Short-Term Exposure to High Sucrose Levels near Weaning Has a Similar Long-Lasting Effect on Hypertension as a Long-Term Exposure in Rats

 , , ,
, , ,  and
and 
Abstract
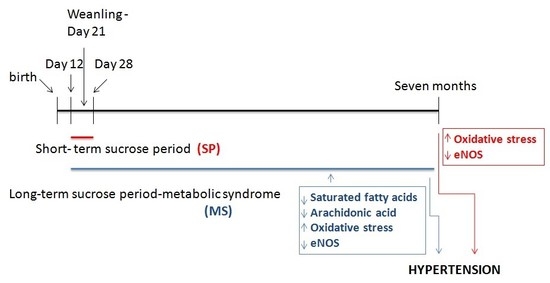
:
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Groups
2.2. Thoracic Aorta Homogenization
2.3. Biochemical Measurements
2.3.1. Glucose, Insulin and HOMA-Index
2.3.2. Lipidic Profile
2.3.3. Lipoperoxidation (LPO)
2.3.4. Total Antioxidant Capacity
2.4. Endothelial Nitric Oxide Synthase and SOD1 and 2 Immunoblotting
2.5. Determination of NO
2.6. Determination of Endothelin-1
2.7. Statistical Analysis
3. Results
3.1. Changes in Body Weight, Abdominal Fat, Glucose, Insulin, HOMA-IR
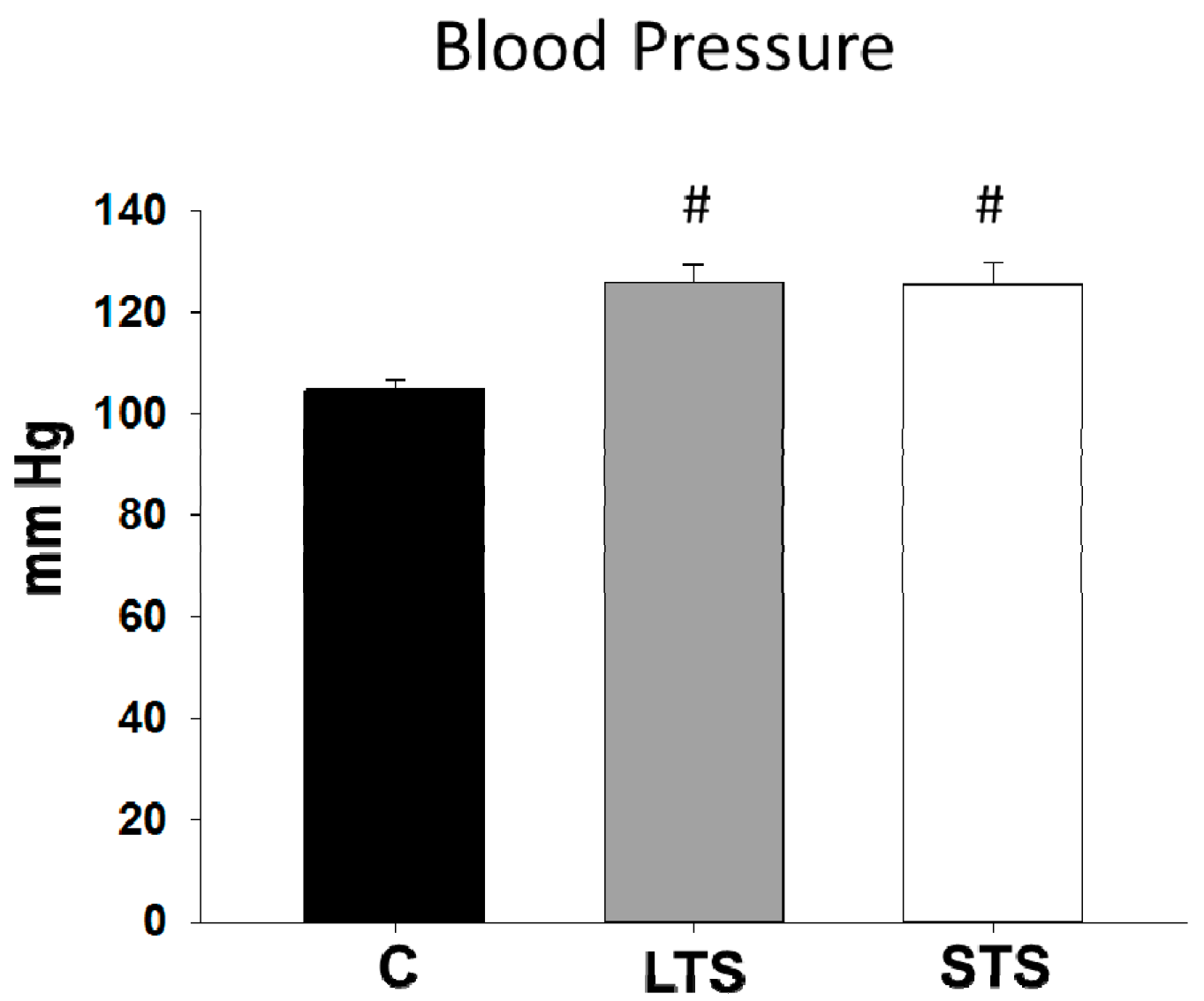
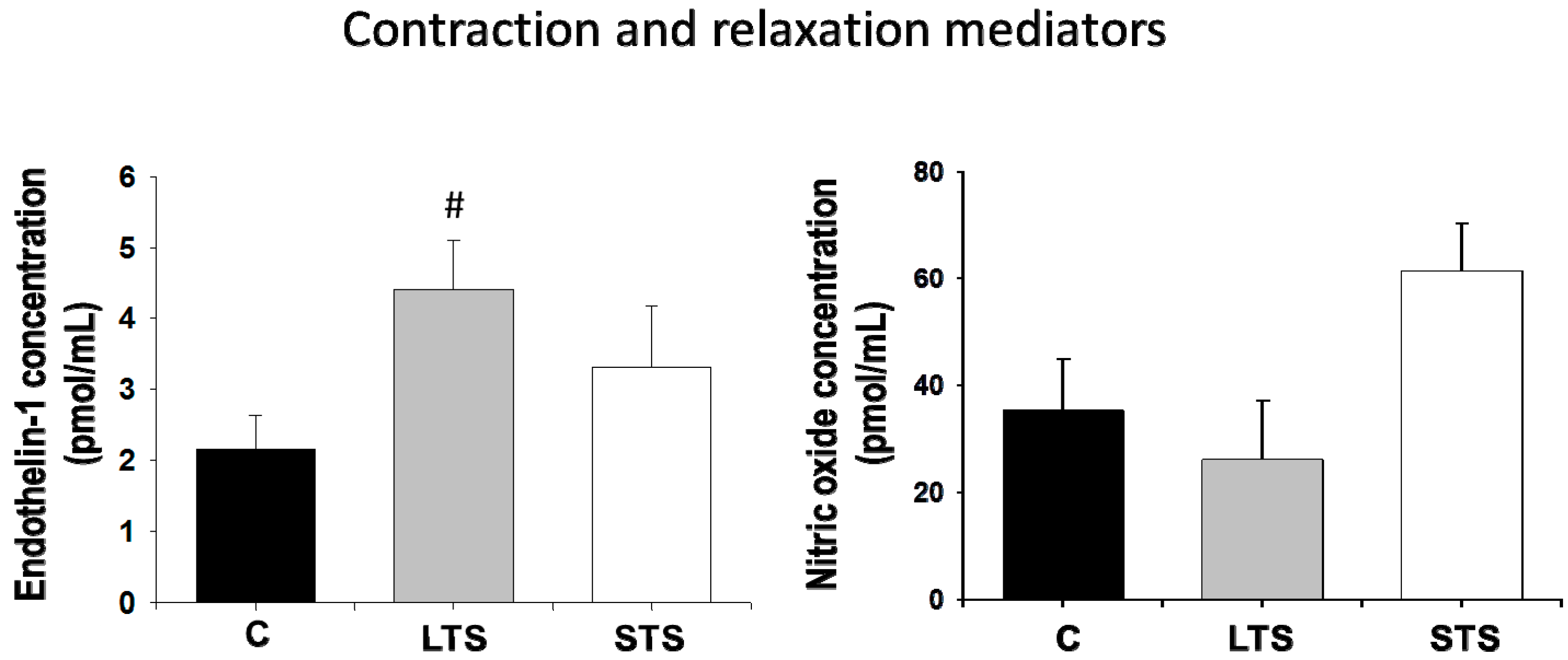
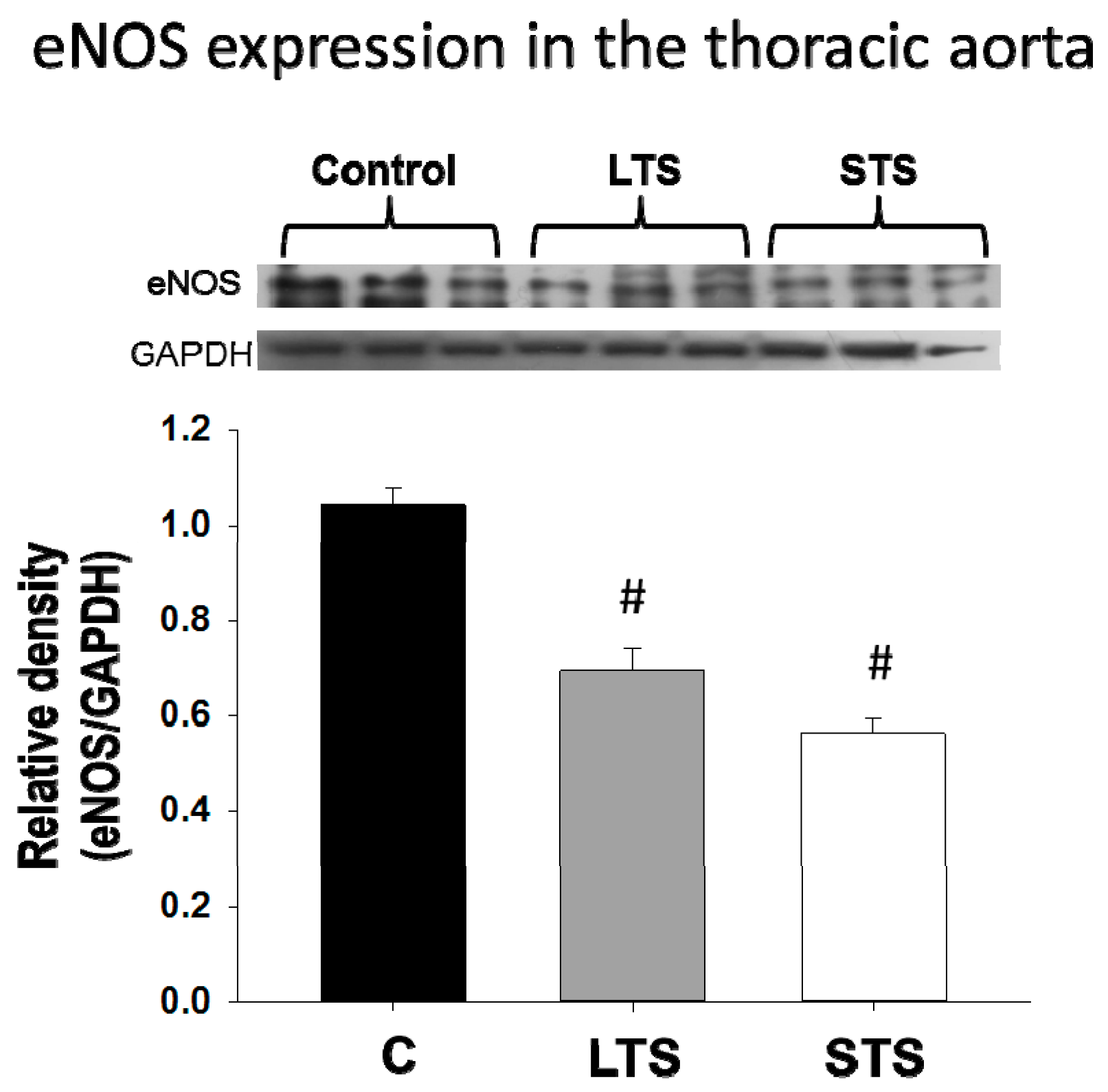
3.2. Changes in Blood Pressure, NO and Endothelin-1 Levels and eNOs Expression
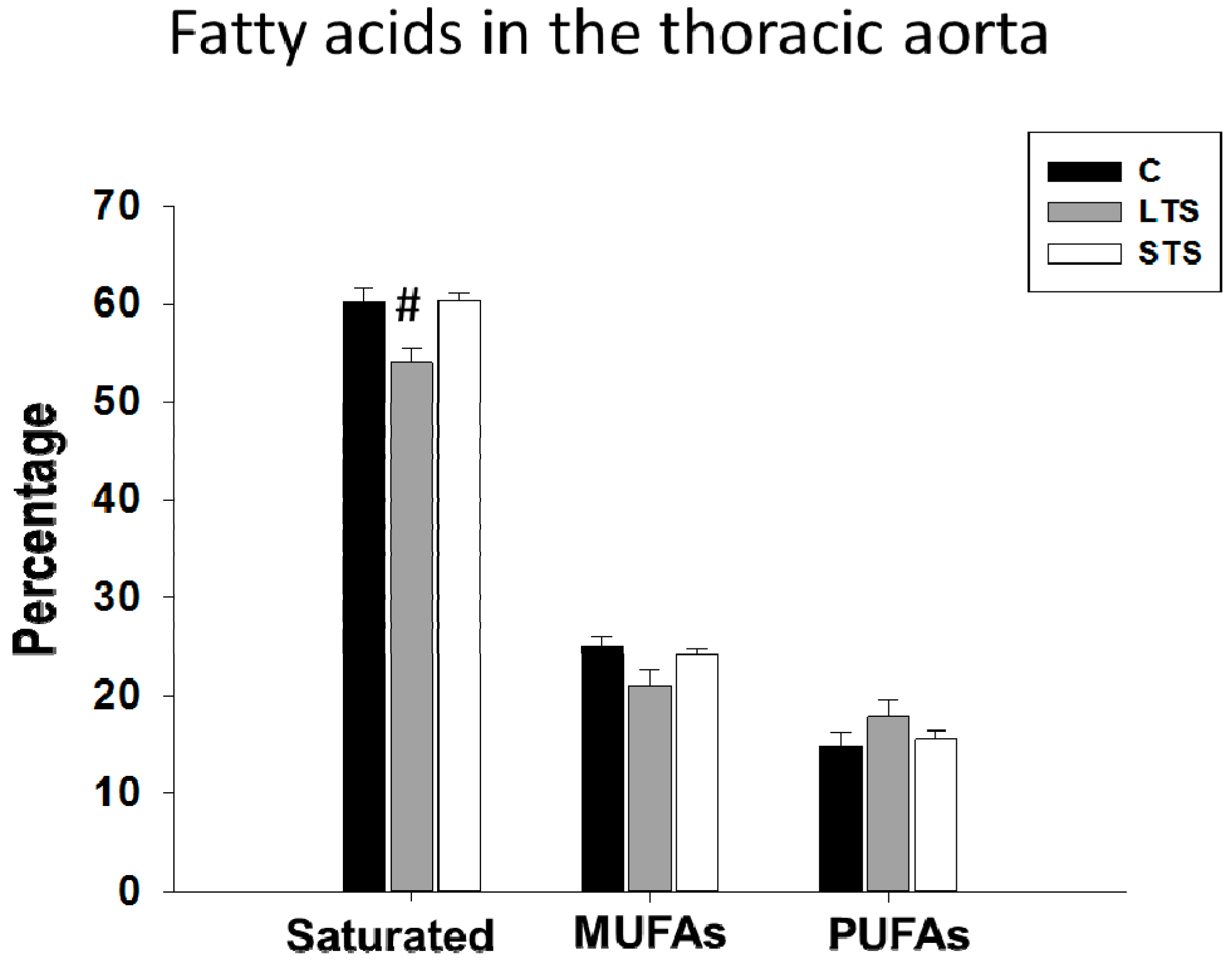
3.3. Lipidic Profile and Free Fatty Acid Levels
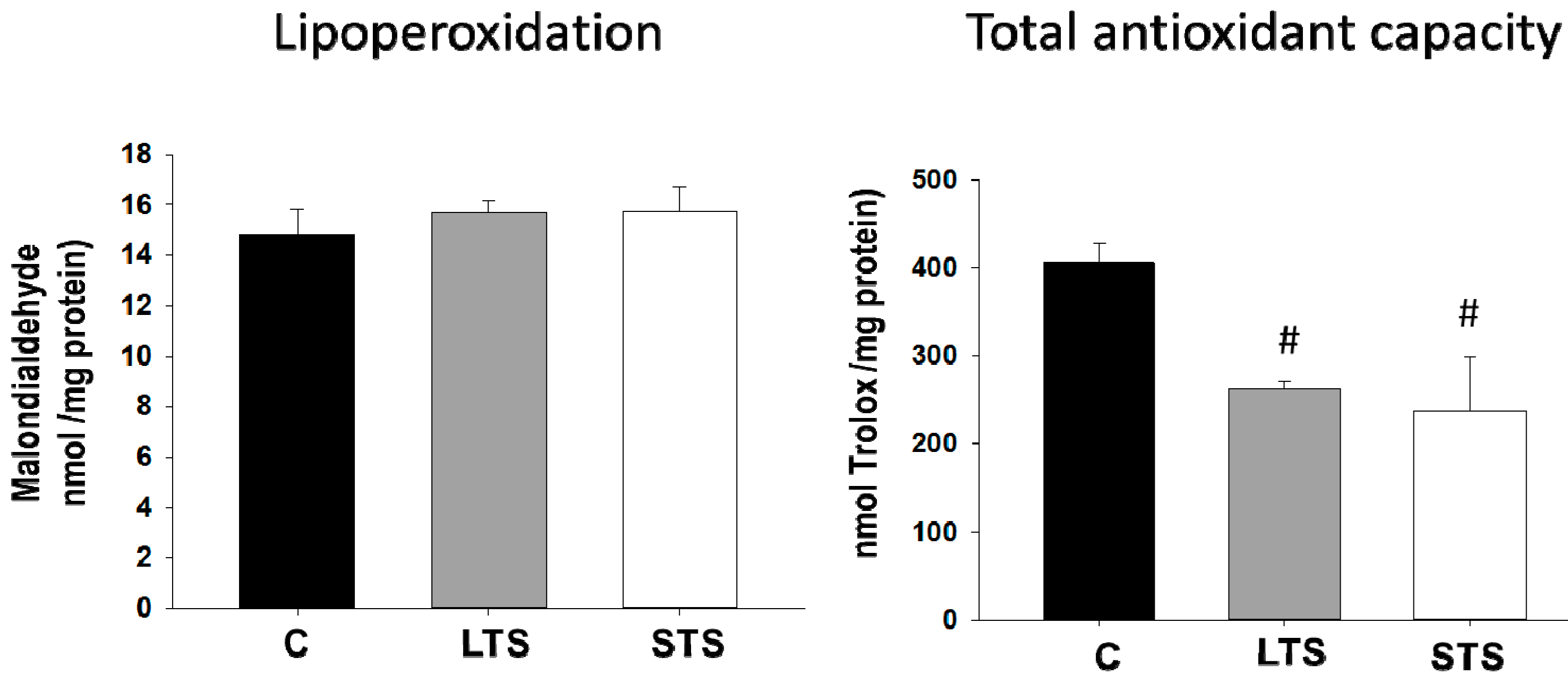
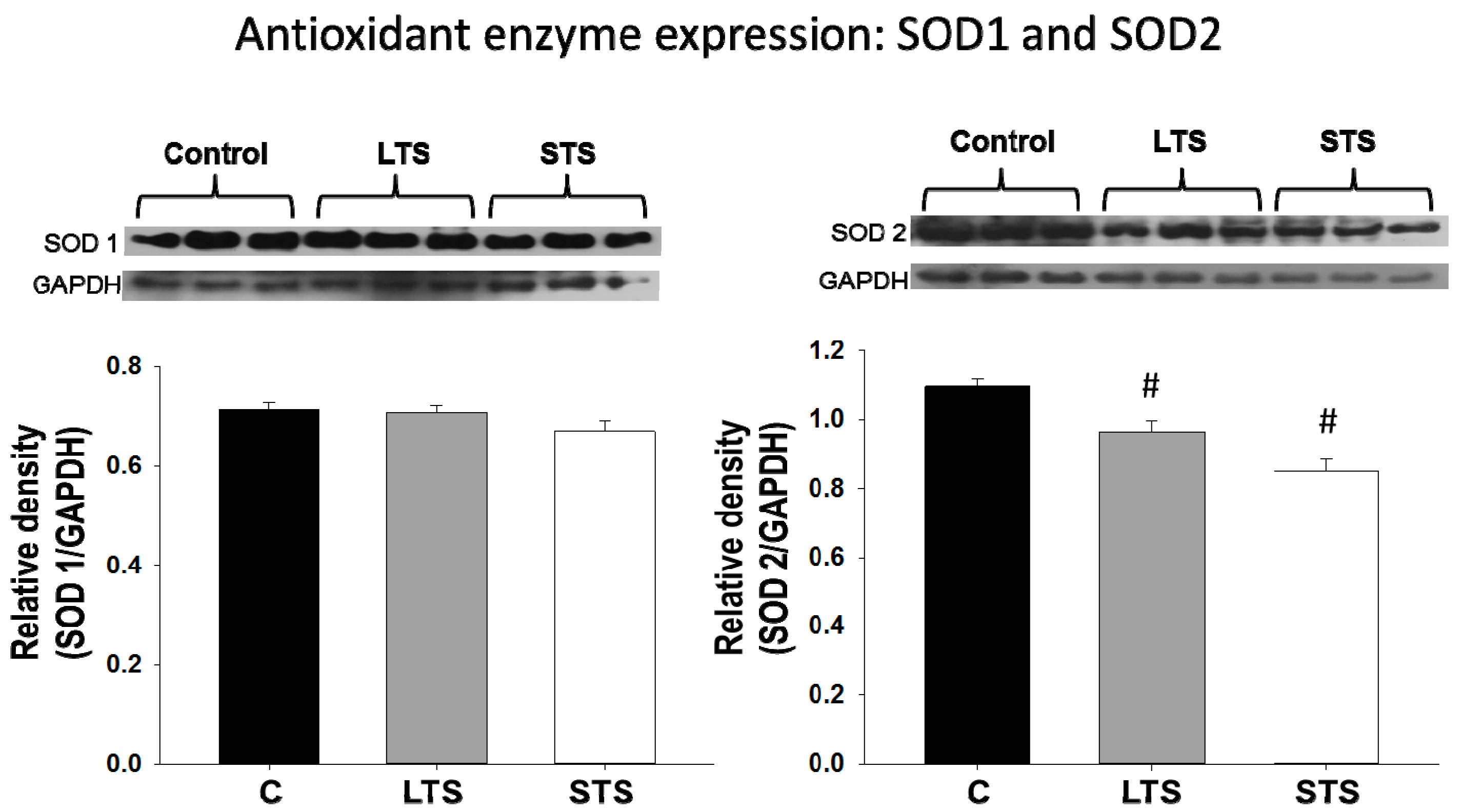
3.4. LPO, Total Antioxidant Capacity, SOD1, and SOD2 Expression
4. Discussion
4.1. Effect of the Short- and Long-Term Exposure to Sucrose on Body Variables
4.2. Nitric Oxide and Endothelin-1 Levels and Expression of eNOS. Regulation by Insulin
4.3. Lipidic Profile, Effect of Free Fatty Acids, Mainly Oleic Acid Diminishing eNOS Activity
4.4. Effect of Oxidative Stress Diminishing the Bioavailability of NO
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| BP | blood pressure |
| CZE | capillary zone electrophoresis |
| ERK | extracellular signal–regulated kinases |
| eNOS | endothelial nitric oxide synthase |
| ETA | endothelin-1 receptor A |
| ETb | endothelin-1 receptor B |
| FA | fatty acids |
| HDL | high-density lipoproteins |
| HOMA-IR | homeostasis model assessment of insulin resistance |
| IDL | intermediate-density lipoproteins |
| iNOS | inducible nitric oxide synthase |
| LDL | low-density lipoproteins |
| LPO | lipoperoxidation |
| LTS | long-term sucrose |
| MAPK | mitogen activated protein kinase |
| MS | metabolic syndrome |
| MUFA | monounsaturated fatty acids |
| NF-kB | necrosis factor kB |
| NAD+ | nicotinamide adenine dinucleotide |
| NO | nitric oxide |
| OA | oleic acid |
| OS | oxidative stress |
| PI3K | phosphatidyl inositol 3 phosphate |
| PKB, AKT | protein kinase B |
| PCG-1α | peroxisome proliferator-activated receptor gamma coactivator 1-α |
| PPAR α | peroxisome proliferator-activated receptor α |
| PUFA | polyunsaturated fatty acids |
| RIA | radioimmunoassay |
| SEM | standard errors of the mean |
| SOD1 | superoxide dismutase 1; Cu-Zn superoxide dismutase |
| SOD2 | superoxide dismutase 2; Mn superoxide dismutase |
| STS | short-term sucrose |
| TC | total cholesterol |
| TGs | triglycerides |
| VLDL | very-low-density lipoproteins |
References
- Lucas, A. Programming by early nutrition. An experimental approach. J. Nutr. 1998, 128, 401S–406S. [Google Scholar] [CrossRef] [PubMed]
- Girard, J. Metabolic adaptations to change of nutrition at birth. Biol. Neonate 1990, 58 (Suppl. 1), 3–15. [Google Scholar] [CrossRef] [PubMed]
- Ozanne, S.E.; Hales, C.N. Early programming of glucosa-insulin metabolism. Trends Endocrinol. Metab. 2002, 13, 368–373. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Srinivasan, M.; Kalhan, S.C.; Laychock, S.G.; Patel, M.S. A dietary intervention (high carbohydrate) during the neonatal period causes islet dysfunction in rats. Am. J. Physiol. 1999, 277, E1061–E1069. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Srinivasan, M.; Aalinkeel, R.; Patel, M.S. Use of a cDNA array for the identification of genes induced in islets of suckling rats by a high-carbohydrate nutritional intervention. Diabetes 2001, 50, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Aalinkeel, R.; Song, F.; Mitrani, P.; Pandya, J.D.; Strutt, B.; Hill, D.J.; Patel, M.S. Maternal hyperinsulinemia predisposes rat fetuses for hyperinsulinemia, and adult-onset obesity and maternal mild food restriction reverses this phenotype. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E129–E134. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Smith, G.D. Early life determinants of adult blood pressure. Curr. Opin. Nephrol. Hypertens. 2005, 14, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Langley, S.C.; Jackson, A.A. Increased systolic blood pressure in adult rats induced by fetal exposure to maternal low-protein diets. Clin. Sci. 1994, 86, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.L.; Weeks, D.L.; Rasch, R. Programming of adult blood pressure by maternal protein restriction: Role of nephrogenesis. Kidney Int. 2004, 65, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.T. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension 2003, 41, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Schreuder, M.F.; van Wijk, J.A.E.; Delemarre-van de Waal, H.A. Intrauterine growth restriction increases blood pressure and central pulse pressure measured with telemetry in aging rats. J. Hypertens. 2006, 24, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M.; Matthews, P.A.; Jansen, E.; Taylor, P.D.; Poston, L. Sucrose feeding in mouse pregnancy leads to hypertension, and sex-linked obesity and insulin resistance in female offspring. Front. Physiol. 2013, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.J.; Wong, D.L.; Henderson, R.; Curtis, K.S.; Smith, J.C. High dietary NaCl early in development enhances mean arterial pressure of adult rats. Physiol. Behav. 2000, 71, 173–181. [Google Scholar] [CrossRef]
- Porter, J.P.; King, S.H.; Honeycut, A.D. Prenatal high-salt diet in the Sprague-Dawley rat programs blood pressure and heart rate hyperresponsiveness to stress in adult female offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R334–R342. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Sanchez-Soto, C.; Godinez-Puig, V.; Gutierrez-Ospina, G.; Hiriart, M. Restructuring of pancreatic islets and insulin secretion in a postnatal critical window. PLoS ONE 2006, 1, e35. [Google Scholar] [CrossRef] [PubMed]
- Nava, P.; Collados, M.T.; Massó, F.; Guarner, V. Endothelin-1 mediation of insulin and glucose induced changes in vascular contractility. Hypertension 1997, 30, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Ruiz, M.E.; Vargas-Gonzalez, A.; Monter-Garrido, M.; Guarner-Lans, V. Aortic vaso-reactivity during a postnatal critical window of the pancreas in rats. Heart Vessel. 2010, 25, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaughnessy, D.T.; McAllister, K.; Worth, L.; Haugen, A.C.; Meyer, J.N.; Domann, F.E.; Van Houten, B.; Mostoslavsky, R.; Bultman, S.J.; Baccarelli, A.A.; et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ. Health Perspect. 2014, 122, 1271–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Díaz, A.; Arana-Martínez, J.C.; Carbó, R.; Espinosa-Cervantes, R.; Sánchez-Muñoz, F. Omentin: Role in insulin resistance, inflammation and cardiovascular protection. Arch. Cardiol. Mex. 2016, 86, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Zúñiga Muñoz, A.; Beltrán-Rodríguez, U.; Díaz-Díaz, E.; Martínez-Memije, R.; Guarner Lans, V. Modification of the liver fatty acids by Hibiscus sabdariffa Linnaeus (Malvaceae) infusion, its possible effect on vascular reactivity in a metabolic syndrome model. Clin. Exp. Hypertens. 2014, 36, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Baños, G.; Carvajal, K.; Cardoso, G.; Zamora, J.; Franco, M. Vascular reactivity and effect of serum in a rat model of hypertriglyceridemia and hypertension. Am. J. Hypertens. 1997, 10, 379–388. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Rubio-Ruíz, M.E.; Del Valle-Mondragón, L.; Castrejón-Tellez, V.; Carreón-Torres, E.; Díaz-Díaz, E.; Guarner-Lans, V. Angiotensin II and 1–7 during aging in metabolic syndrome rats. Expression of AT1, AT2 and Mas receptors in abdominal white adipose tissue. Peptides 2014, 57, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Pérez Méndez, O.; García Hernández, L. High-Density Lipoproteins (HDL) size and composition are modified in the rat by a diet supplemented with ‘Hass’ avocado (Persea americana Miller). Arch. Cardiol. Mex. 2007, 77, 17–24. [Google Scholar] [PubMed]
- Akondi, R.N.; Trexler, R.V.; Pfiffner, S.M.; Mouser, P.J.; Sharma, S. Modified Lipid Extraction Methods for Deep Subsurface Shale. Front. Microbiol. 2017, 8, 1408. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Roque, P.; El Hafidi, M.; Diaz-Diaz, E.; Baños, G. Association of renal damage and oxidative stress in a rat model of metabolic syndrome. Influence of gender. Free Radic. Res. 2009, 43, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, F.A.; del Valle, L.; Gustavo Pastelín, H. Validación de un método analítico espectrofotométrico para la cuantificación de metabolitos estables de óxido nítrico en fluidos biológicos. Rev. Mex. Cienc. Farm. 2005, 36, 31–41. [Google Scholar]
- Kumarathasan, P.; Goegan, P.; Vincent, R. An automated high-performance liquid chromatography fluorescence method for the analyses of endothelin-1s in plasma samples. Anal. Biochem. 2001, 299, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Raitakari, O.T.; Juonala, M.; Viikari, J.S. Obesity in childhood and vascular changes in adulthood: Insights into the Cardiovascular Risk in Young Finns Study. Int. J. Obes. 2005, 29 (Suppl. 2), S101–S104. [Google Scholar] [CrossRef]
- Li, L.; Law, C.; Power, C. Body mass index throughout the life-course and blood pressure in mid-adult life: A birth cohort study. J. Hypertens. 2007, 25, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.; Wadsworth, M.E.; Langenberg, C.; Kuh, D. Birthweight, childhood growth, and blood pressure at 43 years in a British birth cohort. Int. J. Epidemiol. 2004, 33, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, J.J.; Liu, Z.Q.; Yang, J.; Ren, J.; Liu, W.M.; Xu, X.L.; Xiong, S.E. Association between higher blood pressure level in children and adult blood pressure: 17 years follow-up results. Zhonghua Xin Xue Guan Bing Za Zhi 2008, 36, 229–231. [Google Scholar] [PubMed]
- Osmond, C.; Barker, D.J. Fetal, infant, and childhood growth are predictors of coronary heart disease, diabetes, and hypertension in adult men and women. Environ. Health Perspect. 2000, 108 (Suppl. 3), 545–553. [Google Scholar] [CrossRef] [PubMed]
- Berenson, G.S.; Srinivasan, S.R.; Hunter, S.M.; Nicklas, T.A.; Freedman, D.S.; Shear, C.L.; Webber, L.S. Risk factors in early life as predictors of adult heart disease: The Bogalusa Heart Study. Am. J. Med. Sci. 1989, 298, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Grobbee, D.E.; van Hooft, I.M.; de Man, S.A. Determinants of blood pressure in the first decades of life. J. Cardiovasc. Pharmacol. 1990, 16 (Suppl. 7), S71–S74. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Williams, L.; Vuguin, P.M.; Charron, M.J. Minireview: Epigenetic programming of diabetes and obesity: Animal models. Endocrinology 2012, 153, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, P.C.; Pippin, J.J. Of Rodents and Men: Species-Specific Glucose Regulation and Type 2 Diabetes Research. Altex 2014, 31, 157–176. [Google Scholar] [CrossRef] [PubMed]
- Ventura, E.E.; Davis, J.N.; Goran, M.I. Sugar content of popular sweetened beverages based on objective laboratory analysis: Focus on fructose content. Obesity 2011, 19, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.; Sreeramanan, S.; Diwakar, A.; Sivagnanam, G.; Sethuraman, K.R. Soft Drinks and Hard Facts: A Health Perspective. ASEAN Food J. 2007, 14, 69–81. [Google Scholar]
- Goran, M.I.; Martin, A.A.; Alderete, T.L.; Fujiwara, H.; Fields, D.A. Fructose in Breast Milk Is Positively Associated with Infant Body Composition at 6 Months of Age. Nutrients 2017, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Feng, Q.; Zhang, Q.; Wang, T.; Xiao, X. Early Life Fructose Exposure and Its Implications for Long-Term Cardiometabolic Health in Offspring. Nutrients 2016, 8, 685. [Google Scholar] [CrossRef] [PubMed]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. PNAS 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turgeon, P.J.; Sukumar, A.N.; Marsden, P.A. Epigenetics of Cardiovascular Disease–A New “Beat” in Coronary Artery Disease. Med. Epigenet. 2014, 2, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Farghali, H.; Kutinová Canoová, N.; Lekic, N. Resveratrol and Related Compounds as Antioxidants with an Allosteric Mechanism of Action in Epigenetic Drug Targets. Physiol. Res. 2013, 62, 1–13. [Google Scholar] [PubMed]
- Plagemann, A.; Harder, T.; Franke, K.; Kohlhoff, R. Long-term impact of neonatal breast-feeding on body weight and glucose tolerance in children of diabetic mothers. Diabetes Care 2002, 25, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Sampson, D.A.; Hunsaker, H.A.; Jansen, G.R. Dietary protein quality, protein quantity and food intake: Effects on lactation and on protein synthesis and tissue composition in mammary tissue and liver in rats. J. Nutr. 1986, 116, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Rulquin, H.; Rigout, S.; Lemosquet, S.; Bach, A. Infusion of glucose directs circulating amino acids to the mammary gland in well-fed dairy cows. J. Dairy Sci. 2004, 87, 340–349. [Google Scholar] [CrossRef]
- Lemosquet, S.; Delamaire, E.; Lapierre, H.; Blum, J.W.; Peyraud, J.L. Effects of glucose, propionic acid, and nonessential amino acids on glucose metabolism and milk yield in Holstein dairy cows. J. Dairy Sci. 2009, 92, 3244–3257. [Google Scholar] [CrossRef] [PubMed]
- Geursen, A.; Carne, A.; Grigor, M.R. Protein synthesis in mammary acini isolated from lactating rats: Effect of maternal diet. J. Nutr. 1987, 117, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Villegas, L.A.; Tovar, A.R.; López-Barradas, A.M.; Torres, N. The dietary protein/carbohydrate ratio differentially modifies lipogenesis and protein synthesis in the mammary gland, liver and adipose tissue during gestation and lactation. PLoS ONE 2013, 8, e69338. [Google Scholar] [CrossRef] [PubMed]
- El-Hafidi, M.; Franco, M.; Ruiz Ramírez, A.; Sosa, J.S.; Flores, J.A.P.; Acosta, O.L.; Salgado, M.C.; Cardoso-Saldaña, G. Glycine Increases Insulin Sensitivity and Glutathione Biosynthesis and Protects against Oxidative Stress in a Model of Sucrose-Induced Insulin Resistance. Oxid. Med. Cell. Longev. 2018, 2018, 2101562. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Kröller-Schön, S.; Li, C.; Kant, S.; Cai, S.; Chen, K.; Contractor, M.M.; Pei, Y.; Schulz, E.; Keaney, J.F., Jr. PGC-1α dictates endothelial function through regulation of eNOS expression. Sci. Rep. 2016, 6, 38210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nigris, V.; Pujadas, G.; La Sala, L.; Testa, R.; Genovese, S.; Ceriello, A. Short-term high glucose exposure impairs insulin signaling in endothelial cells. Cardiovasc. Diabetol. 2015, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Guo, Y.; Xu, L.; Hou, N.; Han, F.; Sun, X. Induction of haemeoxygenase-1 Directly Improves Endothelial Function in Isolated Aortas from Obese Rats through the Ampk-Pi3k/Akt-Enos Pathway. Cell. Physiol. Biochem. 2015, 36, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- El Hafidi, M.; Valdez, R.; Baños, G. Possible relationship between altered fatty acid composition of serum, platelets, and aorta and hypertension induced by sugar feeding in rats. Clin. Exp. Hypertens. 2000, 22, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ramírez, M.A.; Chávez-Salgado, J.; Peñeda-Flores, A.; Zapata, E.; Masso, F.; El-Hafidi, M. High-sucrose diet increases ROS generation, FFA accumulation, UCP2 level, and proton leak in liver mitochondria. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1198–E1207. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Fidge, N.H.; Gallon-Beaumier, G.; Kemp, B.E.; Kingwell, B.A. High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc. Natl. Acad. Sci. USA 2004, 101, 6999–7004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priante, G.; Musacchio, E.; Pagnin, E.; Calò, L.A.; Baggio, B. Specific effect to arachidonic acid on inducible nitric oxide synthase mRNA expression in human osteoblastic cells. Clin. Sci. 2005, 109, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Verlengia, R.; Gorjão, R.; Kanunfre, C.C.; Bordin, S.; de Lima, T.M.; Newsholme, P.; Curi, R. Genes regulated by arachidonicand oleic acids in Raji cells. Lipids 2003, 38, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Rupp, H.; Turcani, M.; Ohkubo, T.; Maisch, B.; Brilla, C.G. Dietary linolenic acid mediated increasein vascular prostacyclin formation. Mol. Cell. Biochem. 1996, 162, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. A defect in D6 and D5 desaturases may be a factor in the initiation and progression of insulin resistance, the metabolic syndrome and ischemic heart disease in South Asians. Lipids Health Dis. 2010, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Garaulet, M.; Hernandez-Morante, J.J.; Tebar, F.J.; Zamora, S. Relation between degree of obesity and site-specific adipose tissue fatty acid composition in a Mediterranean population. Nutrition 2011, 27, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Vila, L. Cyclooxygenase and 5-lipoxygenase pathways in the vessel wall: Role in atherosclerosis. Med. Res. Rev. 2004, 24, 399–424. [Google Scholar] [CrossRef] [PubMed]
- Høstmark, A.T.; Haug, A. Percentages of oleic acid and arachidonic acid are inversely related in phospholipids of human sera. Lipids Health Dis. 2013, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lima, T.M.; de Sa Lima, L.; Scavone, C.; Curi, R. Fatty acid control of nitric oxide production by macrophages. FEBS Lett. 2006, 580, 3287–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cury-Boaventura, M.F.; Gorjão, R.; de Lima, T.M.; Newsholme, P.; Curi, R. Comparative toxicity of oleic and linoleic acid on human lymphocytes. Life Sci. 2006, 78, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, Y.M.; Song, H.S.; Park, K.Y.; Kim, Y.M.; Kim, M.S.; Pak, Y.K.; Lee, I.K.; Lee, J.D.; Park, S.J.; Lee, K.U. Oleic acid induces endothelin-1-1 expression through activation of protein kinase C and NF-kappa B. Biochem. Biophys. Res. Commun. 2003, 303, 891–995. [Google Scholar] [CrossRef]
- Di Marzo, V. Arachidonic acid and eicosanoids as targets and effectors in second messenger interactions. Prostaglandins Leukot. Essent. Fat. Acids 1995, 53, 239–524. [Google Scholar] [CrossRef]
- Xia, Y.; Tsai, A.L.; Berka, V.; Zweier, J.L. Superoxide generation from endothelial nitric-oxide synthase: A Ca2+/calmodulin dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem. 1998, 273, 25804–25808. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.B.; Siwik, D.A.; Xiao, L.; Pimentel, D.R.; Singh, K.; Colucci, W.S. Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell. Cardiol. 2002, 34, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.O.; Etienne-Selloum, N.; Madeira, S.V.F.; Sarr, M.; Walter, A.; Dal-Ros, S.; Schott, C.; Chataigneau, T.; Schini-Kerth, V.B. Endothelium-derived contracting factors mediate the Ang II induced endothelial dysfunction in the rat aorta: Preventive effect of red wine polyphenols. Pflugers Arch. Eur. J. Physiol. 2010, 459, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Serpillon, S.; Floyd, B.C.; Gupte, R.S.; George, S.; Kozicky, M.; Neito, V.; Recchia, F.; Stanley, W.; Wolin, M.S.; Gupte, S.A. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose- 6-phosphate dehydrogenase-derived NADPH. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H153–H162. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Jeroudi, M.O.; Patel, B.S.; Aruoma, O.I.; Halliwell, B.; Lai, E.K.; McCay, P.B. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion: Evidence that myocardial ‘stunning’ is a manifestation of reperfusion injury. Circ. Res. 1989, 65, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Charniot, J.C.; Bonnefont-Rousselot, D.; Albertini, J.P.; Zerhouni, K.; Dever, S.; Richard, I.; Nataf, P.; Pavie, A.; Monsuez, J.J.; Delattre, J.; Artigou, J.E. Oxidative stress implication in a new ex-vivo cardiac concordant xenotransplantation model. Free Radic. Res. 2007, 41, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Lasségue, B.; Ushio-Fukai, M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minhas, K.M.; Saraiva, R.M.; Schuleri, K.H.; Lehrke, S.; Zheng, M.; Saliaris, A.P.; Berry, C.E.; Vandegaer, K.M.; Li, D.; Hare, J.M. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ. Res. 2006, 98, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.I.; Egashira, K.; Takeshita, A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; Van Der Laarse, A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol. Cell. Biochem. 2010, 333, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Investig. 2005, 115, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, W.F.; Paolocci, N.; St. John, M.E.; Skaf, M.W.; Stewart, G.C.; Xie, J.S.; Harrison, R.W.; Zeichner, J.; Mudrick, D.; Marbán, E.; et al. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechano energetic uncoupling in the failing heart. Circ. Res. 2002, 90, 297–304. [Google Scholar] [PubMed]
- White, C.N.; Liu, C.C.; Garcia, A.; Hamilton, E.J.; Chia, K.K.; Figtree, G.A.; Rasmussen, H.H. Activation of cAMP dependent signaling induces oxidative modification of the cardiac Na+-K+ pump and inhibits its activity. J. Biol. Chem. 2010, 285, 13712–13720. [Google Scholar] [CrossRef] [PubMed]
- Belik, J.; Jankov, R.P.; Pan, J.; Yi, M.; Pace-Asciak, C.R.; Tanswell, A.K. Effect of 8-isoprostaglandin F2α on the newborn rat pulmonary arterialmuscle and endothelium. J. Appl. Physiol. 2003, 95, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Guerra, R.C.; Zúñiga-Muñoz, A.; Guarner-Lans, V.; Díaz-Díaz, E.; Tena-Betancourt, C.A.; Pérez-Torres, I. Modulation of the activities of cabalase, Cu-Zn, Mn Superoxide dismutase and glutathione peroxidase in adipocytes from ovariectomized female rats with metabolic syndrome. Int. J. Endocrinol. 2014, 2014, 175080. [Google Scholar] [CrossRef] [PubMed]
- Constantini, D.; Verhulst, S. Does high antioxidant capacity indicate low oxidative stress? Funct. Ecol. 2009, 23, 506–509. [Google Scholar] [CrossRef] [Green Version]
- Rubbo, H.; Parthasarathy, S.; Barnes, S.; Kirk, M.; Kalyanaraman, B.; Freeman, B.A. Nitric oxide inhibition of lipoxygenase-dependent liposome and low- density lipoprotein oxidation: Termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives. Arch. Biochem. Biophys. 1995, 324, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, H.; Freeman, B.A. Nitric oxide regulation of lipid oxidation reactions: Formation and analysis of nitrogen-containing oxidized lipid derivatives. Methods Enzymol. 1996, 269, 385–394. [Google Scholar] [PubMed]
- Rubbo, H.; Radi, R.; Anselmi, D.; Kirk, M.; Barnes, S.; Eiserich, J.; Freeman, B.A. Nitric oxide reaction with lipid peroxyl radicals spares α-tocopherol during lipid peroxidation: Greater oxidant protection from the pair nitric oxide/α-tocopherol than α-tocopherol/ascorbate. J. Biol. Chem. 2000, 275, 10812–10818. [Google Scholar] [CrossRef] [PubMed]
- Trostchansky, A.; Batthyány, C.; Botti, H.; Radi, R.; Denicola, A.; Rubbo, H. Formation of lipid-protein adducts in low-density lipoprotein by fluxes of peroxynitrite and its inhibition by nitric oxide. Arch. Biochem. Biophys. 2001, 395, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Ryan, M.J.; Didion, L.A.; Fegan, P.E.; Sigmund, C.D.; Faraci, F.M. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ. Res. 2002, 91, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Stevenson, M.D.; Alahari, S.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. Attenuated Superoxide Dismutase 2 Activity Induces Atherosclerotic Plaque Instability During Aging in Hyperlipidemic Mice. J. Am. Heart Assoc. 2017, 6, e006775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapata-Martín del Campo, C.M.; Martínez-Rosas, M.; Guarner-Lans, V. Epigenetics of Subcellular Structure Functioning in the Origin of Risk or Resilience to Comorbidity of Neuropsychiatric and Cardiometabolic Disorders. Int. J. Mol. Sci. 2018, 19, E1456. [Google Scholar] [CrossRef] [PubMed]
- Mármol, F.; Sánchez, J.; López, D.; Martinez, N.; Rosello-Catafau, J.; Mitjavila, M.T.; Puig-Parellada, P. Loss of adaptation to oxidative stress as a mechanism for aortic damage in aging rats. J. Physiol. Biochem. 2007, 63, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; DiCecco, P.; Zucchelli, G.C. Role of superoxide dismutase in vascular inflammation and in coronary artery disease. Clin. Exp. Med. 2006, 6, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | LTS | STS | |
|---|---|---|---|
| Weight (g) | 509.20 ± 3.68 | 419.67 ± 5.24 | 496.00 ± 5.00 |
| Visceral adipose tissue (g) | 4.08 ± 0.44 | 9.82 ± 1.16 # | 4.23 ± 0.41 |
| Systolic pressure (mm Hg) | 105.4 ± 0.75 | 125.81 ± 1.13 # | 125.47 ± 1.07 # |
| Glucose (mg/dL) | 110.63 ± 7.6 | 100.73 ± 5.61 | 93.36 ± 3.5 |
| Insulin (µU/mL) | 7.61 ± 0.73 | 13.62 ± 1.18 # | 9.66 ± 0.52 |
| HOMA-IR | 1.36 ± 0.16 | 2.48 ± 0.29 # | 2.03 ± 0.17 |
| Control | LTS | STS | |
|---|---|---|---|
| Triglycerides (mg/dL) | 66.48 ± 9.55 | 126.95 ± 8.10 # | 47.29 ± 4.98 |
| Total cholesterol (mg/dL) | 51.34 ± 4.11 | 54.77 ± 4.97 | 62.40 ± 3.94 |
| Cholesterol-HDL (mg/dL) | 26.32 ± 1.96 | 19.76 ± 1.65 | 34.28 ± 2.6 |
| Cholesterol non-HDL (mg/dL) | 23.54 ± 1.60 | 31.59 ± 3.72 | 26.25 ± 1.92 |
| Fatty Acid (%) | Control | LTS | STS |
|---|---|---|---|
| Palmitic | 27.21 ± 0.67 | 33.53 ± 0.57 | 27.22 ± 0.62 |
| Stearic | 32.98 ± 1.03 | 20.48 ± 1.40 # | 33.15 ± 0.75 |
| Oleic | 24.94 ± 0.96 | 20.93 ± 1.52 | 24.24 ± 0.42 |
| Linoleic | 2.47 ± 0.29 | 10.46 ± 1.20 # | 5.29 ± 0.59 |
| Arachidonic | 11.27 ± 0.90 | 8.33 ± 0.82 # | 10.20 ± 0.53 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villegas-Romero, M.; Castrejón-Téllez, V.; Pérez-Torres, I.; Rubio-Ruiz, M.E.; Carreón-Torres, E.; Díaz-Díaz, E.; Del Valle-Mondragón, L.; Guarner-Lans, V. Short-Term Exposure to High Sucrose Levels near Weaning Has a Similar Long-Lasting Effect on Hypertension as a Long-Term Exposure in Rats. Nutrients 2018, 10, 728. https://doi.org/10.3390/nu10060728
Villegas-Romero M, Castrejón-Téllez V, Pérez-Torres I, Rubio-Ruiz ME, Carreón-Torres E, Díaz-Díaz E, Del Valle-Mondragón L, Guarner-Lans V. Short-Term Exposure to High Sucrose Levels near Weaning Has a Similar Long-Lasting Effect on Hypertension as a Long-Term Exposure in Rats. Nutrients. 2018; 10(6):728. https://doi.org/10.3390/nu10060728
Chicago/Turabian StyleVillegas-Romero, Mariana, Vicente Castrejón-Téllez, Israel Pérez-Torres, María Esther Rubio-Ruiz, Elizabeth Carreón-Torres, Eulises Díaz-Díaz, Leonardo Del Valle-Mondragón, and Verónica Guarner-Lans. 2018. "Short-Term Exposure to High Sucrose Levels near Weaning Has a Similar Long-Lasting Effect on Hypertension as a Long-Term Exposure in Rats" Nutrients 10, no. 6: 728. https://doi.org/10.3390/nu10060728