydia, Coxiella, Mycobacterium, and Salmonella infection (from left to right); (iii) immune modulation and lipid inflammatory mediator production from lipid droplets seen during Chlamydia, Coxiella, and Mycobacterium infection (from left to right); (iv) acquirement of host fatty acid β-oxidation (FAO) metabolites for energy production by Mycobacterium and modulation of host FAO regulators by Chlamydia; (v) regulation of bacterial stage transition of Legionella initiated by host intracellular lipid composition. The legend on the bottom left-hand side of the model shows a representative image of the structures used in the panels describing each pathogens’ utilization of host lipids. Toll-like receptor 2 (TLR2); NOD-like receptor protein 3 (NLRP3); peroxisome proliferation activation receptor α (PPARα); peroxisome proliferation activation receptor ɣ (PPARɣ); adipose triglyceride lipase (ATGL); fatty acid binding protein (FABP4); patatin-like phospholipase domain containing protein 2 (PNPLA2); transcription factor EB (TFEB); perilipin 2 (plin2).

Figure 2.
Potential host lipid pathways altered during cytosolic Rickettsia infection. A representative cytosolic, intracellular bacteria species, Rickettsia conorii, requires host fatty acid synthase, and therefore, free fatty acids for successful replication. Mammalian infection with rickettsial pathogens also alters host prostaglandin production to stimulate the presentation of disease. Metabolically, Rickettsia spp. are not self-sufficient and require the host for survival; however, the host pathways required for energy production and subsequent bacterial survival are largely unknown. Evidence suggests that host glycolysis is downregulated leaving other metabolic pathways, such as fatty acid β-oxidation (FAO), as likely candidates for carbon source generation required for oxidative phosphorylation and ATP production. Early studies also suggest the alteration and incorporation of host phosphatidylcholine into the rickettsial membrane, thus indicating a likely requirement of host lipid pathways for bacterial membrane structure during infection. Red arrows indicate speculated host processes and functions of host metabolites during cytosolic infection. Blue figures and words indicate known alterations that occur during infection. Black arrows indicate common pathways and functions of host metabolites. The legend on the right-hand side of the model shows a representative image of the structures use to describe Rickettsia utilization of host lipids within infected host cells.
Figure 2.
Potential host lipid pathways altered during cytosolic Rickettsia infection. A representative cytosolic, intracellular bacteria species, Rickettsia conorii, requires host fatty acid synthase, and therefore, free fatty acids for successful replication. Mammalian infection with rickettsial pathogens also alters host prostaglandin production to stimulate the presentation of disease. Metabolically, Rickettsia spp. are not self-sufficient and require the host for survival; however, the host pathways required for energy production and subsequent bacterial survival are largely unknown. Evidence suggests that host glycolysis is downregulated leaving other metabolic pathways, such as fatty acid β-oxidation (FAO), as likely candidates for carbon source generation required for oxidative phosphorylation and ATP production. Early studies also suggest the alteration and incorporation of host phosphatidylcholine into the rickettsial membrane, thus indicating a likely requirement of host lipid pathways for bacterial membrane structure during infection. Red arrows indicate speculated host processes and functions of host metabolites during cytosolic infection. Blue figures and words indicate known alterations that occur during infection. Black arrows indicate common pathways and functions of host metabolites. The legend on the right-hand side of the model shows a representative image of the structures use to describe Rickettsia utilization of host lipids within infected host cells.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Allen, P.E.; Martinez, J.J. Modulation of Host Lipid Pathways by Pathogenic Intracellular Bacteria. Pathogens 2020, 9, 614. https://doi.org/10.3390/pathogens9080614
AMA Style
Allen PE, Martinez JJ. Modulation of Host Lipid Pathways by Pathogenic Intracellular Bacteria. Pathogens. 2020; 9(8):614. https://doi.org/10.3390/pathogens9080614
Chicago/Turabian StyleAllen, Paige E., and Juan J. Martinez. 2020. "Modulation of Host Lipid Pathways by Pathogenic Intracellular Bacteria" Pathogens 9, no. 8: 614. https://doi.org/10.3390/pathogens9080614
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.