
A Multi-Laboratory Comparison of Methods for Detection and Quantification of African Swine Fever Virus

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Detection and Quantification of Viral DNA by qPCR
2.2. Detection and Quantification of Virus Infectivity in Cells
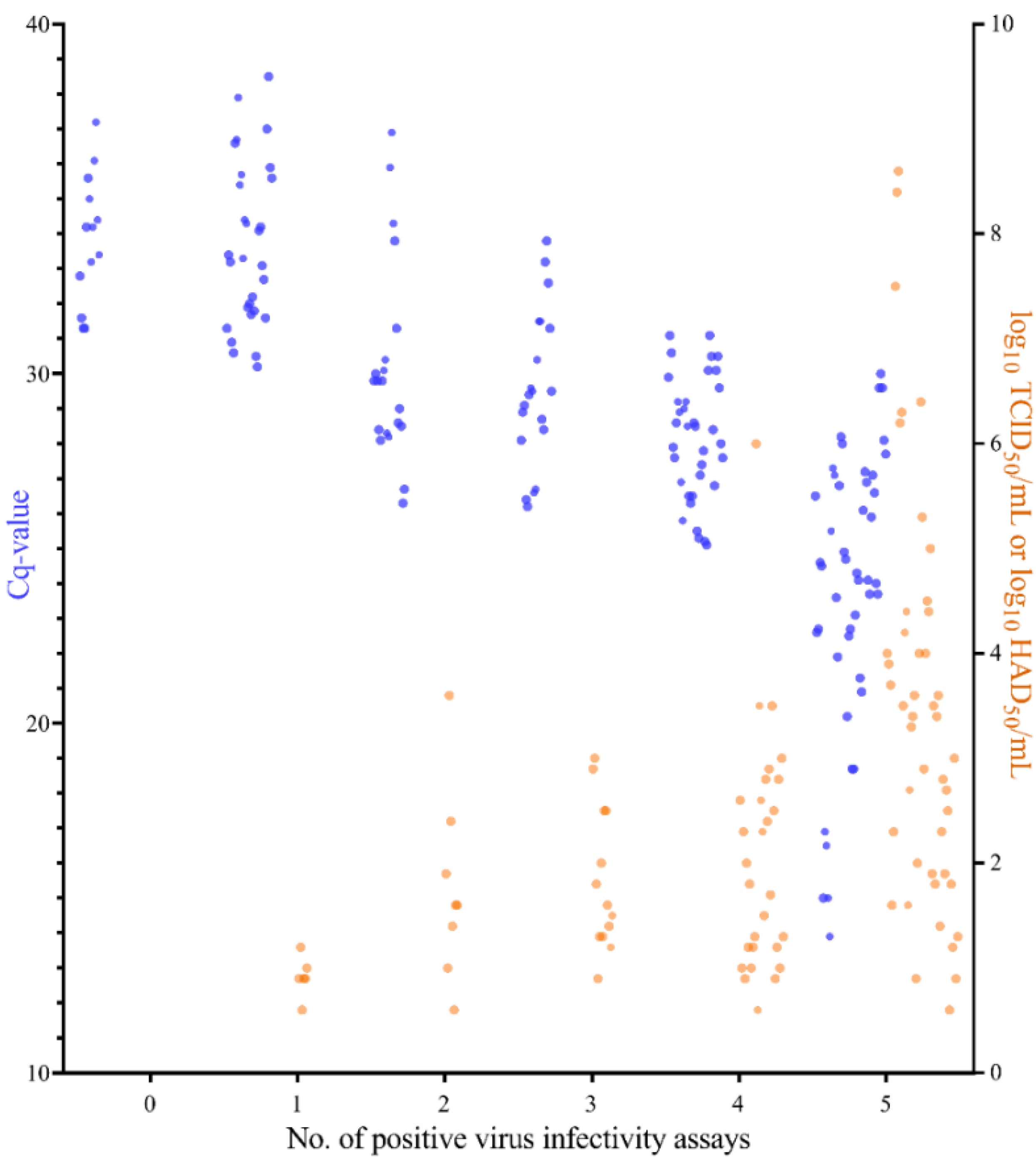
2.3. Comparison between qPCR and Virus Infectivity Assays
3. Discussion
4. Materials and Methods
4.1. Viruses
4.2. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.2.1. DTU
4.2.2. FLI
4.2.3. WBVR
4.3. Virus Infectivity Assays in Cells
4.3.1. DTU
4.3.2. FLI
4.3.3. WBVR
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alonso, C.; Borca, M.; Dixon, L.; Revilla, Y.; Rodriguez, F.; Escribano, J.M. ICTV Report Consortium. ICTV Virus Taxon. Profile Asfarviridae. J. Gen. Virol. 2018, 99, 613–614. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Sun, H.; Roberts, H. African swine fever. Antivir. Res. 2019, 165, 34–41. [Google Scholar] [CrossRef] [PubMed]
- OIE WAHIS Interface. Available online: https://wahis.oie.int/#/dashboards/country-or-disease-dashboard (accessed on 2 December 2021).
- Carrascosa, A.L.; Bustos, M.J.; de León, P. Methods for growing and titrating African swine fever virus: Field and laboratory samples. Curr. Protoc. Cell Biol. 2011, 53, 26.14.1–26.14.25. [Google Scholar] [CrossRef]
- De León, P.; Bustos, M.J.; Carrascosa, A.L. Laboratory methods to study African swine fever virus. Virus Res. 2013, 173, 168–179. [Google Scholar] [CrossRef]
- Enjuanes, L.; Carrascosa, A.L.; Moreno, M.A.; Viñuela, E. Titration of African swine fever (ASF) virus. J. Gen. Virol. 1976, 32, 471–477. [Google Scholar] [CrossRef]
- Hurtado, C.; Bustos, M.J.; Carrascosa, A.L. The use of COS-1 cells for studies of field and laboratory African swine fever virus samples. J. Virol. Methods 2010, 164, 131–134. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho Ferreira, H.C.; Weesendorp, E.; Elbers, A.; Bouma, A.; Quak, S.; Stegeman, J.; Loeffen, W. African swine fever virus excretion patterns in persistently infected animals: A quantitative approach. Vet. Microbiol. 2012, 160, 327–340. [Google Scholar] [CrossRef]
- Rai, A.; Pruitt, S.; Ramirez-Medina, E.; Vuono, E.A.; Silva, E.; Velazquez-Salinas, L.; Carrillo, C.; Borca, M.V.; Gladue, D.P. Identification of a continuously stable and commercially available cell line for the identification of infectious African swine fever virus in clinical samples. Viruses 2020, 12, 820. [Google Scholar] [CrossRef]
- Keil, G.M.; Giesow, K.; Portugal, R. A novel bromodeoxyuridine-resistant wild boar lung cell line facilitates generation of African swine fever virus recombinant. Arch. Virol. 2014, 159, 2421–2428. [Google Scholar] [CrossRef]
- Portugal, R.; Martins, C.; Keil, G.M. Novel approach for the generation of recombinant African swine fever virus from a field isolate using GFP expression and 5-bromo-2′-deoxyuridine selection. J. Virol. Methods 2012, 183, 86–89. [Google Scholar] [CrossRef]
- Weingartl, H.M.; Sabara, M.; Pasick, J.; van Moorlehem, E.; Babiuk, L. Continuous porcine cell lines developed from alveolar macrophages: Partial characterization and virus susceptibility. J. Virol. Methods 2002, 104, 203–216. [Google Scholar] [CrossRef]
- Portugal, R.; Goatley, L.C.; Husmann, R.; Zuckermann, F.A.; Dixon, L.K. A porcine macrophage cell line that supports high levels of replication of OURT88/3, an attenuated strain of African swine fever virus. Emerg. Microbes Infect. 2020, 9, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Masujin, K.; Kitamura, T.; Kameyama, K.; Okadera, K.; Nishi, T.; Takenouchi, T.; Kitan, H.; Kokuho, T. An immortalized porcine macrophage cell line competent for the isolation of African swine fever virus. Sci. Rep. 2021, 11, 4759. [Google Scholar] [CrossRef]
- Malmquist, W.A.; Hay, D. Hemadsorption and cytopathic effect produced by African swine fever virus in swine bone marrow and buffy coat cultures. Am. J. Vet. Res. 1960, 21, 104–108. [Google Scholar] [PubMed]
- Heuschele, W.P.; Hess, W.R. Diagnosis of African swine fever by immunofluorescence. Trop. Anim. Health Prod. 1973, 5, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Wensvoort, G.; Terpstra, C.; Bloemraad, M. Detection of antibodies against African swine fever virus using infected monolayers and monoclonal antibodies. Vet. Rec. 1988, 122, 536–539. [Google Scholar] [CrossRef] [PubMed]
- King, D.P.; Reid, S.M.; Hutchings, G.H.; Grierson, S.S.; Wilkinson, P.J.; Dixon, L.K.; Bastos, A.D.S.; Drew, T.W. Development of a TaqMan® PCR assay with internal amplification control for the detection of African swine fever virus. J. Virol. Methods 2003, 107, 53–61. [Google Scholar] [CrossRef]
- Steiger, Y.; Ackermann, M.; Mettraux, C.; Kihm, U. Rapid and biologically safe diagnosis of African swine fever virus infection by using polymerase chain reaction. J. Clin. Microbiol. 1992, 30, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Tignon, M.; Gallardo, C.; Iscaro, C.; Hutet, E.; Van der Stede, Y.; Kolbasov, D.; Mario de Mia, G.; Le Potier, M.-F.; Bishop, R.P.; Arias, M.; et al. Development and inter-laboratory validation study of an improved new real-time PCR assay with internal control for detection and laboratory diagnosis of African swine fever virus. J. Virol. Methods 2011, 178, 161–170. [Google Scholar] [CrossRef]
- Pfaffl, M.W. Quantification strategies in real-time PCR. In A-Z of Quantitative PCR; Pfaffl, S.A., Ed.; International University Line (IUL): La Jolla, CA, USA, 2004; pp. 87–111. [Google Scholar]
- Koo, T.K.; Li, M.Y. A guideline of selecting and reporting intraclass correlation coefficients for reliability research. J. Chiropr. Med. 2016, 15, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Olesen, A.S.; Lohse, L.; Boklund, A.; Halasa, T.; Belsham, G.J.; Rasmussen, T.B.; Bøtner, A. Short time window for transmissibility of African swine fever virus from a contaminated environment. Transbound. Emerg. Dis. 2018, 65, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Olesen, A.S. (Department of Virus & Microbiological Special Diagnostics, Statens Serum Institut, DK-2300 Copenhagen S, Denmark); Bøtner, A. (Department of Virus & Microbiological Special Diagnostics, Statens Serum Institut, DK-2300 Copenhagen S, Denmark). Unpublished work. 2015. [Google Scholar]
- Rodríguez, J.M.; Moreno, L.T.; Alejo, A.; Lacasta, A.; Rodríguez, F.; Salas, M.L. Genome sequence of African swine fever virus BA71, the virulent parental strain of the nonpathogenic and tissue-culture adapted BA71V. PLoS ONE 2015, 10, e0142889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Torres, C.; Gómez-Puertas, P.; Góomez-del-Moral, M.; Alonso, F.; Escribano, J.M.; Ezquerra, A.; Domínguez, J. Expression of porcine CD163 on monocytes/macrophages correlates with permissiveness to African swine fever infection. Arch. Virol. 2003, 148, 2307–2323. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.C.; Basta, S.; Knötig, S.; Gerber, H.; Schaffner, R.; Kim, Y.B.; Saalmüller, A. Intermediate stages in monocyte–macrophage differentiation modulate phenotype and susceptibility to virus infection. Immunology 1999, 98, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Fernández-Pinero, J.; Arias, M. African swine fever (ASF) diagnosis, an essential tool in the epidemiological investigation. Virus Res. 2019, 271, 197676. [Google Scholar] [CrossRef]
- Suspéne, R.; Aynaud, M.-M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.-P.; Meyerhans, A.; Wain-Hobson, S. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef] [Green Version]
- Döhner, K.; Radtke, K.; Schmidt, S.; Sodeik, B. Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein VP26. J. Virol. 2006, 80, 8211–8224. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, J.E.; Henderson, E.P.; Grose, C. Enumeration of an extremely high particle-to-PFU ratio for Varicella-zoster virus. J. Virol. 2009, 83, 6917–6921. [Google Scholar] [CrossRef] [Green Version]
- Bourry, O.; Hutet, E.; Le Dimna, M.; Paboeuf, F.; The Potter, M.F. Evaluation of ASF virus inactivation. In Proceedings of the Workshop on Laboratory Diagnostics and Control of CSF and ASF, Hannover, Germany, 29–30 May 2018. [Google Scholar]
- Petrini, S.; Feliziani, F.; Casciari, C.; Giammarioli, M.; Torresi, C.; Mario De Mia, G. Survival of African swine fever virus (ASFV) in various traditional Italian dry-cured meat products. Prev. Vet. Med. 2019, 162, 126–130. [Google Scholar] [CrossRef]
- Olesen, A.S.; Lohse, L.; Hansen, M.F.; Boklund, A.; Halasa, T.; Belsham, G.J.; Rasmussen, T.B.; Bøtner, A.; Bødker, R. Infection of pigs with African swine fever virus via ingestion of stable flies (Stomoxys calcitrans). Transbound. Emerg. Dis. 2018, 65, 1152–1157. [Google Scholar] [CrossRef] [Green Version]
- Olesen, A.S.; Hansen, M.F.; Rasmussen, T.B.; Belsham, G.J.; Bødker, R.; Bøtner, A. Survival and localization of African swine fever virus in stable flies (Stomoxys calcitrans) after feeding on viremic blood using a membrane feeder. Vet. Microbiol. 2018, 222, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Mebus, C.A.; Dardiri, A.H.; Hamdy, F.M.; Ferris, D.H.; Hess, W.R.; Callis, J.J. Some characteristics of African swine fever viruses isolated from Brazil and the Dominican Republic. Proc. Annu. Meet. US Anim. Health Assoc. 1978, 82, 232–236. [Google Scholar]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Lubisi, B.A.; Bastos, A.D.S.; Dwarka, R.M.; Vosloo, W. Molecular epidemiology of African swine fever in East Africa. Arch. Virol. 2005, 150, 2439–2452. [Google Scholar] [CrossRef]
- Simulundu, E.; Lubaba, C.H.; van Heerden, J.; Kajihara, M.; Mataa, L.; Chambaro, H.M.; Sinkala, Y.; Munjita, S.M.; Munang’andu, H.M.; Nalubamba, K.S.; et al. The epidemiology of African swine fever in “nonendemic” regions of Zambia (1989–2015): Implications for disease prevention and control. Viruses 2017, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, P.J.; Lawman, M.J.; Johnston, R.S. African swine fever in Malta, 1978. Vet. Rec. 1980, 106, 94–97. [Google Scholar] [CrossRef]
- Terpstra, C.; Wensvoort, G. African swine fever in the Netherlands. Tijdschr. voor Diergeneeskd. 1986, 111, 389–392. [Google Scholar]
- Boinas, F.S.; Hutchings, G.H.; Dixon, L.K.; Wilkinson, P.J. Characterization of pathogenic and non-pathogenic African swine fever virus isolates from Ornithodoros erraticus inhabiting pig premises in Portugal. J. Gen. Virol. 2004, 85, 2177–2187. [Google Scholar] [CrossRef]
- Bøtner, A.; Nielsen, J.; Bille-Hansen, V. Isolation of porcine reproductive and respiratory syndrome (PRRS) virus in a Danish swine herd and experimental infection of pregnant gilts with the virus. Vet. Microbiol. 1994, 40, 351–360. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method for estimating fifty percent endpoints. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]
- Pietschmann, J.; Guinat, C.; Beer, M.; Pronin, V.; Tauscher, K.; Petrov, A.; Keil, G.; Blome, S. Course and transmission characteristics of oral low-dose infection of domestic pigs and European wild boar with a caucasian African swine fever virus isolate. Arch. Virol. 2015, 60, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Wensvoort, G.; Terpstra, C.; Pol, J.M.A.; ter Lak, E.A.; Bloemraad, M.; de Kluyver, E.P.; Kragten, C.; van Buiten, L.; den Besten, A.; Wagenaar, F.; et al. Mystery swine disease in the Netherlands: The isolation of Lelystad virus. Vet. Q. 1991, 13, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Revelle, W. Psych: Procedures for Personality and Psychological Research, Version 2.0.8.; Northwestern University: Evanston, IL, USA, 2020; Available online: https://CRAN.R-project.org/package=psych (accessed on 1 August 2020).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 1 August 2020).



{kind=link}
{kind=link}
| Test | Method/Assay | Laboratory | TP | FN | Probability of Detecting ASFV % |
|---|---|---|---|---|---|
| qPCR | 1 | DTU | 80 | 0 | 100 |
| 1 | DTU | 80 | 0 | 100 | |
| 2 | FLI | 80 | 0 | 100 | |
| 3 | FLI | 77 | 3 | 96 | |
| 4 | WBVR | 79 | 1 | 99 | |
| 4 | WBVR | 80 | 0 | 100 | |
| 5 | WBVR | 78 | 2 | 98 | |
| 5 | WBVR | 80 | 0 | 100 | |
| VI | PBMCs | FLI | 60 | 20 | 75 |
| PAMs | WBVR | 72 | 8 | 90 | |
| PAMs | DTU | 67 | 13 | 84 | |
| WSL | FLI | 54 | 26 | 68 | |
| MARC145 | WBVR | 59 | 21 | 74 | |
| MARC145 | DTU | 63 | 17 | 79 | |
| MARC145 | DTU | 68 | 12 | 85 |
| VI Assay Cell Type | qPCR Quantification Method | ||
|---|---|---|---|
| Method 1 (DTU) | Method 4 (WBVR) | Method 5 (WBVR) | |
| PBMCs (FLI) | 0.93 (0.90–0.95) | 0.82 (0.75–0.87) | 0.86 (0.81–0.90) |
| PAMs (WBVR) | 0.91 (0.87–0.93) | 0.79 (0.71–0.85) | 0.81 (0.74–0.87) |
| PAMs (DTU) | 0.91 (0.87–0.93) | 0.79 (0.71–0.85) | 0.85 (0.79–0.90) |
| WSL (FLI) | 0.82 (0.75–0.87) | 0.77 (0.68–0.83) | 0.80 (0.73–0.86) |
| MARC (WBVR) | 0.91 (0.87–0.94) | 0.80 (0.72–0.86) | 0.86 (0.80–0.90) |
| MARC (DTU) | 0.95 (0.93–0.96) | 0.89 (0.84–0.92) | 0.89 (0.84–0.92) |
| Legend | |||
| excellent (ICC > 0.90) | |||
| good to excellent (ICC 0.75–0.90) | |||
| good (ICC 0.75–0.90) | |||
| moderate to good (ICC < 0.75–0.90) | |||
| Virus | Country of Origin | Species of Origin | P72 Genotype | Reference |
|---|---|---|---|---|
| Brazil’78 | Brazil | Suidae | I | [36] |
| Georgia 2007/1 | Georgia 2007/1 | Suidae | II | [37] |
| LIV 13/33 | Zambia | Ornithodorus | I | [38,39] |
| Malta’78 | Malta | Suidae | I | [40] |
| Netherlands’86 | The Netherlands | Suidae | I | [41] |
| OUR T88/1 | Portugal | Ornithodorus | I | [42] |
| DTU Method 1 | FLI Method 2 | FLI Method 3 | WBVR Method 4 | WBVR Method 5 | |
|---|---|---|---|---|---|
| Extraction | |||||
| Input | 200 µL | 140 µL | 140 µL | 200 µL | 200 µL |
| Extraction kit | MagNA Pure 96 DNA/Viral NA S.V. 2.0 Kit (Roche, Basel, Switzerland) | QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) | QIAamp Viral RNA Mini Kit (Qiagen) | MagNA Pure LC Total Nucleic Acid Isolation Kit (Roche) | MagNA Pure LC Total Nucleic Acid Isolation Kit (Roche) |
| Platform | MagNA Pure 96 instrument (Roche) | na | na | MagNA Pure LC instrument (Roche) | MagNA Pure LC instrument (Roche) |
| Protocol | Viral NA Plasma extern lysis S.V. 3.1. | na | na | na | na |
| Elution volume | 50 µL | 50 µL | 50 µL | 100 µL | 100 µL |
| qPCR | |||||
| Input | 5 µL | 5 µL | 5 µL | 10 µL | 10 µL |
| Primers | ASF-P72 (1) | ASF-P72 (1) | ASF-P72 (2) | ASFV-p72p3 and ASFV-p72p4 (3) | ASFV-p72p3 and ASFV-p72p4 (3) |
| Probes | ASF-P72-FAM (1) | ASF-P72-FAM (1) | ASF-P72-FAM (2) | ASFV-p72 LC-FL and ASFV-p72 LC-Red640 (3) | ASFV-p72 LC-FL and ASFV-p72 LC-Red640 (3) |
| PCR kit | QuantiTectTM Multiplex PCR kit (Qiagen) (1) | QuantiTectTM Multiplex PCR kit (Qiagen) (1) | QuantiTectTM Multiplex PCR kit (Qiagen) (2) | DNA kit (Roche) | Quantifast Probe RT-PCR kit (Qiagen) |
| Platform | Mx3005P qPCR system (Agilent Technologies, Santa Clara, CA, USA) | CFX 96 Real-Time System (Bio-Rad, Hercules, CA, USA) | CFX 96 Real-Time System (Bio-Rad) | LightCycler® 480 (Roche) | LightCycler® 480 (Roche) |
| Cycle conditions | 2 min–15 min–1 min–1 min (last two steps 45 cycles) | 15 min–1 min–1 min (last two steps 45 cycles) | 15 min–1 min–1 min (last two steps 45 cycles) | 10 min–1 sec–10 sec–10 sec (last three steps 45 cycles) | 10 min–1 sec–10 sec–10 sec (last three steps 45 cycles) |
| Cycle temperature | 50–95–94–60 °C | 95–95–60 °C | 95–95–60 °C | 95–95–59–72 °C | 95–95–59–72 °C |
| DTU | DTU | FLI | FLI | WBVR | WBVR | |
|---|---|---|---|---|---|---|
| Cells | PAMs | MARC145 | PBMCs | WSL | PAMs | MARC145 |
| Plates | 96 well | 96 well | 96 well | 96 well | 24 well | 96 well |
| Cells/well | 2 × 105 | 4 × 104 | 5 × 104 | 1.5 × 105 | 1 × 106 | 2.1 × 105 |
| Detection method | IPMA | IPMA | HAT | IFA | HAT | IPMA |
| Sample amount/well | 50 µL | 50 µL | 100 µL | 100 µL | 125 µL | 100 µL |
| Incubation | 2 days | 3 days | 3 days | 3 days | 3–7 days | 3 days |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olesen, A.S.; Bruun Rasmussen, T.; Saxmose Nielsen, S.; Belsham, G.J.; Boklund, A.; Ploegaert, T.; Moonen-Leusen, B.; Blome, S.; Bøtner, A. A Multi-Laboratory Comparison of Methods for Detection and Quantification of African Swine Fever Virus. Pathogens 2022, 11, 325. https://doi.org/10.3390/pathogens11030325
Olesen AS, Bruun Rasmussen T, Saxmose Nielsen S, Belsham GJ, Boklund A, Ploegaert T, Moonen-Leusen B, Blome S, Bøtner A. A Multi-Laboratory Comparison of Methods for Detection and Quantification of African Swine Fever Virus. Pathogens. 2022; 11(3):325. https://doi.org/10.3390/pathogens11030325
Chicago/Turabian StyleOlesen, Ann Sofie, Thomas Bruun Rasmussen, Søren Saxmose Nielsen, Graham J. Belsham, Anette Boklund, Tosca Ploegaert, Bernie Moonen-Leusen, Sandra Blome, and Anette Bøtner. 2022. "A Multi-Laboratory Comparison of Methods for Detection and Quantification of African Swine Fever Virus" Pathogens 11, no. 3: 325. https://doi.org/10.3390/pathogens11030325