1. Background
A diverse range of small noncoding RNA (small ncRNA, 17–200 nucleotides) species have been shown to contribute to human development and diseases [
1,
2,
3]. However, many existing large-scale research efforts, such as The Cancer Genome Atlas (TCGA), have generated data sets that enrich for small RNAs with lengths less than 35 nucleotides (nt) [
4], which resulted in many small RNA species greater than 36 nt being under-represented. While this led to the development of tools that dramatically advanced the microRNA field [
5,
6,
7], much remains to be understood about the biology of RNA species within the 36–200 nt range (
Figure S1). To address this, we have optimized a method to enrich and sequence reads of 17 to 200 nt to capture the full diversity of small RNA species. This method enabled us to discover previously unannotated and differentially expressed RNAs in acute myeloid leukemia (AML) [
8].
The ability to sequence variable read lengths can provide important insights concerning a class of small noncoding RNAs, as exemplified by tRNA fragments of varying lengths [
9]. However, we were unable to apply the majority of existing small RNA analysis tools to our deep-sequenced data due to their inability to analyze sequence reads of varying lengths, inability to process larger quantities of sequence reads, or their lack of support for diverse small RNA species. A few tools capable of processing such data struggle with false positives and accurate classification of small RNA transcripts (
Table S1). To overcome these limitations, we developed a new tool named DANSR, which can be broadly applied to large-scale sequencing data with variable read lengths to discover and quantify different classes of small noncoding RNAs.
3. Discussion
Our study shows DANSR is a novel tool that represents a valuable addition to existing methods in the area of small ncRNA detection, quantification, and discovery. DANSR implements the algorithms that have been empirically established through our prior ncRNA discovery studies, making these powerful methods easily available to users [
8]. Further, we have demonstrated improved performance of DANSR through its boundary optimization algorithm, multi-node cluster network model, and decision tree model for discovering and classifying candidate small RNA read clusters (
Figure 1). The boundary optimization method used by DANSR greatly increases the Jaccard scores of known small RNA species in relation to their annotation (
Figure S2B–D), increasing its ability to accurately annotate known small ncRNAs. It also produces accurate size estimates for known and novel small ncRNAs discovered among the RNA-seq data, facilitating classification and eventual functional analysis of these novel clusters (
Figure 6A). Using a multi-node cluster network model allowed for accurate interpretation of the significant number of multi-mapped reads due to homology among small ncRNAs. False positive clusters consisting solely of multi-mapped reads that have alternate alignments within clusters containing uniquely aligned reads are rejected; the multi-mapped reads are instead assigned to the proper genomic locus of RNA expression with unique read support. In addition, small ncRNAs with multiple copies throughout the genome are counted only a single time when calculating expression levels. Finally, the application of a carefully calibrated decision tree model (
Figure S3) to each cluster within a graph created by the network model ensures that low-quality read clusters are rejected, reducing false positives, while clusters of acceptable quality are accurately classified as annotated, in close proximity to known annotation, or unannotated small ncRNAs.
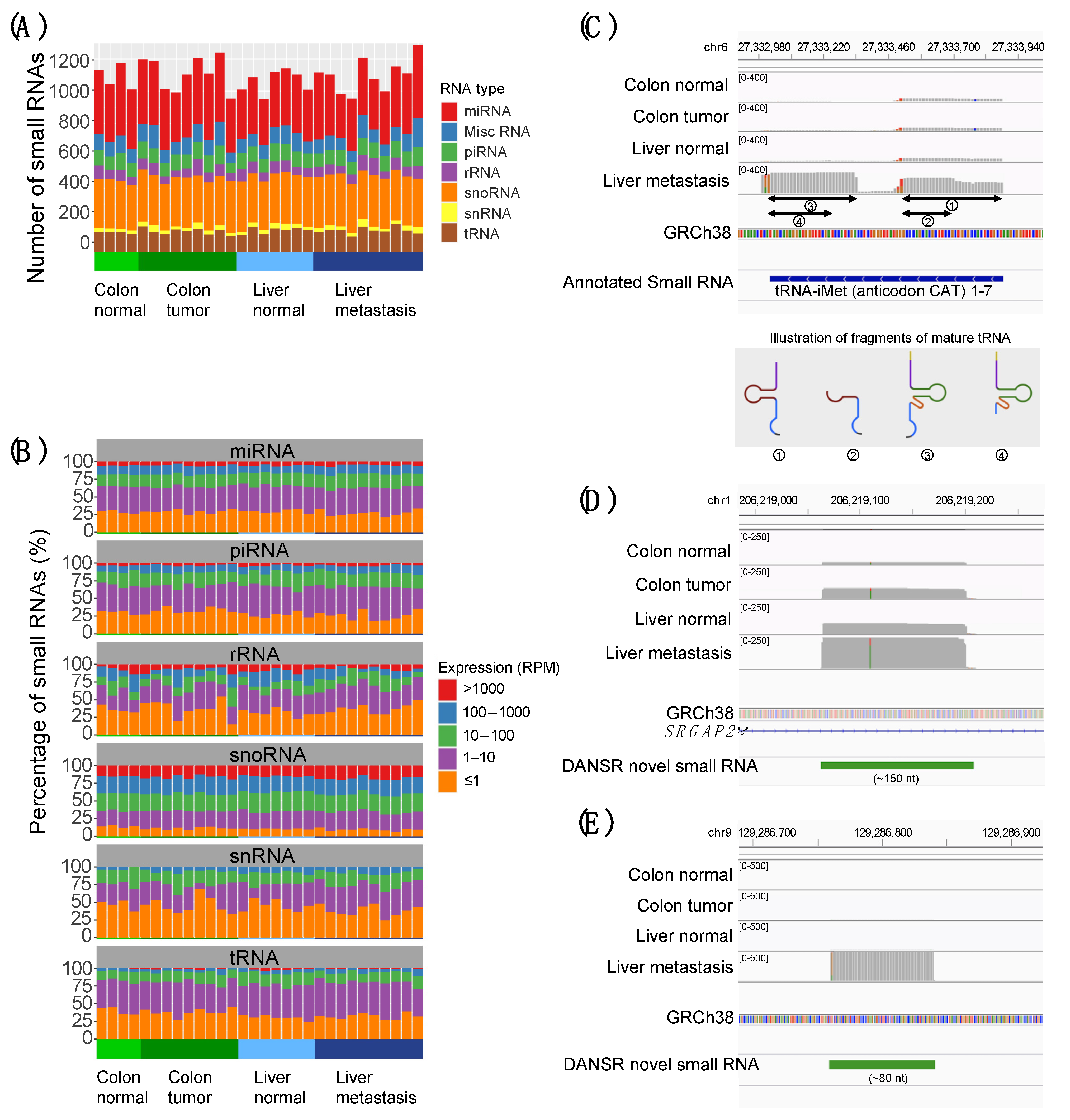
We have demonstrated the effectiveness of the DANSR model by applying it to a deep-sequenced CRC cohort consisting of colon tumor, matched normal, liver metastasis, and liver normal samples whose library preparation was designed specifically to facilitate the discovery of both short (17–35 nt) and mid-length (36–200 nt) ncRNAs (
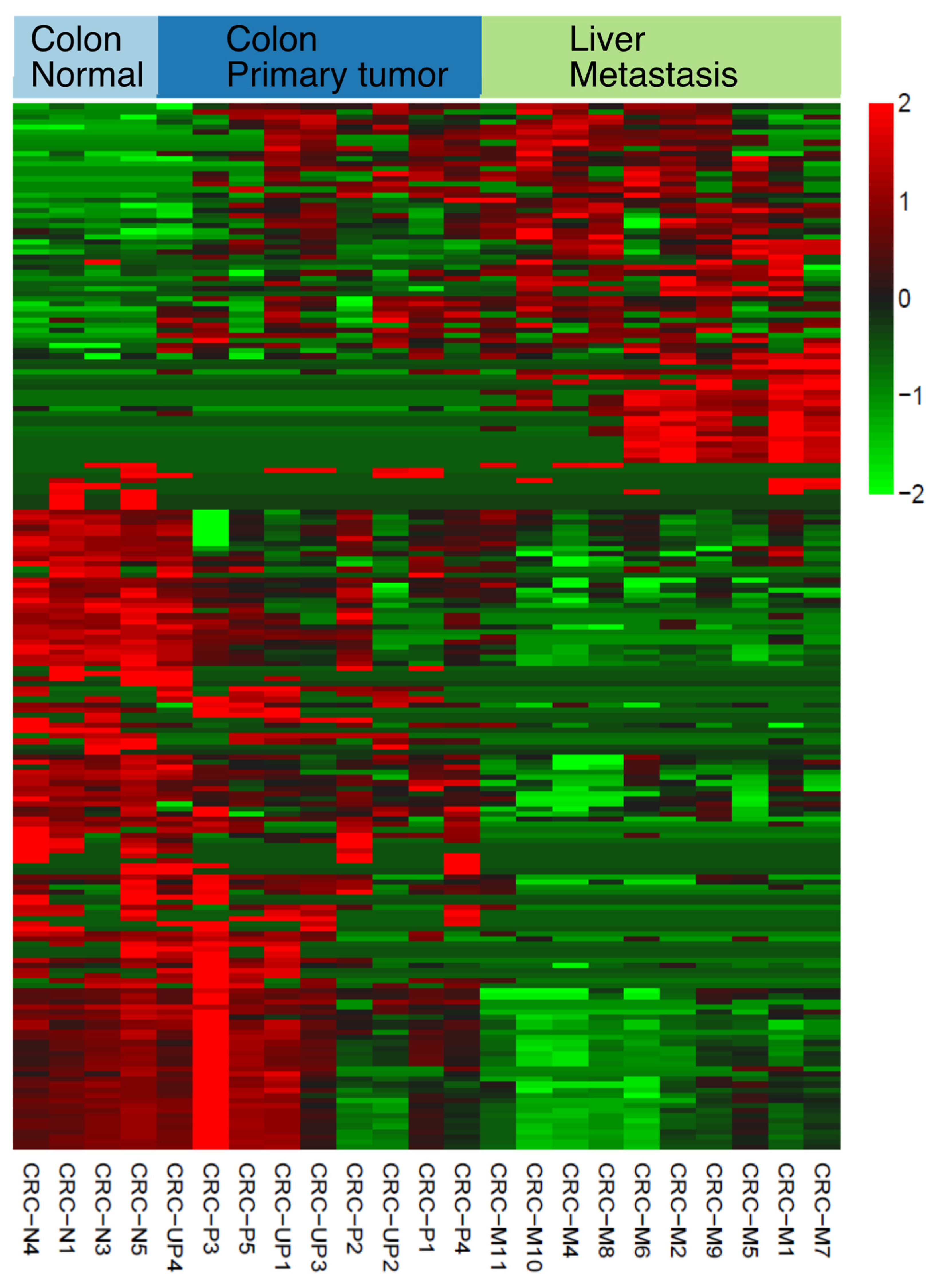
Figure 2). DANSR was able to detect over 7700 small ncRNA clusters within the deep-sequenced CRC cohort, including 204 small RNAs with significant differential expression in at least two of the pairwise comparisons between colon normal-colon tumor, colon tumor-liver metastasis, and colon normal-liver metastasis (
Figure 3), thereby illustrating DANSR’s utility in exploring the role of ncRNAs in cancer biology. In addition, we highlighted one of many examples in which DANSR detected different fragments of a particular tRNA in different tissue types from a single patient (
Figure 2C), demonstrating our tool’s potential contribution to another area of ongoing research interest. While we applied DANSR to a cancer related study in this paper, DANSR is broadly applicable to any small RNA dataset to study different phenotypes users are interested in.
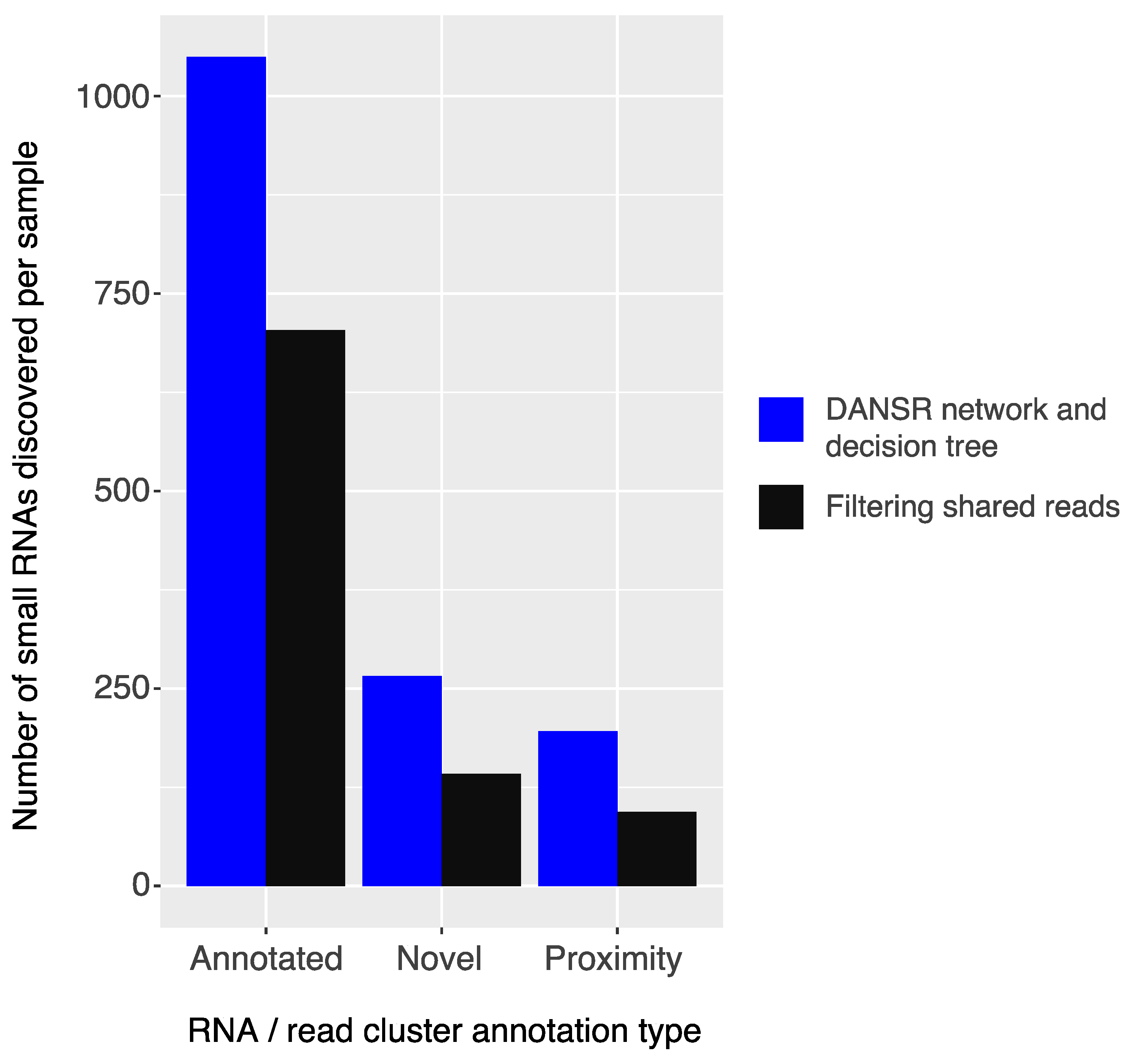
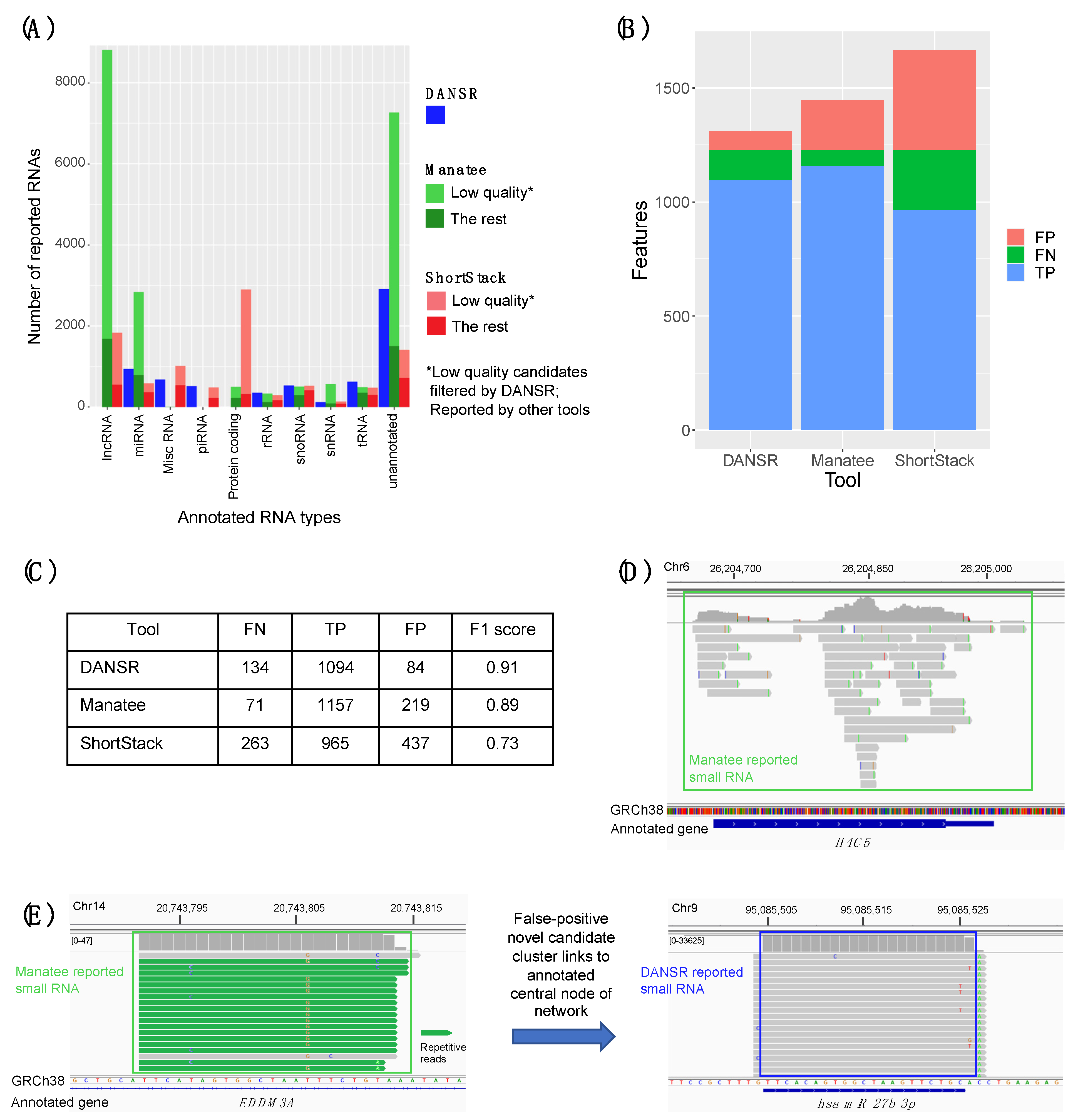
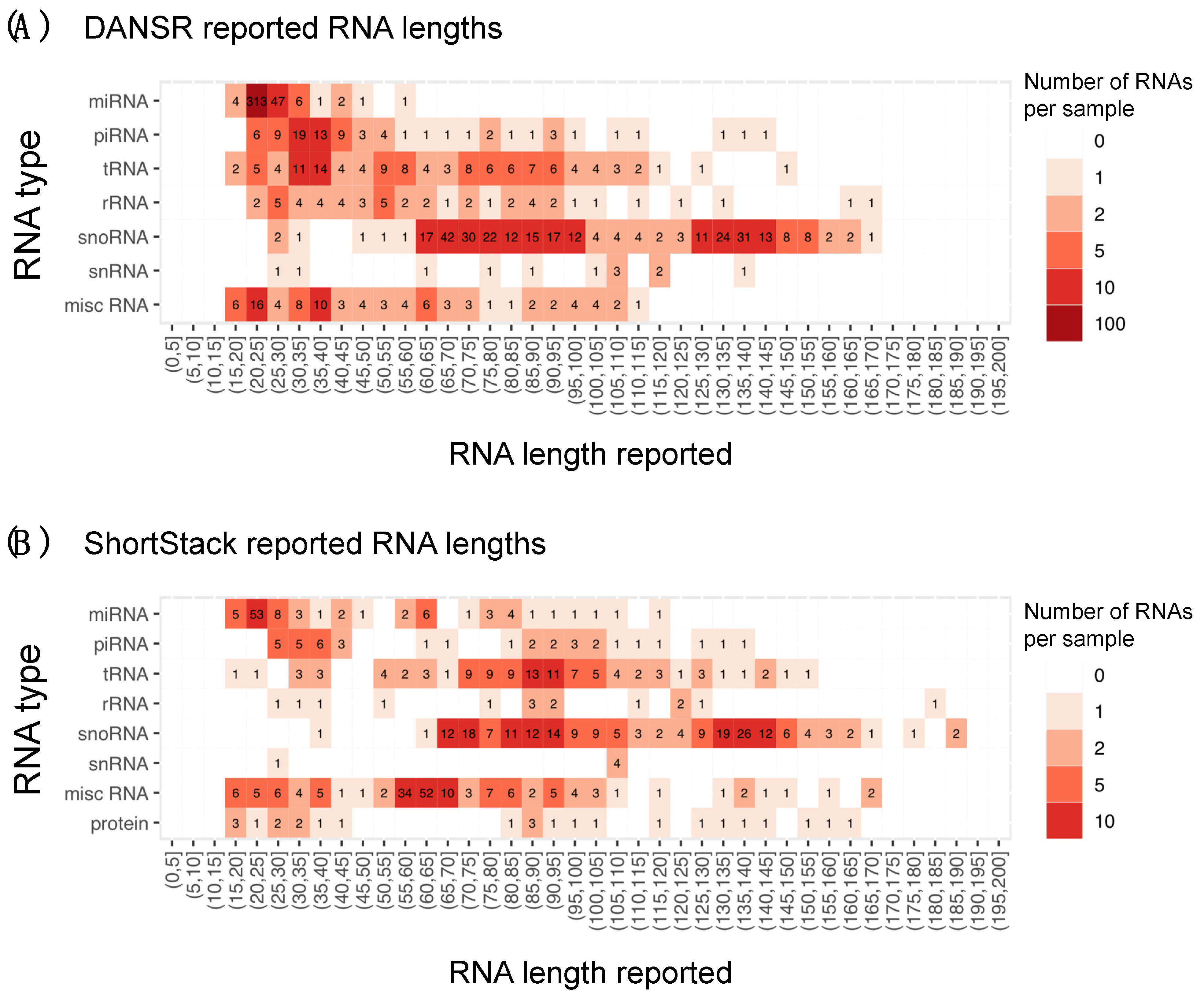
We examined the results of a comparison between DANSR and two alternative tools, Manatee and ShortStack. In addition to DANSR’s more complete output, with full annotation and cluster range information, we saw that both tools returned many thousands of results that DANSR’s algorithm rejected as low-quality (
Figure 5A). Manual inspection of such clusters confirms that they are false positives of a variety of types, including misreported expression of protein-coding exons and sub-optimal clusters of multi-aligned reads (
Figure 5D,E). In contrast,
Figure S4 demonstrates DANSR’s ability to identify true novel and annotated small ncRNAs that overlap protein coding and lncRNA genes without reporting large numbers of false positive results consisting of protein coding and lncRNA transcripts. Excluding such low-quality clusters, DANSR detects more small RNAs across all species types, as well as a roughly equal number of novel clusters, when compared with the other tools. This result highlights its superior performance in sensitively detecting true small RNA clusters while filtering false positives. The potential for novel small ncRNA discovery using DANSR is illustrated in
Figure S5, which shows novel miRNAs with hairpin precursors identified using DANSR’s unannotated candidate clusters for deep-sequenced CRC samples. DANSR’s superior ability to sensitively detect true positive clusters while controlling false negatives was confirmed by simulated data (
Figure 5B,C).
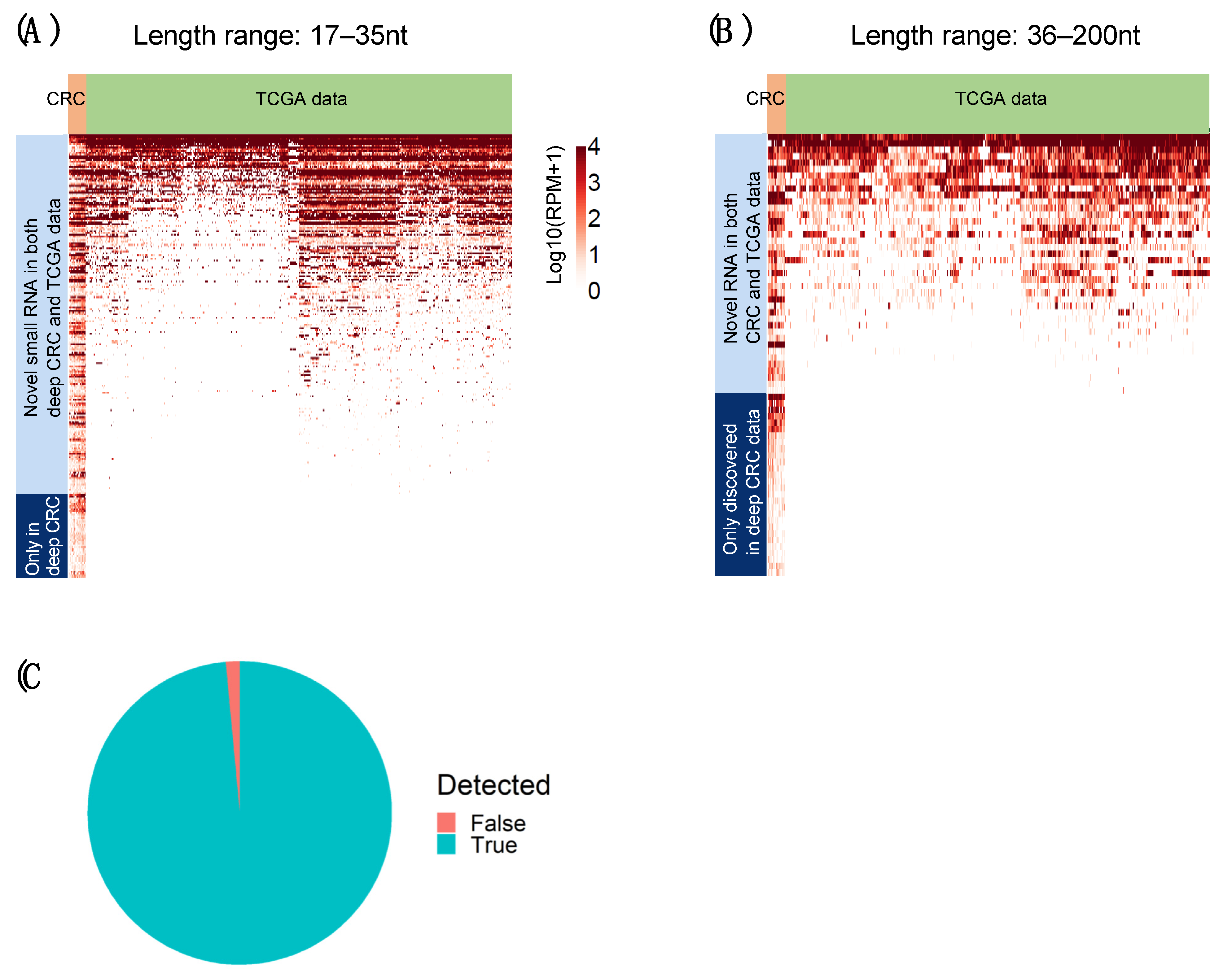
Finally, we used data from the TCGA-COAD and TCGA-READ studies as a positive control to benchmark DANSR’s ability to detect known and novel small ncRNAs. We ran DANSR directly on these data and detected, as expected, over 99% of the known miRNAs identified by TCGA (
Figure 7C). This finding demonstrated that DANSR can be applied to existing miRNA data to report high quality results, while further benefiting from DANSR’s novel modules. Moreover, we used the TCGA miRNA-seq cohorts to validate the common novel small RNA species detected by DANSR within >50% of samples in the deep-sequenced CRC cohort. These analyses demonstrated the reliability of DANSR’s novel discoveries. A large majority (82%) of the novel small RNAs commonly discovered in the cohort at the short end of the length range (17–35 nt) were also detected in TCGA samples, indicating that these novel small RNAs are real and common across individuals with this cancer type (
Figure 7A). The advantage of the greater sequencing depth applied to our CRC cohort is demonstrated by the additional 48 novel clusters present in >50% of samples in our cohort at lower levels of expression, which were not detectable among the TCGA miRNA-seq data. In the mid-size range of 36–200 nt, 42% of common novel small RNAs could not be detected within TCGA samples (
Figure 7B). The presence of these novel clusters in a majority of deep-sequenced individuals creates confidence that such calls are true positives, again demonstrating the advantage of a deep-sequenced cohort optimized to facilitate discovery within the full 17–200 nt length range. These common mid-sized ncRNAs could have biological implications in these diseases, making the ability to detect them important.
4. Methods
4.1. Implementation of DANSR Tool
The DANSR tool is implemented in the Python and C++ programming languages and employs six main steps (
Figure 1A): (1) adapter trimming, (2) read alignment, (3) cluster identification, (4) boundary optimization, (5) identification of single and multi-node clusters, and (6) identification of annotated and unannotated small RNAs. Each step in this pipeline and the default values of its associated parameters were implemented based on the lessons learned in overcoming the challenges to small RNA discovery in our previous study [
8].
4.2. Standard Data Input/Output Format and Small RNA Annotation
In the optional first step, DANSR trims user-supplied adapter sequences from the submitted reads using Cutadapt [
11], retaining reads of at least 17 nt (the minimum length of small RNAs). DANSR accepts small RNA sequencing data in the FASTQ/FASTA format.
The next step aligns the reads to the human reference genome using BWA ver. 0.7.17 [
11]. The aligned reads are output in SAM/BAM format. Although alignment parameters can be defined by the user, use of the default values (-q 5 -k 1 -l 17), which were found to produce excellent results in earlier efforts to discover small noncoding RNAs [
8], is highly recommended.
In the third step, clusters are identified based on the overlap between read alignments, with several tunable quality filters applied to the reads before clustering. First, reads are filtered on their CIGAR string such that only insertions or deletions of lengths no more than 5 are allowed, and all indels must be flanked by alignments of lengths of at least 5 (these restrictions may be tuned using the parameter file cigars_allow.txt). Second, aligned reads are grouped into uniquely mapped reads, multi-mapped reads, and repeat reads. Unique and multi-mapped reads are retained, while repeat reads are excluded from further analysis; the number of alignments above which a read is designated as repeat and discarded is controlled by the number-hits parameter (default 5). To form clusters of reads, the retained reads are first converted into BED format and overlapped alignments are merged into read clusters using BEDTools (2.27.0 or later) [
12]. Clusters containing fewer than the minimum allowable number of reads (defined by the number-reads parameter, default 5) are then excluded from further analysis.
4.3. Optimize Small RNA Boundaries
In the fourth step, a heuristic algorithm is employed to optimize the boundaries of each read cluster identified in the third step to more accurately define the range of the small RNA cluster and split it into sub-clusters if it represents more than one feature. (
Figure 1B and
Figure S2). This algorithm works by assigning a weight to each read based on the level of its contribution to the original cluster. The read weight is computed by first walking through the cluster bases and computing the read depth of each base (number of reads covering the base). An individual read’s weight is defined as the total coverage of the bases it spans divided by the read length. Thus, if
represents the position of the
ith base in a read of length
n, the read weight can be represented by the following equation:
where
cov = total number of reads covering the base.
Once all read weights are computed, reads within the cluster are ordered by weight, and a number of the lowest-weight reads equal to a proportion of the total number of reads (defined by the percent-cur parameter, default 0.3) in the original cluster, designated
, are provisionally removed. The updated cluster (
) is then split into sub-clusters, each of which starts at a transition from 0 read coverage to coverage > 0 and ends at a transition from coverage > 0 to 0.
is also subdivided into sections at the midpoint of each gap between sub-clusters in
. For each candidate sub-cluster, the average coverage depth is compared to that of the corresponding section of
. If the ratio of coverage depths
, where cutoff is the input parameter (default 0.3), the reads and boundaries of the new sub-cluster in
are accepted; otherwise, the initial cluster reads and boundary from the corresponding section of
are retained. Consecutive retained sections of
are merged into single clusters.
Figure S2 shows a sketch illustrating this process.
4.4. Identify Single- and Multi-Node Clusters
In the fifth step, a network model is built based on uniquely aligned and multi-mapped reads to identify single-node read clusters and multi-node read clusters (i.e., a group of clusters that share multiply-aligned reads between them) (
Figure 1C), which are used in the next step to identify low quality clusters caused by repetitive reads. The network consists of a graph in which nodes represent clusters and edges represent the shared reads between clusters. Thus, if a cluster is comprised of only uniquely mapped reads, the cluster is called a single-node cluster. All single-node clusters are reported as results and do not need to be processed by the decision tree model in the sixth step. In its general construction, the network is a graph consisting of a large number of subnetworks. To assess the quality of the clusters, all multi-node clusters are subjected to a quality check in the next step.
Each node of the network is a cluster of read alignments, and two nodes are connected by a link if >50% of the alignments from at least one node have a hit in the other node. Fully connected sub-networks come in two shapes: star-like and clique. A star-like sub-network represents the case in which a central small RNA node generates false positive read clusters at multiple locations, which are difficult to filter in the absence of network analysis. A clique shape represents a small non-coding RNA that has multiple copies on the human genome. The network analysis ensures that, on the one hand, no copy of the same small non-coding RNA is missed and, on the other hand, that the expression level of the small non-coding RNA is calculated accurately by including reads from repetitive small RNAs only once.
At the same time as each cluster is processed by the network model, it is annotated using a human reference annotation and one or more small RNA annotation sets (all in GTF format). DANSR provides a GRCh38/hg38 small RNA library that includes annotations for >187 k features and is sourced from RNAcentral [
20]. A curated GRCh37/hg19 small noncoding library collected from multiple resources by our team to cover any small RNA genes missing from the standard human genome annotation in a prior study [
8] is also provided; users may employ either annotation file or provide their own. Clusters that overlap one or more features in the provided annotation files are provisionally annotated with each of those features.
4.5. Identify Annotated and Unannotated Small RNAs
To evaluate the quality of clusters and reduce the false positive rate, a decision tree model has been constructed to classify clusters (graph nodes) into three main categories: (1) annotated, (2) unannotated, and (3) low-quality clusters (
Figure 1D). The decision tree model works by traversing each subgraph of the network and asking questions (
Figure S3) based on the properties of each cluster to determine its class. The largest node in each cluster is classified as annotated if it overlaps one or more regions in the small RNA annotation file; otherwise, it is classified as unannotated or low-quality based on whether the number of reads within the cluster exceeds the cutoff value for acceptance (number-reads parameter, default 5). Each of the smaller nodes in a graph are then processed. First, a check is carried out to determine whether the number of reads within the cluster falls below the cutoff value for acceptance; if so, the cluster is classified as low quality. Then, the proportion of reads shared with the largest cluster of the graph is checked; if it exceeds a threshold (ov-with-largest parameter, default 0.75), the cluster is classified as low quality. Otherwise, the proportion of unique reads within the cluster is checked; if it exceeds the cutoff (percent-uniq parameter, default 0.5), the cluster is classified either as annotated (if it overlaps one or more annotations) or unannotated. Otherwise, the proportion of reads shared with other clusters is checked; if it is less than 50%, the cluster is classified either as annotated (if it overlaps annotations) or unannotated. Otherwise, the number of uniquely aligned reads within the cluster is counted; if it exceeds a cutoff value (unique-reads parameter, default 2), the cluster is classified either as annotated (if it overlaps annotations) or unannotated, and otherwise as low-quality.
Once all clusters are classified, low-quality clusters are written to the output file of rejected candidates and the remaining read clusters are further refined. Among annotated clusters (including single-node clusters that did not pass through the decision tree), if the highest Jaccard score among its provisional annotations is greater than a cutoff value (jaccard-index parameter, default 0.3), that annotation is applied to the cluster and it is written to the annotated output file. Clusters with scores below this value that fall within the length range 17–200 nt and that do not overlap with protein coding exons are designated as falling in close proximity to a known annotation and written to the close proximity output file; otherwise, the clusters are written to the rejected file. Clusters identified as unannotated are filtered for length and written to the unannotated output file.
4.6. Sequencing Protocol for Small RNAs in the 17–200 Nucleotide Range
In this paper’s application, we have improved the protocol [
8] using Blue Pippin (Sage Science, Beverly, MA, USA) to enrich for library fragments in the full length range of 17–200 nt. Total RNA was isolated from snap-frozen tissue samples using TRIzol™ Reagent (ThermoFisher Scientific, Waltham, MA, USA). Working on dry ice, 2–5 mm Tissue Lyser beads (QIAGEN, Hilden, Germany) and 1.0 mL of TRIzol™ Reagent were added to a 100 mg tissue sample in a 2.0 mL Safe-lock Tube (Eppendorf, Hamburg, Germany). The sample was homogenized using the Tissue LyserLT (QIAGEN, Hilden, Germany) at top speed for 10 min at room temperature. For every 1.0 mL of TRIzol™ Reagent used, 0.2 mL of chloroform (Millipore Sigma, St. Louis, MO, USA) was added. The capped sample was vigorously shaken for 30 s. Following a 3 min room temperature incubation, the sample was centrifuged at 14,000 rpm for 15 min at 4 °C. The aqueous layer was removed and placed in a new 1 mL tube. For every 1.0 mL of TRIzol™ Reagent that was used for homogenization, 0.5 mL of 100% isopropanol (Millipore Sigma, St. Louis, MO, USA) was added to the aqueous layer. The sample was vortexed briefly. Following a 10 min incubation at room temperature, the sample was centrifuged at 14,000 rpm for 15 min at 4 °C. The supernatant was removed and the RNA pellet was washed with 1 mL of 75% ethanol (Millipore Sigma, St. Louis, MO, USA). The sample was briefly vortexed and then centrifuged at 14,000 rpm for 15 min at 4 °C. The wash was discarded. The RNA pellet was allowed to air dry for 5–10 min. The RNA pellet was gently resuspended in 50 μL of Nuclease-Free Water.
The isolated RNA was purified using the RNA Clean and Concentrator kit (Zymo Research, Irvine, CA, USA) per the manufacturer’s instructions, including the in-column DNase I treatment. The concentration of the RNA was determined using the Qubit RNA BR Assay kit (ThermoFisher Scientific, Waltham, MA, USA). Yields ranged from 500 ng–2 μg. The quality of the RNA was determined using the RNA 6000 Nano kit (Agilent Technologies, Santa Clara, CA, USA). The RNA samples had an RNA Integrity Number (RIN) of 4 or higher.
Small RNA libraries were generated from 1 μg inputs of purified RNA using the NEBNext® Multiplex Small RNA Library Prep Set for Illumina (New England BioLabs, Ipswich, MA, USA) following the manufacturer’s protocol. During PCR amplification, a unique index was added to each sample. Each amplified sample was purified using a QIAQuick PCR Purification column (QIAGEN, Hilden, Germany). The purified DNA was eluted from the column in 32 μL of Nuclease-Free Water. The concentration of the purified DNA was measured using the Qubit dsDNA HS assay kit (ThermoFisher Scientific, Waltham, MA, USA). The size distribution was determined by running an aliquot of each library on the Agilent Bioanalyzer High Sensitivity DNA Analysis assay (Agilent Technologies, Santa Clara, CA, USA). The remaining library sample was then size-selected on a Blue Pippin 3% Agarose Gel Cassette, dye-free, with internal standards (Sage Science, Beverly, MA, USA) to enrich for library fragments 17–200 nt in length. The concentration of the enriched fragments was determined using the Qubit dsDNA HS assay kit (ThermoFisher Scientific, Waltham, MA, USA). The fragment size distribution was verified by running an aliquot of the enriched fragments library on the Agilent Bioanalyzer High Sensitivity DNA Analysis assay (Agilent Technologies, Santa Clara, CA, USA).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






