

Type I Interferon Signaling Controls Gammaherpesvirus Latency In Vivo
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Primary MHV-68 Infection Induces an IFN Response In Vivo
2.2. IFN Signaling Is Required to Prevent MHV-68 Dissemination upon In Vivo Reactivation
2.3. MHV-68 In Vivo Reactivation Induces an IFN Response
2.4. Type I IFN Signaling Does Not Prevent Low-Level Cell-to-Cell Transmission of MHV-68 during Latency
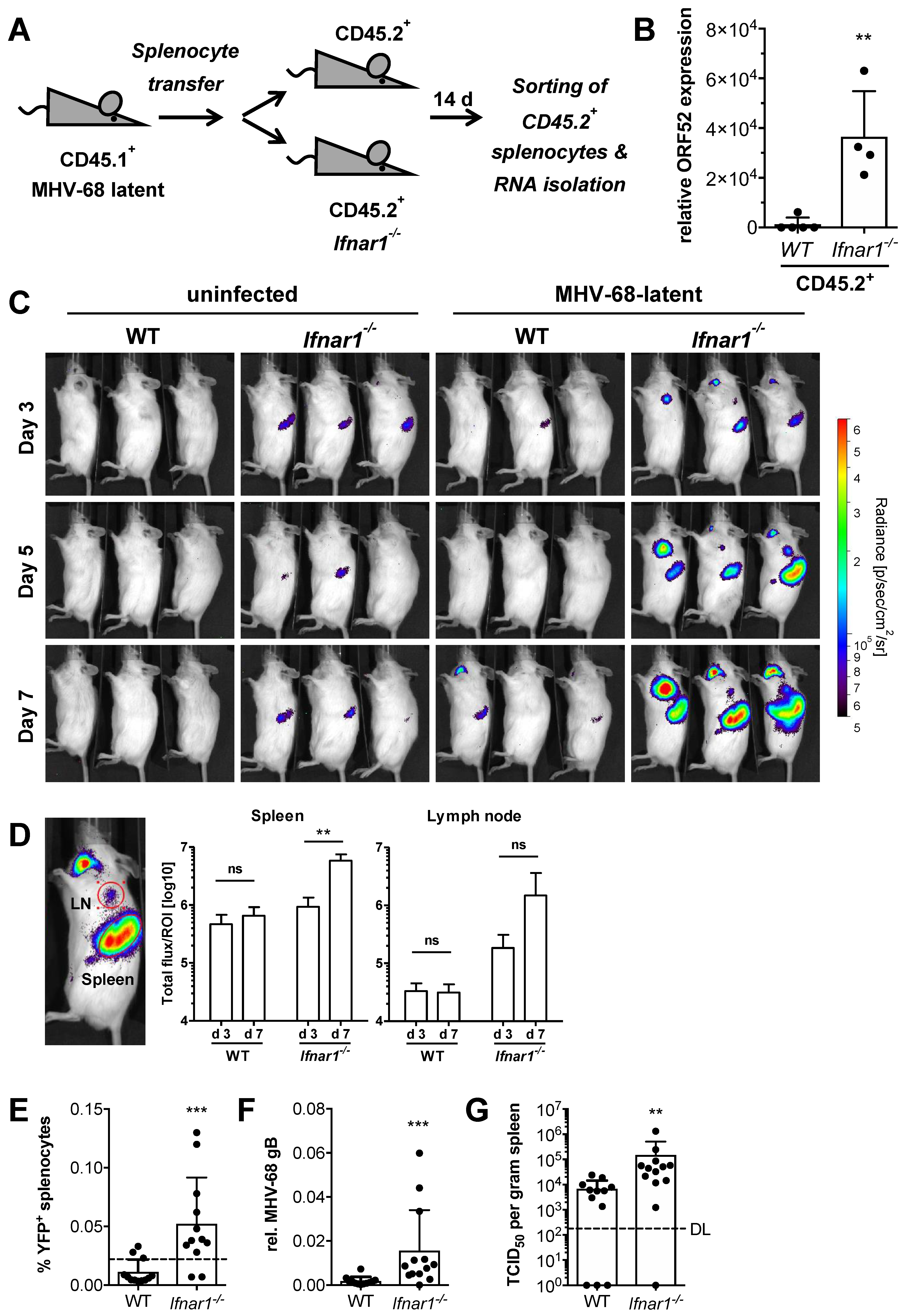
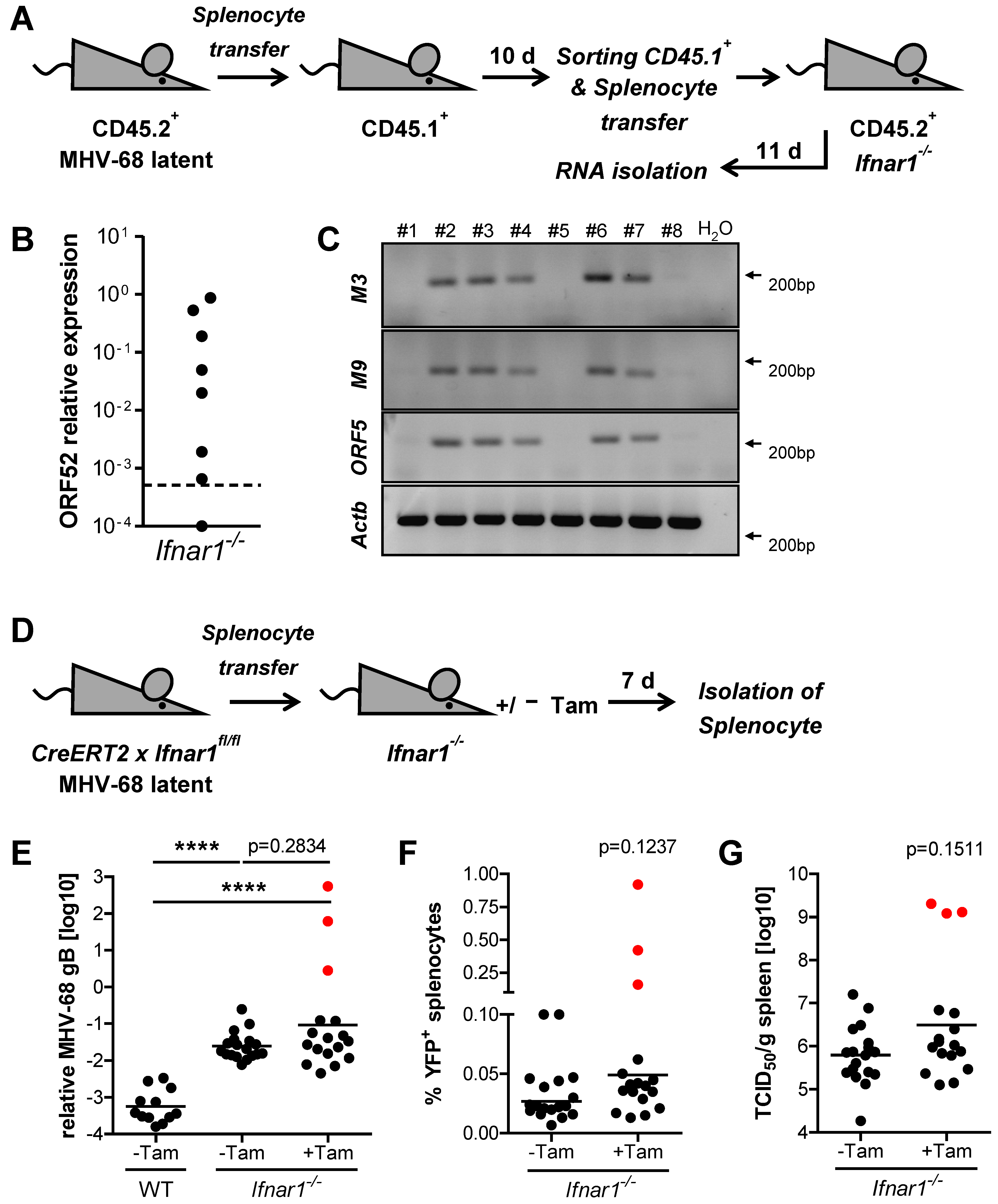
2.5. Type I IFN Signaling Controls MHV-68 Latency by Acting on Latently Infected Splenocytes
3. Discussion
4. Methods
4.1. Mice and Ethics Statement
4.2. Viruses, Infection, and Tamoxifen Treatment of Mice
4.3. Adoptive Transfer and Sorting of Splenocytes
4.4. Quantification of Luciferase Activity
4.5. Quantification of Virus Burden from Tissue Samples
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thorley-Lawson, D.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Minarovits, J. Epigenotypes of latent herpesvirus genomes. Curr. Top. Microbiol. Immunol. 2006, 310, 61–80. [Google Scholar] [PubMed]
- Souza, T.A.; Stollar, B.D.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Influence of EBV on the peripheral blood memory B cell compartment. J. Immunol. 2007, 179, 3153–3160. [Google Scholar] [CrossRef] [Green Version]
- Simas, J.P.; Efstathiou, S. Murine gammaherpesvirus 68: A model for the study of gammaherpesvirus pathogenesis. Trends Microbiol. 1998, 6, 276–282. [Google Scholar] [CrossRef]
- Speck, S.H.; Virgin, H.W. Host and viral genetics of chronic infection: A mouse model of gamma-herpesvirus pathogenesis. Curr. Opin. Microbiol. 1999, 2, 403–409. [Google Scholar] [CrossRef]
- Virgin, H.W.; Latreille, P.; Wamsley, P.; Hallsworth, K.; Weck, K.E.; Dal Canto, A.J.; Speck, S.H. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 1997, 71, 5894–5904. [Google Scholar] [CrossRef] [Green Version]
- Sunil-Chandra, N.P.; Arno, J.; Fazakerley, J.; Nash, A.A. Lymphoproliferative disease in mice infected with murine gammaherpesvirus 68. Am. J. Pathol. 1994, 145, 818–826. [Google Scholar]
- Flaño, E.; Woodland, D.L.; Blackman, M.A. A mouse model for infectious mononucleosis. Immunol. Res. 2002, 25, 201–217. [Google Scholar] [CrossRef]
- Martinez-Guzman, D.; Rickabaugh, T.; Wu, T.T.; Brown, H.; Cole, S.; Song, M.J.; Tong, L.; Sun, R. Transcription program of murine gammaherpesvirus 68. J. Virol. 2003, 77, 10488–10503. [Google Scholar] [CrossRef] [Green Version]
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the gamma-herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Flaño, E.; Husain, S.M.; Sample, J.T.; Woodland, D.L.; Blackman, M.A. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 2000, 165, 1074–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willer, D.O.; Speck, S.H. Long-term latent murine Gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 2003, 77, 8310–8321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardin, R.D.; Brooks, J.W.; Sarawar, S.R.; Doherty, P.C. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 1996, 184, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Freeman, M.L.; Burkum, C.E.; Yager, E.J.; Woodland, D.L.; Blackman, M.A. De Novo infection of B cells during murine gammaherpesvirus 68 latency. J. Virol. 2011, 85, 10920–10925. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.J.; Flaño, E.; Woodland, D.L.; Blackman, M.A. Antibody-mediated control of persistent gamma-herpesvirus infection. J. Immunol. 2002, 168, 3958–3964. [Google Scholar] [CrossRef] [Green Version]
- McClellan, K.B.; Gangappa, S.; Speck, S.H.; Virgin, H.W., IV. Antibody-independent control of γ-herpesvirus latency via B cell induction of anti-viral T cell responses. PLoS Pathog. 2006, 2, 578–590. [Google Scholar] [CrossRef]
- Weck, K.E.; Kim, S.S.; Virgin, H.W.; Speck, S.H. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 1999, 73, 4651–4661. [Google Scholar] [CrossRef] [Green Version]
- Barton, E.; Mandal, P.; Speck, S.H. Pathogenesis and host control of gammaherpesviruses: Lessons from the mouse. Annu. Rev. Immunol. 2011, 29, 351–397. [Google Scholar] [CrossRef]
- Jondle, C.N.; Tarakanova, V.L. Innate immunity and alpha/gammaherpesviruses: First impressions last a lifetime. Curr. Opin. Virol. 2020, 44, 81–89. [Google Scholar] [CrossRef]
- Usherwood, E.J.; Meadows, S.K.; Crist, S.G.; Bellfy, S.C.; Sentman, C.L. Control of murine gammaherpesvirus infection is independent of NK cells. Eur. J. Immunol. 2005, 35, 2956–2961. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.C.; Petrik, J.; Nash, A.A.; Dutia, B.M. Expansion and activation of NK cell populations in a gammaherpesvirus infection. Scand. J. Immunol. 2008, 67, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Adler, H.; Steer, B.; Juskewitz, E.; Kammerer, R. To the editor: Murine gammaherpesvirus 68 (MHV-68) escapes from NK-cell-mediated immune surveillance by a CEACAM1-mediated immune evasion mechanism. Eur. J. Immunol. 2014, 44, 2521–2522. [Google Scholar] [CrossRef] [PubMed]
- Lawler, C.; Tan, C.S.; Simas, J.P.; Stevenson, P.G. Type I Interferons and NK Cells Restrict Gammaherpesvirus Lymph Node Infection. J. Virol. 2016, 90, 9046–9057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, E.S.; Lutzke, M.L.; Rochford, R.; Virgin, H.W., IV. Alpha/Beta Interferons Regulate Murine Gammaherpesvirus Latent Gene Expression and Reactivation from Latency. J. Virol. 2005, 79, 14149–14160. [Google Scholar] [CrossRef] [Green Version]
- Sadler, A.J.; Williams, B.R.G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef]
- Dutia, B.M.; Allen, D.J.; Dyson, H.; Nash, A.A. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology 1999, 261, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Paden, C.R.; Forrest, J.C.; Moorman, N.J.; Speck, S.H. Murine gammaherpesvirus 68 LANA is essential for virus reactivation from splenocytes but not long-term carriage of viral genome. J. Virol. 2010, 84, 7214–7224. [Google Scholar] [CrossRef] [Green Version]
- Bennion, B.G.; Ingle, H.; Ai, T.L.; Miner, C.A.; Platt, D.J.; Smith, A.M.; Baldridge, M.T.; Miner, J.J. A Human Gain-of-Function STING Mutation Causes Immunodeficiency and Gammaherpesvirus-Induced Pulmonary Fibrosis in Mice. J. Virol. 2019, 93, e01806-18. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Kim, K.S.; Flano, E.; Wu, T.T.; Tong, L.M.; Park, A.N.; Song, M.J.; Sanchez, D.J.; O’Connell, R.M.; Cheng, G.; et al. Conserved Herpesviral Kinase Promotes Viral Persistence by Inhibiting the IRF-3-Mediated Type I Interferon Response. Cell Host Microbe 2009, 5, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.R.; Cheong, W.C.; Park, J.E.; Ryu, S.; Cho, H.J.; Youn, H.; Ahn, J.H.; Song, M.J. Murine gammaherpesvirus 68 encoding open reading frame 11 targets TANK binding kinase 1 to negatively regulate the host type I interferon response. J. Virol. 2014, 88, 6832–6846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.; Krueger, B.E.; Oldenburg, D.; Andry, K.A.; Beard, R.S.; White, D.W.; Barton, E.S. A gammaherpesvirus cooperates with interferon-alpha/beta-induced IRF2 to halt viral replication, control reactivation, and minimize host lethality. PLoS Pathog. 2011, 7, e1002371. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Schattgen, S.A.; Pisitkun, P.; Jorgensen, J.P.; Hilterbrand, A.T.; Wang, L.J.; West, J.A.; Hansen, K.; Horan, K.A.; Jakobsen, M.R.; et al. Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J. Immunol. 2015, 194, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Lienenklaus, S.; Cornitescu, M.; Zietara, N.; Łyszkiewicz, M.; Gekara, N.; Jabłónska, J.; Edenhofer, F.; Rajewsky, K.; Bruder, D.; Hafner, M.; et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J. Immunol. 2009, 183, 3229–3236. [Google Scholar] [CrossRef] [Green Version]
- Bortz, E.; Wang, L.; Jia, Q.; Wu, T.T.; Whitelegge, J.P.; Deng, H.; Zhou, Z.H.; Sun, R. Murine gammaherpesvirus 68 ORF52 encodes a tegument protein required for virion morphogenesis in the cytoplasm. J. Virol. 2007, 81, 10137–10150. [Google Scholar] [CrossRef] [Green Version]
- Pulverer, J.E.; Rand, U.; Lienenklaus, S.; Kugel, D.; Zietara, N.; Kochs, G.; Naumann, R.; Weiss, S.; Staeheli, P.; Hauser, H.; et al. Temporal and spatial resolution of type I and III interferon responses in vivo. J. Virol. 2010, 84, 8626–8638. [Google Scholar] [CrossRef] [Green Version]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-lambda Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890.e876. [Google Scholar] [CrossRef]
- Lopušná, K.; Benkóczka, T.; Lupták, J.; Matúšková, R.; Lukáčiková, Ľ.; Ovečková, I.; Režuchová, I. Murine gammaherpesvirus targets type I IFN receptor but not type III IFN receptor early in infection. Cytokine 2016, 83, 158–170. [Google Scholar] [CrossRef]
- Collins, C.M.; Speck, S.H. Tracking murine gammaherpesvirus 68 infection of germinal center B cells in vivo. PLoS ONE 2012, 7, e33230. [Google Scholar] [CrossRef] [Green Version]
- Virgin, H.W.; Speck, S.H. Unraveling immunity to gamma-herpesviruses: A new model for understanding the role of immunity in chronic virus infection. Curr. Opin. Immunol. 1999, 11, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Belz, G.T.; Doherty, P.C. Virus-specific and bystander CD8+ T-cell proliferation in the acute and persistent phases of a gammaherpesvirus infection. J. Virol. 2001, 75, 4435–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaño, E.; Kim, I.J.; Moore, J.; Woodland, D.L.; Blackman, M.A. Differential gamma-herpesvirus distribution in distinct anatomical locations and cell subsets during persistent infection in mice. J. Immunol. 2003, 170, 3828–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameyer, D.; Loonstra, A.; Eshkind, L.; Schmitt, S.; Antunes, C.; Groen, A.; Bindels, E.; Jonkers, J.; Krimpenfort, P.; Meuwissen, R.; et al. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol. Genom. 2007, 31, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Kamphuis, E.; Junt, T.; Waibler, Z.; Forster, R.; Kalinke, U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 2006, 108, 3253–3261. [Google Scholar] [CrossRef]
- Bhushal, S.; Wolfsmüller, M.; Selvakumar, T.A.; Kemper, L.; Wirth, D.; Hornef, M.W.; Hauser, H.; Köster, M. Cell Polarization and Epigenetic Status Shape the Heterogeneous Response to Type III Interferons in Intestinal Epithelial Cells. Front. Immunol. 2017, 8, 671. [Google Scholar] [CrossRef] [Green Version]
- Kugel, D.; Pulverer, J.E.; Köster, M.; Hauser, H.; Staeheli, P. Novel nonviral bioassays for mouse type I and type III interferon. J. Interferon Cytokine Res. 2011, 31, 345–349. [Google Scholar] [CrossRef]
- Schwerk, J.; Köster, M.; Hauser, H.; Rohde, M.; Fulde, M.; Hornef, M.W.; May, T. Generation of Mouse Small Intestinal Epithelial Cell Lines That Allow the Analysis of Specific Innate Immune Functions. PLoS ONE 2013, 8, e72700. [Google Scholar] [CrossRef] [Green Version]
- Rand, U.; Rinas, M.; Schwerk, J.; Nöhren, G.; Linnes, M.; Kröger, A.; Flossdorf, M.; Kály-Kullai, K.; Hauser, H.; Höfer, T.; et al. Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol. Syst. Biol. 2012, 8, 584. [Google Scholar] [CrossRef]
- Taniguchi, T.; Takaoka, A. A weak signal for strong responses: Interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol. 2001, 2, 378–386. [Google Scholar] [CrossRef]
- Salinas, E.; Gupta, A.; Sifford, J.M.; Oldenburg, D.G.; White, D.W.; Forrest, J.C. Conditional mutagenesis in vivo reveals cell type- and infection stage-specific requirements for LANA in chronic MHV68 infection. PLoS Pathog. 2018, 14, e1006865. [Google Scholar] [CrossRef] [PubMed]
- Sfogliarini, C.; Pepe, G.; Dolce, A.; Della Torre, S.; Cesta, M.C.; Allegretti, M.; Locati, M.; Vegeto, E. Tamoxifen Twists Again: On and off-Targets in Macrophages and Infections. Front. Pharmacol. 2022, 13, 879020. [Google Scholar] [CrossRef]
- Behjati, S.; Frank, M.H. The effects of tamoxifen on immunity. Curr. Med. Chem. 2009, 16, 3076–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussey, K.A.; Reimer, E.; Todt, H.; Denker, B.; Gallo, A.; Konrad, A.; Ottinger, M.; Adler, H.; Stürzl, M.; Brune, W.; et al. The gammaherpesviruses Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 modulate the Toll-like receptor-induced proinflammatory cytokine response. J. Virol. 2014, 88, 9245–9259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, L.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwerk, J.; Kemper, L.; Bussey, K.A.; Lienenklaus, S.; Weiss, S.; Čičin-Šain, L.; Kröger, A.; Kalinke, U.; Collins, C.M.; Speck, S.H.; et al. Type I Interferon Signaling Controls Gammaherpesvirus Latency In Vivo. Pathogens 2022, 11, 1554. https://doi.org/10.3390/pathogens11121554
Schwerk J, Kemper L, Bussey KA, Lienenklaus S, Weiss S, Čičin-Šain L, Kröger A, Kalinke U, Collins CM, Speck SH, et al. Type I Interferon Signaling Controls Gammaherpesvirus Latency In Vivo. Pathogens. 2022; 11(12):1554. https://doi.org/10.3390/pathogens11121554
Chicago/Turabian StyleSchwerk, Johannes, Lucas Kemper, Kendra A. Bussey, Stefan Lienenklaus, Siegfried Weiss, Luka Čičin-Šain, Andrea Kröger, Ulrich Kalinke, Christopher M. Collins, Samuel H. Speck, and et al. 2022. "Type I Interferon Signaling Controls Gammaherpesvirus Latency In Vivo" Pathogens 11, no. 12: 1554. https://doi.org/10.3390/pathogens11121554