Early Life Vitamin C Deficiency Does Not Alter Morphology of Hippocampal CA1 Pyramidal Neurons or Markers of Synaptic Plasticity in a Guinea Pig Model

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Biochemistry
2.3. Protein Extraction
2.4. ELISA
2.5. Western Blotting
2.6. Golgi-Staining and Section Processing
2.7. Image Acquisition and Analysis
2.8. Statistical Analysis
3. Results
3.1. Animals
3.2. Biochemistry
3.3. ELISA and Western Blotting
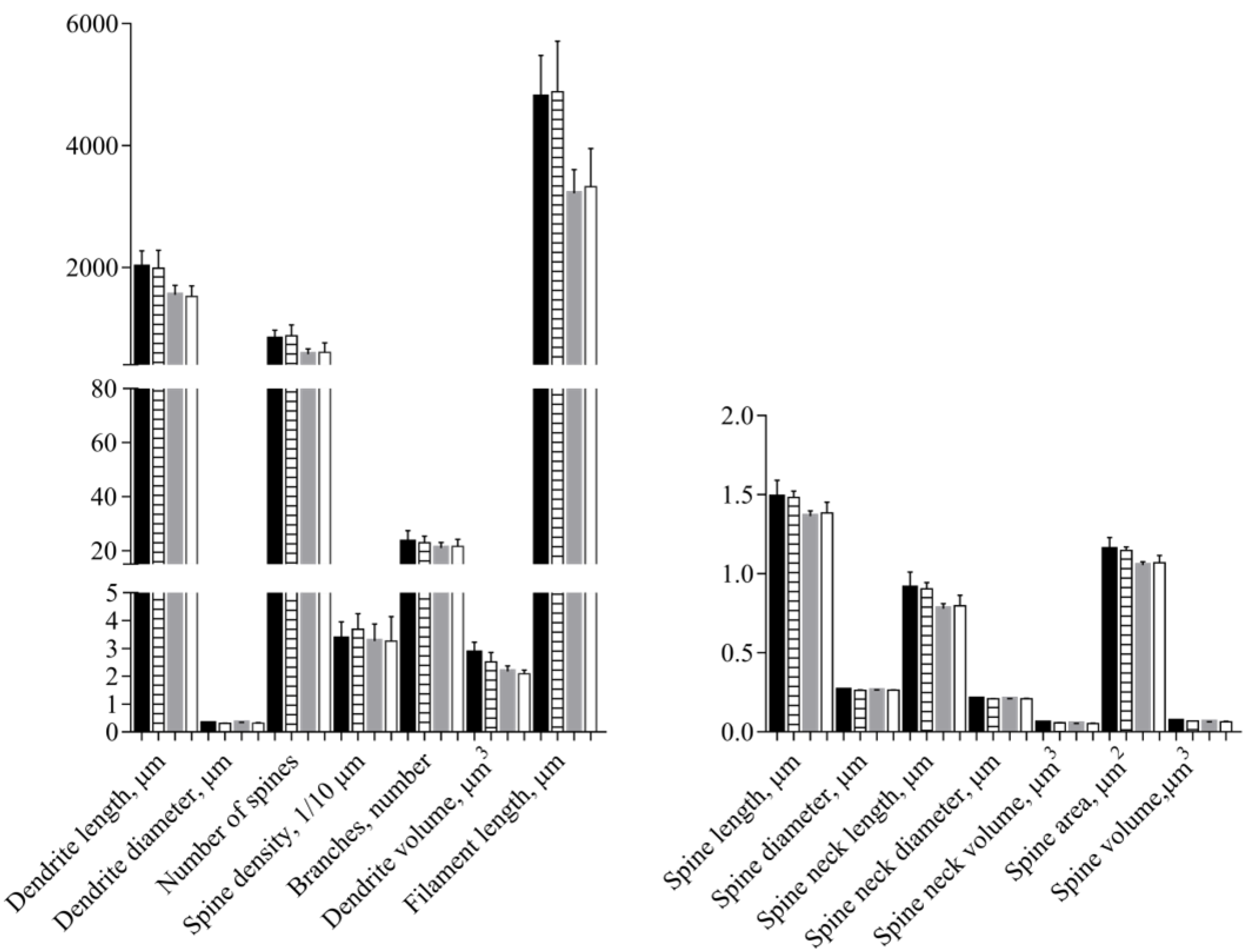
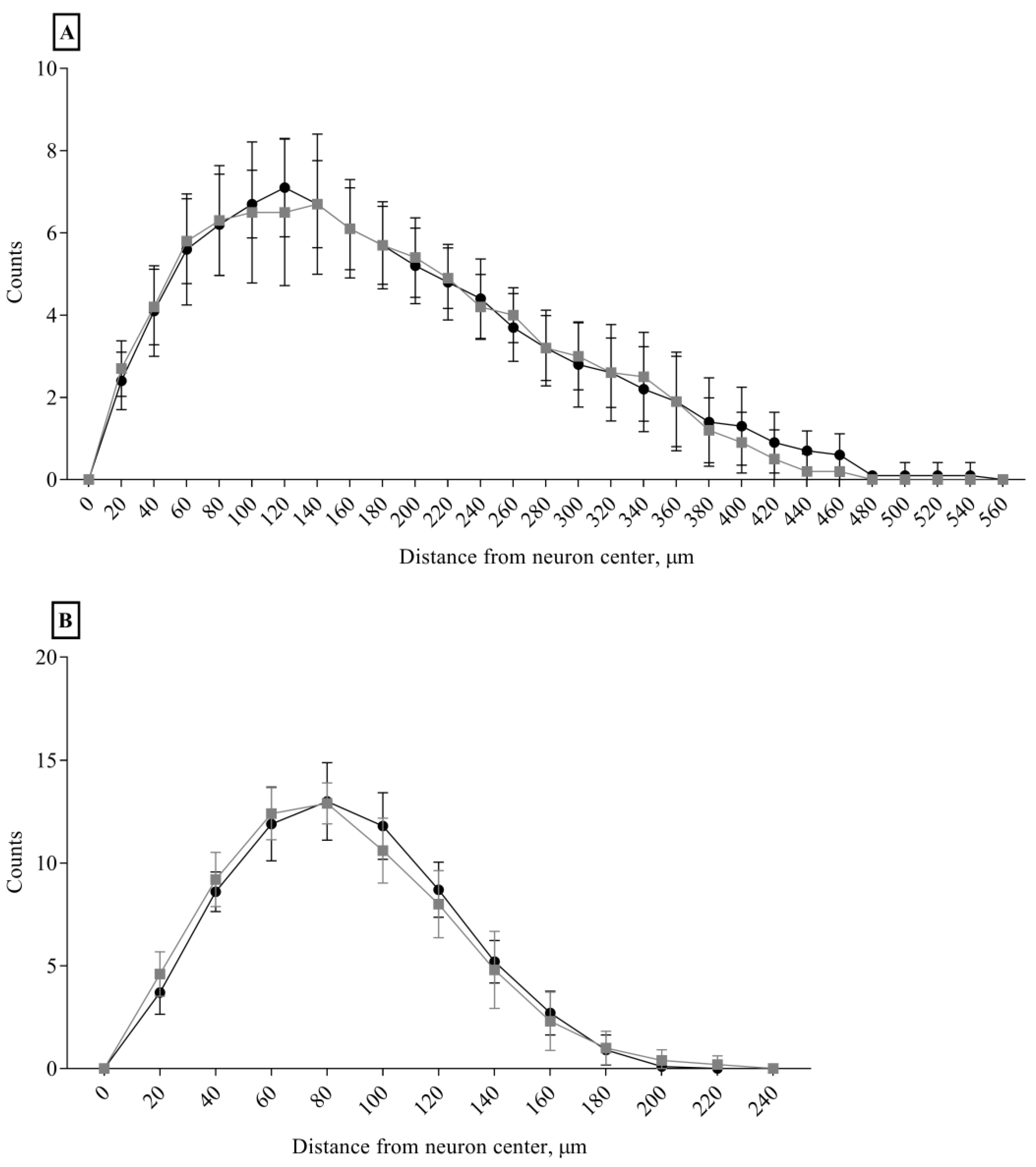
3.4. Morphological Data
4. Discussion
5. Conclusions
Author Contributions
Ethical Approval
Acknowledgments
Conflicts of Interest
References
- Dobbing, J. Later growth of the brain and its vulnerability. Pediatrics 1974, 53, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Kaindl, A.M. Neuronal death and oxidative stress in the developing brain. Antioxid. Redox Signal. 2011, 14, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, F.; Pozzi, D.; Benfenati, F.; Fornasiero, E.F. The synapsins: Multitask modulators of neuronal development. Semin. Cell Dev. Biol. 2011, 22, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Tolsa, C.B.; Zimine, S.; Warfield, S.K.; Freschi, M.; Rossignol, A.S.; Lazeyras, F.; Hanquinet, S.; Pfizenmaier, M.; Hüppi, P.S. Early alteration of structural and functional brain development in premature infants born with intrauterine growth restriction. Pediatr. Res. 2004, 56, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Morgane, P.J.; Mokler, D.J.; Galler, J.R. Effects of prenatal protein malnutrition on the hippocampal formation. Neurosci. Biobehav. Rev. 2002, 26, 471–483. [Google Scholar] [CrossRef]
- Akitake, Y.; Katsuragi, S.; Hosokawa, M.; Mishima, K.; Ikeda, T.; Miyazato, M.; Hosoda, H. Moderate maternal food restriction in mice impairs physical growth, behavior, and neurodevelopment of offspring. Nutr. Res. 2015, 35, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Mosdøl, A.; Erens, B.; Brunner, E.J. Estimated prevalence and predictors of vitamin C deficiency within UK’s low-income population. J. Public Health 2008, 30, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Langlois, K.; Cooper, M.; Colapinto, C.K. Vitamin C status of Canadian adults: Findings from the 2012/2013 Canadian Health Measures Survey. Health Rep. 2016, 27, 3–10. [Google Scholar] [PubMed]
- Schüpbach, R.; Wegmüller, R.; Berguerand, C.; Bui, M.; Herter-Aeberli, I. Micronutrient status and intake in omnivores, vegetarians and vegans in Switzerland. Eur. J. Nutr. 2017, 56, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Juhl, B.; Lauszus, F.F.; Lykkesfeldt, J. Poor vitamin C status late in pregnancy is associated with increased risk of complications in type 1 diabetic women: A cross-sectional study. Nutrients 2017, 9, 186. [Google Scholar] [CrossRef] [PubMed]
- Scaife, A.R.; McNeill, G.; Campbell, D.M.; Martindale, S.; Devereux, G.; Seaton, A. Maternal intake of antioxidant vitamins in pregnancy in relation to maternal and fetal plasma levels at delivery. Br. J. Nutr. 2006, 95, 771–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, A.M.; Rondo, P.H.D.; Barros, S.B.D. Concentrations of ascorbic acid in the plasma of pregnant smokers and nonsmokers and their newborns. Int. J. Vitam. Nutr. Res. 2004, 74, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Distribution of vitamin C is tissue specific with early saturation of the brain and adrenal glands following differential oral dose regimens in guinea pigs. Br. J. Nutr. 2015, 113, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.E.; Hurley, R.J.; Jones, P.R. Retention of ascorbic acid by guinea pig tissues. Br. J. Nutr. 1971, 26, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Trueba, G.P.; Poulsen, H.E.; Christen, S. Vitamin C deficiency in weanling guinea pigs: Differential expression of oxidative stress and DNA repair in liver and brain. Br. J. Nutr. 2007, 98, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Qu, Z.-C.; Meredith, M.E. Mechanisms of ascorbic acid stimulation of norepinephrine synthesis in neuronal cells. Biochem. Biophys. Res. Commun. 2012, 426, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebec, G.V.; Pierce, R.C. A vitamin as a neuromodulator—Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutaminergic transmission. Prog. Neurobiol. 1994, 43, 537–565. [Google Scholar] [CrossRef]
- Fitzpatrick, P.F. Tetrahydropterin-dependent amino acid hydroxylases. Annu. Rev. Biochem. 1999, 68, 355–381. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Johansen, L.K.; Raida, Z.; Villumsen, C.K.; Larsen, J.O.; Lykkesfeldt, J. Vitamin C deficiency in early postnatal life impairs spatial memory and reduces the number of hippocampal neurons in guinea pigs. Am. J. Clin. Nutr. 2009, 90, 540–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS ONE 2012, 7, e48488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paidi, M.D.; Schjoldager, J.G.; Lykkesfeldt, J.; Tveden-Nyborg, P. Prenatal vitamin C deficiency results in differential levels of oxidative stress during late gestation in foetal guinea pig brains. Redox Biol. 2014, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.H.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, F.E.; Dawes, S.M.; Meredith, M.E.; Babaev, V.R.; Li, L.; May, J.M. Low vitamin C and increased oxidative stress and cell death in mice that lack the sodium-dependent vitamin C. transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Yu, S.S.; Van Den Bossche, K.L.; Li, L.Y.; May, J.M.; McDonald, M.P. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J. Neurochem. 2008, 106, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and monoamine changes following severe vitamin C deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Beutler, L.R.; Eldred, K.C.; Quintana, A.; Keene, C.D.; Rose, S.E.; Postupna, N.; Montine, T.J.; Palmiter, R.D. Severely impaired learning and altered neuronal morphology in mice lacking NMDA receptors in medium spiny neurons. PLoS ONE 2011, 6, e28168. [Google Scholar] [CrossRef] [PubMed]
- Groc, L.; Gustafsson, B.; Hanse, E. In vivo evidence for an activity-independent maturation of AMPA/NMDA signaling in the developing hippocampus. Neuroscience 2003, 121, 65–72. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.-E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, V.A.; Firestein, B.L. The dendritic tree and brain disorders. Mol. Cell. Neurosci. 2012, 50, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.M.; Barber, V.S.; Griffiths, H.R. The presence of ascorbate induces expression of brain derived neurotrophic factor in SH-SY5Y neuroblastoma cells after peroxide insult, which is associated with increased survival. Proteomics 2005, 5, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, Y.; Bae, S.; Lim, S.H.; Jang, M.; Choi, J.; Jeon, J.; Hwang, Y.I.; Kang, J.S.; Lee, W.J. Vitamin C deficiency causes severe defects in the development of the neonatal cerebellum and in the motor behaviors of Gulo−/− mice. Antioxid. Redox Signal. 2015, 23, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Brambilla, R.; Thomas, K.L. A simple role for BDNF in learning and memory? Front. Mol. Neurosci. 2010, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G.M. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef] [PubMed]
- He, X.-B.; Kim, M.; Kim, S.-Y.; Yi, S.-H.; Rhee, Y.-H.; Kim, T.; Lee, E.-H.; Park, C.-H.; Dixit, S.; Harrison, F.E.; et al. Vitamin C facilitates dopamine neuron differentiation in fetal midbrain through TET1- and JMJD3-dependent epigenetic control manner. Stem Cells 2015, 33, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Li, L.; Weeber, E.J.; May, J.M. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J. Neurosci. Res. 2007, 85, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Salazar, K.; Martinez, M.; Ulloa, V.; Bertinat, R.; Martinez, F.; Jara, N.; Espinoza, F.; Bongarzone, E.R.; Nualart, F. SVCT2 overexpression in neuroblastoma cells induces cellular branching that is associated with ERK signaling. Mol. Neurobiol. 2016, 53, 6668–6679. [Google Scholar] [CrossRef] [PubMed]
- Balleine, B.W.; Delgado, M.R.; Hikosaka, O. The role of the dorsal striatum in reward and decision-making. J. Neurosci. 2007, 27, 8161–8165. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.M. Executive frontal functions. Exp. Brain Res. 2000, 133, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Burgess, N.; Maguire, E.A.; O’Keefe, J. The human hippocampus and spatial and episodic memory. Neuron 2002, 35, 625–641. [Google Scholar] [CrossRef]
- Sanhueza, M.; Fernandez, G. CaMKII: A master functional and structural molecule in synaptic plasticity and memory. In Novel Mechanisms of Memory; Giese, K.P., Radwanska, K., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 43–66. [Google Scholar]
- Lee, S.G.; Yoo, D.Y.; Jung, H.Y.; Nam, S.M.; Kim, J.W.; Choi, J.H.; Yi, S.S.; Won, M.-H.; Yoon, Y.S.; Hwang, I.K.; et al. Neurons in the hippocampal CA1 region, but not the dentate gyrus, are susceptible to oxidative stress in rats with streptozotocin-induced type 1 diabetes. Neural Regen. Res. 2015, 10, 451–456. [Google Scholar] [PubMed]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Goodrich-Hunsaker, N.J.; Hunsaker, M.R.; Kesner, R.P. The interactions and dissociations of the dorsal hippocampus subregions: How the dentate gyrus, CA3, and CA1 process spatial information. Behav. Neurosci. 2008, 122, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Tsien, J.Z.; Huerta, P.T.; Tonegawa, S. The essential role of hippocampal CA1 NMDA receptor–dependent synaptic plasticity in spatial memory. Cell 1996, 87, 1327–1338. [Google Scholar] [CrossRef]
- Schjoldager, J.G.; Paidi, M.D.; Lindblad, M.M.; Birck, M.M.; Kjærgaard, A.B.; Dantzer, V.; Lykkesfeldt, J.; Tveden-Nyborg, P. Maternal vitamin C deficiency during pregnancy results in transient fetal and placental growth retardation in guinea pigs. Eur. J. Nutr. 2014, 54, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G. The Rat Brain in Stereotaxic Coordinates, 4th ed.; Academic Press: San Diego, CA, USA; London, UK, 1998. [Google Scholar]
- Lykkesfeldt, J. Determination of ascorbic acid and dehydroascorbic acid in biological samples by high-performance liquid chromatography using subtraction methods: Reliable reduction with tris[2-carboxyethyl]phosphine hydrochloride. Anal. Biochem. 2000, 282, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J. Measurement of ascorbic acid and dehydroascorbic acid in biological samples. Curr. Protoc. Toxicol. 2002, 12. [Google Scholar] [CrossRef]
- Mortensen, A.; Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Guinea pig ascorbate status predicts tetrahydrobiopterin plasma concentration and oxidation ratio in vivo. Nutr. Res. 2013, 33, 859–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Søgaard, D.; Lindblad, M.M.; Paidi, M.D.; Hasselholt, S.; Lykkesfeldt, J.; Tveden-Nyborg, P. In vivo vitamin C deficiency in guinea pigs increases ascorbate transporters in liver but not kidney and brain. Nutr. Res. 2014, 34, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; du Jardin, K.G.; Waller, J.A.; Sanchez, C.; Nyengaard, J.R.; Wegener, G. Vortioxetine promotes early changes in dendritic morphology compared to fluoxetine in rat hippocampus. Eur. Neuropsychopharmacol. 2016, 26, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, V.; Rajkumar, A.P.; Nyengaard, J.R.; Børglum, A.D.; Grove, J.; Christensen, J.H. The importance of data structure in statistical analysis of dendrite spine morphology. J. Neurosci. Methods 2017, 296, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Hering, H.; Sheng, M. Dentritic spines: Structure, dynamics and regulation. Nat. Rev. Neurosci. 2001, 2, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Fiala, J.C.; Spacek, J.; Harris, K.M. Dendritic spine pathology: Cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 2002, 39, 29–54. [Google Scholar] [CrossRef]
- Da Silva, W.C.N.; Köhler, C.C.; Radiske, A.; Cammarota, M. D1/D5 dopamine receptors modulate spatial memory formation. Neurobiol. Learn. Mem. 2012, 97, 271–275. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.G.; Tejero-Cantero, Á.; Trouche, S.; Campo-Urriza, N.; Dupret, D. Dopaminergic neurons promote hippocampal reactivation and spatial memory persistence. Nat. Neurosci. 2014, 17, 1658–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murchison, C.F.; Zhang, X.-Y.; Zhang, W.-P.; Ouyang, M.; Lee, A.; Thomas, S.A. A distinct role for norepinephrine in memory retrieval. Cell 2004, 117, 131–143. [Google Scholar] [CrossRef]
- Murchison, C.F.; Schutsky, K.; Jin, S.H.; Thomas, S.A. Norepinephrine and ß1-adrenergic signaling facilitate activation of hippocampal CA1 pyramidal neurons during contextual memory retrieval. Neuroscience 2011, 181, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meredith, M.E.; May, J.M. Regulation of embryonic neurotransmitter and tyrosine hydroxylase protein levels by ascorbic acid. Brain Res. 2013, 1539, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Annu. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, H.; Liu, J.; Ouyang, Y.; Wang, D.; Bao, W.; Liu, L. Association between the NF-E2 related factor 2 gene polymorphism and oxidative stress, anti-oxidative status, and newly-diagnosed type 2 diabetes mellitus in a Chinese population. Int. J. Mol. Sci. 2015, 16, 16483–16496. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.; Fassett, R.G.; Geraghty, D.P.; Kunde, D.A.; Ball, M.J.; Robertson, I.K.; Coombes, J.S. Relationships between single nucleotide polymorphisms of antioxidant enzymes and disease. Gene 2012, 501, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.; Bhowmick, S.; Jahan, S.; Rozario, L.; Sarkar, M.; Islam, S.; Basunia, M.A.; Rahman, A.; Choudhury, B.K.; Shahjalal, H. Maternal lead exposure decreases the levels of brain development and cognition-related proteins with concomitant upsurges of oxidative stress, inflammatory response and apoptosis in the offspring rats. Neurotoxicology 2016, 56, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Blum, S.; Moore, A.N.; Adams, F.; Dash, P.K. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J. Neurosci. 1999, 19, 3535–3544. [Google Scholar] [CrossRef] [PubMed]
- Crivello, N.A.; Rosenberg, I.H.; Shukitt-Hale, B.; Bielinski, D.; Dallal, G.E.; Joseph, J.A. Aging modifies brain region-specific vulnerability to experimental oxidative stress induced by low dose hydrogen peroxide. Age 2007, 29, 191–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovic, J.N.; Czernik, A.J.; Fienberg, A.A.; Greengard, P.; Sihra, T.S. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat. Neurosci. 2000, 3, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Cesca, F.; Baldelli, P.; Valtorta, F.; Benfenati, F. The synapsins: Key actors of synapse function and plasticity. Prog. Neurobiol. 2010, 91, 313–348. [Google Scholar] [CrossRef] [PubMed]
- Brunette, K.E.; Tran, P.V.; Wobken, J.D.; Carlson, E.S.; Georgieff, M.K. Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev. Neurosci. 2010, 32, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Janthakhin, Y.; Rincel, M.; Costa, A.-M.; Darnaudéry, M.; Ferreira, G. Maternal high-fat diet leads to hippocampal and amygdala dendritic remodeling in adult male offspring. Psychoneuroendocrinology 2017, 83, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Wang, Y.; Dong, J.; Wang, Y.; Yu, Y.; Shan, Z.; Xi, Q.; Teng, W.; Chen, J. Effects of maternal marginal iodine deficiency on dendritic morphology in the hippocampal CA1 pyramidal neurons in rat offspring. Neuromol. Med. 2016, 18, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Copf, T. Impairments in dendrite morphogenesis as etiology for neurodevelopmental disorders and implications for therapeutic treatments. Neurosci. Biobehav. Rev. 2016, 68, 946–978. [Google Scholar] [CrossRef] [PubMed]
- Tata, D.A.; Anderson, B.J. The effects of chronic glucocorticoid exposure on dendritic length, synapse numbers and glial volume in animal models: Implications for hippocampal volume reductions in depression. Physiol. Behav. 2010, 99, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.C.; Milatovic, S.; Dettbarn, W.-D.; Aschner, M.; Milatovic, D. Neuronal oxidative injury and dendritic damage induced by carbofuran: Protection by memantine. Toxicol. Appl. Pharmacol. 2007, 219, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Yoo, B.H.; Kauppinen, T.M.; Choi, B.Y.; Kim, J.H.; Jang, B.G.; Lee, M.W.; Sohn, M.; Liu, J.; Swanson, R.A.; et al. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J. Neuroinflammation 2012, 9, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Cruz, C.; Nolte, M.W.; van Gaalen, M.M.; Rustay, N.R.; Termont, A.; Tanghe, A.; Kirchhoff, F.; Ebert, U. Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 3926–3934. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.-Z.; He, Y.-C.; Ma, X.K.; Li, S.-T.; Chen, D.-J.; Gao, M.; Qiu, S.-F.; Yin, J.-X.; Shi, J.; Wu, J. Hippocampal synaptic and neural network deficits in young mice carrying the human APOE4 gene. CNS. Neurosci. Ther. 2017, 23, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Steffenach, H.A.; Sloviter, R.S.; Moser, E.I.; Moser, M.B. Impaired retention of spatial memory after transection of longitudinally oriented axons of hippocampal CA3 pyramidal cells. Proc. Natl. Acad. Sci. USA 2002, 99, 3194–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesiger, M.I.; Cressey, J.C.; Boublil, B.; Koenig, J.; Melvin, N.R.; Leutgeb, J.K.; Leutgeb, S. Hippocampal activation during the recall of remote spatial memories in radial maze tasks. Neurobiol. Learn. Mem. 2013, 106, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Jessberger, S.; Clark, R.E.; Broadbent, N.J.; Clemenson, G.D.; Consiglio, A.; Lie, D.C.; Squire, L.R.; Gage, F.H. Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn. Mem. 2009, 16, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, J.S.; Hong, N.S.; McDonald, R.J.; Wojtowicz, J.M. A role for adult neurogenesis in spatial long-term memory. Neuroscience 2005, 130, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Nualart, F.; Mack, L.; Garcia, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martinez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C transporters, recycling and the bystander effect in the nervous system: SVCT2 versus GLUTs. J. Stem Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef] [PubMed]
- Ben Menachem-Zidon, O.; Avital, A.; Ben-Menahem, Y.; Goshen, I.; Kreisel, T.; Shmueli, E.M.; Segal, M.; Ben Hur, T.; Yirmiya, R. Astrocytes support hippocampal-dependent memory and long-term potentiation via interleukin-1 signaling. Brain Behav. Immun. 2011, 25, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Zanetti, A.T.; Hallock, R.M. The role of astrocytes in the regulation of synaptic plasticity and memory formation. Neural Plast. 2013, 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Volterra, A.; Trotti, D.; Tromba, C.; Floridi, S.; Racagni, G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J. Neurosci. 1994, 14, 2924–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowe, E.P.; Tuzer, F.; Gregory, B.D.; Donahue, G.; Gosai, S.J.; Cohen, J.; Leung, Y.Y.; Yetkin, E.; Nativio, R.; Wang, L.-S.; et al. Changes in the transcriptome of human astrocytes accompanying oxidative stress-induced senescence. Front. Aging Neurosci. 2016, 8, 208. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.X.; Peters, C.E.; Sitar, S.M.; Daoust, P.; Gelb, A.W. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000, 858, 61–66. [Google Scholar] [CrossRef]
- Daskalopoulos, R.; Korcok, J.; Tao, L.; Wilson, J.X. Accumulation of intracellular ascorbate from dehydroascorbic acid by astrocytes is decreased after oxidative stress and restored by propofol. Glia 2002, 39, 124–132. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Explanation | Expected Outcome of Vitamin C Deficiency |
|---|---|---|
| Dendrite length | The sum of the lengths of all dendrites on a neuron | ↓ |
| Dendrite diameter | The mean diameter within a dendrite | ↔ or ↓ |
| Dendrite volume | The volume of a dendrite | ↔ or ↓ |
| Branch numbers | The number of dendrite branches on an neuron | ↓ |
| Spine number | The number of spines on the entire neuron | ↓ |
| Spine density | The number of spines per 10 μm dendrite | ↓ |
| Filament length | The sum of dendrite length and spine length | ↓ |
| Spine length | The length of the spines | ↑ or ↓ |
| Spine diameter | The mean of the spine diameter | ↓ |
| Spine area | The surface area of the spine | ↓ |
| Spine volume | The volume of the spine | ↓ |
| Spine neck length | The length of the spine neck | ↓ |
| Spine neck diameter | The diameter of the spine neck | ↓ |
| Spine neck volume | The volume of the spine neck | ↓ |
| Group/Marker | Ctrl (n = 19) | Def (n = 19) | Sev_Def (n = 19) | One-Way ANOVA or Kruskal-Wallis Test | |
|---|---|---|---|---|---|
| Plasma | VitC (μM) * | 43.3 ± 8.5 a | 4.4 ± 0.91 b | 2.2 ± 0.46 c | p < 0.0001 |
| DHA (%) # | 1.9 ± 2.0 | 3.0 ± 3.5 | 2.8 ± 3.5 | NS | |
| BH4 (nM) | 68.3 ± 8.3 a | 47.5 ± 10.1 b | 40.4 ± 6.4 c | p < 0.0001 | |
| BH2/BH4 ¤ | 0.18(0.16; 0.22) a | 0.31(0.25; 0.43) b | 0.36(0.33; 0.41) b | p < 0.0001 | |
| α-tocopherol (μM) | 2.7 ± 0.8 | 2.5 ± 0.7 | 2.5 ± 0.7 | NS | |
| γ-tocopherol (μM) | 0.1 ± 0.06 | 0.1 ± 0.05 | 0.09 ± 0.04 | NS | |
| MDA(μM) | 1.0 ± 0.1 | 1.1 ± 0.2 | 1.1 ± 0.2 | NS | |
| Residual cerebralcortex | VitC (nmol/g) | 1320 ± 254.6 a | 753.2 ± 70.7 b | 393.6 ± 91.3 c | p < 0.0001 |
| DHA (%) ¤ # | 2.2(1.6; 2.6) | 1.6(0.1; 1.9) b | 0.0(0; 0.3) c | p < 0.0001 | |
| GSH (nmol/g) | 1244 ± 113.7 | 1247 ± 79.0 | 1244 ± 76.4 | NS | |
| GSSG (%) | 3.7 ± 1.9 | 4.0 ± 1.8 | 3.9 ± 1.1 | NS | |
| SOD (U/g) | 235 ± 54.0 | 232 ± 63.3 | 229 ± 79.9 | NS | |
| MDA (nmol/g) ¤ | 165.9(135.1; 200.1) | 169.5(140.7; 203.6) | 200.8(152.5; 254.9) | NS | |
| α-tocopherol (nmol/g) | 12.6 ± 3.1 | 11.9 ± 3.3 | 11.9 ± 1.6 | NS | |
| γ-tocopherol (nmol/g) | 0.6 ± 0.2 | 0.6 ± 0.2 | 0.6 ± 0.2 | NS | |
| Adrenal glands | VitC (nmol/g) | 9571 ± 1951 a | 3353 ± 1606 b | 2554 ± 2344 b | p < 0.0001 |
| DHA (%) | 3.5 ± 1.9 a | 4.8 ± 2.0 a b | 5.7 ± 2.1 b | p = 0.0052 | |
| GSH (nmol/g) | 1531 ± 107 a | 1670 ± 172 b | 1745 ± 180 b | p = 0.0004 | |
| GSSG (%) | 2.1 ± 1.5 | 1.8 ± 1.5 | 2.1 ± 1.7 | NS | |
| SOD (U/g) | 180 ± 55.6 | 158 ± 52.0 | 173 ± 57.3 | NS |
| Group/Marker | Ctrl | Def | Sev_Def | One-Way ANOVA | |
|---|---|---|---|---|---|
| Frontal cortex | p-synapsin 1 | 0.64 ± 0.20 | 0.54 ± 0.15 | 0.52 ± 0.15 | NS |
| synapsin 1 | 1.41 ± 0.41 | 1.66 ± 0.80 | 1.36 ± 0.51 | NS | |
| p-synapsin 1/synapsin 1 | 0.47 ± 0.11 | 0.41 ± 0.26 | 0.47 ± 0.29 | NS | |
| p-CAMKII | 0.08 ± 0.05 | 0.05 ± 0.03 | 0.06 ± 0.03 | NS | |
| CAMKII | 0.39 ± 0.07 | 0.38 ± 0.09 | 0.35 ± 0.05 | NS | |
| p-CAMKII/CAMKII | 0.23 ± 0.13 | 0.14 ± 0.08 | 0.18 ± 0.09 | NS | |
| Hippocampus | p-synapsin 1 | 1.24 ± 0.54 | 1.19 ± 0.74 | 1.11 ± 0.48 | NS |
| synapsin 1 | 1.74 ± 0.46 | 1.55 ± 0.41 | 1.51 ± 0.48 | NS | |
| p-synapsin 1/synapsin 1 | 0.71 ± 0.26 | 0.77 ± 0.39 | 0.80 ± 0.43 | NS | |
| p-CAMKII | 0.21 ± 0.12 | 0.17 ± 0.06 | 0.16 ± 0.05 | NS | |
| CAMKII | 0.31 ± 0.09 | 0.33 ± 0.09 | 0.26 ± 0.09 | NS | |
| p-CAMKII/CAMKII | 0.71 ± 0.40 | 0.56 ± 0.27 | 0.63 ± 0.20 | NS | |
| Striatum | p-synapsin 1 | 0.42 ± 0.09 | 0.41 ± 0.13 | 0.38 ± 0.10 | NS |
| synapsin 1 | 2.60 ± 0.92 | 2.23 ± 0.73 | 2.33 ± 0.37 | NS | |
| p-synapsin 1/synapsin 1 | 0.18 ± 0.05 | 0.20 ± 0.11 | 0.17 ± 0.05 | NS | |
| p-CAMKII | ND | ND | ND | - | |
| CAMKII | 0.06 ± 0.04 | 0.06 ± 0.04 | 0.04 ± 0.04 | NS | |
| p-CAMKII/CAMKII | - | - | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, S.N.; Jørgensen, J.M.B.; Nyengaard, J.R.; Lykkesfeldt, J.; Tveden-Nyborg, P. Early Life Vitamin C Deficiency Does Not Alter Morphology of Hippocampal CA1 Pyramidal Neurons or Markers of Synaptic Plasticity in a Guinea Pig Model. Nutrients 2018, 10, 749. https://doi.org/10.3390/nu10060749
Hansen SN, Jørgensen JMB, Nyengaard JR, Lykkesfeldt J, Tveden-Nyborg P. Early Life Vitamin C Deficiency Does Not Alter Morphology of Hippocampal CA1 Pyramidal Neurons or Markers of Synaptic Plasticity in a Guinea Pig Model. Nutrients. 2018; 10(6):749. https://doi.org/10.3390/nu10060749
Chicago/Turabian StyleHansen, Stine N., Jane M. Bjørn Jørgensen, Jens R. Nyengaard, Jens Lykkesfeldt, and Pernille Tveden-Nyborg. 2018. "Early Life Vitamin C Deficiency Does Not Alter Morphology of Hippocampal CA1 Pyramidal Neurons or Markers of Synaptic Plasticity in a Guinea Pig Model" Nutrients 10, no. 6: 749. https://doi.org/10.3390/nu10060749