
The Core Human Microbiome: Does It Exist and How Can We Find It? A Critical Review of the Concept

 , ,
, ,  , , ,
, , ,  , , ,
, , ,
Abstract
:1. Introduction
2. Defining the Core Microbiome
3. The Data: Mostly Genomics, Western-Focused, and Fecal-Based
3.1. Technology: Mostly Genomics
3.2. Population: Western-Focused
3.3. Sample Source: Mostly Fecal
4. The Methods: Low-Resolution Amplicon Studies, Challenging Metagenomics Bioinformatics, and Difficult to Mine Public Databases
4.1. 16S rRNA Gene Surveys: Established Bioinformatics, and Low-Resolution and Partial Information
4.2. Metagenomics: High-Resolution Taxonomic and Functional Data, Challenging Bioinformatics
4.3. Public Microbiome Datasets Are Often Hardly Interoperable and Reusable
5. Beyond Prokaryotic Omics: Interactions of the Core Human Microbiome with Its Host, and with Other Members of the Microbiome
5.1. Specific Effects of Diet on the Microbiome
5.2. The Immune System and Its Interactions with the Microbiome
5.3. The Less Explored Members of the Microbiome
5.3.1. The Role of Eukaryotes in the Human Microbiome
5.3.2. The Host Eukaryotic Virome and the Gut Microbiome
5.3.3. The Prokaryotic Virome (Phageome) and Its Relation to the Gut Microbiome
6. Discussion
7. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krajmalnik-Brown, R.; Ilhan, Z.-E.; Kang, D.-W.; DiBaise, J.K. Effects of gut microbes on nutrient absorption and energy regulation. Nutr. Clin. Pract. 2012, 27, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Iacob, S.; Iacob, D.G.; Luminos, L.M. Intestinal microbiota as a host defense mechanism to infectious threats. Front. Microbiol. 2018, 9, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Cenit, M.C.; Sanz, Y.; Codoñer-Franch, P. Influence of gut microbiota on neuropsychiatric disorders. World J. Gastroenterol. 2017, 23, 5486–5498. [Google Scholar] [CrossRef]
- Integrative HMP (iHMP) Research Network Consortium The integrative human microbiome project. Nature 2019, 569, 641–648. [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- The MetaHIT Consortium; Ehrlich, S.D. Metahit: The european union project on metagenomics of the human intestinal tract. In Metagenomics of the Human Body; Nelson, K.E., Ed.; Springer: New York, NY, USA, 2011; pp. 307–316. ISBN 978-1-4419-7088-6. [Google Scholar]
- Pitlik, S.D.; Koren, O. How holobionts get sick-toward a unifying scheme of disease. Microbiome 2017, 5, 64. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Gordon, J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, F.; Gossmann, T.I.; Frioux, C.; Özkurt, E.; Myers, P.N.; Ferretti, P.; Kuhn, M.; Bahram, M.; Nielsen, H.B.; Bork, P. Dispersal strategies shape persistence and evolution of human gut bacteria. Cell Host Microbe 2021, 29, 1167–1176.e9. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The long-term stability of the human gut microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [Green Version]
- Yassour, M.; Jason, E.; Hogstrom, L.J.; Arthur, T.D.; Tripathi, S.; Siljander, H.; Selvenius, J.; Oikarinen, S.; Hyöty, H.; Virtanen, S.M.; et al. Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe 2018, 24, 146–154.e4. [Google Scholar] [CrossRef] [Green Version]
- Zaura, E.; Keijser, B.J.F.; Huse, S.M.; Crielaard, W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009, 9, 259. [Google Scholar] [CrossRef] [Green Version]
- Björk, J.R.; O’Hara, R.B.; Ribes, M.; Coma, R.; Montoya, J.M. The dynamic core microbiome: Structure, stability and resistance. BioRxiv 2018, 137855. [Google Scholar] [CrossRef]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Risely, A. Applying the core microbiome to understand host-microbe systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Fraser, C.M.; Ringel, Y.; Sanders, M.E.; Sartor, R.B.; Sherman, P.M.; Versalovic, J.; Young, V.; Finlay, B.B. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 2012, 12, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 2021, 70, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Lemanceau, P.; Blouin, M.; Muller, D.; Moënne-Loccoz, Y. Let the core microbiota be functional. Trends Plant Sci. 2017, 22, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Tap, J.; Mondot, S.; Levenez, F.; Pelletier, E.; Caron, C.; Furet, J.-P.; Ugarte, E.; Muñoz-Tamayo, R.; Paslier, D.L.E.; Nalin, R.; et al. Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 2009, 11, 2574–2584. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Bihan, M.; Methé, B.A. Analyses of the stability and core taxonomic memberships of the human microbiome. PLoS ONE 2013, 8, e63139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Ze, X.; Duncan, S.H.; Louis, P.; Flint, H.J. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012, 6, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, N.; Garrido, D. Species Deletions from Microbiome Consortia Reveal Key Metabolic Interactions between Gut Microbes. mSystems 2019, 4, e00185-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Sharma, A.K.; Petrzelkova, K.; Pafco, B.; Jost Robinson, C.A.; Fuh, T.; Wilson, B.A.; Stumpf, R.M.; Torralba, M.G.; Blekhman, R.; White, B.; et al. Traditional human populations and nonhuman primates show parallel gut microbiome adaptations to analogous ecological conditions. mSystems 2020, 5, e00815-20. [Google Scholar] [CrossRef]
- Xiao, L.; Feng, Q.; Liang, S.; Sonne, S.B.; Xia, Z.; Qiu, X.; Li, X.; Long, H.; Zhang, J.; Zhang, D.; et al. A catalog of the mouse gut metagenome. Nat. Biotechnol. 2015, 33, 1103–1108. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Y.; Zhu, B.; Gao, G.F.; Guo, Y.; Hu, Y. Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Commun. Biol. 2021, 4, 1305. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- De Filippis, F.; Pasolli, E.; Ercolini, D. Newly explored faecalibacterium diversity is connected to age, lifestyle, geography, and disease. Curr. Biol. 2020, 30, 4932–4943.e4. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashiardes, S.; Zilberman-Schapira, G.; Elinav, E. Use of metatranscriptomics in microbiome research. Bioinform. Biol. Insights 2016, 10, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdill, R.J.; Adamowicz, E.M.; Blekhman, R. Public human microbiome data are dominated by highly developed countries. PLoS Biol. 2022, 20, e3001536. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium A framework for human microbiome research. Nature 2012, 486, 215–221. [CrossRef] [Green Version]
- Zijlema, W.L.; Smidt, N.; Klijs, B.; Morley, D.W.; Gulliver, J.; de Hoogh, K.; Scholtens, S.; Rosmalen, J.G.M.; Stolk, R.P. The LifeLines Cohort Study: A resource providing new opportunities for environmental epidemiology. Arch. Public Health 2016, 74, 32. [Google Scholar] [CrossRef] [Green Version]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Tyakht, A.V.; Alexeev, D.G.; Popenko, A.S.; Kostryukova, E.S.; Govorun, V.M. Rural and urban microbiota: To be or not to be? Gut Microbes 2014, 5, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Saarenpää, M.; Roslund, M.I.; Puhakka, R.; Grönroos, M.; Parajuli, A.; Hui, N.; Nurminen, N.; Laitinen, O.H.; Hyöty, H.; Cinek, O.; et al. The Adele Research Group Do Rural Second Homes Shape Commensal Microbiota of Urban Dwellers? A Pilot Study among Urban Elderly in Finland. Int. J. Environ. Res. Public Health 2021, 18, 3742. [Google Scholar] [CrossRef] [PubMed]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Schnorr, S.L.; Consolandi, C.; Turroni, S.; Severgnini, M.; Peano, C.; Brigidi, P.; Crittenden, A.N.; Henry, A.G.; Candela, M. Metagenome Sequencing of the Hadza Hunter-Gatherer Gut Microbiota. Curr. Biol. 2015, 25, 1682–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iebba, V.; Santangelo, F.; Totino, V.; Pantanella, F.; Monsia, A.; Di Cristanziano, V.; Di Cave, D.; Schippa, S.; Berrilli, F.; D’Alfonso, R. Gut microbiota related to Giardia duodenalis, Entamoeba spp. and Blastocystis hominis infections in humans from Côte d’Ivoire. J. Infect. Dev. Ctries. 2016, 10, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Gomez, A.; Petrzelkova, K.J.; Burns, M.B.; Yeoman, C.J.; Amato, K.R.; Vlckova, K.; Modry, D.; Todd, A.; Jost Robinson, C.A.; Remis, M.J.; et al. Gut microbiome of coexisting baaka pygmies and bantu reflects gradients of traditional subsistence patterns. Cell Rep. 2016, 14, 2142–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, I.; Stegen, J.C.; Maldonado-Gómez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The gut microbiota of rural papua new guineans: Composition, diversity patterns, and ecological processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [Green Version]
- Das, B.; Ghosh, T.S.; Kedia, S.; Rampal, R.; Saxena, S.; Bag, S.; Mitra, R.; Dayal, M.; Mehta, O.; Surendranath, A.; et al. Analysis of the gut microbiome of rural and urban healthy indians living in sea level and high altitude areas. Sci. Rep. 2018, 8, 10104. [Google Scholar] [CrossRef]
- Ayeni, F.A.; Biagi, E.; Rampelli, S.; Fiori, J.; Soverini, M.; Audu, H.J.; Cristino, S.; Caporali, L.; Schnorr, S.L.; Carelli, V.; et al. Infant and Adult Gut Microbiome and Metabolome in Rural Bassa and Urban Settlers from Nigeria. Cell Rep. 2018, 23, 3056–3067. [Google Scholar] [CrossRef]
- Obregon-Tito, A.J.; Tito, R.Y.; Metcalf, J.; Sankaranarayanan, K.; Clemente, J.C.; Ursell, L.K.; Zech Xu, Z.; Van Treuren, W.; Knight, R.; Gaffney, P.M.; et al. Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun. 2015, 6, 6505. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippo, C.; Di Paola, M.; Ramazzotti, M.; Albanese, D.; Pieraccini, G.; Banci, E.; Miglietta, F.; Cavalieri, D.; Lionetti, P. Diet, environments, and gut microbiota. A preliminary investigation in children living in rural and urban burkina faso and italy. Front. Microbiol. 2017, 8, 1979. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, J.; Yamamoto, A.; Palermo-Conde, L.A.; Higashi, K.; Sonomoto, K.; Tan, J.; Lee, Y.-K. Impact of westernized diet on gut microbiota in children on leyte island. Front. Microbiol. 2017, 8, 197. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmora, N.; Zilberman-Schapira, G.; Suez, J.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Kotler, E.; Zur, M.; Regev-Lehavi, D.; Brik, R.B.-Z.; et al. Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics Is Associated with Unique Host and Microbiome Features. Cell 2018, 174, 1388–1405.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Stearns, J.C.; Lynch, M.D.J.; Senadheera, D.B.; Tenenbaum, H.C.; Goldberg, M.B.; Cvitkovitch, D.G.; Croitoru, K.; Moreno-Hagelsieb, G.; Neufeld, J.D. Bacterial biogeography of the human digestive tract. Sci. Rep. 2011, 1, 170. [Google Scholar] [CrossRef] [Green Version]
- Zoetendal, E.G.; Raes, J.; van den Bogert, B.; Arumugam, M.; Booijink, C.C.G.M.; Troost, F.J.; Bork, P.; Wels, M.; de Vos, W.M.; Kleerebezem, M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012, 6, 1415–1426. [Google Scholar] [CrossRef]
- Albenberg, L.; Esipova, T.V.; Judge, C.P.; Bittinger, K.; Chen, J.; Laughlin, A.; Grunberg, S.; Baldassano, R.N.; Lewis, J.D.; Li, H.; et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 2014, 147, 1055–1063.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espey, M.G. Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic. Biol. Med. 2013, 55, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Georgieva, D.C.; Egli, D.; Wang, K. NanoMod: A computational tool to detect DNA modifications using Nanopore long-read sequencing data. BMC Genom. 2019, 20, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepage, P.; Seksik, P.; Sutren, M.; de la Cochetière, M.-F.; Jian, R.; Marteau, P.; Doré, J. Biodiversity of the mucosa-associated microbiota is stable along the distal digestive tract in healthy individuals and patients with IBD. Inflamm. Bowel Dis. 2005, 11, 473–480. [Google Scholar] [CrossRef]
- Green, G.L.; Brostoff, J.; Hudspith, B.; Michael, M.; Mylonaki, M.; Rayment, N.; Staines, N.; Sanderson, J.; Rampton, D.S.; Bruce, K.D. Molecular characterization of the bacteria adherent to human colorectal mucosa. J. Appl. Microbiol. 2006, 100, 460–469. [Google Scholar] [CrossRef]
- Altomare, A.; Putignani, L.; Del Chierico, F.; Cocca, S.; Angeletti, S.; Ciccozzi, M.; Tripiciano, C.; Dalla Piccola, B.; Cicala, M.; Guarino, M.P.L. Gut mucosal-associated microbiota better discloses inflammatory bowel disease differential patterns than faecal microbiota. Dig. Liver Dis. 2019, 51, 648–656. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [Green Version]
- Lo Presti, A.; Zorzi, F.; Del Chierico, F.; Altomare, A.; Cocca, S.; Avola, A.; De Biasio, F.; Russo, A.; Cella, E.; Reddel, S.; et al. Fecal and mucosal microbiota profiling in irritable bowel syndrome and inflammatory bowel disease. Front. Microbiol. 2019, 10, 1655. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Hylemon, P.B.; Ridlon, J.M.; Heuman, D.M.; Daita, K.; White, M.B.; Monteith, P.; Noble, N.A.; Sikaroodi, M.; Gillevet, P.M. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G675–G685. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; Ramírez-Puebla, S.T.; Borisy, G.G. Oral Microbiome Geography: Micron-Scale Habitat and Niche. Cell Host Microbe 2020, 28, 160–168. [Google Scholar] [CrossRef]
- Nearing, J.T.; DeClercq, V.; Van Limbergen, J.; Langille, M.G.I. Assessing the Variation within the Oral Microbiome of Healthy Adults. mSphere 2020, 5, e00451-20. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Vannini, L.; La Storia, A.; Laghi, L.; Piombino, P.; Stellato, G.; Serrazanetti, D.I.; Gozzi, G.; Turroni, S.; Ferrocino, I.; et al. The same microbiota and a potentially discriminant metabolome in the saliva of omnivore, ovo-lacto-vegetarian and Vegan individuals. PLoS ONE 2014, 9, e112373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, T.; Kageyama, S.; Furuta, M.; Tsuboi, H.; Takeuchi, K.; Shibata, Y.; Shimazaki, Y.; Akifusa, S.; Ninomiya, T.; Kiyohara, Y.; et al. Bacterial diversity in saliva and oral health-related conditions: The Hisayama Study. Sci. Rep. 2016, 6, 22164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Quinque, D.; Horz, H.-P.; Li, M.; Rzhetskaya, M.; Raff, J.A.; Hayes, M.G.; Stoneking, M. Comparative analysis of the human saliva microbiome from different climate zones: Alaska, Germany, and Africa. BMC Microbiol. 2014, 14, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Ercolini, D. High-throughput sequencing and metagenomics: Moving forward in the culture-independent analysis of food microbial ecology. Appl. Environ. Microbiol. 2013, 79, 3148–3155. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, F.; Parente, E.; Ercolini, D. Recent past, present, and future of the food microbiome. Annu. Rev. Food Sci. Technol. 2018, 9, 589–608. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The impact of next-generation sequencing on genomics. J. Genet. Genomics 2011, 38, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Plummer, E.; Twin, J. A Comparison of Three Bioinformatics Pipelines for the Analysis of Preterm Gut Microbiota using 16S rRNA Gene Sequencing Data. J. Proteom. Bioinform. 2015, 8, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [Green Version]
- Fuks, G.; Elgart, M.; Amir, A.; Zeisel, A.; Turnbaugh, P.J.; Soen, Y.; Shental, N. Combining 16S rRNA gene variable regions enables high-resolution microbial community profiling. Microbiome 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, K.; Rosenthal, C.A.; Sengupta, D.; Owens, J.; Cookson, B.T.; Hoffman, N.G.; Salipante, S.J. Improved Species-Level Clinical Identification of Enterobacteriaceae through Broad-Range dnaJ PCR and Sequencing. J. Clin. Microbiol. 2019, 57, e00986-19. [Google Scholar] [CrossRef] [PubMed]
- Větrovský, T.; Baldrian, P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE 2013, 8, e57923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.D.; Auffret, M.D.; Snelling, T.J.; Roehe, R.; Watson, M. MAGpy: A reproducible pipeline for the downstream analysis of metagenome-assembled genomes (MAGs). Bioinformatics 2019, 35, 2150–2152. [Google Scholar] [CrossRef] [Green Version]
- Quijada, N.M.; Hernández, M.; Rodríguez-Lázaro, D. High-throughput sequencing and food microbiology. Adv. Food Nutr. Res. 2020, 91, 275–300. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Tamames, J.; Cobo-Simón, M.; Puente-Sánchez, F. Assessing the performance of different approaches for functional and taxonomic annotation of metagenomes. BMC Genom. 2019, 20, 960. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Milanese, A.; Mende, D.R.; Paoli, L.; Salazar, G.; Ruscheweyh, H.-J.; Cuenca, M.; Hingamp, P.; Alves, R.; Costea, P.I.; Coelho, L.P.; et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019, 10, 1014. [Google Scholar] [CrossRef]
- Pignatelli, M.; Aparicio, G.; Blanquer, I.; Hernandez, V.; Moya, A.; Tamames, J. Metagenomics reveals our incomplete knowledge of global diversity. Bioinformatics 2008, 24, 2124–2125. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; Wilmes, P. Human gut microbiome: Function matters. Trends Microbiol. 2018, 26, 563–574. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Sharon, I.; Banfield, J.F. Microbiology. Genomes from metagenomics. Science 2013, 342, 1057–1058. [Google Scholar] [CrossRef]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2020, 39, 105–114. [Google Scholar] [CrossRef]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A new genomic blueprint of the human gut microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiergeist, A.; Reischl, U.; Priority Program 1656 Intestinal Microbiota Consortium/quality assessment participants; Gessner, A. Multicenter quality assessment of 16S ribosomal DNA-sequencing for microbiome analyses reveals high inter-center variability. Int. J. Med. Microbiol. 2016, 306, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomaeus, T.U.P.; Birkner, T.; Bartolomaeus, H.; Löber, U.; Avery, E.G.; Mähler, A.; Weber, D.; Kochlik, B.; Balogh, A.; Wilck, N.; et al. Quantifying technical confounders in microbiome studies. Cardiovasc. Res. 2021, 117, 863–875. [Google Scholar] [CrossRef]
- Fouhy, F.; Clooney, A.G.; Stanton, C.; Claesson, M.J.; Cotter, P.D. 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol. 2016, 16, 123. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.S.; He, S.; Woyke, T.; Lee, J.; Chen, F.; Dangl, J.L.; Tringe, S.G. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef] [Green Version]
- Duvallet, C. Meta-analysis generates and prioritizes hypotheses for translational microbiome research. Microb. Biotechnol. 2018, 11, 273–276. [Google Scholar] [CrossRef] [Green Version]
- Harrison, P.W.; Ahamed, A.; Aslam, R.; Alako, B.T.F.; Burgin, J.; Buso, N.; Courtot, M.; Fan, J.; Gupta, D.; Haseeb, M.; et al. The european nucleotide archive in 2020. Nucleic Acids Res. 2021, 49, D82–D85. [Google Scholar] [CrossRef]
- Kodama, Y.; Shumway, M.; Leinonen, R. International Nucleotide Sequence Database Collaboration The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, P.; Kottmann, R.; Field, D.; Knight, R.; Cole, J.R.; Amaral-Zettler, L.; Gilbert, J.A.; Karsch-Mizrachi, I.; Johnston, A.; Cochrane, G.; et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 2011, 29, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Buttigieg, P.L.; Morrison, N.; Smith, B.; Mungall, C.J.; Lewis, S.E. ENVO Consortium The environment ontology: Contextualising biological and biomedical entities. J. Biomed. Semant. 2013, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Degtyarenko, K.; de Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcántara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008, 36, D344–D350. [Google Scholar] [CrossRef] [PubMed]
- Schriml, L.M.; Arze, C.; Nadendla, S.; Chang, Y.-W.W.; Mazaitis, M.; Felix, V.; Feng, G.; Kibbe, W.A. Disease Ontology: A backbone for disease semantic integration. Nucleic Acids Res. 2012, 40, D940–D946. [Google Scholar] [CrossRef] [Green Version]
- Gralka, M.; Szabo, R.; Stocker, R.; Cordero, O.X. Trophic interactions and the drivers of microbial community assembly. Curr. Biol. 2020, 30, R1176–R1188. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.A.; Azcarate-Peril, M.A.; Cadenas, M.B.; Butz, N.; Paster, B.J.; Chen, T.; Bair, E.; Arnold, R.R. The oral bacterial microbiome of occlusal surfaces in children and its association with diet and caries. PLoS ONE 2017, 12, e0180621. [Google Scholar] [CrossRef] [PubMed]
- Adler, C.J.; Dobney, K.; Weyrich, L.S.; Kaidonis, J.; Walker, A.W.; Haak, W.; Bradshaw, C.J.A.; Townsend, G.; Sołtysiak, A.; Alt, K.W.; et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat. Genet. 2013, 45, 450–455e1. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Elinav, E. Diet-microbiota interactions and personalized nutrition. Nat. Rev. Microbiol. 2019, 17, 742–753. [Google Scholar] [CrossRef]
- Charbonneau, M.R.; O’Donnell, D.; Blanton, L.V.; Totten, S.M.; Davis, J.C.C.; Barratt, M.J.; Cheng, J.; Guruge, J.; Talcott, M.; Bain, J.R.; et al. Sialylated Milk Oligosaccharides Promote Microbiota-Dependent Growth in Models of Infant Undernutrition. Cell 2016, 164, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laursen, M.F.; Bahl, M.I.; Michaelsen, K.F.; Licht, T.R. First foods and gut microbes. Front. Microbiol. 2017, 8, 356. [Google Scholar] [CrossRef] [Green Version]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Ostan, R.; Candela, M.; Biagi, E.; Brigidi, P.; Capri, M.; Franceschi, C. Gut microbiota changes in the extreme decades of human life: A focus on centenarians. Cell. Mol. Life Sci. 2018, 75, 129–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, W.; Zheng, H.-M.; Li, P.; McDonald, D.; Sheng, H.-F.; Chen, M.-X.; Chen, Z.-H.; Ji, G.-Y.; Zheng, Z.-D.-X.; et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018, 24, 1532–1535. [Google Scholar] [CrossRef]
- Tarallo, S.; Ferrero, G.; De Filippis, F.; Francavilla, A.; Pasolli, E.; Panero, V.; Cordero, F.; Segata, N.; Grioni, S.; Pensa, R.G.; et al. Stool microRNA profiles reflect different dietary and gut microbiome patterns in healthy individuals. Gut 2021, 71, 1302–1314. [Google Scholar] [CrossRef]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- Meslier, V.; Laiola, M.; Roager, H.M.; De Filippis, F.; Roume, H.; Quinquis, B.; Giacco, R.; Mennella, I.; Ferracane, R.; Pons, N.; et al. Mediterranean diet intervention in overweight and obese subjects lowers plasma cholesterol and causes changes in the gut microbiome and metabolome independently of energy intake. Gut 2020, 69, 1258–1268. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, E.D.; Sonnenburg, J.L. Starving our microbial self: The deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014, 20, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Wegner, K.; Just, S.; Gau, L.; Mueller, H.; Gérard, P.; Lepage, P.; Clavel, T.; Rohn, S. Rapid analysis of bile acids in different biological matrices using LC-ESI-MS/MS for the investigation of bile acid transformation by mammalian gut bacteria. Anal. Bioanal. Chem. 2017, 409, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, D.W.; Koropatkin, N.M. Polysaccharide degradation by the intestinal microbiota and its influence on human health and disease. J. Mol. Biol. 2016, 428, 3230–3252. [Google Scholar] [CrossRef] [PubMed]
- Campos-Perez, W.; Martinez-Lopez, E. Effects of short chain fatty acids on metabolic and inflammatory processes in human health. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158900. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Turroni, S.; Brigidi, P.; Cavalli, A.; Candela, M. Microbiota-Host Transgenomic Metabolism, Bioactive Molecules from the Inside. J. Med. Chem. 2018, 61, 47–61. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, F.; Pasolli, E.; Tett, A.; Tarallo, S.; Naccarati, A.; De Angelis, M.; Neviani, E.; Cocolin, L.; Gobbetti, M.; Segata, N.; et al. Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe 2019, 25, 444–453.e3. [Google Scholar] [CrossRef] [Green Version]
- Tett, A.; Huang, K.D.; Asnicar, F.; Fehlner-Peach, H.; Pasolli, E.; Karcher, N.; Armanini, F.; Manghi, P.; Bonham, K.; Zolfo, M.; et al. The Prevotella copri Complex Comprises Four Distinct Clades Underrepresented in Westernized Populations. Cell Host Microbe 2019, 26, 666–679.e7. [Google Scholar] [CrossRef] [Green Version]
- Pasolli, E.; De Filippis, F.; Mauriello, I.E.; Cumbo, F.; Walsh, A.M.; Leech, J.; Cotter, P.D.; Segata, N.; Ercolini, D. Large-scale genome-wide analysis links lactic acid bacteria from food with the gut microbiome. Nat. Commun. 2020, 11, 2610. [Google Scholar] [CrossRef]
- Dethlefsen, L.; McFall-Ngai, M.; Relman, D.A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 2007, 449, 811–818. [Google Scholar] [CrossRef]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Schell, S.L.; Schneider, A.M.; Nelson, A.M. Yin and Yang: A disrupted skin microbiome and an aberrant host immune response in hidradenitis suppurativa. Exp. Dermatol. 2021, 30, 1453–1470. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, G.; Banerjee, N.; Liang, Y.; Du, X.; Boor, P.J.; Hoffman, K.L.; Khan, M.F. Aberrant gut microbiome contributes to intestinal oxidative stress, barrier dysfunction, inflammation and systemic autoimmune responses in mrl/lpr mice. Front. Immunol. 2021, 12, 651191. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Takeda, K. Host-microbiota interactions in rheumatoid arthritis. Exp. Mol. Med. 2019, 51, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Main, B.S.; Minter, M.R. Microbial Immuno-Communication in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 151. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Meade, K.G.; O’Farrelly, C. β-Defensins: Farming the Microbiome for Homeostasis and Health. Front. Immunol. 2018, 9, 3072. [Google Scholar] [CrossRef]
- Zong, X.; Fu, J.; Xu, B.; Wang, Y.; Jin, M. Interplay between gut microbiota and antimicrobial peptides. Anim. Nutr. 2020, 6, 389–396. [Google Scholar] [CrossRef]
- Pabst, O.; Slack, E. IgA and the intestinal microbiota: The importance of being specific. Mucosal Immunol. 2020, 13, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The host shapes the gut microbiota via fecal microrna. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.; Hasnain, S.Z.; Huat, L.Z.; Koh, D. Human diseases, immunity and the oral microbiota—Insights gained from metagenomic studies. Oral Science International 2017, 14, 27–32. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Elinav, E. Metabolites: Messengers between the microbiota and the immune system. Genes Dev. 2016, 30, 1589–1597. [Google Scholar] [CrossRef]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-Modulated Metabolites at the Interface of Host Immunity. J. Immunol. 2017, 198, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Postler, T.S.; Ghosh, S. Understanding the holobiont: How microbial metabolites affect human health and shape the immune system. Cell Metab. 2017, 26, 110–130. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Blacher, E.; Elinav, E. Microbiome, metabolites and host immunity. Curr. Opin. Microbiol. 2017, 35, 8–15. [Google Scholar] [CrossRef]
- Geva-Zatorsky, N.; Sefik, E.; Kua, L.; Pasman, L.; Tan, T.G.; Ortiz-Lopez, A.; Yanortsang, T.B.; Yang, L.; Jupp, R.; Mathis, D.; et al. Mining the human gut microbiota for immunomodulatory organisms. Cell 2017, 168, 928–943.e11. [Google Scholar] [CrossRef] [Green Version]
- Parfrey, L.W.; Walters, W.A.; Knight, R. Microbial eukaryotes in the human microbiome: Ecology, evolution, and future directions. Front. Microbiol. 2011, 2, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogitsh, B.J.; Carter, C.E.; Oeltmann, T.N. Human Parasitology; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Huseyin, C.E.; O’Toole, P.W.; Cotter, P.D.; Scanlan, P.D. Forgotten fungi-the gut mycobiome in human health and disease. FEMS Microbiol. Rev. 2017, 41, 479–511. [Google Scholar] [CrossRef] [Green Version]
- Stensvold, C.R.; van der Giezen, M. Associations between gut microbiota and common luminal intestinal parasites. Trends Parasitol. 2018, 34, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, J.; Kuchta, R.; Scholz, T.; Pomajbíková, K. (Self-) infections with parasites: Re-interpretations for the present. Trends Parasitol. 2014, 30, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Durkin, L.; Jansson, T.; Sanchez, M.; Khomich, M.; Ryberg, M.; Kristiansson, E.; Nilsson, R.H. When mycologists describe new species, not all relevant information is provided (clearly enough). MycoKeys 2020, 72, 109–128. [Google Scholar] [CrossRef]
- Hofstetter, V.; Buyck, B.; Eyssartier, G.; Schnee, S.; Gindro, K. The unbearable lightness of sequenced-based identification. Fungal Divers. 2019, 96, 243–284. [Google Scholar] [CrossRef] [Green Version]
- Chin, V.K.; Yong, V.C.; Chong, P.P.; Amin Nordin, S.; Basir, R.; Abdullah, M. Mycobiome in the gut: A multiperspective review. Mediat. Inflamm. 2020, 2020, 9560684. [Google Scholar] [CrossRef] [Green Version]
- Pérez, J.C. Fungi of the human gut microbiota: Roles and significance. Int. J. Med. Microbiol. 2021, 311, 151490. [Google Scholar] [CrossRef]
- Huseyin, C.E.; Rubio, R.C.; O’Sullivan, O.; Cotter, P.D.; Scanlan, P.D. The fungal frontier: A comparative analysis of methods used in the study of the human gut mycobiome. Front. Microbiol. 2017, 8, 1432. [Google Scholar] [CrossRef] [Green Version]
- Kounosu, A.; Murase, K.; Yoshida, A.; Maruyama, H.; Kikuchi, T. Improved 18S and 28S rDNA primer sets for NGS-based parasite detection. Sci. Rep. 2019, 9, 15789. [Google Scholar] [CrossRef] [Green Version]
- Wylezich, C.; Höper, D. Meta-Ribosomalomics: RNA Sequencing Is an Unbiased Method for Parasite Detection of Different Sample Types. Front. Microbiol. 2021, 12, 614553. [Google Scholar] [CrossRef] [PubMed]
- Wylezich, C.; Caccio, S.M.; Walochnik, J.; Beer, M.; Höper, D. Untargeted metagenomics shows a reliable performance for synchronous detection of parasites. Parasitol. Res. 2020, 119, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Mirisho, R.; Neizer, M.L.; Sarfo, B. Prevalence of intestinal helminths infestation in children attending princess marie louise children’s hospital in accra, ghana. J. Parasitol. Res. 2017, 2017, 8524985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhotská, Z.; Jirků, M.; Hložková, O.; Brožová, K.; Jirsová, D.; Stensvold, C.R.; Kolísko, M.; Jirků Pomajbíková, K. A Study on the Prevalence and Subtype Diversity of the Intestinal Protist Blastocystis sp. in a Gut-Healthy Human Population in the Czech Republic. Front. Cell. Infect. Microbiol. 2020, 10, 544335. [Google Scholar] [CrossRef]
- Ogilvie, L.A.; Jones, B.V. The human gut virome: A multifaceted majority. Front. Microbiol. 2015, 6, 918. [Google Scholar] [CrossRef] [Green Version]
- Lecuit, M.; Eloit, M. The human virome: New tools and concepts. Trends Microbiol. 2013, 21, 510–515. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, M.; Zhang, C.; Du, H.; Wei, G.; Pang, X.; Zhou, H.; Liu, B.; Zhao, L. Pattern extraction of structural responses of gut microbiota to rotavirus infection via multivariate statistical analysis of clone library data. FEMS Microbiol. Ecol. 2009, 70, 21–29. [Google Scholar] [CrossRef]
- Harper, A.; Vijayakumar, V.; Ouwehand, A.C.; Ter Haar, J.; Obis, D.; Espadaler, J.; Binda, S.; Desiraju, S.; Day, R. Viral infections, the microbiome, and probiotics. Front. Cell. Infect. Microbiol. 2020, 10, 596166. [Google Scholar] [CrossRef]
- Kesika, P.; Sivamaruthi, B.S.; Thangaleela, S.; Chaiyasut, C. The antiviral potential of probiotics—a review on scientific outcomes. Appl. Sci. 2021, 11, 8687. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.-Y.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Lv, Z.; Xiong, D.; Shi, J.; Long, M.; Chen, Z. The interaction between viruses and intestinal microbiota: A review. Curr. Microbiol. 2021, 78, 3597–3608. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Woods Acevedo, M.A.; McCune, B.T.; Pfeiffer, J.K. Related enteric viruses have different requirements for host microbiota in mice. J. Virol. 2019, 93, e01339-19. [Google Scholar] [CrossRef] [PubMed]
- Yaron, J.R.; Ambadapadi, S.; Zhang, L.; Chavan, R.N.; Tibbetts, S.A.; Keinan, S.; Varsani, A.; Maldonado, J.; Kraberger, S.; Tafoya, A.M.; et al. Immune protection is dependent on the gut microbiome in a lethal mouse gammaherpesviral infection. Sci. Rep. 2020, 10, 2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monedero, V.; Buesa, J.; Rodríguez-Díaz, J. The Interactions between Host Glycobiology, Bacterial Microbiota, and Viruses in the Gut. Viruses 2018, 10, 96. [Google Scholar] [CrossRef] [Green Version]
- Neu, U.; Mainou, B.A. Virus interactions with bacteria: Partners in the infectious dance. PLoS Pathog. 2020, 16, e1008234. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive expansion of human gut bacteriophage diversity. Cell 2021, 184, 1098–1109.e9. [Google Scholar] [CrossRef]
- Moreno-Gallego, J.L.; Chou, S.-P.; Di Rienzi, S.C.; Goodrich, J.K.; Spector, T.D.; Bell, J.T.; Youngblut, N.D.; Hewson, I.; Reyes, A.; Ley, R.E. Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins. Cell Host Microbe 2019, 25, 261–272.e5. [Google Scholar] [CrossRef] [Green Version]
- Devoto, A.E.; Santini, J.M.; Olm, M.R.; Anantharaman, K.; Munk, P.; Tung, J.; Archie, E.A.; Turnbaugh, P.J.; Seed, K.D.; Blekhman, R.; et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 2019, 4, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, K.L. Phages tune in to host cell quorum sensing. Cell 2019, 176, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Leigh, B.A. Cooperation among Conflict: Prophages Protect Bacteria from Phagocytosis. Cell Host Microbe 2019, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.; Diard, M. Crowd Controlled-Host Quorum Sensing Drives Phage Decision. Cell Host Microbe 2019, 25, 179–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jancheva, M.; Böttcher, T. A Metabolite of Pseudomonas Triggers Prophage-Selective Lysogenic to Lytic Conversion in Staphylococcus aureus. J. Am. Chem. Soc. 2021, 143, 8344–8351. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M. Phage diversity in the human gut microbiome: A taxonomist’s perspective. mSystems 2021, 6, e0079921. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ng, Q.X.; Zhang, H.; Zhang, B.; Ong, C.N.; He, Y. Metabolite changes behind faster growth and less reproduction of Daphnia similis exposed to low-dose silver nanoparticles. Ecotoxicol. Environ. Saf. 2018, 163, 266–273. [Google Scholar] [CrossRef]
- Nagana Gowda, G.A.; Raftery, D. Can NMR solve some significant challenges in metabolomics? J. Magn. Reson. 2015, 260, 144–160. [Google Scholar] [CrossRef] [Green Version]
- Callejón-Leblic, B.; Selma-Royo, M.; Collado, M.C.; Gómez-Ariza, J.L.; Abril, N.; García-Barrera, T. Untargeted Gut Metabolomics to Delve the Interplay between Selenium Supplementation and Gut Microbiota. J. Proteome Res. 2022, 21, 758–767. [Google Scholar] [CrossRef]
- Wang, L.-M.; Wang, P.; Teka, T.; Zhang, Y.-C.; Yang, W.-Z.; Zhang, Y.; Wang, T.; Liu, L.-X.; Han, L.-F.; Liu, C.-X. 1H NMR and UHPLC/Q-Orbitrap-MS-Based Metabolomics Combined with 16S rRNA Gut Microbiota Analysis Revealed the Potential Regulation Mechanism of Nuciferine in Hyperuricemia Rats. J. Agric. Food Chem. 2020, 68, 14059–14070. [Google Scholar] [CrossRef]
- Lai, Y.; Liu, C.-W.; Yang, Y.; Hsiao, Y.-C.; Ru, H.; Lu, K. High-coverage metabolomics uncovers microbiota-driven biochemical landscape of interorgan transport and gut-brain communication in mice. Nat. Commun. 2021, 12, 6000. [Google Scholar] [CrossRef]
- Peisl, B.Y.L.; Schymanski, E.L.; Wilmes, P. Dark matter in host-microbiome metabolomics: Tackling the unknowns-A review. Anal. Chim. Acta 2018, 1037, 13–27. [Google Scholar] [CrossRef]
- Ghannam, R.B.; Techtmann, S.M. Machine learning applications in microbial ecology, human microbiome studies, and environmental monitoring. Comput. Struct. Biotechnol. J. 2021, 19, 1092–1107. [Google Scholar] [CrossRef]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Loncar Turukalo, T.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of machine learning in human microbiome studies: A review on feature selection, biomarker identification, disease prediction and treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]


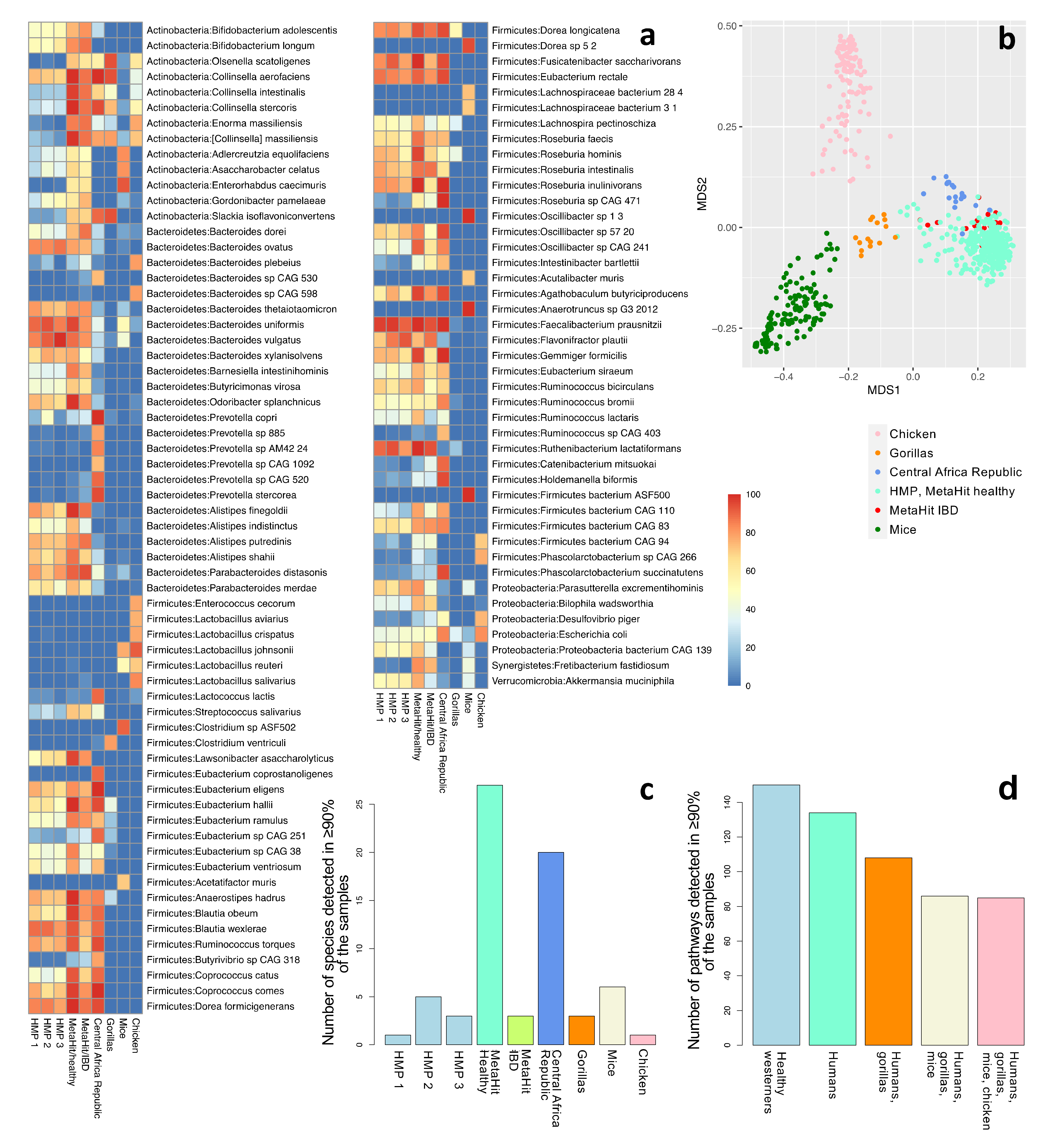{kind=link}
| Approach | Pros | Cons | Examples |
|---|---|---|---|
| Community composition: the core microbiome is described in terms of shared taxa | Relatively simple to implement; can be applied to amplicon studies | Common taxa are usually identified at high taxonomic levels only | [17,28,29,30,31] |
| Functional profile: definition relies on a set of common functions | Captures the contribution of the core human microbiome to the host and the community | It is difficult to distinguish between human-specific and broad core functions | [9,12,13,26] |
| Ecology: include taxon abundance, interactions, co-occurrence, and other community-level patterns | Can capture complex patterns in community structure; may be more realistic than community composition alone | Less clear which patterns should be considered; no standard methods and programs are available | [19] |
| Stability: consider factors that maintain community stability and resilience | Stability is a critical characteristic of the core microbiome that is not captured through community composition alone | Definition is vague; there are no widely accepted methods for evaluating stability and resilience | [32] |
| Data (Section 3) | |
| Technology | Hundreds of thousands of human-associated 16S and metagenomics samples. Only thousands of other meta-omics samples |
| Population | Mostly Westerners. Agricultural and traditional populations are significantly underrepresented |
| Body part | Mostly fecal samples. The gut environment consists of multiple niches, each may have its own core microbiome |
| Methods (Section 4) | |
| 16S rRNA surveys | Hundreds of thousands of public 16S samples are available. The method is inexpensive with established lab procedures and bioinformatics pipelines. The data provides only low taxonomic-resolution community composition and no functional information |
| Metagenomics bioinformatics | Tens of thousands of public human-associated metagenomes available. The data can provide strain-level and functional information. Bioinformatics analysis is complex, reference databases still lack a significant portion of human-associated microbial species. |
| Difficult to mine public databases | Available metadata is typically partial, performing meta-analysis requires significant manual effort |
| Beyond prokaryotic omics (Section 5) | |
| Diet | Both diet and the immune system shape the gut microbiome and are related to the functions it provides. Small amounts of data are available for both, with no high-throughput methods available for data collection |
| Microbiome–immune system interactions | |
| Non-prokaryotic members of the microbiome | Eukaryotes and viruses may be either part of the core human microbiome or related to the functions it provides. Both groups are invisible in 16S studies, current metagenomics bioinformatics mostly ignores them |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharon, I.; Quijada, N.M.; Pasolli, E.; Fabbrini, M.; Vitali, F.; Agamennone, V.; Dötsch, A.; Selberherr, E.; Grau, J.H.; Meixner, M.; et al. The Core Human Microbiome: Does It Exist and How Can We Find It? A Critical Review of the Concept. Nutrients 2022, 14, 2872. https://doi.org/10.3390/nu14142872
Sharon I, Quijada NM, Pasolli E, Fabbrini M, Vitali F, Agamennone V, Dötsch A, Selberherr E, Grau JH, Meixner M, et al. The Core Human Microbiome: Does It Exist and How Can We Find It? A Critical Review of the Concept. Nutrients. 2022; 14(14):2872. https://doi.org/10.3390/nu14142872
Chicago/Turabian StyleSharon, Itai, Narciso Martín Quijada, Edoardo Pasolli, Marco Fabbrini, Francesco Vitali, Valeria Agamennone, Andreas Dötsch, Evelyne Selberherr, José Horacio Grau, Martin Meixner, and et al. 2022. "The Core Human Microbiome: Does It Exist and How Can We Find It? A Critical Review of the Concept" Nutrients 14, no. 14: 2872. https://doi.org/10.3390/nu14142872