The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review


Department of Pediatrics, Division of Neonatology, University of Oklahoma Health Sciences Center, 1200 North Everett Dr., ETNP7504, Oklahoma City, OK 73104, USA
*
Author to whom correspondence should be addressed.
Nutrients 2020, 12(2), 546; https://doi.org/10.3390/nu12020546
Submission received: 21 January 2020
/
Revised: 14 February 2020
/
Accepted: 16 February 2020
/
Published: 20 February 2020
(This article belongs to the Special Issue Neonatal Nutrition for Inflammatory Disorders and Necrotizing Enterocolitis)
Abstract
:Necrotizing enterocolitis, a potentially fatal intestinal inflammatory disorder affecting primarily premature infants, is a significant cause of morbidity and mortality in neonates. While the etiology of the disease is, as yet, unknown, a number of risk factors for the development of necrotizing enterocolitis have been identified. One such risk factor, formula feeding, has been shown to contribute to both increased incidence and severity of the disease. The protective influences afforded by breastfeeding are likely attributable to the unique composition of human milk, an extremely potent, biologically active fluid. This review brings together knowledge on the pathogenesis of necrotizing enterocolitis and current thinking on the instrumental role of one of the more prominent classes of bioactive components in human breast milk, glycosaminoglycans.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Necrotizing enterocolitis (NEC) is a common intestinal inflammatory disorder developing during the neonatal period. The disease progresses rapidly from subtle abdominal distension to necrosis, intestinal perforation, multi-organ failure, and, in severe cases, death [1,2]. Because of better survivorship among the smallest premature infants [3], as well as a dearth of treatments for the disease [4], the incidence and health burden of NEC have only grown in recent decades [1]. Mortality rates often approach 30% in infants born less than 1500 g, and range higher in those babies requiring surgical intervention [1]. Infants surviving NEC often suffer from long-term morbidities related to both the disease and its treatment, including neurodevelopmental delays, retinopathy, and short-bowel syndrome [5,6].
Although the exact etiology of the disease is still unclear [7], a number of risk factors have been identified. These include, among others, prematurity and low birthweight status, developmental immaturity of both the intestine and immune system, inappropriate microbial colonization of the gut, and formula feeding [8]. What few advances have been made in NEC treatment recently revolve around the growing understanding of the importance of optimal infant nutrition, particularly that provided by human breast milk (HM), sourced either from the mother or a donor [5,9,10].
HM functions in several critical biological roles in neonates, including support for intestinal development and maturation, protection against pathogens, and basic dietary sustenance for growth [9,11]. In babies fed exclusively HM, enteric infections are reduced by approximately 50% compared to infants fed bovine-based formula [12,13]. In the preterm population specifically, infants fed exclusively HM, as opposed to at least partial bovine-based feedings, experience significant reductions in morbidity and mortality [14]. In very low birthweight (VLBW, <1500 g) infants, feedings composed of at least 50% HM in the first two weeks of life correlate with a six-fold decrease in the incidence of NEC [15]. Additional studies [16,17,18] have also indicated HM, particularly that sourced from the biological mother, reduces the incidence and severity of NEC in preterm infants. Donor milk, while arguably preferable to bovine-based formula [19], loses effectiveness through the pasteurization process, and is also not age-matched to the developmental stage of the infant to whom it is donated [20,21]. Thus, studies utilizing donor HM have shown mixed results when examining utility in protection against preterm pathogens, NEC, and mortality [22,23,24].
2. Pathogenesis of Necrotizing Enterocolitis
The pathogenesis of NEC appears to be highly complex and multifactorial. Data predominantly implicate developmental immaturity of the intestinal immune system [27], as well as an altered microbiome [28,29], in the development of a dysfunctional intestinal barrier [30] in necrotizing enterocolitis. While the sequence of events in NEC etiology remains unclear, the disease is likely initiated by an excessive stimulation of toll-like receptor 4 (TLR4) by Gram-negative bacteria [31] in the ileum of the premature infant. Activation of this receptor [32,33,34,35,36,37,38] leads to extensive inflammation, denoted by apoptosis of enterocytes along the luminal border, impaired replacement of these enterocytes, increased release of proinflammatory cytokines and chemokines, and, in total, breakdown of the intestinal barrier [39,40,41]. This impaired intestinal barrier allows for greater bacterial translocation [42], leading to increased inflammation via direct contact of pathogenic bacterial antigens with the mucosal immune system [43]. Neutrophil recruitment to the intestinal border and production of reactive oxygen species (ROS) further contributes to this inflammation [1,44,45]. TLR4 activation of the underlying endothelium initiates microvascular complications, including a reduction in endothelial nitric oxide synthase (eNOS), resulting in intestinal ischemia and necrosis [38,46,47]. Altogether, a positive feedback loop of inflammation is created, overwhelming any counterregulatory attempts by the host. Inflammation spreads systemically, leading to full-blown NEC and complications in organs as distant as the brain [48]. Our understanding of the clinical picture in NEC is muddied by a lack of appropriate animal models through which researchers can replicate most aspects of the human condition, and subsequent inability to translate findings in these animal models to the bedside. In particular, our limited understanding of the immature innate and adaptive immune systems and developing microbiome, and potential interplay of these two factors, hinders abilities to target effective treatments for NEC.
2.1. Developmental Immaturity
2.1.1. Innate Immune System
The innate immune system in the small intestine is comprised of both a physical barrier of intestinal epithelial cells (IECs) and their biochemical products, and an underlying and complementary immunological barrier [49]. The physical barrier is often considered to include intestinal alkaline phosphatase, a loose layer of mucus [50], tight junctions linking IECs, and antimicrobial proteins (AMPs) released by a specialized lineage of IECs, Paneth cells [51].
A number of developmental differences in IECs and the innate intestinal immune system have been associated with the pathogenesis of NEC. For example, goblet cell numbers and levels of their signature mucin, Muc2, are reduced in both mouse models and premature human infants with NEC [32], likely associated with developmental immaturity of the ileum [52]. Reductions in goblet cell numbers are thought to contribute to increased severity and incidence of NEC [52,53], potentially due to increased levels of bacterial translocation across an epithelium now inadequately guarded by mucus [53,54]. In addition, Paneth cells in premature infants are deficient both in number and function [55], altering the levels of lysozyme and defensins [56,57] and likely contributing to the development of NEC via associated changes in the microbiome [58].
Immune cells in the intestines of premature infants often appear to function differently than those of term neonates and adults, predisposing these infants to the development of NEC. Neutrophils, first responders to tissue injury, demonstrate impaired phagocytic ability, increased oxidative burst products, and variable cytokine production in premature infants compared to term babies [59]. Intestinal macrophages in preterm infants appear to be hyperreactive and produce excessive proinflammatory cytokines [60,61,62]. In addition, dendritic cell morphology and functionality in preterm neonates appears to differ from that of term babies [63], and in a mouse model of experimental NEC, the recruitment of dendritic cells to the luminal border directly disrupts intestinal barrier function [64].
Alterations in inflammatory signaling also predispose premature infants to the development of NEC by creating a host environment hyperreactive to both commensal and pathogenic organisms [33,65]. TLR4, specifically, is thought to play a crucial role in NEC [66] due to its abnormally increased expression in prematurity, both in mice and human infants [67,68]. Increased TLR4 expression appears to precede histological damage of the intestine in mouse NEC models, strongly implicating a role for TLR4 in the pathogenesis of the disease [65,69].
2.1.2. Adaptive Immune System
Adaptive immunity in the small intestine, thought to be less effective than innate immunity in a newborn [70], is dependent upon antigen-presenting cells (APC), primarily dendritic cells and IECs. A number of differences in adaptive immune function exist in infants with NEC, many of which are likely a developmental artifact of prematurity. Levels of secretory immunoglobulin A (sIgA), an antibody produced by lamina propia B cells and recognized for its ability to maintain the microbiome by neutralizing pathogenic bacteria [71], are reduced in premature infants compared to term babies [72,73], with clear implications for the development of NEC. Additionally, a significant role for T cells in the pathogenesis of NEC is becoming increasingly evident. For example, mice lacking functional T and B cells are less susceptible to NEC, but transfer of functional T cells to these animals increases susceptibility to the disease [34].
Intraepithelial lymphocytes (IELs), regulators and initiators of both innate and adaptive immune responses to bacterial invasion [74], are dispersed within the intestinal epithelium [41]. The γδ subset of IELs, created during embryogenesis, are more reactive in the neonate than in the adult [75], and preterm infants with NEC show significantly lower levels of these specialized T cells compared to healthy preterm babies [76]. Regulatory T cells (Tregs), T cells modulating the immune response and promoting tolerogenicity, are decreased in both experimental mouse [34] and human [77] NEC. In neonates, baseline levels of the Treg inhibitor STAT3 (signal transducer and activator of transcription 3) are increased compared to those of adults [34]. In a mouse model of NEC, when a STAT3 inhibitor is introduced, levels of Tregs increase and NEC severity is reduced [34].
Finally, T helper (Th) cell differentiation also appears to be dysregulated during NEC. In particular, Th17 cells, characterized by production of the inflammatory interleukin 17A (IL-17A) cytokine, have been shown to be upregulated in both murine and human NEC, and are thought to play a role in intestinal barrier dysfunction [34]. In an experimental model of NEC, mice treated with all-trans retinoic acid (ATRA), an inhibitor of Th17 differentiation [78], showed lower levels of Th17 cells, increased populations of Tregs, and reduced NEC severity [34,79]. Interestingly, retinoic acid is produced endogenously by IECs, but this production is thought to be largely dictated by luminal commensal bacteria [80].
2.2. Dysbiosis
Colonization of the infant intestine, previously thought to commence at birth, may originate in the placenta [81], where the fetus is possibly surrounded by non-sterile amniotic fluid [82], though this finding has been recently debated [83,84]. The main event responsible for infant intestinal colonization, however, is likely birth [85]. Vaginal delivery of term infants results in initial colonization with predominantly aerobic bacteria, including Streptococcus, Staphylococcus, and Lactobacillus [86]. As these aerobic bacteria consume oxygen, the microbiome shifts to reflect greater populations of facultative anaerobes, followed by strict anaerobes such as Clostridia and Bifidobacteria species [87,88]. These obligate anaerobes produce short-chain fatty acids (SCFAs), anti-inflammatory lipids known to regulate epithelial and immune cell development in the gut [89], and protect against the proliferation of pathogenic bacteria [90]. In preterm infants, the development of the intestinal microbiome following birth appears to follow a reasonably predictable progression from Bacilli to Gammaproteobacteria to Clostridia [91]. The resulting intestinal population in preterm infants is characterized by lower diversity, fewer species numbers, and a greater proportion of pathogenic bacteria, many of which could initiate the TLR4 signaling cascade via lipopolysaccharide (LPS), compared to that of infants born at term [92,93,94,95]. This errant microbiome in the premature infant, together with an immature intestinal immune system, presents a mechanism for hyperinflammation and deterioration of the critical intestinal barrier.
Dysbiosis can refer to improper proportions of microbial species, as well as a lack of diversity and richness of species overall [96]. A skewed microbiome can also result from the gain or loss of critical microbial populations, often negatively affecting the functionality of both the intestine and its interwoven immune system. An appropriate microbiome is thought to be indispensable in triggering the maturation of the mucosal immune system in the gut [97]. Support for the role of dysbiosis in NEC is largely derived from studies in germ-free animals, in which the disease cannot be reproduced [98,99,100]. Additionally, factors indirectly influencing microbial colonization in the infant, such as antibiotic use in the mother [67], can increase NEC development risk in the infant. While a single pathogen is not thought to induce NEC, a series of microbial shifts in the microbiome has been associated with development of the disease [28], and these changes usually precede diagnosis [101], implicating a potential role for dysbiosis in the pathogenesis of NEC. For example, infants with NEC often have reduced populations of Bifidobacteria, Bacteroidetes, and Firmicutes anaerobes, particularly Negativicutes, and increased levels of Proteobacteria and Actinobacteria [28,101,102,103,104,105]. This reduction in anaerobes in NEC leads to a decline in the production of protective SCFAs [7,103,104], a further complication of NEC-associated dysbiosis. Generally, the microbiome of infants developing NEC appears to be characterized by reductions in both species richness and diversity [95,106,107], though not all studies have noted these trends [101,105,108].
A number of factors beyond prematurity can influence the microbial colonization of the infant intestine. The use of antibiotics, rampant in the premature infant population [109,110], is known to increase the risk of NEC development, with risk correlating strongly to duration of treatment [111,112]. Antibiotic exposure in neonates may lead to increases in Proteobacteria, decreases in Actinobacteria, and, as with all antibiotic usage, inadvertent selection for antibiotic-resistant strains [85,113,114,115]. Mode of delivery also strongly influences the development of the infant microbiome. Babies born via caesarean section are often colonized by increased populations of Clostridium and Staphylococcus and decreased levels of Bifidobacterium and Bacteroides compared to infants born vaginally [102,116,117]. Antacid use, particularly histamine-2 (H2) blockers, can disrupt the acid-base balance in the premature intestine [118], predisposing the infant to NEC [119,120] by favoring populations of Proteobacteria over those of Firmicutes [121,122]. Even endogenous factors may affect the relative proportions of intestinal colonizers. For example, Paneth cell lysozyme and defensin secretion patterns, altered in premature infants [56,57], can lead to irregular microbial colonization in infants [58,123]. Finally, mode of feeding can direct the development of the neonatal microbiome. HM contains a microbiome of its own [124], likely specialized for the infant with whom it is associated [125,126], and thus may be uniquely protective. While breastfeeding stimulates the expansion of Bifidobacterium, in particular, and inhibits the growth of pathogenic bacteria [127,128], formula feeding often leads to a slightly more diverse assemblage of Enterobacteriaceae, Bacteroides, Lactobacillus, Prevotella, and, especially, Clostridium [87,129,130,131]. A number of biological components of HM are thought to help shape the development of the infant microbiome, as well as prime intestinal immune development and maturation.
3. Glycosaminoglycans in Milk
HM is a complex mixture of biologically active molecules known to play a role in infant nutrition, protection from pathogens, and development and maturation of the intestinal immune system. The composition of HM is not static, changing over time to meet the needs of a growing infant. Colostrum, the first milk, is high in minerals, vitamins, hormones, and growth factors [132]. Transitional milk replaces colostrum at approximately one week postpartum, and is high in fat and lactose [133]. Finally, mature milk follows at two weeks postpartum, consisting largely of water and nutritional macronutrients necessary for infant growth [134]. All stages of HM, however, contain various compounds necessary for development of the microbiome and protection of the infant from pathogens. For example, oligosaccharides, commonly referred to as human milk oligosaccharides (HMOS), are found in large quantities in HM and are largely unabsorbed, serving as prebiotics for commensal gut bacteria [5], thereby bolstering and developing the innate immune system and microbiome in neonates [135,136]. HM also contains immunoglobulins, such as sIga, which potentially serve as prebiotics capable of assisting in the proper colonization of the newborn gut [137] while inhibiting growth of pathogenic bacteria [138]. Glycosaminoglycans (GAGs), a class of polysaccharides found in the extracellular matrix and outer surface of cells, are also prevalent in HM. Our understanding of the potential functions of this class of molecules is still evolving, but their elevated concentrations in early HM and a number of studies indicating protective capabilities of these molecules against pathogens may indicate importance in the prevention of NEC.
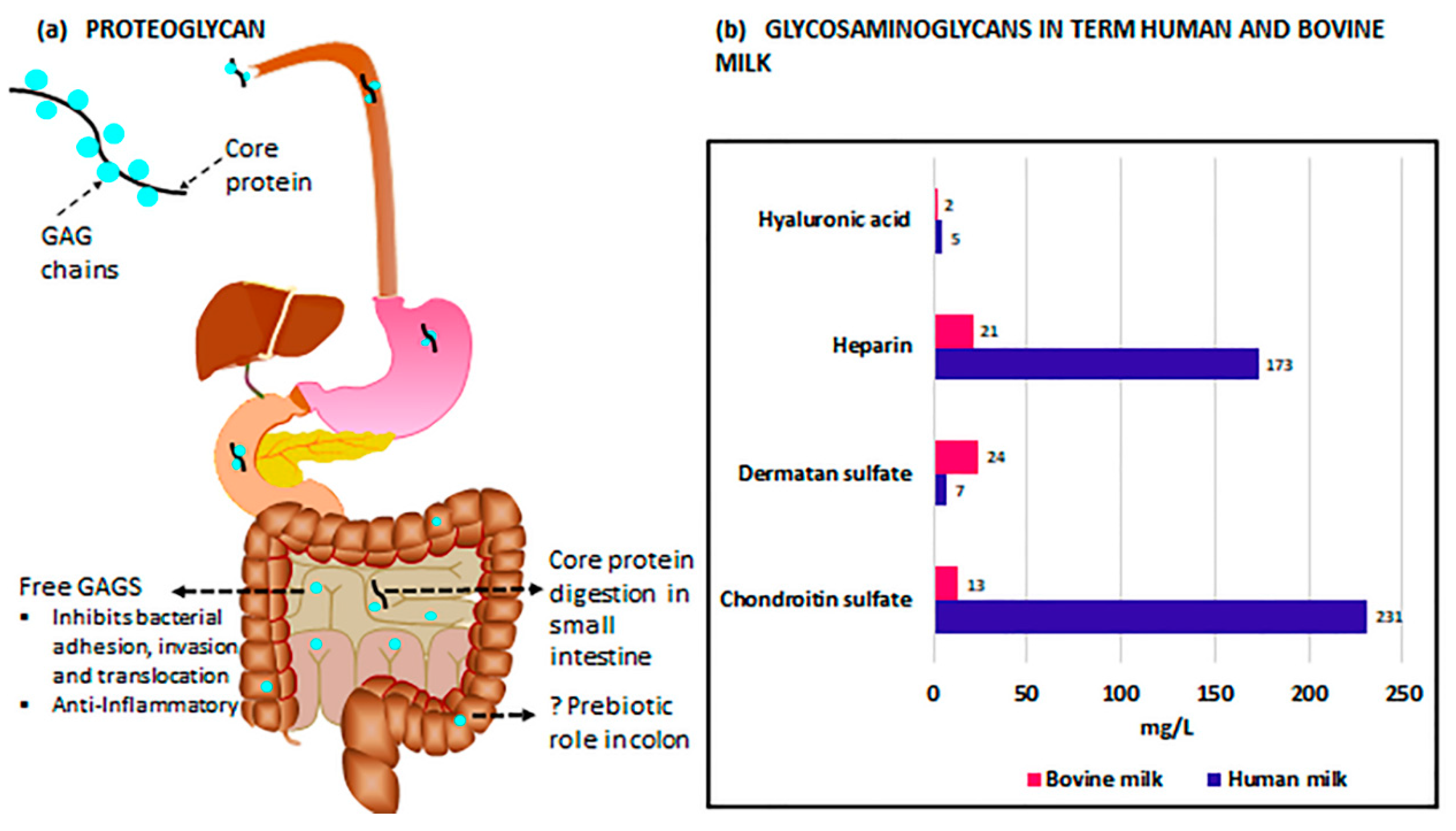
GAGs, molecules composed of repeating, often highly sulfated disaccharides, include heparin, heparan sulfate (HS), keratan sulfate (KS), hyaluronic acid (HA), chondroitin sulfate (CS), and dermatan sulfate (DS) [139]. In HM, these GAGs, with the exception of HA, are bound to a protein core and expressed as proteoglycans [140]. HA, uniquely, is neither sulfated nor assembled bound to a protein core [141]. Once in the small intestine, pancreatic enzymes digest the proteins, resulting in free GAGs (Figure 1a). Due to a lack of endogenous host enzymes in the small intestine capable of further breaking down free GAGs [142], these molecules remain largely undigested through the majority of the gastrointestinal tract [143], with eventual breakdown likely occurring in the cecum or colon [142]. In the case of HA, and potentially other GAGs, the resulting fragment sizes, whether created by endogenous breakdown of the parent molecule or intentional supplementation of a specific molecular weight, may differ vastly in function [144,145].
The composition of GAGs in milk differs greatly depending upon the source. In term HM, GAGs, in sum, are approximately seven times more prevalent than in bovine milk, the basis of most infant formulas [146]. Coppa et al. [146] determined, via a variety of methods, that large differences in GAG relative composition also exist between the two milks (Figure 1b). CS accounts for 55% of the term HM GAGs compared to only 21% of GAGs in bovine milk, but because of the reduced total quantity of GAGs in bovine milk, this amounts to nearly 23 times as much CS in term human compared to bovine milk. Term HM also has substantially higher levels of heparin (173 mg/L compared to 21 mg/L) and HA (5 mg/L compared to 2 mg/L) and lower levels of DS (7 mg/L compared to 24 mg/L) than bovine milk, though, interestingly, bovine milk is higher in HA by percentage (4.5% compared to 1.3%). Additionally, GAGs present in term HM appear to be generally less sulfated compared with those in bovine milk [146], though any impact of this difference on the infant has not been explored.
A disparity in bioavailability also appears to exist between human and bovine milk. Maccari et al. [143] compared the residual GAG content of the feces of term infants fed either HM or formula, and noted significantly lower recovery of HM GAGs compared to those from formula, indicating a much greater utilization of the GAGs derived from HM. In addition, a greater proportion of highly sulfated GAGs, present at higher levels in bovine milk compared to HM, appeared in the feces of both groups, potentially suggesting an inability of distal intestinal bacteria to catabolize these compounds [147]. Differences in milk glycosaminoglycan composition, overall quantity, and bioavailability may underly some of the protective effects of breastfeeding with regard to NEC pathogenesis.
Notably, gestational age of the infant at birth and stage of lactation have prominent influences on the GAG composition of HM. Coppa et al. [148] demonstrated that while preterm and term HM have consistent proportions of the two foremost GAGs, chondroitin sulfate and heparin, the total GAG content is approximately three times higher in preterm milk. The respective percentages of CS and heparin are maintained as total GAG levels vary over the first month of lactation, with peak levels of GAGs on day 4 of colostrum (9.3 g/L and 3.8 g/L in preterm and term HM, respectively) and a subsequent decline to the end of the month (4.3 g/L and 0.4 g/L in preterm and term HM, respectively). Interestingly, 50% (preterm) and 73% (term) of this reduction in GAG content is noted to occur between days 4 and 10 [148]. Wang et al. [149] have established this progressive decrease of HM GAG content occurs through at least the first six months of lactation. Additionally, differences in the degree of sulfation occur during the lactation period, with HS sulfation increasing slightly over the first six months and CS sulfation peaking at day 43 before subsequently declining [149]. While the physiological rationale underlying GAG sulfation variability during the breastfeeding period has not yet been appreciated, these changes in sulfation patterns are likely functionally significant [150].
Maternal characteristics may also alter both the GAG composition of HM and the ability of the infant, indirectly, to break down those GAGs in the distal intestine. While Volpi et al. [151] did not find GAG compositional changes among milk samples from mothers of varying ethnicities, Mannello et al. [152] noted maternal health could directly influence GAG composition, as alterations in the structure and sulfation levels of CS in the milk of a breast affected by invasive carcinoma differed with those in the unaffected breast of the same mother. Finally, Cerdό et al. [153] have established that the microbiome of infants born to obese mothers is more capable of glycosaminoglycan degradation compared with that of infants born to mothers of normal weight, with unspecified effects on the risk of developing NEC. Further investigation into the potential impact of maternal obesity on GAG content of HM and the ability of infants born to obese mothers to utilize HM GAGs would be of interest.
The utility of donor milk as a suitable substitute for formula has often been questioned, particularly given the pasteurization process is known to reduce the bioactivity and concentration of critical immunoglobulins, growth factors, and digestive enzymes [19]. HM contains active glycosidases, enzymes capable of degrading glycoconjugates such as glycosaminoglycans in a time- and temperature-dependent manner; however, these enzymes are unlikely to significantly alter the composition of GAGs in donor milk, as little breakdown in glycoconjugates is seen with storage at 37 °C for up to 16 h [154]. Additionally, Coscia et al. [155] subjected donor milk samples to Holder pasteurization, a method often utilized by milk banks, and noted the concentration and proportions of HM GAGs are not significantly altered by the process. Thus, the current preference for donor milk over formula may be warranted in this context, especially in at-risk, preterm infants, as GAG concentrations and relative proportions remain largely unaffected by common storage and processing techniques.
4. Glycosaminoglycan Protection against NEC
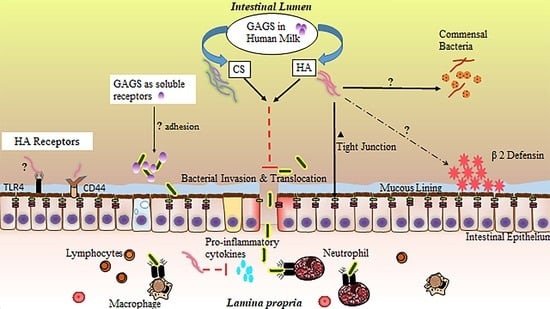
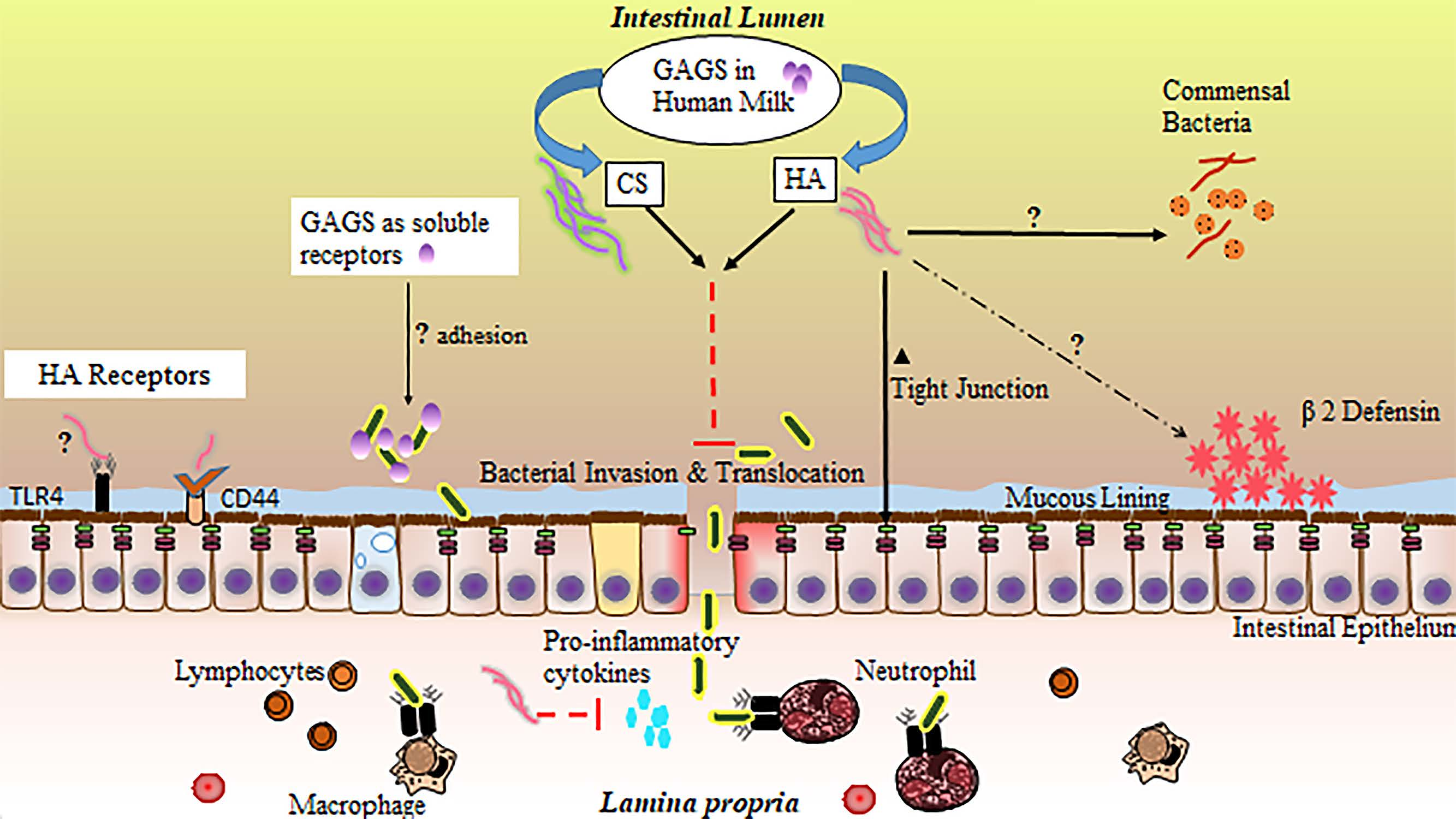
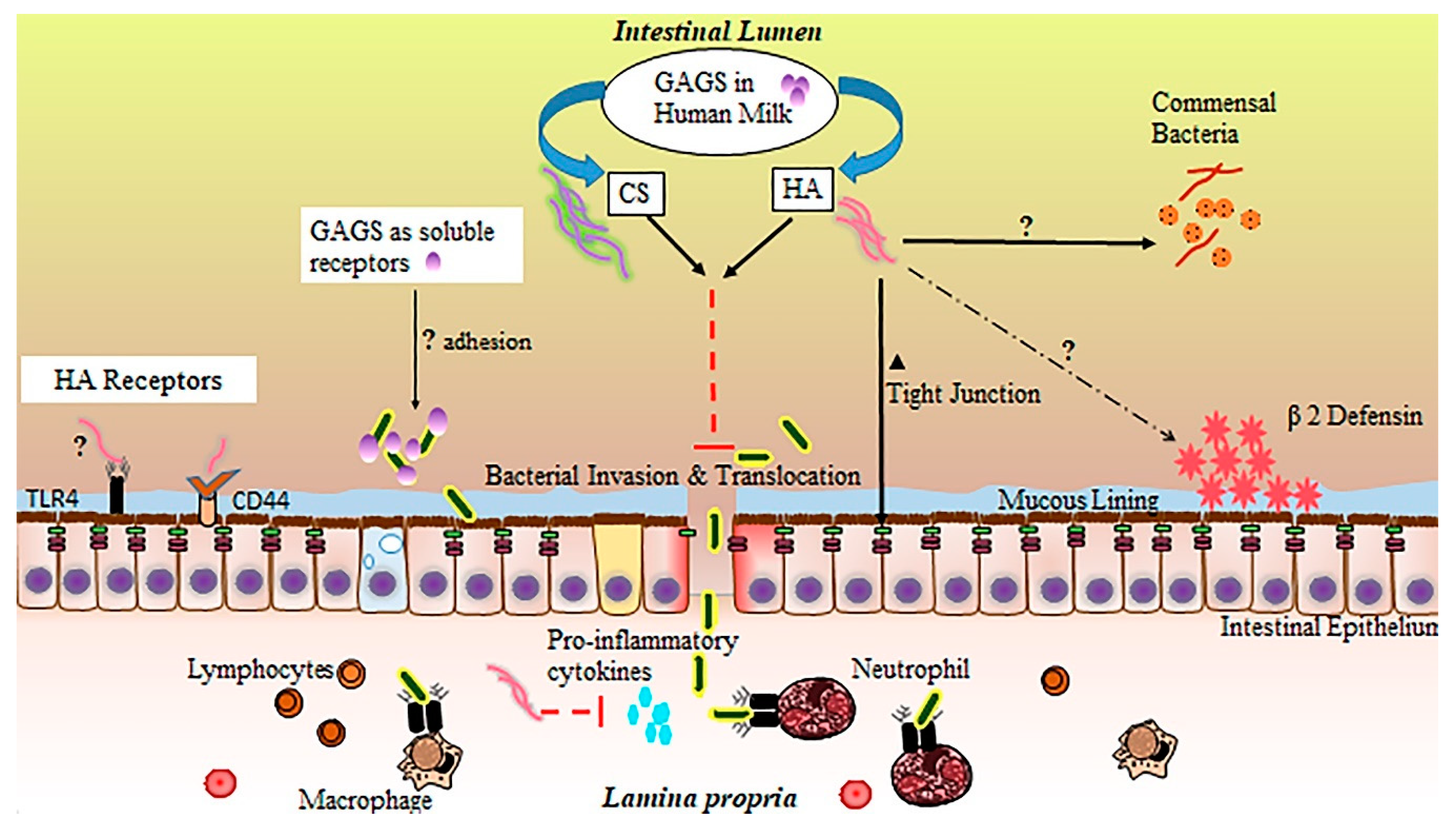
In the small intestine, GAGs are believed to participate in a number of biological processes, including molecular trafficking, maturation, and differentiation of a variety of cell types, modulation of inflammatory events, structural support, and adhesion to bacteria in the intestinal lumen [156,157,158,159]. Importantly, GAG incorporation into the extracellular matrix and epithelial cell surface is thought to be essential to a functional intestinal barrier [160]. Inflammation, particularly driven by proinflammatory cytokine release, has been shown to disrupt endogenous production of sulfated GAGs [161]. In NEC, impaired distribution of intestinal GAGs appears to mirror the patchy, skip lesion nature of the disease [162]. While GAGs are not digested and incorporated into small intestinal tissue [143], their supplementation through HM or other means may still provide significant benefits to the neonatal small intestinal epithelium through their interaction with the epithelial surface or luminal contents [163], especially in the context of their potential loss during inflammation. These extracellular interactions of GAGs with IECs or luminal bacteria likely contribute substantially to the protective effects of HM against NEC. While the precise function of GAGs in HM is incompletely understood (Figure 2), studies attributing protective effects to individual GAGs, sourced either from HM extractions or biosynthetic preparations, are accumulating, with the large majority focusing on CS, HA, and heparin.
Generally, GAGs are believed to exhibit antiviral and antibacterial properties [164]. HA has been demonstrated to inhibit bacterial growth [165,166], and is a common matrix component of bioengineered orthopedic scaffolding because of this bacteriostatic property. HM GAGs may also influence the composition of the neonatal microbiome beyond growth constraints applied to certain microbial species, resulting in a wide variety of potential physiological effects. Recently, several species of human commensal bacteria, including strains associated with common probiotic formulas, have been shown to actively degrade host GAGs in the intestine [167], lending some credence to the idea that HM or formula-supplemented GAGs could act as prebiotics [168], promoting the growth of only those commensal species outfitted with the enzymes necessary to metabolize these compounds. While these prebiotic effects would be far more likely to affect the distal intestine as opposed to the ileum, the ramifications of this potential GAG influence on the microbiome in total may include changes in NEC susceptibility and require further investigation.
GAGs are also known to reduce microbial adhesion to IECs, often the initial step in infection. Sava et al. [169] pretreated Caco-2 colonocytes with a mixture of heparin, HS, and CS, reducing the capacity of Enterococci bacteria to adhere to the host cell surface. Hafez et al. [170] demonstrated similar findings with regard to Staphylococci adhesion to host epithelial cells in the presence of free GAGs. Treatment of HT-29 colorectal cells with heparin, a prominent GAG in HM, has also been shown to reduce internalization of a number of bacterial species via a reduction in cellular adhesion [171]. Antimicrobial characteristics of GAGs have also been shown to extend to those isolated directly from HM. For example, Newburg et al. [172] demonstrated CS or a CS-like compound extracted from HM can inhibit binding of the gp120 human immunodeficiency virus (HIV) envelope protein to its receptor, an essential early step in transmission of the virus. Coppa et al. [140] treated intestinal cell lines with GAGs extracted from HM and demonstrated a reduction in pathogenic bacterial binding of intestinal receptors, while Hill et al. [173] demonstrated the ability of HA 35 (biosynthetic HA of an intermediate 35 kDa size and with qualitatively similar effects to that of the inclusive HM HA fraction) to limit intestinal adhesion of Salmonella Typhimurium.
GAGs present in HM may also work synergistically with the host immune system to both upregulate endogenous defenses and tame the type of destructive, runaway inflammation characteristic of NEC [174]. HA fragments of varied sizes have been shown to have protective effects on the intestinal epithelium. Zheng et al. [175] noted exogenous administration of 750 kDa HA fragments ameliorated disease in a colitis mouse model through a TLR4- and cyclooxygenase-2 (COX-2)-dependent repair of the epithelium, while Riehl et al. [176] found similar protective effects in irradiated small intestinal tissue. HA of 900 kDa size has also been shown to protect the intestinal epithelium of immunocompromised mice through a reduction in inflammatory signaling [177]. Additionally, both HA 35 and a polydisperse HM HA extract upregulate the antimicrobial protein human β-defensin 2 (HβD2) in human intestinal epithelial cells and its ortholog in the murine large intestine [173,178].
Our group recently demonstrated the effectiveness of HA 35 in reducing the incidence and severity of disease in a mouse model of necrotizing enterocolitis. Gunasekaran et al. [179] treated mouse pups (age P14–16) with HA 35 (15 mg/kg or 30 mg/kg) once per day for three days prior to the initiation of NEC. NEC was induced using a two-hit model of intraperitoneal dithizone injection followed by oral administration of Klebsiella pneumoniae [55]. A stark reduction in proinflammatory cytokine release (tumor necrosis factor-alpha (TNF-α), GRO-α (growth-regulated oncogene-alpha), IL-12p70, and IL-6) was seen with HA 35 treatment (either dose) compared to untreated NEC. These changes, coupled with upregulation in tight junction proteins, likely led to the reduction in pathological intestinal permeability and associated bacteremia, ultimately resulting in significantly improved pathology of the ileum, substantially diminished disease severity, and significantly greater pup survival.
CS also exerts a number of well-documented effects on inflammation, including reductions in pro-inflammatory cytokine and NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) levels [180], weakened COX-2 and NOS-2 (nitric oxide synthase-2) activities [181], and the upregulation of a variety of antioxidant enzymes [182]. CS has demonstrated positive impacts in intestinal bowel disease (IBD) [183,184], likely through anti-inflammatory effects [185] and increased epithelial and mucosal tissue repair [183]. The anti-inflammatory effects of heparin, including reductions in pro-inflammatory TNF-α and IL-6 signaling [186], have also been demonstrated in the context of intestinal inflammatory diseases. Often utilized as a first line of treatment in IBD [187], heparin, specifically low-molecular-weight or unfractionated heparin, has been shown to ameliorate disease activity through a combination of anti-inflammatory and anticoagulative effects [188], resulting in increased mucosal healing and improved intestinal barrier function [189].
Altogether, glycosaminoglycan-associated reduction in pathogen binding to host IECs, an upregulation in intestinal defenses by GAGs, and a reduction in excessive inflammatory signaling is likely to lead to an improvement in intestinal barrier function and a significant decline in bacterial invasion and translocation, critical events in the pathogenesis of NEC [190,191]. Hall et al. [192] established a line of goblet-like cells are more susceptible to invasion when bacteria are freely suspended in bovine-based formula compared to HM. Hill et al. [173] demonstrated a polydisperse HM HA extract is capable of protecting colonocytes from Salmonella infection in vitro. Mice treated with HA 35 are protected from Citrobacter rodentium infection via an upregulation in the critical tight junction protein, ZO-1 (zonula occludens-1), resulting in reduced intestinal permeability and inhibited bacterial translocation across colonic epithelium [193], similar to our findings in a murine NEC model [179].
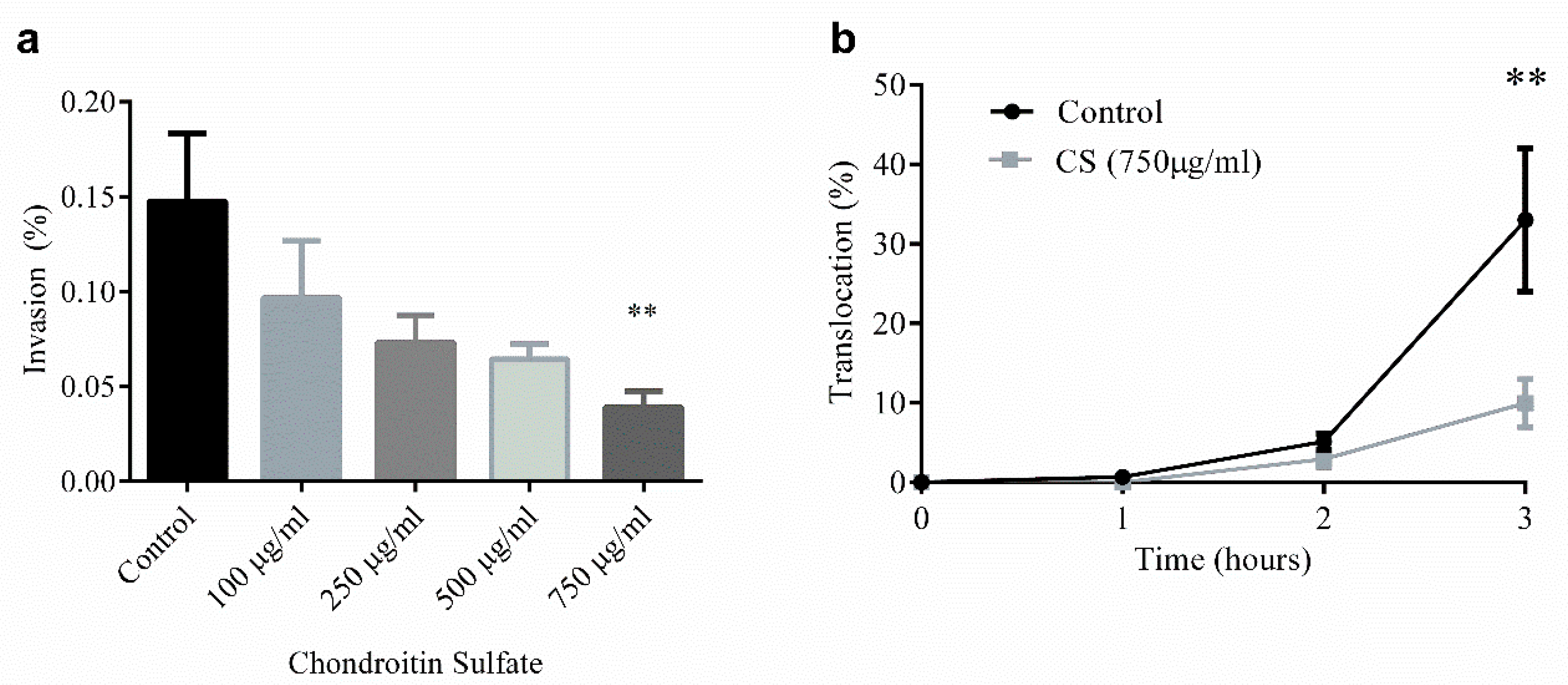
Our group has also directly interrogated the ability of CS, the most common GAG in HM [146], to limit both bacterial invasion (Figure 3a) and translocation (Figure 3b) in T84 colonocyte monolayers, an in vitro model of the intestinal epithelium [194]. CS, at a concentration of 750 µg/mL given prophylactically for 48 h prior to bacterial challenge, reduces invasion and translocation of SCB34 Escherichia coli, an invasive, multi-drug resistant bacterial strain isolated from a neonatal early-onset sepsis case [195], by 75% compared to control. In this study, CS shows no effects on cell viability while also reducing, though not significantly, the production of the inflammatory chemokine, IL-8. Given the potent effects of GAGs on inflammation and prevention of infection, the availability of these compounds in HM, and potentially their supplementation in formula following further systematic review, may be critical to neonatal health in general, and specifically, in the prevention of NEC.
5. Conclusions
In this review, we assessed the GAG composition of sources of neonatal nutrition and relative changes across the duration of infant feeding, as well as summarized the potential protective effects of these GAGs against necrotizing enterocolitis. As common components of HM, GAGs are receiving increased attention because of their demonstrated antimicrobial and anti-inflammatory effects, and their potential to ameliorate intestinal inflammation and associated bacterial translocation of the epithelium. The protective effects on host barrier function, combined with beneficial interactions with, and positive influences on, luminal bacteria, likely serve to strengthen innate defenses against gastrointestinal infection in the neonate. Additional studies are needed to further characterize the effects of HM-derived GAGs on the intestinal epithelium, their interactions with specific bacteria, and their influence on the neonatal intestinal microbiome in full, particularly in the contexts of prematurity and NEC.
Author Contributions
All authors participated in the literature review. K.B. and E.B. wrote the manuscript. A.G. prepared the figures. J.E. and H.C. reviewed, edited, and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from the National Institute of General Medical Sciences, grant number K08GM127308, provided to H.C.
Acknowledgments
The authors acknowledge support from the Division of Neonatology at the University of Oklahoma Health Sciences Center (OUHSC) and the National Institute of General Medical Sciences (K08GM127308).
Conflicts of Interest
The authors declare no conflict of interest. Funding played no role in the preparation of this manuscript.
References
- Neu, J.; Walker, W.A. Necrotizing Enterocolitis. N. Engl. J. Med. 2011, 364, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, S.M.; Berryhill, T.F.; Ellenburg, J.L.; Jilling, T.; Cleveland, D.S.; Lorenz, R.G.; Martin, C.A. Pathogenesis of necrotizing enterocolitis: Modeling the innate immune response. Am. J. Pathol. 2015, 185, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victora, J.D.; Silveira, M.F.; Tonial, C.T.; Victora, C.G.; Barros, F.C.; Horta, B.L.; Santos, I.S.D.; Bassani, D.G.; Garcia, P.C.R.; Scheeren, M.; et al. Prevalence, mortality and risk factors associated with very low birth weight preterm infants: An analysis of 33 years. J. Pediatr. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rich, B.S.; Dolgin, S.E. Necrotizing Enterocolitis. Pediatr. Rev. 2017, 38, 552–559. [Google Scholar] [CrossRef]
- Bering, S.B. Human Milk Oligosaccharides to Prevent Gut Dysfunction and Necrotizing Enterocolitis in Preterm Neonates. Nutrients 2018, 10, 1461. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, J. Human milk and the premature infant. Ann. Nutr. Metab. 2013, 62 (Suppl. 3), 8–14. [Google Scholar] [CrossRef]
- Neu, J.; Pammi, M. Pathogenesis of NEC: Impact of an altered intestinal microbiome. Semin. Perinatol. 2017, 41, 29–35. [Google Scholar] [CrossRef]
- Samuels, N.; van de Graaf, R.A.; de Jonge, R.C.J.; Reiss, I.K.M.; Vermeulen, M.J. Risk factors for necrotizing enterocolitis in neonates: A systematic review of prognostic studies. BMC Pediatr. 2017, 17, 105. [Google Scholar] [CrossRef]
- Cacho, N.T.; Parker, L.A.; Neu, J. Necrotizing Enterocolitis and Human Milk Feeding: A Systematic Review. Clin. Perinatol. 2017, 44, 49–67. [Google Scholar] [CrossRef]
- Miller, J.; Tonkin, E.; Damarell, R.A.; McPhee, A.J.; Suganuma, M.; Suganuma, H.; Middleton, P.F.; Makrides, M.; Collins, C.T. A Systematic Review and Meta-Analysis of Human Milk Feeding and Morbidity in Very Low Birth Weight Infants. Nutrients 2018, 10, 707. [Google Scholar] [CrossRef] [Green Version]
- Andreas, N.J.; Kampmann, B.; Mehring Le-Doare, K. Human breast milk: A review on its composition and bioactivity. Early Hum. Dev. 2015, 91, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Grulee, C.G.; Sanford, H.N.; Herron, P.H. Breast and artificial feeding: Influence on morbidity and mortality of twenty thousand infants. JAMA J. Am. Med. Assoc. 1934, 103, 735–738. [Google Scholar] [CrossRef]
- Howie, P.W.; Forsyth, J.S.; Ogston, S.A.; Clark, A.; Florey, C.D. Protective effect of breast feeding against infection. BMJ 1990, 300, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Abrams, S.A.; Schanler, R.J.; Lee, M.L.; Rechtman, D.J. Greater mortality and morbidity in extremely preterm infants fed a diet containing cow milk protein products. Breastfeed. Med. 2014, 9, 281–285. [Google Scholar] [CrossRef]
- Sisk, P.M.; Lovelady, C.A.; Dillard, R.G.; Gruber, K.J.; O’Shea, T.M. Early human milk feeding is associated with a lower risk of necrotizing enterocolitis in very low birth weight infants. J. Perinatol. 2007, 27, 428–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, A.; Cole, T.J. Breast milk and neonatal necrotising enterocolitis. Lancet 1990, 336, 1519–1523. [Google Scholar] [CrossRef]
- Meinzen-Derr, J.; Poindexter, B.; Wrage, L.; Morrow, A.L.; Stoll, B.; Donovan, E.F. Role of human milk in extremely low birth weight infants’ risk of necrotizing enterocolitis or death. J. Perinatol. 2009, 29, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpeleijn, W.E.; Kouwenhoven, S.M.; Paap, M.C.; van Vliet, I.; Scheerder, I.; Muizer, Y.; Helder, O.K.; van Goudoever, J.B.; Vermeulen, M.J. Intake of own mother’s milk during the first days of life is associated with decreased morbidity and mortality in very low birth weight infants during the first 60 days of life. Neonatology 2012, 102, 276–281. [Google Scholar] [CrossRef]
- Arslanoglu, S.; Corpeleijn, W.; Moro, G.; Braegger, C.; Campoy, C.; Colomb, V.; Decsi, T.; Domellof, M.; Fewtrell, M.; Hojsak, I.; et al. Donor human milk for preterm infants: Current evidence and research directions. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 535–542. [Google Scholar] [CrossRef]
- Li, Y.; Nguyen, D.N.; de Waard, M.; Christensen, L.; Zhou, P.; Jiang, P.; Sun, J.; Bojesen, A.M.; Lauridsen, C.; Lykkesfeldt, J.; et al. Pasteurization Procedures for Donor Human Milk Affect Body Growth, Intestinal Structure, and Resistance against Bacterial Infections in Preterm Pigs. J. Nutr. 2017, 147, 1121–1130. [Google Scholar] [CrossRef] [Green Version]
- Aksu, T.; Atalay, Y.; Turkyilmaz, C.; Gulbahar, O.; Hirfanoglu, I.M.; Demirel, N.; Onal, E.; Ergenekon, E.; Koc, E. The effects of breast milk storage and freezing procedure on interleukine-10 levels and total antioxidant activity. J. Matern. Fetal Neonatal Med. 2015, 28, 1799–1802. [Google Scholar] [CrossRef] [PubMed]
- Corpeleijn, W.E.; de Waard, M.; Christmann, V.; van Goudoever, J.B.; Jansen-van der Weide, M.C.; Kooi, E.M.; Koper, J.F.; Kouwenhoven, S.M.; Lafeber, H.N.; Mank, E.; et al. Effect of Donor Milk on Severe Infections and Mortality in Very Low-Birth-Weight Infants: The Early Nutrition Study Randomized Clinical Trial. JAMA Pediatr. 2016, 170, 654–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canizo Vazquez, D.; Salas Garcia, S.; Izquierdo Renau, M.; Iglesias-Platas, I. Availability of Donor Milk for Very Preterm Infants Decreased the Risk of Necrotizing Enterocolitis without Adversely Impacting Growth or Rates of Breastfeeding. Nutrients 2019, 11, 1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, S.L.; Lohmann, P.; Preidis, G.A.; Gordon, P.S.; O’Donnell, A.; Hagan, J.; Venkatachalam, A.; Balderas, M.; Luna, R.A.; Hair, A.B. Improved feeding tolerance and growth are linked to increased gut microbial community diversity in very-low-birth-weight infants fed mother’s own milk compared with donor breast milk. Am. J. Clin. Nutr. 2019, 109, 1088–1097. [Google Scholar] [CrossRef]
- Baethge, C.; Goldbeck-Wood, S.; Mertens, S. SANRA-a scale for the quality assessment of narrative review articles. Res. Integr. Peer Rev. 2019, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R. Writing narrative style literature reviews. Med Writ. 2015, 24, 230–235. [Google Scholar] [CrossRef]
- Battersby, A.J.; Gibbons, D.L. The gut mucosal immune system in the neonatal period. Pediatr. Allergy Immunol. 2013, 24, 414–421. [Google Scholar] [CrossRef]
- Pammi, M.; Cope, J.; Tarr, P.I.; Warner, B.B.; Morrow, A.L.; Mai, V.; Gregory, K.E.; Kroll, J.S.; McMurtry, V.; Ferris, M.J.; et al. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: A systematic review and meta-analysis. Microbiome 2017, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Denning, N.L.; Prince, J.M. Neonatal intestinal dysbiosis in necrotizing enterocolitis. Mol. Med. 2018, 24, 4. [Google Scholar] [CrossRef]
- Moore, S.A.; Nighot, P.; Reyes, C.; Rawat, M.; McKee, J.; Lemon, D.; Hanson, J.; Ma, T.Y. Intestinal barrier dysfunction in human necrotizing enterocolitis. J. Pediatr. Surg. 2016, 51, 1907–1913. [Google Scholar] [CrossRef] [Green Version]
- Krappmann, D.; Wegener, E.; Sunami, Y.; Esen, M.; Thiel, A.; Mordmuller, B.; Scheidereit, C. The IkappaB kinase complex and NF-kappaB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol. Cell. Biol. 2004, 24, 6488–6500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, C.P.; Neal, M.D.; Siggers, R.; Sho, S.; Ma, C.; Branca, M.F.; Prindle, T., Jr.; Russo, A.M.; Afrazi, A.; Good, M.; et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 2012, 143, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaphart, C.L.; Cavallo, J.; Gribar, S.C.; Cetin, S.; Li, J.; Branca, M.F.; Dubowski, T.D.; Sodhi, C.P.; Hackam, D.J. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 2007, 179, 4808–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, C.E.; Sodhi, C.P.; Good, M.; Lin, J.; Jia, H.; Yamaguchi, Y.; Lu, P.; Ma, C.; Branca, M.F.; Weyandt, S.; et al. Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J. Clin. Investig. 2016, 126, 495–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, M.; Siggers, R.H.; Sodhi, C.P.; Afrazi, A.; Alkhudari, F.; Egan, C.E.; Neal, M.D.; Yazji, I.; Jia, H.; Lin, J.; et al. Amniotic fluid inhibits Toll-like receptor 4 signaling in the fetal and neonatal intestinal epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, 11330–11335. [Google Scholar] [CrossRef] [Green Version]
- Good, M.; Sodhi, C.P.; Egan, C.E.; Afrazi, A.; Jia, H.; Yamaguchi, Y.; Lu, P.; Branca, M.F.; Ma, C.; Prindle, T., Jr.; et al. Breast milk protects against the development of necrotizing enterocolitis through inhibition of Toll-like receptor 4 in the intestinal epithelium via activation of the epidermal growth factor receptor. Mucosal. Immunol. 2015, 8, 1166–1179. [Google Scholar] [CrossRef]
- Neal, M.D.; Jia, H.; Eyer, B.; Good, M.; Guerriero, C.J.; Sodhi, C.P.; Afrazi, A.; Prindle, T., Jr.; Ma, C.; Branca, M.; et al. Discovery and validation of a new class of small molecule Toll-like receptor 4 (TLR4) inhibitors. PLoS ONE 2013, 8, e65779. [Google Scholar] [CrossRef] [Green Version]
- Good, M.; Sodhi, C.P.; Yamaguchi, Y.; Jia, H.; Lu, P.; Fulton, W.B.; Martin, L.Y.; Prindle, T.; Nino, D.F.; Zhou, Q.; et al. The human milk oligosaccharide 2’-fucosyllactose attenuates the severity of experimental necrotising enterocolitis by enhancing mesenteric perfusion in the neonatal intestine. Br. J. Nutr. 2016, 116, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Sodhi, C.P.; Hackam, D.J. Toll-like receptor regulation of intestinal development and inflammation in the pathogenesis of necrotizing enterocolitis. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2014, 21, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Burge, K.; Gunasekaran, A.; Eckert, J.; Chaaban, H. Curcumin and Intestinal Inflammatory Diseases: Molecular Mechanisms of Protection. Int. J. Mol. Sci. 2019, 20, 1912. [Google Scholar] [CrossRef] [Green Version]
- Mara, M.A.; Good, M.; Weitkamp, J.H. Innate and adaptive immunity in necrotizing enterocolitis. Semin. Fetal Neonatal Med. 2018, 23, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Udall, J.N.; Pang, K.; Fritze, L.; Kleinman, R.; Walker, W.A. Development of gastrointestinal mucosal barrier. I. The effect of age on intestinal permeability to macromolecules. Pediatr. Res. 1981, 15, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Managlia, E.; Liu, S.X.L.; Yan, X.; Tan, X.D.; Chou, P.M.; Barrett, T.A.; De Plaen, I.G. Blocking NF-kappaB Activation in Ly6c(+) Monocytes Attenuates Necrotizing Enterocolitis. Am. J. Pathol. 2019, 189, 604–618. [Google Scholar] [CrossRef] [Green Version]
- De Plaen, I.G.; Liu, S.X.; Tian, R.; Neequaye, I.; May, M.J.; Han, X.B.; Hsueh, W.; Jilling, T.; Lu, J.; Caplan, M.S. Inhibition of nuclear factor-kappaB ameliorates bowel injury and prolongs survival in a neonatal rat model of necrotizing enterocolitis. Pediatr. Res. 2007, 61, 716–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markel, T.A.; Crisostomo, P.R.; Wairiuko, G.M.; Pitcher, J.; Tsai, B.M.; Meldrum, D.R. Cytokines in necrotizing enterocolitis. Shock 2006, 25, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Yazji, I.; Sodhi, C.P.; Lee, E.K.; Good, M.; Egan, C.E.; Afrazi, A.; Neal, M.D.; Jia, H.; Lin, J.; Ma, C.; et al. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 9451–9456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.J.; Besner, G.E. The role of the intestinal microcirculation in necrotizing enterocolitis. Semin. Pediatr. Surg. 2013, 22, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoma, C. Preventing brain damage in necrotizing enterocolitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, B.O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. 2019, 7, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ghosh, S.S.; Ghosh, S. Curcumin improves intestinal barrier function: Modulation of intracellular signaling, and organization of tight junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, C438–C445. [Google Scholar] [CrossRef]
- Martin, N.A.; Mount Patrick, S.K.; Estrada, T.E.; Frisk, H.A.; Rogan, D.T.; Dvorak, B.; Halpern, M.D. Active transport of bile acids decreases mucin 2 in neonatal ileum: Implications for development of necrotizing enterocolitis. PLoS ONE 2011, 6, e27191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.A.; Doelle, S.M.; Halpern, M.D.; Saunders, T.A.; Holubec, H.; Dvorak, K.; Boitano, S.A.; Dvorak, B. Intestinal barrier failure during experimental necrotizing enterocolitis: Protective effect of EGF treatment. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G938–G949. [Google Scholar] [CrossRef]
- McElroy, S.J.; Prince, L.S.; Weitkamp, J.H.; Reese, J.; Slaughter, J.C.; Polk, D.B. Tumor necrosis factor receptor 1-dependent depletion of mucus in immature small intestine: A potential role in neonatal necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G656–G666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Sherman, M.P.; Prince, L.S.; Bader, D.; Weitkamp, J.H.; Slaughter, J.C.; McElroy, S.J. Paneth cell ablation in the presence of Klebsiella pneumoniae induces necrotizing enterocolitis (NEC)-like injury in the small intestine of immature mice. Dis. Models Mech. 2012, 5, 522–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markasz, L.; Wanders, A.; Szekely, L.; Lilja, H.E. Diminished DEFA6 Expression in Paneth Cells Is Associated with Necrotizing Enterocolitis. Gastroenterol Res Pract. 2018, 2018, 7345426. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, H.B.; da Mota, H.C.; Coutinho, V.B.; Robalinho, T.I.; Furtado, A.F.; Walker, E.; King, G.; Mahida, Y.R.; Sewell, H.F.; Wakelin, D. Absence of lysozyme (muramidase) in the intestinal Paneth cells of newborn infants with necrotising enterocolitis. J. Clin. Pathol. 1998, 51, 512–514. [Google Scholar] [CrossRef] [Green Version]
- Salzman, N.H.; Bevins, C.L. Dysbiosis--a consequence of Paneth cell dysfunction. Semin. Immunol. 2013, 25, 334–341. [Google Scholar] [CrossRef]
- Strunk, T.; Temming, P.; Gembruch, U.; Reiss, I.; Bucsky, P.; Schultz, C. Differential maturation of the innate immune response in human fetuses. Pediatr. Res. 2004, 56, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Maheshwari, A.; Kelly, D.R.; Nicola, T.; Ambalavanan, N.; Jain, S.K.; Murphy-Ullrich, J.; Athar, M.; Shimamura, M.; Bhandari, V.; Aprahamian, C.; et al. TGF-beta2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 2011, 140, 242–253. [Google Scholar] [CrossRef] [Green Version]
- MohanKumar, K.; Namachivayam, K.; Chapalamadugu, K.C.; Garzon, S.A.; Premkumar, M.H.; Tipparaju, S.M.; Maheshwari, A. Smad7 interrupts TGF-beta signaling in intestinal macrophages and promotes inflammatory activation of these cells during necrotizing enterocolitis. Pediatr. Res. 2016, 79, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namachivayam, K.; Blanco, C.L.; MohanKumar, K.; Jagadeeswaran, R.; Vasquez, M.; McGill-Vargas, L.; Garzon, S.A.; Jain, S.K.; Gill, R.K.; Freitag, N.E.; et al. Smad7 inhibits autocrine expression of TGF-beta2 in intestinal epithelial cells in baboon necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G167–G180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuller, S.S.; Sadeghi, K.; Wisgrill, L.; Dangl, A.; Diesner, S.C.; Prusa, A.R.; Klebermasz-Schrehof, K.; Greber-Platzer, S.; Neumuller, J.; Helmer, H.; et al. Preterm neonates display altered plasmacytoid dendritic cell function and morphology. J. Leukoc. Biol. 2013, 93, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Emami, C.N.; Mittal, R.; Wang, L.; Ford, H.R.; Prasadarao, N.V. Recruitment of dendritic cells is responsible for intestinal epithelial damage in the pathogenesis of necrotizing enterocolitis by Cronobacter sakazakii. J. Immunol. 2011, 186, 7067–7079. [Google Scholar] [CrossRef] [Green Version]
- Jilling, T.; Simon, D.; Lu, J.; Meng, F.J.; Li, D.; Schy, R.; Thomson, R.B.; Soliman, A.; Arditi, M.; Caplan, M.S. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J. Immunol. 2006, 177, 3273–3282. [Google Scholar] [CrossRef] [Green Version]
- Le Mandat Schultz, A.; Bonnard, A.; Barreau, F.; Aigrain, Y.; Pierre-Louis, C.; Berrebi, D.; Peuchmaur, M. Expression of TLR-2, TLR-4, NOD2 and pNF-kappaB in a neonatal rat model of necrotizing enterocolitis. PLoS ONE 2007, 2, e1102. [Google Scholar] [CrossRef] [Green Version]
- Dimmitt, R.A.; Staley, E.M.; Chuang, G.; Tanner, S.M.; Soltau, T.D.; Lorenz, R.G. Role of postnatal acquisition of the intestinal microbiome in the early development of immune function. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Nanthakumar, N.; Meng, D.; Goldstein, A.M.; Zhu, W.; Lu, L.; Uauy, R.; Llanos, A.; Claud, E.C.; Walker, W.A. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: An immature innate immune response. PLoS ONE 2011, 6, e17776. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhu, L.; Fatheree, N.Y.; Liu, X.; Pacheco, S.E.; Tatevian, N.; Rhoads, J.M. Changes in intestinal Toll-like receptors and cytokines precede histological injury in a rat model of necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G442–G450. [Google Scholar] [CrossRef] [Green Version]
- Dowling, D.J.; Levy, O. Ontogeny of early life immunity. Trends Immunol. 2014, 35, 299–310. [Google Scholar] [CrossRef]
- Gutzeit, C.; Magri, G.; Cerutti, A. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev. 2014, 260, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Borges, M.C.; Sesso, M.L.; Roberti, L.R.; de Menezes Oliveira, M.A.; Nogueira, R.D.; Geraldo-Martins, V.R.; Ferriani, V.P. Salivary antibody response to streptococci in preterm and fullterm children: A prospective study. Arch. Oral Biol. 2015, 60, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, R.D.; Sesso, M.L.; Borges, M.C.; Mattos-Graner, R.O.; Smith, D.J.; Ferriani, V.P. Salivary IgA antibody responses to Streptococcus mitis and Streptococcus mutans in preterm and fullterm newborn children. Arch. Oral Biol. 2012, 57, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, A.S.; Behrendt, C.L.; Hooper, L.V. Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J. Immunol. 2009, 182, 3047–3054. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Haque, S.F.; Silberzahn, T.; Hamilton, K.; Langford, C.; Ellis, P.; Carr, R.; Hayday, A.C. Neonates harbour highly active gammadelta T cells with selective impairments in preterm infants. Eur. J. Immunol. 2009, 39, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Weitkamp, J.H.; Rosen, M.J.; Zhao, Z.; Koyama, T.; Geem, D.; Denning, T.L.; Rock, M.T.; Moore, D.J.; Halpern, M.D.; Matta, P.; et al. Small intestinal intraepithelial TCRgammadelta+ T lymphocytes are present in the premature intestine but selectively reduced in surgical necrotizing enterocolitis. PLoS ONE 2014, 9, e99042. [Google Scholar] [CrossRef] [PubMed]
- Weitkamp, J.H.; Koyama, T.; Rock, M.T.; Correa, H.; Goettel, J.A.; Matta, P.; Oswald-Richter, K.; Rosen, M.J.; Engelhardt, B.G.; Moore, D.J.; et al. Necrotising enterocolitis is characterised by disrupted immune regulation and diminished mucosal regulatory (FOXP3)/effector (CD4, CD8) T cell ratios. Gut 2013, 62, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Elias, K.M.; Laurence, A.; Davidson, T.S.; Stephens, G.; Kanno, Y.; Shevach, E.M.; O’Shea, J.J. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 2008, 111, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Nino, D.F.; Sodhi, C.P.; Egan, C.E.; Zhou, Q.; Lin, J.; Lu, P.; Yamaguchi, Y.; Jia, H.; Martin, L.Y.; Good, M.; et al. Retinoic Acid Improves Incidence and Severity of Necrotizing Enterocolitis by Lymphocyte Balance Restitution and Repopulation of LGR5+ Intestinal Stem Cells. Shock 2017, 47, 22–32. [Google Scholar] [CrossRef]
- Grizotte-Lake, M.; Zhong, G.; Duncan, K.; Kirkwood, J.; Iyer, N.; Smolenski, I.; Isoherranen, N.; Vaishnava, S. Commensals Suppress Intestinal Epithelial Cell Retinoic Acid Synthesis to Regulate Interleukin-22 Activity and Prevent Microbial Dysbiosis. Immunity 2018, 49, 1103–1115. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinson, L.F.; Boyce, M.C.; Payne, M.S.; Keelan, J.A. The Not-so-Sterile Womb: Evidence That the Human Fetus Is Exposed to Bacteria Prior to Birth. Front. Microbiol. 2019, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Leiby, J.S.; McCormick, K.; Sherrill-Mix, S.; Clarke, E.L.; Kessler, L.R.; Taylor, L.J.; Hofstaedter, C.E.; Roche, A.M.; Mattei, L.M.; Bittinger, K.; et al. Lack of detection of a human placenta microbiome in samples from preterm and term deliveries. Microbiome 2018, 6, 196. [Google Scholar] [CrossRef] [PubMed]
- de Goffau, M.C.; Lager, S.; Sovio, U.; Gaccioli, F.; Cook, E.; Peacock, S.J.; Parkhill, J.; Charnock-Jones, D.S.; Smith, G.C.S. Human placenta has no microbiome but can contain potential pathogens. Nature 2019, 572, 329–334. [Google Scholar] [CrossRef]
- Patel, R.M.; Denning, P.W. Intestinal microbiota and its relationship with necrotizing enterocolitis. Pediatr. Res. 2015, 78, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [Green Version]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Chassard, C. New insights in gut microbiota establishment in healthy breast fed neonates. PLoS ONE 2012, 7, e44595. [Google Scholar] [CrossRef]
- Elgin, T.G.; Kern, S.L.; McElroy, S.J. Development of the Neonatal Intestinal Microbiome and Its Association with Necrotizing Enterocolitis. Clin. Ther. 2016, 38, 706–715. [Google Scholar] [CrossRef] [Green Version]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- La Rosa, P.S.; Warner, B.B.; Zhou, Y.; Weinstock, G.M.; Sodergren, E.; Hall-Moore, C.M.; Stevens, H.J.; Bennett, W.E., Jr.; Shaikh, N.; Linneman, L.A.; et al. Patterned progression of bacterial populations in the premature infant gut. Proc. Natl. Acad. Sci. USA 2014, 111, 12522–12527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, M.K.; Wang, B.; Ahmadi, S.; Burnham, C.A.; Tarr, P.I.; Warner, B.B.; Dantas, G. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol. 2016, 1, 16024. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.V.; Scholz, M.; Zolfo, M.; Taft, D.H.; Schibler, K.R.; Tett, A.; Segata, N.; Morrow, A.L. Metagenomic Sequencing with Strain-Level Resolution Implicates Uropathogenic E. coli in Necrotizing Enterocolitis and Mortality in Preterm Infants. Cell Rep. 2016, 14, 2912–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlisle, E.M.; Morowitz, M.J. The intestinal microbiome and necrotizing enterocolitis. Curr. Opin. Pediatr. 2013, 25, 382–387. [Google Scholar] [CrossRef]
- Wang, Y.; Hoenig, J.D.; Malin, K.J.; Qamar, S.; Petrof, E.O.; Sun, J.; Antonopoulos, D.A.; Chang, E.B.; Claud, E.C. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009, 3, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef]
- Afrazi, A.; Sodhi, C.P.; Richardson, W.; Neal, M.; Good, M.; Siggers, R.; Hackam, D.J. New insights into the pathogenesis and treatment of necrotizing enterocolitis: Toll-like receptors and beyond. Pediatr. Res. 2011, 69, 183–188. [Google Scholar] [CrossRef]
- Musemeche, C.A.; Kosloske, A.M.; Bartow, S.A.; Umland, E.T. Comparative effects of ischemia, bacteria, and substrate on the pathogenesis of intestinal necrosis. J. Pediatr. Surg. 1986, 21, 536–538. [Google Scholar] [CrossRef]
- Morowitz, M.J.; Poroyko, V.; Caplan, M.; Alverdy, J.; Liu, D.C. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics 2010, 125, 777–785. [Google Scholar] [CrossRef]
- Mai, V.; Young, C.M.; Ukhanova, M.; Wang, X.; Sun, Y.; Casella, G.; Theriaque, D.; Li, N.; Sharma, R.; Hudak, M.; et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS ONE 2011, 6, e20647. [Google Scholar] [CrossRef] [PubMed]
- Torrazza, R.M.; Neu, J. The altered gut microbiome and necrotizing enterocolitis. Clin. Perinatol. 2013, 40, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Warner, B.B.; Deych, E.; Zhou, Y.; Hall-Moore, C.; Weinstock, G.M.; Sodergren, E.; Shaikh, N.; Hoffmann, J.A.; Linneman, L.A.; Hamvas, A.; et al. Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: A prospective case-control study. Lancet 2016, 387, 1928–1936. [Google Scholar] [CrossRef] [Green Version]
- Neu, J.; Pammi, M. Necrotizing enterocolitis: The intestinal microbiome, metabolome and inflammatory mediators. Semin. Fetal Neonatal Med. 2018, 23, 400–405. [Google Scholar] [CrossRef]
- Torrazza, R.M.; Ukhanova, M.; Wang, X.; Sharma, R.; Hudak, M.L.; Neu, J.; Mai, V. Intestinal microbial ecology and environmental factors affecting necrotizing enterocolitis. PLoS ONE 2013, 8, e83304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbler, P.T.; Procianoy, R.S.; Mai, V.; Silveira, R.C.; Corso, A.L.; Rojas, B.S.; Roesch, L.F.W. Low Microbial Diversity and Abnormal Microbial Succession Is Associated with Necrotizing Enterocolitis in Preterm Infants. Front. Microbiol. 2017, 8, 2243. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishna, K.P.; Macadangdang, B.R.; Rogers, M.B.; Tometich, J.T.; Firek, B.A.; Baker, R.; Ji, J.; Burr, A.H.P.; Ma, C.; Good, M.; et al. Maternal IgA protects against the development of necrotizing enterocolitis in preterm infants. Nat. Med. 2019, 25, 1110–1115. [Google Scholar] [CrossRef]
- Morrow, A.L.; Lagomarcino, A.J.; Schibler, K.R.; Taft, D.H.; Yu, Z.; Wang, B.; Altaye, M.; Wagner, M.; Gevers, D.; Ward, D.V.; et al. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome 2013, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Bizzarro, M.J. Avoiding Unnecessary Antibiotic Exposure in Premature Infants: Understanding When (Not) to Start and When to Stop. JAMA Netw. Open 2018, 1, e180165. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, R.G.; Chowdhury, D.; Hansen, N.I.; Smith, P.B.; Stoll, B.J.; Sanchez, P.J.; Das, A.; Puopolo, K.M.; Mukhopadhyay, S.; Higgins, R.D.; et al. Prolonged duration of early antibiotic therapy in extremely premature infants. Pediatr. Res. 2019, 85, 994–1000. [Google Scholar] [CrossRef]
- Cotten, C.M. Adverse consequences of neonatal antibiotic exposure. Curr. Opin. Pediatr. 2016, 28, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, V.N.; Northrup, V.; Bizzarro, M.J. Antibiotic exposure in the newborn intensive care unit and the risk of necrotizing enterocolitis. J. Pediatr. 2011, 159, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouhy, F.; Guinane, C.M.; Hussey, S.; Wall, R.; Ryan, C.A.; Dempsey, E.M.; Murphy, B.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C.; et al. High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob. Agents Chemother. 2012, 56, 5811–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Kobayashi, T.; Songjinda, P.; Tateyama, A.; Tsubouchi, M.; Kiyohara, C.; Shirakawa, T.; Sonomoto, K.; Nakayama, J. Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota. FEMS Immunol. Med. Microbiol. 2009, 56, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, C.; Morrow, A.L.; Lagomarcino, A.J.; Altaye, M.; Taft, D.H.; Yu, Z.; Newburg, D.S.; Ward, D.V.; Schibler, K.R. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J. Pediatr. 2014, 165, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Biasucci, G.; Rubini, M.; Riboni, S.; Morelli, L.; Bessi, E.; Retetangos, C. Mode of delivery affects the bacterial community in the newborn gut. Early Hum. Dev. 2010, 86 (Suppl. 1), 13–15. [Google Scholar] [CrossRef]
- Singh, N.; Dhayade, A.; Mohamed, A.L.; Chaudhari, T.V. Morbidity and Mortality in Preterm Infants following Antacid Use: A Retrospective Audit. Int. J. Pediatr. 2016, 2016, 9649162. [Google Scholar] [CrossRef] [Green Version]
- Terrin, G.; Passariello, A.; De Curtis, M.; Manguso, F.; Salvia, G.; Lega, L.; Messina, F.; Paludetto, R.; Canani, R.B. Ranitidine is associated with infections, necrotizing enterocolitis, and fatal outcome in newborns. Pediatrics 2012, 129, e40–e45. [Google Scholar] [CrossRef] [Green Version]
- Bilali, A.; Galanis, P.; Bartsocas, C.; Sparos, L.; Velonakis, E. H2-blocker therapy and incidence of necrotizing enterocolitis in preterm infants: A case-control study. Pediatr. Neonatol. 2013, 54, 141–142. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.W.; Tran, L.; Norori, J.; Ferris, M.J.; Eren, A.M.; Taylor, C.M.; Dowd, S.E.; Penn, D. Histamine-2 receptor blockers alter the fecal microbiota in premature infants. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Lueschow, S.R.; Stumphy, J.; Gong, H.; Kern, S.L.; Elgin, T.G.; Underwood, M.A.; Kalanetra, K.M.; Mills, D.A.; Wong, M.H.; Meyerholz, D.K.; et al. Loss of murine Paneth cell function alters the immature intestinal microbiome and mimics changes seen in neonatal necrotizing enterocolitis. PLoS ONE 2018, 13, e0204967. [Google Scholar] [CrossRef] [PubMed]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; Adisetiyo, H.; Zabih, S.; Lincez, P.J.; Bittinger, K.; et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr. 2017, 171, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.M.; Foster, J.A.; Forney, L.J.; Schutte, U.M.; Beck, D.L.; Abdo, Z.; Fox, L.K.; Williams, J.E.; McGuire, M.K.; McGuire, M.A. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS ONE 2011, 6, e21313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, Z.T.; Totten, S.M.; Smilowitz, J.T.; Popovic, M.; Parker, E.; Lemay, D.G.; Van Tassell, M.L.; Miller, M.J.; Jin, Y.S.; German, J.B.; et al. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 2015, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Putignani, L.; Del Chierico, F.; Petrucca, A.; Vernocchi, P.; Dallapiccola, B. The human gut microbiota: A dynamic interplay with the host from birth to senescence settled during childhood. Pediatr. Res. 2014, 76, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Oozeer, R.; van Limpt, K.; Ludwig, T.; Ben Amor, K.; Martin, R.; Wind, R.D.; Boehm, G.; Knol, J. Intestinal microbiology in early life: Specific prebiotics can have similar functionalities as human-milk oligosaccharides. Am. J. Clin. Nutr. 2013, 98, 561s–571s. [Google Scholar] [CrossRef]
- Gomez-Llorente, C.; Plaza-Diaz, J.; Aguilera, M.; Munoz-Quezada, S.; Bermudez-Brito, M.; Peso-Echarri, P.; Martinez-Silla, R.; Vasallo-Morillas, M.I.; Campana-Martin, L.; Vives-Pinera, I.; et al. Three main factors define changes in fecal microbiota associated with feeding modality in infants. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 461–466. [Google Scholar] [CrossRef]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; van den Brandt, P.A.; Stobberingh, E.E. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Penders, J.; Vink, C.; Driessen, C.; London, N.; Thijs, C.; Stobberingh, E.E. Quantification of Bifidobacterium spp.; Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiol. Lett. 2005, 243, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, S.; Aydin, S.; Ozkan, Y.; Kumru, S. Ghrelin is present in human colostrum, transitional and mature milk. Peptides 2006, 27, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Michalski, M.C.; Briard, V.; Michel, F.; Tasson, F.; Poulain, P. Size distribution of fat globules in human colostrum, breast milk, and infant formula. J. Dairy Sci. 2005, 88, 1927–1940. [Google Scholar] [CrossRef] [Green Version]
- Ballard, O.; Morrow, A.L. Human milk composition: Nutrients and bioactive factors. Pediatr. Clin. N. Am. 2013, 60, 49–74. [Google Scholar] [CrossRef] [Green Version]
- Coppa, G.V.; Gabrielli, O.; Zampini, L.; Galeazzi, T.; Ficcadenti, A.; Padella, L.; Santoro, L.; Soldi, S.; Carlucci, A.; Bertino, E.; et al. Oligosaccharides in 4 different milk groups, Bifidobacteria, and Ruminococcus obeum. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 80–87. [Google Scholar] [CrossRef]
- Morrow, A.L.; Ruiz-Palacios, G.M.; Altaye, M.; Jiang, X.; Guerrero, M.L.; Meinzen-Derr, J.K.; Farkas, T.; Chaturvedi, P.; Pickering, L.K.; Newburg, D.S. Human milk oligosaccharides are associated with protection against diarrhea in breast-fed infants. J. Pediatr. 2004, 145, 297–303. [Google Scholar] [CrossRef]
- Goldman, A.S. Modulation of the gastrointestinal tract of infants by human milk. Interfaces and interactions. An evolutionary perspective. J. Nutr. 2000, 130, 426s–431s. [Google Scholar] [CrossRef]
- Santaolalla, R.; Fukata, M.; Abreu, M.T. Innate immunity in the small intestine. Curr. Opin. Gastroenterol. 2011, 27, 125–131. [Google Scholar] [CrossRef]
- Ricard-Blum, S. Glycosaminoglycans: Major biological players. Glycoconj. J. 2017, 34, 275–276. [Google Scholar] [CrossRef] [Green Version]
- Coppa, G.V.; Facinelli, B.; Magi, G.; Marini, E.; Zampini, L.; Mantovani, V.; Galeazzi, T.; Padella, L.; Marchesiello, R.L.; Santoro, L.; et al. Human milk glycosaminoglycans inhibit in vitro the adhesion of Escherichia coli and Salmonella fyris to human intestinal cells. Pediatr. Res. 2016, 79, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Barthe, L.; Woodley, J.; Lavit, M.; Przybylski, C.; Philibert, C.; Houin, G. In vitro intestinal degradation and absorption of chondroitin sulfate, a glycosaminoglycan drug. Arzneimittelforschung 2004, 54, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Maccari, F.; Mantovani, V.; Gabrielli, O.; Carlucci, A.; Zampini, L.; Galeazzi, T.; Galeotti, F.; Coppa, G.V.; Volpi, N. Metabolic fate of milk glycosaminoglycans in breastfed and formula fed newborns. Glycoconj. J. 2016, 33, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic Acid in the Third Millennium. Polymers 2018, 10, 701. [Google Scholar] [CrossRef] [Green Version]
- Lee-Sayer, S.S.; Dong, Y.; Arif, A.A.; Olsson, M.; Brown, K.L.; Johnson, P. The where, when, how, and why of hyaluronan binding by immune cells. Front. Immunol. 2015, 6, 150. [Google Scholar] [CrossRef] [Green Version]
- Coppa, G.V.; Gabrielli, O.; Buzzega, D.; Zampini, L.; Galeazzi, T.; Maccari, F.; Bertino, E.; Volpi, N. Composition and structure elucidation of human milk glycosaminoglycans. Glycobiology 2011, 21, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Volpi, N.; Gabrielli, O.; Carlucci, A.; Zampini, L.; Santoro, L.; Padella, L.; Marchesello, R.L.; Maccari, F.; Coppa, G.V. Human milk glycosaminoglycans in feces of breastfed newborns: Preliminary structural elucidation and possible biological role. Breastfeed. Med. Off. J. Acad. Breastfeed. Med. 2014, 9, 105–106. [Google Scholar] [CrossRef]
- Coppa, G.V.; Gabrielli, O.; Zampini, L.; Galeazzi, T.; Maccari, F.; Buzzega, D.; Galeotti, F.; Bertino, E.; Volpi, N. Glycosaminoglycan content in term and preterm milk during the first month of lactation. Neonatology 2012, 101, 74–76. [Google Scholar] [CrossRef]
- Wang, C.; Lang, Y.; Li, Q.; Jin, X.; Li, G.; Yu, G. Glycosaminoglycanomic profiling of human milk in different stages of lactation by liquid chromatography-tandem mass spectrometry. Food Chem. 2018, 258, 231–236. [Google Scholar] [CrossRef]
- Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Volpi, N.; Maccari, F.; Galeotti, F.; Peila, C.; Coscia, A.; Zampini, L.; Monachesi, C.; Gabrielli, O.; Coppa, G. Human milk glycosaminoglycan composition from women of different countries: A pilot study. J. Matern. Fetal Neonatal Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mannello, F.; Maccari, F.; Santinelli, A.; Volpi, N. Chondroitin sulfate structure is modified in human milk produced by breast affected by invasive carcinoma. Breast 2011, 20, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Cerdo, T.; Ruiz, A.; Jauregui, R.; Azaryah, H.; Torres-Espinola, F.J.; Garcia-Valdes, L.; Teresa Segura, M.; Suarez, A.; Campoy, C. Maternal obesity is associated with gut microbial metabolic potential in offspring during infancy. J. Physiol. Biochem. 2018, 74, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Wiederschain, G.Y.; Newburg, D.S. Glycoconjugate stability in human milk: Glycosidase activities and sugar release. J. Nutr. Biochem. 2001, 12, 559–564. [Google Scholar] [CrossRef]
- Coscia, A.; Peila, C.; Bertino, E.; Coppa, G.V.; Moro, G.E.; Gabrielli, O.; Zampini, L.; Galeazzi, T.; Maccari, F.; Volpi, N. Effect of holder pasteurisation on human milk glycosaminoglycans. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Cartmell, A.; Lowe, E.C.; Basle, A.; Firbank, S.J.; Ndeh, D.A.; Murray, H.; Terrapon, N.; Lombard, V.; Henrissat, B.; Turnbull, J.E.; et al. How members of the human gut microbiota overcome the sulfation problem posed by glycosaminoglycans. Proc. Natl. Acad. Sci. USA 2017, 114, 7037–7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Liu, X.; Lyu, Z.; Gu, H.; Li, D.; Chen, H. Glycosaminoglycans (GAGs) and GAG mimetics regulate the behavior of stem cell differentiation. Colloids Surf. B Biointerfaces 2017, 150, 175–182. [Google Scholar] [CrossRef]
- Poterucha, T.J.; Libby, P.; Goldhaber, S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb. Haemost. 2017, 117, 437–444. [Google Scholar] [CrossRef]
- Pomin, V.H. Sulfated glycans in inflammation. Eur. J. Med. Chem. 2015, 92, 353–369. [Google Scholar] [CrossRef]
- Bode, L.; Salvestrini, C.; Park, P.W.; Li, J.-P.; Esko, J.D.; Yamaguchi, Y.; Murch, S.; Freeze, H.H. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J. Clin. Investig. 2008, 118, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Klein, N.J.; Shennan, G.I.; Heyderman, R.S.; Levin, M. Alteration in glycosaminoglycan metabolism and surface charge on human umbilical vein endothelial cells induced by cytokines, endotoxin and neutrophils. J. Cell Sci. 1992, 102, 821–832. [Google Scholar] [PubMed]
- Ade-Ajayi, N.; Spitz, L.; Kiely, E.; Drake, D.; Klein, N. Intestinal glycosaminoglycans in neonatal necrotizing enterocolitis. Br. J. Surg. 1996, 83, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Nakase, H.; Matsuura, M.; Honzawa, Y.; Matsumura, K.; Uza, N.; Yamaguchi, Y.; Mizoguchi, E.; Chiba, T. Heparan sulfate on intestinal epithelial cells plays a critical role in intestinal crypt homeostasis via Wnt/beta-catenin signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G241–G249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozin, A.P.; Goldstein, M.; Sprecher, H. Antibacterial activity of glucosamine sulfate and chondroitine sulfate? Clin. Exp. Rheumatol. 2008, 26, 509–510. [Google Scholar]
- Carlson, G.A.; Dragoo, J.L.; Samimi, B.; Bruckner, D.A.; Bernard, G.W.; Hedrick, M.; Benhaim, P. Bacteriostatic properties of biomatrices against common orthopaedic pathogens. Biochem. Biophys. Res. Commun. 2004, 321, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Pirnazar, P.; Wolinsky, L.; Nachnani, S.; Haake, S.; Pilloni, A.; Bernard, G.W. Bacteriostatic effects of hyaluronic acid. J. Periodontol. 1999, 70, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Kamochi, R.; Oiki, S.; Murata, K.; Hashimoto, W. Probiotics in human gut microbiota can degrade host glycosaminoglycans. Sci. Rep. 2018, 8, 10674. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, M.; Monedero, V.; Yebra, M.J. Utilization of Host-Derived Glycans by Intestinal Lactobacillus and Bifidobacterium Species. Front. Microbiol. 2018, 9, 1917. [Google Scholar] [CrossRef] [Green Version]
- Sava, I.G.; Zhang, F.; Toma, I.; Theilacker, C.; Li, B.; Baumert, T.F.; Holst, O.; Linhardt, R.J.; Huebner, J. Novel interactions of glycosaminoglycans and bacterial glycolipids mediate binding of enterococci to human cells. J. Biol. Chem. 2009, 284, 18194–18201. [Google Scholar] [CrossRef] [Green Version]
- Hafez, M.M.; Aboulwafa, M.M.; Yassien, M.A.; Hassouna, N.A. Role of different classes of mammalian cell surface molecules in adherence of coagulase positive and coagulase negative staphylococci. J. Basic Microbiol. 2008, 48, 353–362. [Google Scholar] [CrossRef]
- Henry-Stanley, M.J.; Hess, D.J.; Erlandsen, S.L.; Wells, C.L. Ability of the heparan sulfate proteoglycan syndecan-1 to participate in bacterial translocation across the intestinal epithelial barrier. Shock 2005, 24, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Newburg, D.S.; Linhardt, R.J.; Ampofo, S.A.; Yolken, R.H. Human milk glycosaminoglycans inhibit HIV glycoprotein gp120 binding to its host cell CD4 receptor. J. Nutr. 1995, 125, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.R.; Rho, H.K.; Kessler, S.P.; Amin, R.; Homer, C.R.; McDonald, C.; Cowman, M.K.; de la Motte, C.A. Human milk hyaluronan enhances innate defense of the intestinal epithelium. J. Biol. Chem. 2013, 288, 29090–29104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Plaen, I.G. Inflammatory signaling in necrotizing enterocolitis. Clin. Perinatol. 2013, 40, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Riehl, T.E.; Stenson, W.F. Regulation of colonic epithelial repair in mice by Toll-like receptors and hyaluronic acid. Gastroenterology 2009, 137, 2041–2051. [Google Scholar] [CrossRef] [Green Version]
- Riehl, T.E.; Foster, L.; Stenson, W.F. Hyaluronic acid is radioprotective in the intestine through a TLR4 and COX-2-mediated mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G309–G316. [Google Scholar] [CrossRef] [Green Version]
- Asari, A.; Kanemitsu, T.; Kurihara, H. Oral administration of high molecular weight hyaluronan (900 kDa) controls immune system via Toll-like receptor 4 in the intestinal epithelium. J. Biol. Chem. 2010, 285, 24751–24758. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.R.; Kessler, S.P.; Rho, H.K.; Cowman, M.K.; de la Motte, C.A. Specific-sized hyaluronan fragments promote expression of human beta-defensin 2 in intestinal epithelium. J. Biol. Chem. 2012, 287, 30610–30624. [Google Scholar] [CrossRef] [Green Version]
- Gunasekaran, A.; Eckert, J.; Burge, K.; Zheng, W.; Yu, Z.; Kessler, S.; de la Motte, C.; Chaaban, H. Hyaluronan 35 kDa enhances epithelial barrier function and protects against the development of murine necrotizing enterocolitis. Pediatr. Res. 2019. [Google Scholar] [CrossRef]
- Stabler, T.V.; Huang, Z.; Montell, E.; Verges, J.; Kraus, V.B. Chondroitin sulphate inhibits NF-kappaB activity induced by interaction of pathogenic and damage associated molecules. Osteoarthr. Cartil. 2017, 25, 166–174. [Google Scholar] [CrossRef] [Green Version]
- du Souich, P.; Garcia, A.G.; Verges, J.; Montell, E. Immunomodulatory and anti-inflammatory effects of chondroitin sulphate. J. Cell Mol. Med. 2009, 13, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Garcia, A.G.; Verges, J.; Montell, E.; Lopez, M.G. Antioxidant, antiinflammatory and neuroprotective actions of chondroitin sulfate and proteoglycans. Osteoarthr. Cartil. 2010, 18 (Suppl. 1), S24–S27. [Google Scholar] [CrossRef] [Green Version]
- Linares, P.M.; Chaparro, M.; Algaba, A.; Roman, M.; Moreno Arza, I.; Abad Santos, F.; Ochoa, D.; Guerra, I.; Bermejo, F.; Gisbert, J.P. Effect of Chondroitin Sulphate on Pro-Inflammatory Mediators and Disease Activity in Patients with Inflammatory Bowel Disease. Digestion 2015, 92, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Hoshino, J.; Yamazaki, C.; Sekiguchi, T.; Miyauchi, S.; Horie, K. Effects of chondroitin sulfate on colitis induced by dextran sulfate sodium in rats. Jpn. J. Pharm. 2001, 85, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segarra, S.; Martinez-Subiela, S.; Cerda-Cuellar, M.; Martinez-Puig, D.; Munoz-Prieto, A.; Rodriguez-Franco, F.; Rodriguez-Bertos, A.; Allenspach, K.; Velasco, A.; Ceron, J. Oral chondroitin sulfate and prebiotics for the treatment of canine Inflammatory Bowel Disease: A randomized, controlled clinical trial. BMC Vet. Res 2016, 12, 49. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Cao, J.; Jiang, X.; Cui, H. Effect of low molecular weight heparin rectal suppository on experimental ulcerative colitis in mice. Biomed. Pharm. 2010, 64, 441–445. [Google Scholar] [CrossRef]
- Zezos, P.; Kouklakis, G.; Saibil, F. Inflammatory bowel disease and thromboembolism. World J. Gastroenterol. 2014, 20, 13863–13878. [Google Scholar] [CrossRef]
- Mousavi, S.; Moradi, M.; Khorshidahmad, T.; Motamedi, M. Anti-Inflammatory Effects of Heparin and Its Derivatives: A Systematic Review. Adv. Pharm. Sci. 2015, 2015, 507151. [Google Scholar] [CrossRef] [Green Version]
- Lean, Q.Y.; Gueven, N.; Eri, R.D.; Bhatia, R.; Sohal, S.S.; Stewart, N.; Peterson, G.M.; Patel, R.P. Heparins in ulcerative colitis: Proposed mechanisms of action and potential reasons for inconsistent clinical outcomes. Exp. Rev. Clin. Pharm. 2015, 8, 795–811. [Google Scholar] [CrossRef]
- Remon, J.I.; Amin, S.C.; Mehendale, S.R.; Rao, R.; Luciano, A.A.; Garzon, S.A.; Maheshwari, A. Depth of bacterial invasion in resected intestinal tissue predicts mortality in surgical necrotizing enterocolitis. J. Perinatol. 2015, 35, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Heida, F.H.; Harmsen, H.J.; Timmer, A.; Kooi, E.M.; Bos, A.F.; Hulscher, J.B. Identification of bacterial invasion in necrotizing enterocolitis specimens using fluorescent in situ hybridization. J. Perinatol. 2017, 37, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Dymock, D.; Corfield, A.P.; Weaver, G.; Woodward, M.; Berry, M. Bacterial invasion of HT29-MTX-E12 monolayers: Effects of human breast milk. J. Pediatr. Surg. 2013, 48, 353–357, discussion 357–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kessler, S.P.; Obery, D.R.; Homer, C.R.; McDonald, C.; de la Motte, C.A. Hyaluronan 35 kDa treatment protects mice from Citrobacter rodentium infection and induces epithelial tight junction protein ZO-1 in vivo. Matrix Biol. 2017, 62, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Burge, K.Y.; Hannah, L.; Eckert, J.V.; Gunasekaran, A.; Chaaban, H. The Protective Influence of Chondroitin Sulfate, a Component of Human Milk, on Intestinal Bacterial Invasion and Translocation. J. Hum. Lact. 2019, 35, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Bueno, S.; Day, M.W.; Toby, I.T.; Akins, D.R.; Dyer, D.W. Genome Sequence of SCB34, a Sequence Type 131 Multidrug-Resistant Escherichia coli Isolate Causing Neonatal Early-Onset Sepsis. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Glycosaminoglycans, found in much greater quantities in human breast milk (HM), traverse the intestines largely untouched: (a) Digestion and potential function of glycosaminoglycan-associated proteoglycans in milk. (b) Comparison of total glycosaminoglycan content of term human and bovine milk. GAG: glycosaminoglycan; GAGS: glycosaminoglycans.
Figure 1.
Glycosaminoglycans, found in much greater quantities in human breast milk (HM), traverse the intestines largely untouched: (a) Digestion and potential function of glycosaminoglycan-associated proteoglycans in milk. (b) Comparison of total glycosaminoglycan content of term human and bovine milk. GAG: glycosaminoglycan; GAGS: glycosaminoglycans.

Figure 2.
Schematic of potential glycosaminoglycan mechanisms of protection in necrotizing enterocolitis (NEC). CS: chondroitin sulfate; HA: hyaluronic acid; GAGS: glycosaminoglycans.
Figure 2.
Schematic of potential glycosaminoglycan mechanisms of protection in necrotizing enterocolitis (NEC). CS: chondroitin sulfate; HA: hyaluronic acid; GAGS: glycosaminoglycans.

Figure 3.
The effects of chondroitin sulfate on E. coli invasion and translocation of T84 colonocyte monolayers: (a) chondroitin sulfate (CS) was given prophylactically for 48 h prior to infection. A dose-dependent reduction in bacterial invasion occurred with CS (M ± SEM), with 750 µg/mL showing significantly lower bacterial invasion compared to control (** p = 0.0071; eta-squared = 0.0944); (b) CS at 750 µg/mL was significantly protective by the third hour of inoculation (M ± SEM, ** p = 0.0018). (Reprinted with permission of authors and SAGE Publishing [194]).
Figure 3.
The effects of chondroitin sulfate on E. coli invasion and translocation of T84 colonocyte monolayers: (a) chondroitin sulfate (CS) was given prophylactically for 48 h prior to infection. A dose-dependent reduction in bacterial invasion occurred with CS (M ± SEM), with 750 µg/mL showing significantly lower bacterial invasion compared to control (** p = 0.0071; eta-squared = 0.0944); (b) CS at 750 µg/mL was significantly protective by the third hour of inoculation (M ± SEM, ** p = 0.0018). (Reprinted with permission of authors and SAGE Publishing [194]).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Burge, K.; Bergner, E.; Gunasekaran, A.; Eckert, J.; Chaaban, H. The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review. Nutrients 2020, 12, 546. https://doi.org/10.3390/nu12020546
AMA Style
Burge K, Bergner E, Gunasekaran A, Eckert J, Chaaban H. The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review. Nutrients. 2020; 12(2):546. https://doi.org/10.3390/nu12020546
Chicago/Turabian StyleBurge, Kathryn, Erynn Bergner, Aarthi Gunasekaran, Jeffrey Eckert, and Hala Chaaban. 2020. "The Role of Glycosaminoglycans in Protection from Neonatal Necrotizing Enterocolitis: A Narrative Review" Nutrients 12, no. 2: 546. https://doi.org/10.3390/nu12020546
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.