Simultaneous Nasal Carriage by Methicillin-Resistant and Methicillin Susceptible Staphylococcus aureus of Lineage ST398 in a Live Pig Transporter

 and
and
Abstract
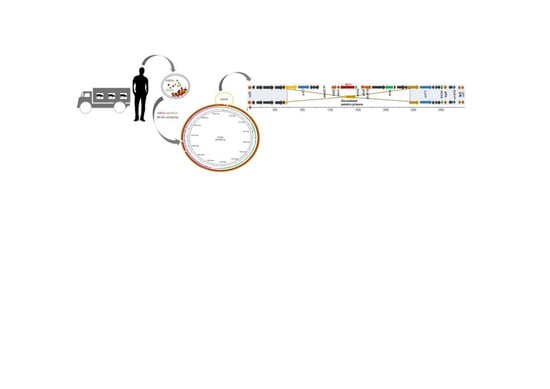
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Identification of the Isolates, Molecular Typing, and Antibiotic Resistance Phenotype
2.2. Whole Genome Sequencing Results
2.3. Virulence and Antimicrobial Resistance Genotype
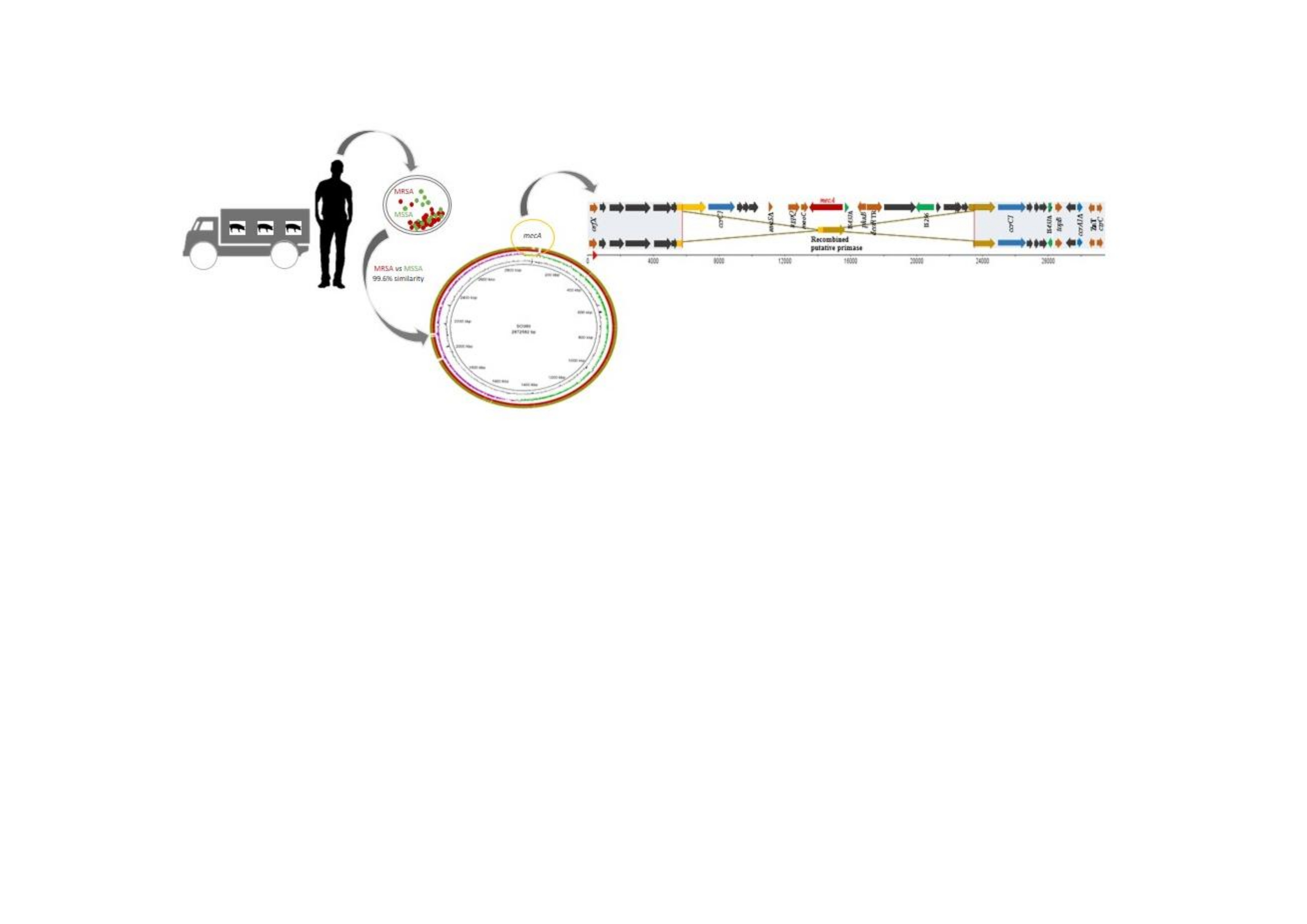
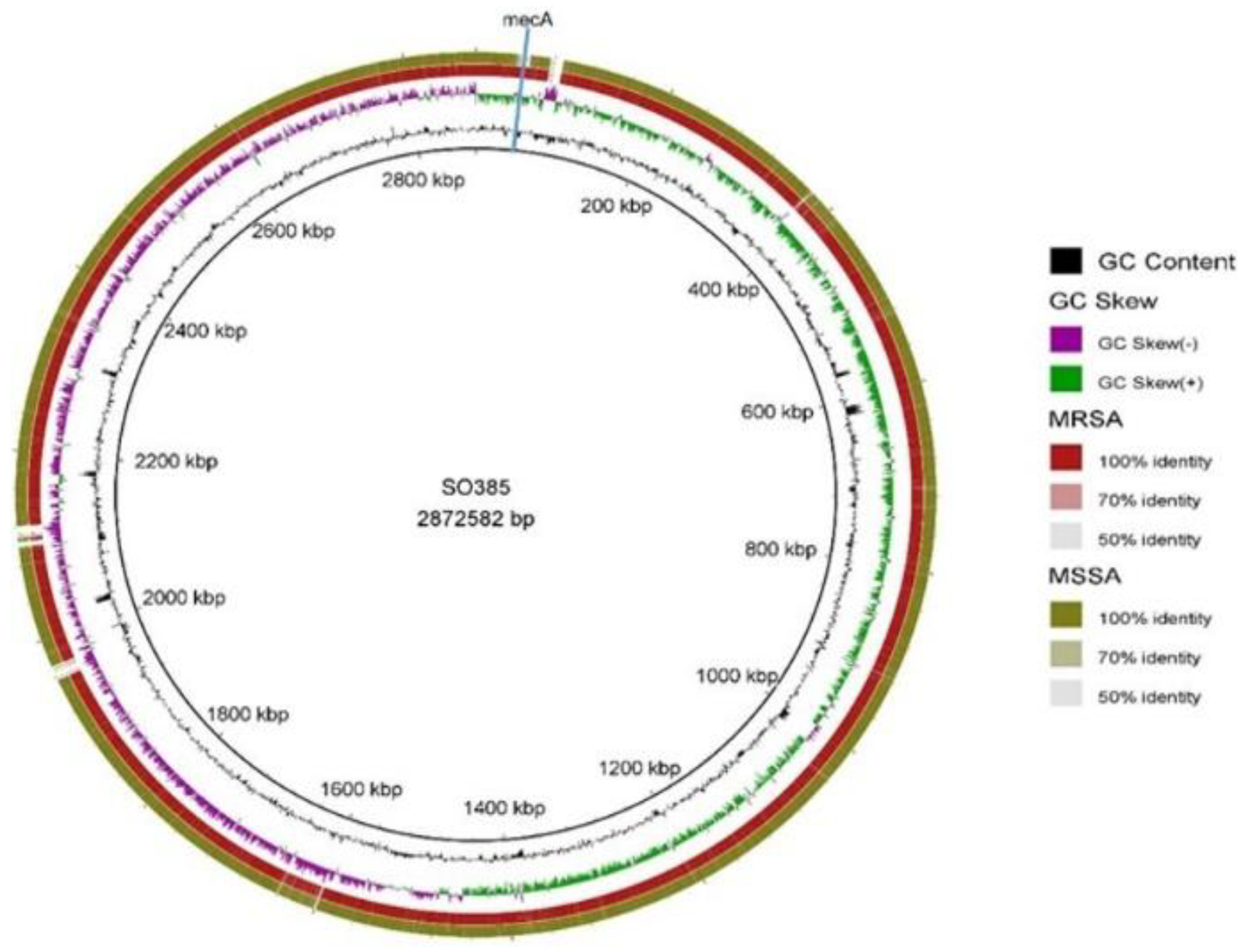
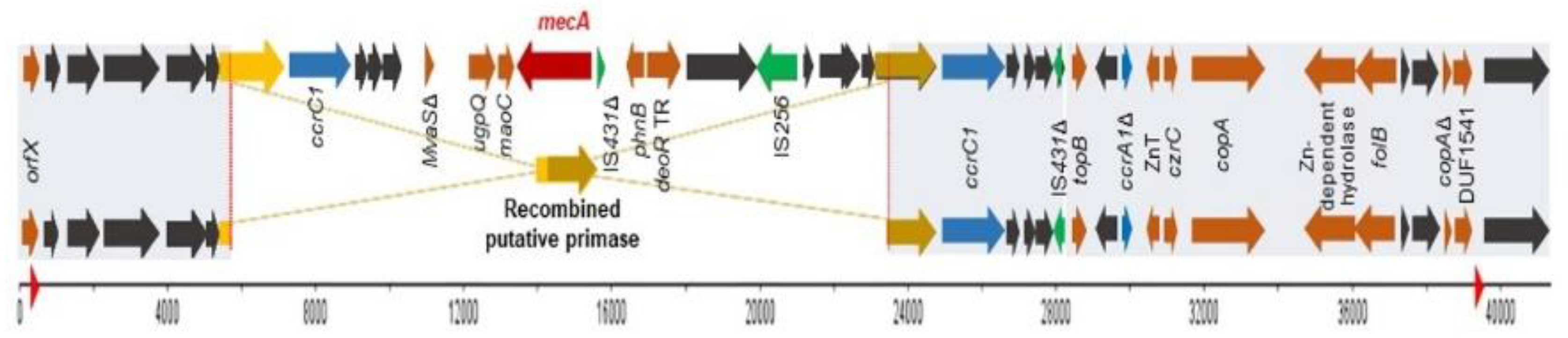
2.4. Comparison between Both Strains
3. Materials and Methods
3.1. Sample Collection, Isolation and Identification
3.2. Molecular Typing and Antimicrobial Susceptibility Testing
3.3. Whole Genome Sequencing and Analysis of Sequences
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vanderhaeghen, W.; Hermans, K.; Haesebrouck, F.; Butaye, P. Methicillin-resistant Staphylococcus aureus (MRSA) in food production animals. Epidemiol. Infect. 2010, 138, 606–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, L.B.; Stegger, M.; Hasman, H.; Aziz, M.; Larsen, J.; Andersen, P.S.; Pearson, T.; Waters, A.E.; Foster, J.T.; Schupp, J.; et al. Staphylococcus aureus CC398: Host adaptation and emergence of methicillin resistance in livestock. mBio 2012, 3, e00305-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegger, M.; Liu, C.M.; Larsen, J.; Soldanova, K.; Aziz, M.; Contente-Cuomo, T.; Petersen, A.; Vandendriessche, S.; Jiménez, J.N.; Mammina, C.; et al. Rapid Differentiation between Livestock-Associated and Livestock-Independent Staphylococcus aureus CC398 Clades. PLoS ONE 2013, 8, e79645. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.J.; Witney, A.A.; Gould, K.A.; Moodley, A.; Guardabassi, L.; Voss, A.; Denis, O.; Broens, E.M.; Hinds, J.; Lindsay, J.A. The distribution of mobile genetic elements (MGEs) in MRSA CC398 is associated with both host and country. Genome Biol. Evol. 2011, 3, 1164–1174. [Google Scholar] [CrossRef] [Green Version]
- Koreen, L.; Ramaswamy, S.V.; Naidich, S.; Koreen, I.V.; Graff, G.R.; Graviss, E.A.; Kreiswirth, B.N. Comparative sequencing of the serine-aspartate repeat-encoding region of the clumping factor B gene (clfB) for resolution within clonal groups of Staphylococcus aureus. J. Clin. Microbiol. 2005, 43, 3985–3994. [Google Scholar] [CrossRef] [Green Version]
- Sabat, A.; Melles, D.C.; Martirosian, G.; Grundmann, H.; van Belkum, A.; Hryniewicz, W. Distribution of the Serine-Aspartate Repeat Protein-Encoding sdr Genes among Nasal-Carriage and Invasive Staphylococcus aureus Strains. J. Clin. Microbiol. 2006, 44, 1135–1138. [Google Scholar] [CrossRef] [Green Version]
- Sharp, J.A.; Echague, C.G.; Hair, P.S.; Ward, M.D.; Nyalwidhe, J.O.; Geoghegan, J.A.; Foster, T.J.; Cunnion, K.M. Staphylococcus aureus surface protein SdrE binds complement regulator factor H as an immune evasion tactic. PLoS ONE 2012, 7, e38407. [Google Scholar] [CrossRef]
- Chlebowicz, M.A.; Nganou, K.; Kozytska, S.; Arends, J.P.; Engelmann, S.; Grundmann, H.; Ohlsen, K.; van Dijl, J.M.; Buist, G. Recombination between ccrC genes in a type V (5C2&5) staphylococcal cassette chromosome mec (SCCmec) of Staphylococcus aureus ST398 leads to conversion from methicillin resistance to methicillin susceptibility in vivo. Antimicrob. Agents Chemother. 2010, 54, 783–791. [Google Scholar]
- Vandendriessche, S.; Vanderhaeghen, W.; Larsen, J.; de Mendonça, R.; Hallin, M.; Butaye, P.; Hermans, K.; Haesebrouck, F.; Denis, O. High genetic diversity of methicillin-susceptible Staphylococcus aureus (MSSA) from humans and animals on livestock farms and presence of SCCmec remnant DNA in MSSA CC398. J. Antimicrob. Chemother. 2014, 69, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Noto, M.J.; Fox, P.M.; Archer, G.L. Spontaneous deletion of the methicillin resistance determinant, mecA, partially compensates for the fitness cost associated with high-level vancomycin resistance in Staphylococcus aureus. J. Antimicrob. Chemother. 2008, 52, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Shore, A.C.; Rossney, A.S.; O’Connell, B.; Herra, C.M.; Sullivan, D.J.; Humphreys, H.; Coleman, D.C. Detection of staphylococcal cassette chromosome mec-associated DNA segments in multiresistant methicillin-susceptible Staphylococcus aureus (MSSA) and identification of Staphylococcus epidermidis ccrAB4 in both methicillin-resistant S. aureus and MSSA. Antimicrob. Agents Chemother. 2008, 52, 4407–4419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Griethuysen, A.; van Loo, I.; van Belkum, A.; Vandenbroucke-Grauls, C.; Wannet, W.; van Keulen, P.; Kluytmans, J. Loss of the mecA gene during storage of methicillin-resistant Staphylococcus aureus strains. J. Clin. Microbiol. 2005, 43, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Zhou, K.; Liu, Y.; Yang, L.; Zhang, Q.; Guan, J.; Zhong, N.; Zhuo, C. A potential role of transposon IS431 in the loss of mecA gene. Sci. Rep. 2017, 7, 41237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, K.L.; Pedersen, T.M.; Udekwu, K.I.; Petersen, A.; Skov, R.L.; Hansen, L.H.; Hughes, D.; Frimodt-Møller, N. Fitness cost: A bacteriological explanation for the demise of the first international methicillin-resistant Staphylococcus aureus epidemic. J. Antimicrob. Chemother. 2012, 67, 1325–1332. [Google Scholar] [CrossRef]
- Sasaki, T.; Tsubakishita, S.; Tanaka, Y.; Sakusabe, A.; Ohtsuka, M.; Hirotaki, S.; Kawakami, T.; Fukata, T.; Hiramatsu, K. Multiplex-PCR method for species identification of coagulase-positive staphylococci. J. Clin. Microbiol. 2010, 48, 765–769. [Google Scholar] [CrossRef] [Green Version]
- Argudín, M.A.; Fetsch, A.; Tenhagen, B.A.; Hammerl, J.A.; Hertwig, S.; Kowall, J.; Rodicio, M.R.; Käsbohrer, A.; Helmuth, R.; Schroeter, A.; et al. High heterogeneity within methicillin-resistant Staphylococcus aureus ST398 isolates, defined by Cfr9I macrorestriction-pulsed-field gel electrophoresis profiles and spa and SCCmec types. Appl. Environ. Microbiol. 2010, 76, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Quail, M.A.; Kozarewa, I.; Smith, F.; Scally, A.; Stephens, P.J.; Durbin, R.; Swerdlow, H.; Turner, D.J. A large genome center’s improvements to the Illumina sequencing system. Nat. Methods. 2008, 5, 1005–1010. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; De Silva, N.; Hunt, M.; Quail, M.A.; Parkhill, J.; Harris, S.R.; Otto, T.D.; Keane, J.A. Robust high-throughput prokaryote de novo assembly and improvement pipeline for Illumina data. Microb. Genom. 2016, 2, e000083. [Google Scholar] [CrossRef]
- Rissman, A.I.; Mau, B.; Biehl, B.S.; Darling, A.E.; Glasner, J.D.; Perna, N.T. Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics 2009, 25, 2071–2073. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Stamatakis, A.; Ludwig, T.; Meier, H. RAxML-III: A fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 2005, 21, 456–463. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez, P.; Aspiroz, C.; Hadjirin, N.F.; Benito, D.; Zarazaga, M.; Torres, C.; Holmes, M.A. Simultaneous Nasal Carriage by Methicillin-Resistant and Methicillin Susceptible Staphylococcus aureus of Lineage ST398 in a Live Pig Transporter. Pathogens 2020, 9, 401. https://doi.org/10.3390/pathogens9050401
Gómez P, Aspiroz C, Hadjirin NF, Benito D, Zarazaga M, Torres C, Holmes MA. Simultaneous Nasal Carriage by Methicillin-Resistant and Methicillin Susceptible Staphylococcus aureus of Lineage ST398 in a Live Pig Transporter. Pathogens. 2020; 9(5):401. https://doi.org/10.3390/pathogens9050401
Chicago/Turabian StyleGómez, Paula, Carmen Aspiroz, Nazreen F. Hadjirin, Daniel Benito, Myriam Zarazaga, Carmen Torres, and Mark A. Holmes. 2020. "Simultaneous Nasal Carriage by Methicillin-Resistant and Methicillin Susceptible Staphylococcus aureus of Lineage ST398 in a Live Pig Transporter" Pathogens 9, no. 5: 401. https://doi.org/10.3390/pathogens9050401