
A Cellular GWAS Approach to Define Human Variation in Cellular Pathways Important to Inflammation

{kind=link}
Abstract
:1. Introduction
2. Cellular GWAS Approach to Understand Pathways in Human Disease
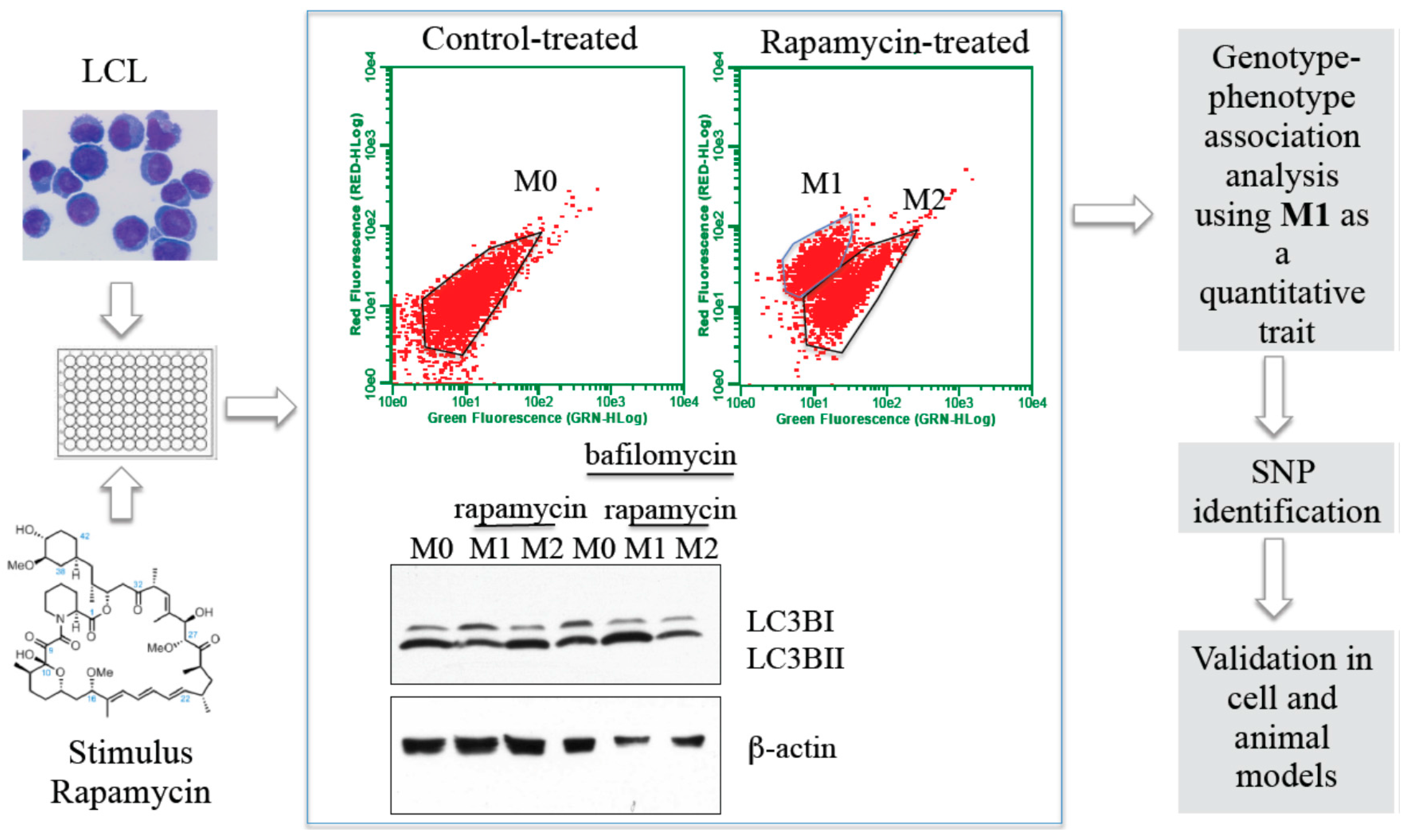
Cellular GWAS: A complementary approach to GWAS of disease
- Reduces environmental variation due to differences in dose, infectious strain, medical treatment and other concomitant factors.
- Provides clues to mechanism.
- Allows for experimental follow-up in the same cells used for the screen.
- Allows for controlled, reproducible measurements of phenotype in the laboratory.
- Not restricted to diseases that have large, currently accessible patient populations.
- Accessible to small labs.
- Study cellular phenotypes of relevance to multiple diseases.
2.1. Hapmap Cells Provide a Relevant Model to Study Immune Responses
2.2. Immune Phenotypes Studied in Hi-HOST
2.2.1. The Inflammasome
2.2.2. Autophagy
2.3. Identification of Inherited Genetic Factors That Contribute to Phenotypic Variation in Inflammatory Pathways Using Hi-HOST
3. Expression of Non-Functional CARD8 Allows for More Robust Pyroptosis in Response to S. typhimurium Infection [4]
4. A Role for Methionine Salvage Enzyme Apaf-1 (Apoptotic Protease Activating Factor 1) Interacting Protein (APIP) in Pyroptosis [5]
5. A Role for Microtubule Stability in Pathogen-Mediated Pyroptosis [44]
6. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014, 15, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D. Gene-environment-wide association studies: Emerging approaches. Nat. Rev. Genet. 2010, 11, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Schneider, D.S. Balancing resistance and infection tolerance through metabolic means. Proc. Natl. Acad. Sci. USA 2012, 109, 13886–13887. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.C.; Shukla, K.P.; Fong, C.; Wasnick, M.; Brittnacher, M.J.; Wurfel, M.M.; Holden, T.D.; O’Keefe, G.E.; van Yserloo, B.; Akey, J.M.; et al. A genome-wide in vitro bacterial-infection screen reveals human variation in the host response associated with inflammatory disease. Am. J. Hum. Genet. 2009, 85, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.C.; Gamazon, E.R.; Shukla, K.P.; Pfuetzner, R.A.; Whittington, D.; Holden, T.D.; Brittnacher, M.J.; Fong, C.; Radey, M.; Ogohara, C.; et al. Functional genetic screen of human diversity reveals that a methionine salvage enzyme regulates inflammatory cell death. Proc. Natl. Acad. Sci. USA 2012, 109, E2343–E2352. [Google Scholar] [CrossRef] [PubMed]
- The International HapMap Consortium. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar]
- Stranger, B.E.; Nica, A.C.; Forrest, M.S.; Dimas, A.; Bird, C.P.; Beazley, C.; Ingle, C.E.; Dunning, M.; Flicek, P.; Koller, D.; et al. Population genomics of human gene expression. Nat. Genet. 2007, 39, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.S.; Duan, S.; Bleibel, W.K.; Kistner, E.O.; Zhang, W.; Clark, T.A.; Chen, T.X.; Schweitzer, A.C.; Blume, J.E.; Cox, N.J.; et al. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc. Natl. Acad. Sci. USA 2007, 104, 9758–9763. [Google Scholar] [CrossRef] [PubMed]
- Loeuillet, C.; Deutsch, S.; Ciuffi, A.; Robyr, D.; Taffe, P.; Munoz, M.; Beckmann, J.S.; Antonarakis, S.E.; Telenti, A. In vitro whole-genome analysis identifies a susceptibility locus for HIV-1. PLoS Biol. 2008, 6, e32. [Google Scholar] [CrossRef] [PubMed]
- Rauch, P.J.; Chudnovskiy, A.; Robbins, C.S.; Weber, G.F.; Etzrodt, M.; Hilgendorf, I.; Tiglao, E.; Figueiredo, J.L.; Iwamoto, Y.; Theurl, I.; et al. Innate response activator B cells protect against microbial sepsis. Science 2012, 335, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Scumpia, K.M.; Scumpia, P.O.; Weinstein, J.S.; Delano, M.J.; Cuenca, A.G.; Nacionales, D.C.; Wynn, J.L.; Lee, P.Y.; Kumagai, Y.; Efron, P.A.; et al. B cells enhance early innate immune responses during bacterial sepsis. J. Exp. Med. 2011, 208, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, J.M.; Copple, A.; Batson, L.P.; Landers, C.D.; Yannelli, J.R. Cell surface phenotyping and cytokine production of Epstein-Barr Virus (EBV)-transformed lymphoblastoid cell lines (LCLs). J. Immunol. Methods 2002, 264, 19–28. [Google Scholar] [CrossRef]
- Prentice, A.M. Starvation in humans: Evolutionary background and contemporary implications. Mech. Ageing Dev. 2005, 126, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Sironi, M.; Pozzoli, U.; Ferrer-Admetlla, A.; Pattini, L.; Nielsen, R. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 2011, 7, e1002355. [Google Scholar] [CrossRef]
- Henckaerts, L.; Nielsen, K.R.; Steffensen, R.; van Steen, K.; Mathieu, C.; Giulietti, A.; Wouters, P.J.; Milants, I.; Vanhorebeek, I.; Langouche, L.; et al. Polymorphisms in innate immunity genes predispose to bacteremia and death in the medical intensive care unit. Crit. Care Med. 2009, 37, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Carratala, J.; Gudiol, F.; Pallares, R.; Dorca, J.; Verdaguer, R.; Ariza, J.; Manresa, F. Risk factors for nosocomial Legionella pneumophila pneumonia. Am. J. Respir. Crit. Care Med. 1994, 149, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Fowke, K.R.; Nagelkerke, N.J.; Kimani, J.; Simonsen, J.N.; Anzala, A.O.; Bwayo, J.J.; MacDonald, K.S.; Ngugi, E.N.; Plummer, F.A. Resistance to HIV-1 infection among persistently seronegative prostitutes in Nairobi, Kenya. Lancet 1996, 348, 1347–1351. [Google Scholar] [CrossRef]
- Wattanathum, A.; Manocha, S.; Groshaus, H.; Russell, J.A.; Walley, K.R. Interleukin-10 haplotype associated with increased mortality in critically ill patients with sepsis from pneumonia but not in patients with extrapulmonary sepsis. Chest 2005, 128, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Westendorp, R.G.; Langermans, J.A.; Huizinga, T.W.; Elouali, A.H.; Verweij, C.L.; Boomsma, D.I.; Vandenbroucke, J.P. Genetic influence on cytokine production and fatal meningococcal disease. Lancet 1997, 349, 170–173. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Witzenrath, M.; Pache, F.; Lorenz, D.; Koppe, U.; Gutbier, B.; Tabeling, C.; Reppe, K.; Meixenberger, K.; Dorhoi, A.; Ma, J.; et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J. Immunol. 2011, 187, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Ayres, J.S.; Trinidad, N.J.; Vance, R.E. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat. Med. 2012, 18, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Dolinay, T.; Kim, Y.S.; Howrylak, J.; Hunninghake, G.M.; An, C.H.; Fredenburgh, L.; Massaro, A.F.; Rogers, A.; Gazourian, L.; Nakahira, K.; et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am. J. Respir. Crit. Care Med. 2012, 185, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Raupach, B.; Peuschel, S.K.; Monack, D.M.; Zychlinsky, A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 2006, 74, 4922–4926. [Google Scholar] [CrossRef] [PubMed]
- LaRock, D.L.; Chaudhary, A.; Miller, S.I. Salmonellae interactions with host processes. Nat. Rev. Microbiol. 2015, 13, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Sellin, M.E.; Muller, A.A.; Felmy, B.; Dolowschiak, T.; Diard, M.; Tardivel, A.; Maslowski, K.M.; Hardt, W.D. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 2014, 16, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Kastner, D.L.; Aksentijevich, I.; Goldbach-Mansky, R. Autoinflammatory Disease Reloaded: A Clinical Perspective. Cell 2010, 140, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Yoshimori, T.; Suzuki, T.; Sagara, H.; Mizushima, N.; Sasakawa, C. Escape of intracellular Shigella from autophagy. Science 2005, 307, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, C.L.; Smith, A.C.; Bakowski, M.A.; Yoshimori, T.; Brumell, J.H. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 2006, 281, 11374–11383. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, E.; Muenzer, J.T.; Hawkins, W.G.; Davis, C.G.; Dixon, D.J.; McDunn, J.E.; Brackett, D.J.; Lerner, M.R.; Swanson, P.E.; Hotchkiss, R.S. Sepsis induces extensive autophagic vacuolization in hepatocytes: A clinical and laboratory-based study. Lab. Investig. 2009, 89, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mendonsa, G.R.; Symington, J.W.; Zhang, Q.; Cadwell, K.; Virgin, H.W.; Mysorekar, I.U. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 11008–11013. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; de la Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Klionsky, D.J. Autophagy and human disease. Cell Cycle 2007, 6, 1837–1849. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era–concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Fontalba, A.; Martinez-Taboada, V.; Gutierrez, O.; Pipaon, C.; Benito, N.; Balsa, A.; Blanco, R.; Fernandez-Luna, J.L. Deficiency of the NF-kappaB inhibitor caspase activating and recruitment domain 8 in patients with rheumatoid arthritis is associated with disease severity. J. Immunol. 2007, 179, 4867–4873. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Marioni, J.C.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 2010, 464, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Salinas, R.E.; Ogohara, C.; Thomas, M.I.; Shukla, K.P.; Miller, S.I.; Ko, D.C. A cellular genome-wide association study reveals human variation in microtubule stability and a role in inflammatory cell death. Mol. Biol. Cell 2014, 25, 76–86. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, S.I.; Chaudhary, A. A Cellular GWAS Approach to Define Human Variation in Cellular Pathways Important to Inflammation. Pathogens 2016, 5, 39. https://doi.org/10.3390/pathogens5020039
Miller SI, Chaudhary A. A Cellular GWAS Approach to Define Human Variation in Cellular Pathways Important to Inflammation. Pathogens. 2016; 5(2):39. https://doi.org/10.3390/pathogens5020039
Chicago/Turabian StyleMiller, Samuel I., and Anu Chaudhary. 2016. "A Cellular GWAS Approach to Define Human Variation in Cellular Pathways Important to Inflammation" Pathogens 5, no. 2: 39. https://doi.org/10.3390/pathogens5020039