Large Improvement in the Mechanical Properties of Polyurethane Nanocomposites Based on a Highly Concentrated Graphite Nanoplate/Polyol Masterbatch


Abstract
:
1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of Graphite Nanoplates (GNPs)
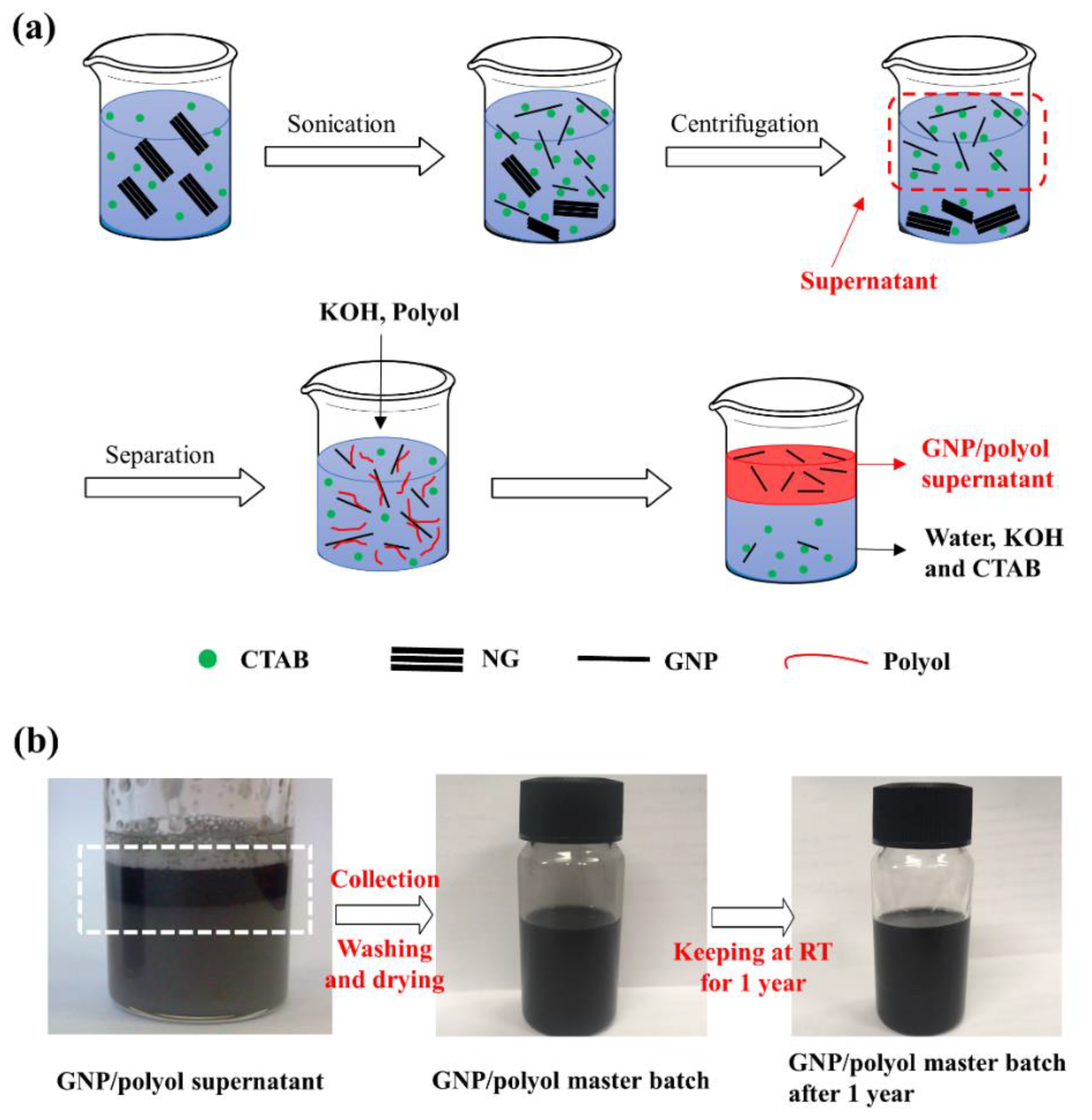
2.3. Preparation of GNP/Polyol Masterbatch
2.4. Polymerization of Polyurethane (PU)/GNP Nanocomposites
2.5. Characterization
3. Results and Discussion
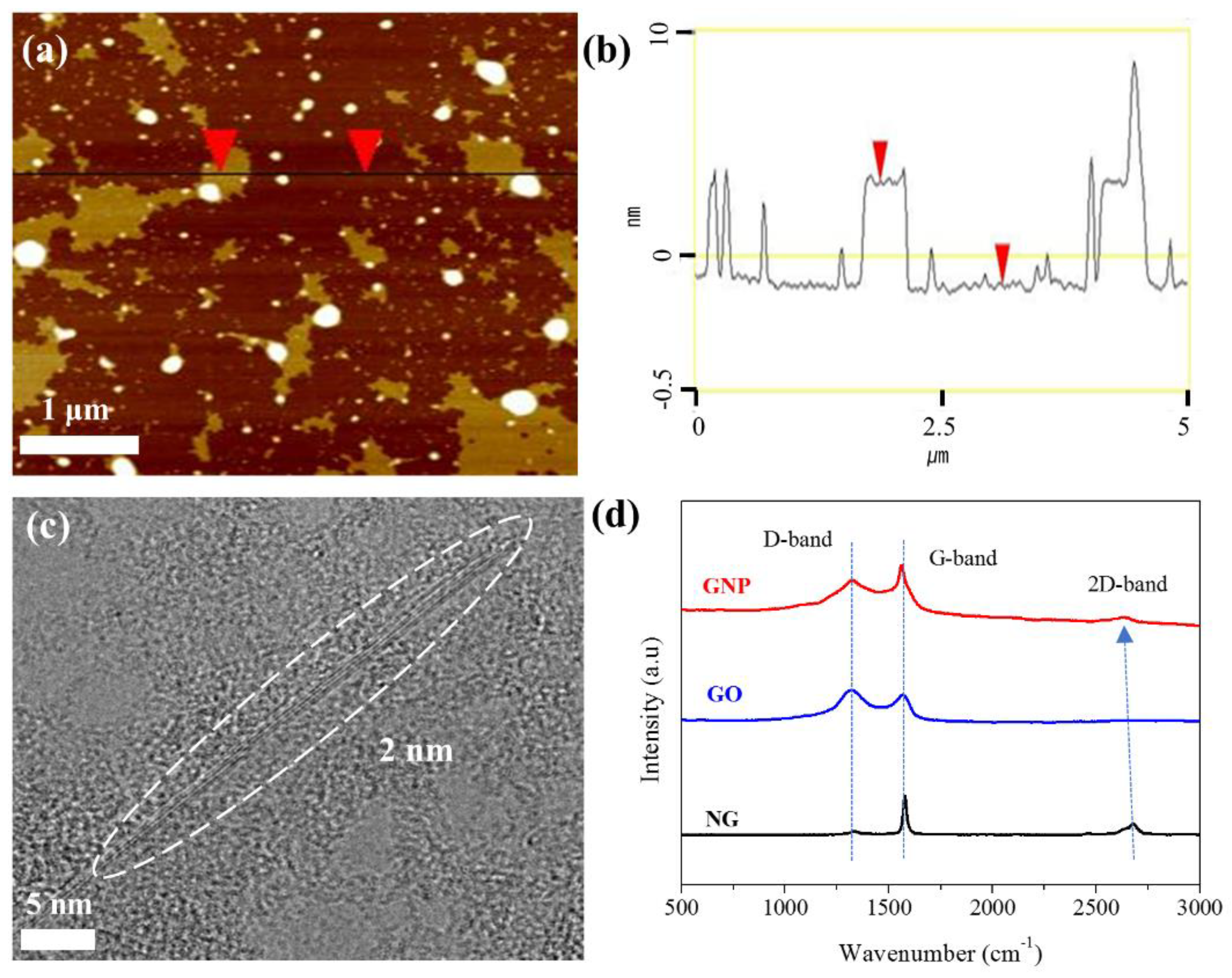
3.1. Characterization of GNPs
3.2. Preparation of GNP/Polyol Masterbatch
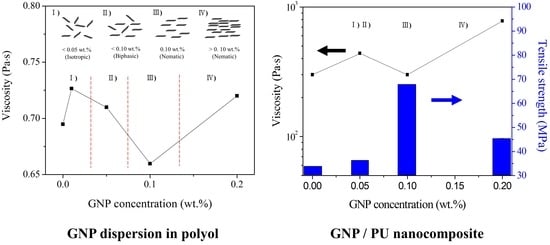
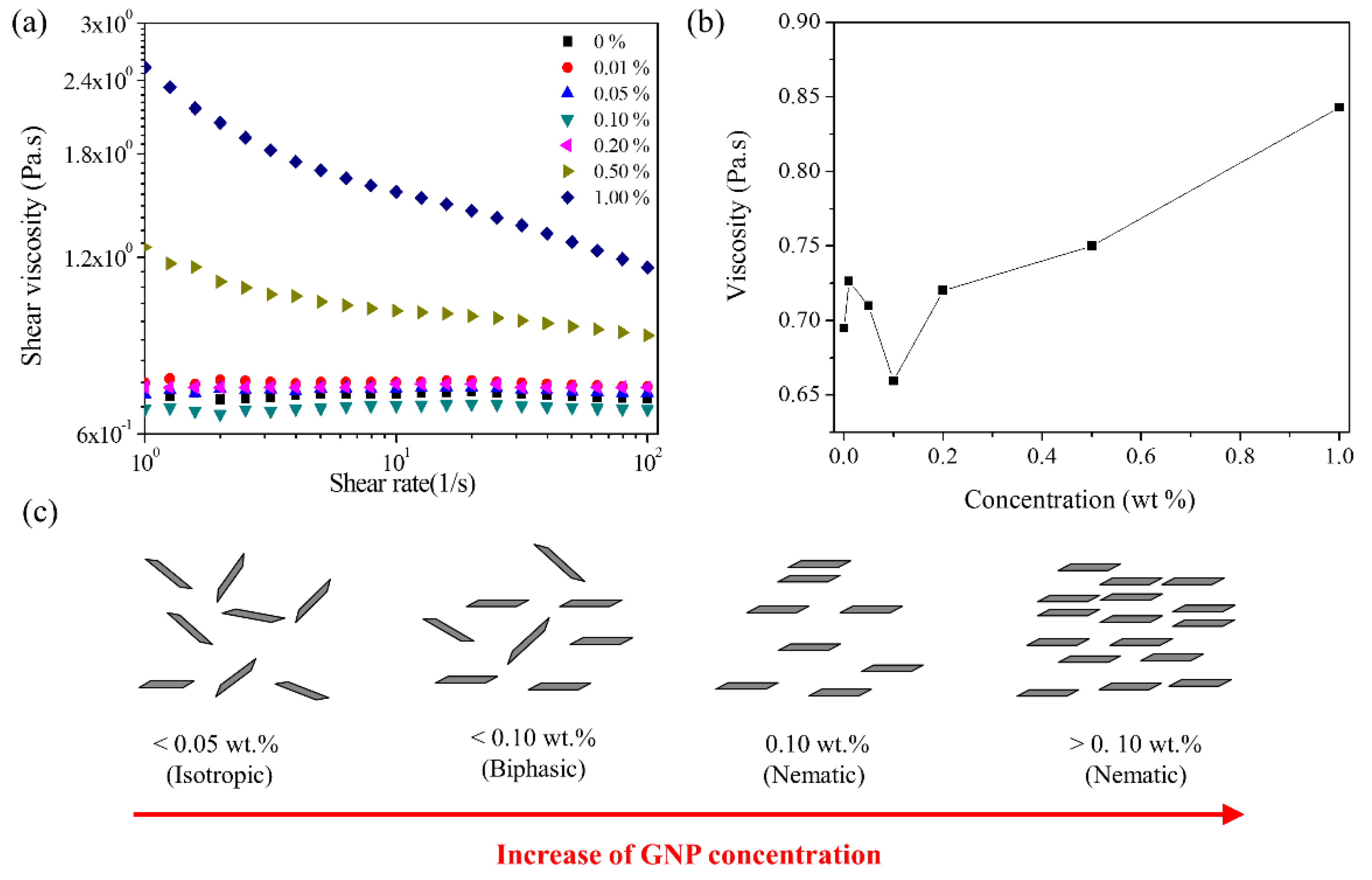
3.3. Lyotropic Liquid Crystalline (LLC) Behavior of GNPs in Polyol
3.4. PU/GNP Nanocomposites
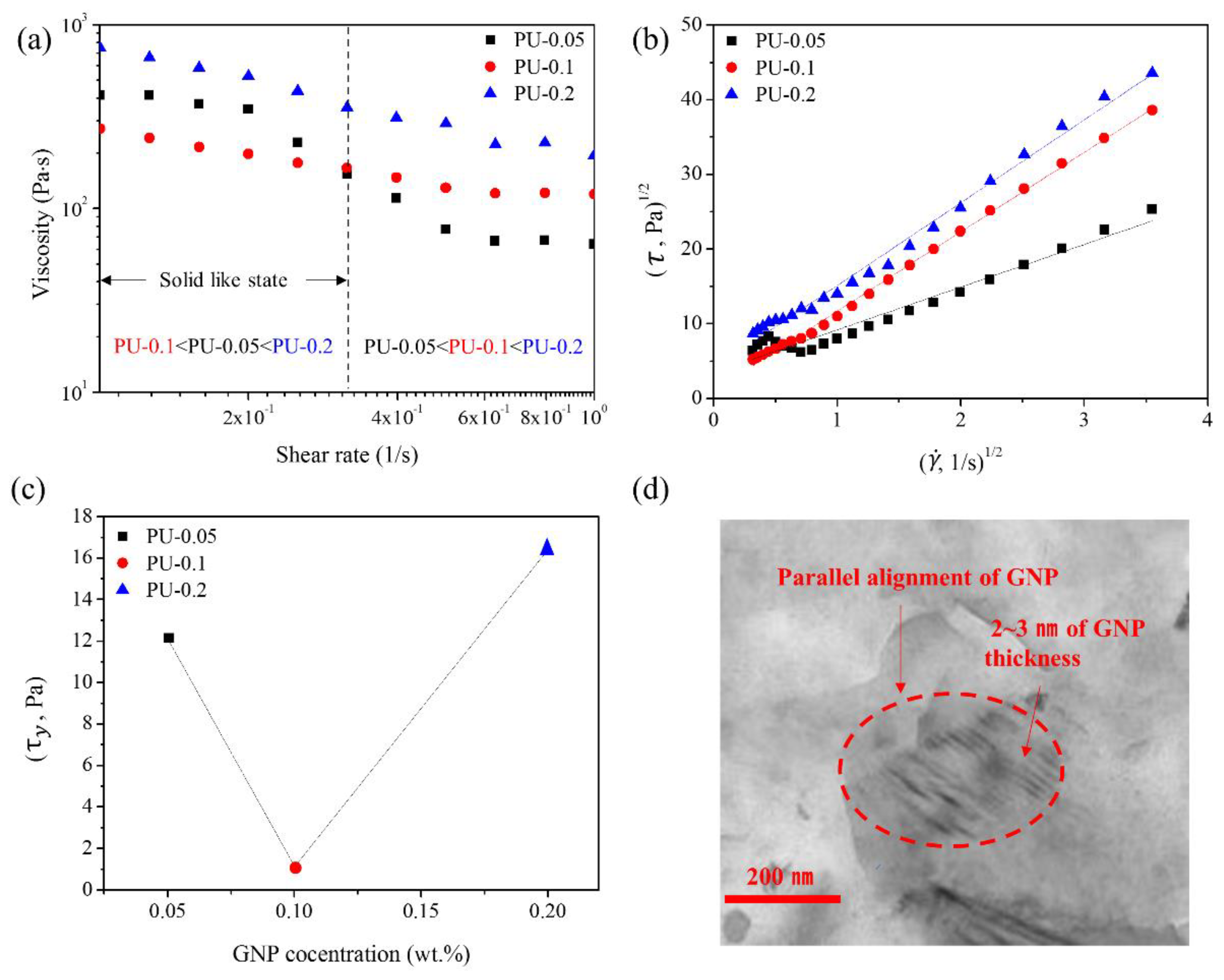
3.4.1. LLC Behavior of GNPs in PU Nanocomposites
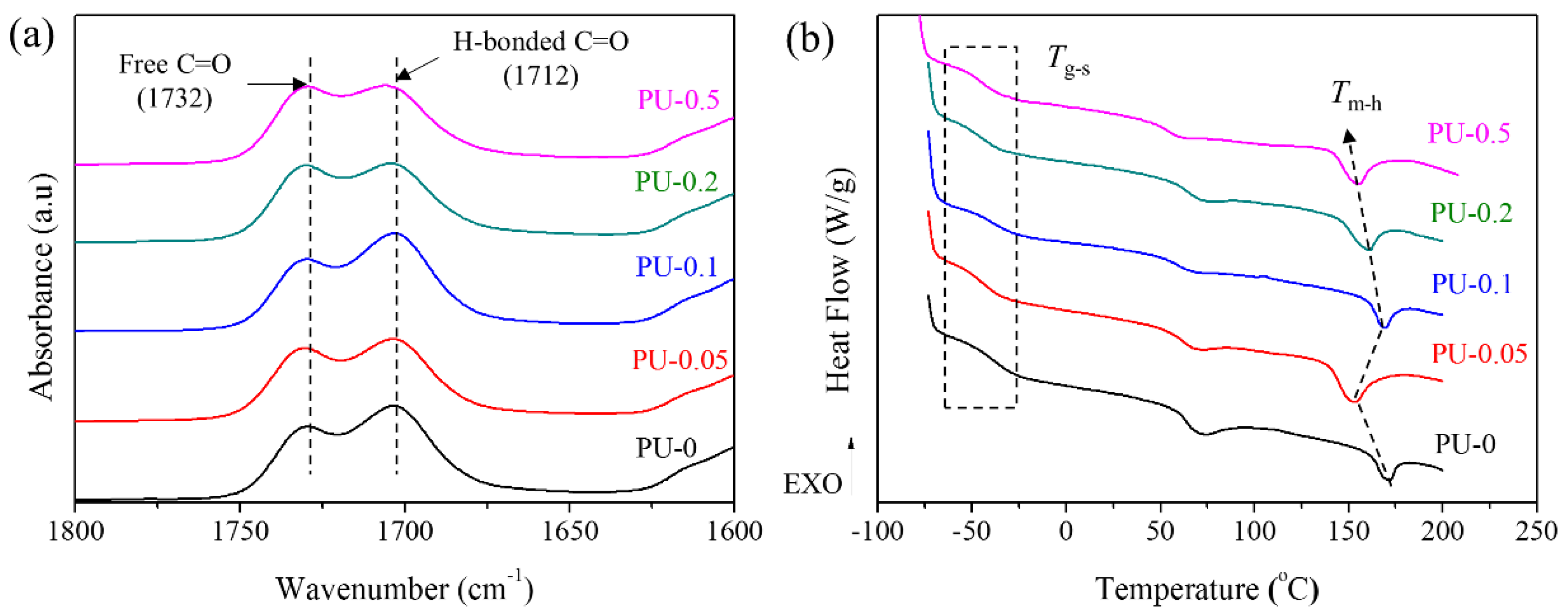
3.4.2. Degree of Hydrogen Bonding and Microphase Separation of Nanocomposites
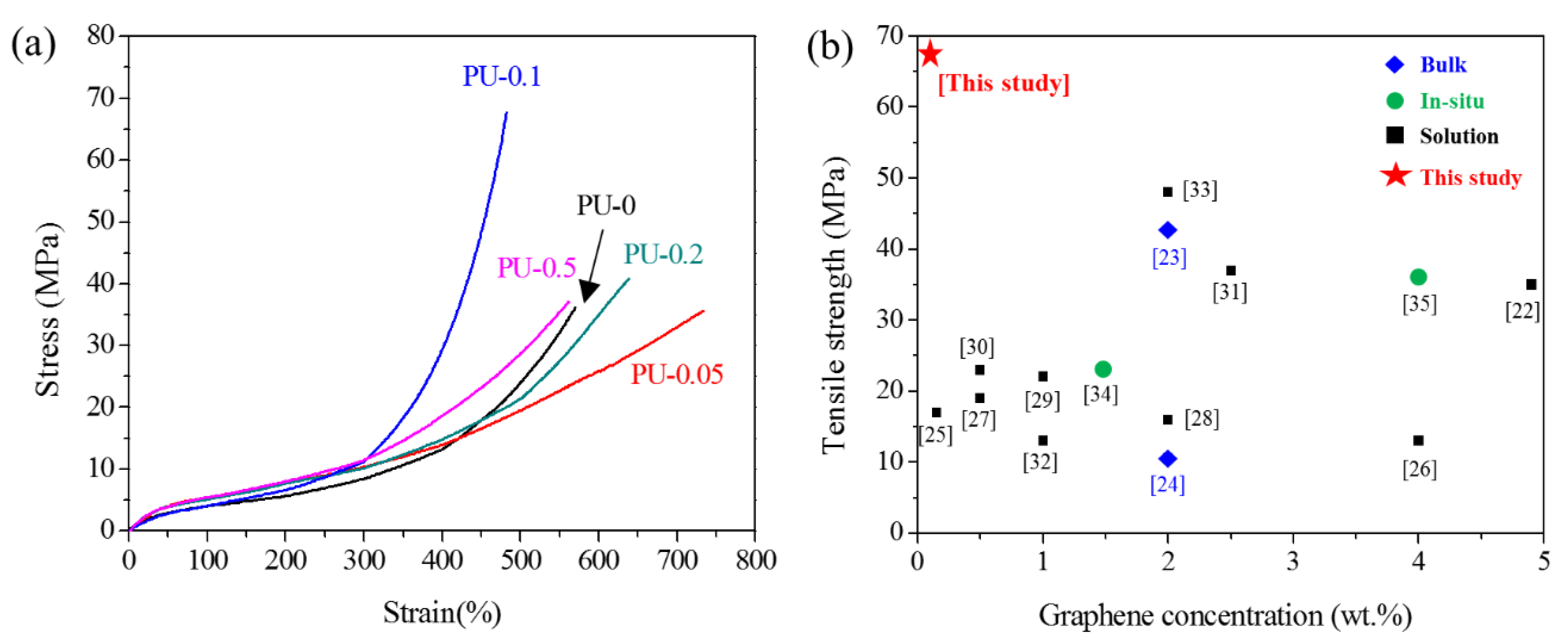
3.4.3. Mechanical Properties of GNP/PU Nanocomposites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Camargo, P.H.C.; Satyanarayana, K.G.; Wypych, F. Nanocomposites: Synthesis, structure, properties and new application opportunities. Mater. Res. 2009, 12, 1–39. [Google Scholar] [CrossRef]
- Lu, H.; Yao, Y.; Huang, W.M.; Leng, J.; Hui, D. Significantly improving infrared light-induced shape recovery behavior of shape memory polymeric nanocomposite via a synergistic effect of carbon nanotube and boron nitride. Compos. Part B Eng. 2014, 62, 256–261. [Google Scholar] [CrossRef]
- Liu, W.; Lu, G.; Chen, H.; Huang, X.; Li, Y.; Cao, F.; Jiang, X. Thermo-Responsive Graphene Oxide/Poly(Ethyl Ethylene Phosphate) Nanocomposite via Ring Opening Polymerization. Nanomaterials 2019, 9, 207. [Google Scholar]
- Fakhrullin, R.; Milioto, S.; Parisi, F.; Lisuzzo, L.; Cavallaro, G.; Lazzara, G. Core/Shell Gel Beads with Embedded Halloysite Nanotubes for Controlled Drug Release. Coatings 2019, 9, 70. [Google Scholar]
- Ahmad, I.; Kim, H.-K.; Deveci, S.; Kumar, R. Non-Isothermal Crystallisation Kinetics of Carbon Black-Graphene-Based Multimodal-Polyethylene Nanocomposites. Nanomaterials 2019, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, G.; Lazzara, G.; Lisuzzo, L.; Milioto, S.; Parisi, F. Filling of mater-BI with nanoclays to enhance the biofilm rigidity. J. Funct. Biomater. 2018, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.K.; Kessler, M.R. Self-healing polymer nanocomposite materials: A review. Polymer 2015, 69, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Karakeçili, A.; Messina, G.M.L.; Yurtsever, M.Ç.; Gümüşderelioğlu, M.; Marletta, G. Impact of selective fibronectin nanoconfinement on human dental pulp stem cells. Colloids Surf. B Biointerfaces 2014, 123, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Beecroft, L.L.; Ober, C.K. Nanocomposite Materials for Optical Applications. Chem. Mater. 1997, 9, 1302–1317. [Google Scholar] [CrossRef]
- Zhao, L.; Pang, X.; Adhikary, R.; Petrich, J.W.; Lin, Z. Semiconductor anisotropic nanocomposites obtained by directly coupling conjugated polymers with quantum rods. Angew. Chem. Int. Ed. 2011, 50, 3958–3962. [Google Scholar] [CrossRef] [PubMed]
- McKee, J.R.; Appel, E.A.; Seitsonen, J.; Kontturi, E.; Scherman, O.A.; Ikkala, O. Healable, stable and stiff hydrogels: Combining conflicting properties using dynamic and selective three-component recognition with reinforcing cellulose nanorods. Adv. Funct. Mater. 2014, 24, 2706–2713. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, J.; Chen, M.; Ma, L.; Zhao, X.; Dang, Z.M.; Wang, Z. Improved self-healing of polyethylene/carbon black nanocomposites by their shape memory effect. J. Phys. Chem. B 2013, 117, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Huang, H.X. Simultaneously toughening and reinforcing poly(lactic acid)/thermoplastic polyurethane blend via enhancing interfacial adhesion by hydrophobic silica nanoparticles. Polym. Test. 2015, 45, 107–113. [Google Scholar] [CrossRef]
- Kang, D.J.; Park, G.U.; Im, H.G.; Park, H.Y.; Jin, J. Silica nanoparticle-embedded urethane acrylate nanohybrid thermosets for photo-patternable transparent hard coating. Polymer 2016, 105, 19–24. [Google Scholar] [CrossRef]
- Zhai, T.; Li, D.; Fei, G.; Xia, H. Piezoresistive and compression resistance relaxation behavior of water blown carbon nanotube/polyurethane composite foam. Compos. Part A Appl. Sci. Manuf. 2015, 72, 108–114. [Google Scholar] [CrossRef]
- Zeng, Z.; Chen, M.; Jin, H.; Li, W.; Xue, X.; Zhou, L.; Pei, Y.; Zhang, H.; Zhang, Z. Thin and flexible multi-walled carbon nanotube/waterborne polyurethane composites with high-performance electromagnetic interference shielding. Carbon 2016, 96, 768–777. [Google Scholar] [CrossRef]
- Cai, W.; Feng, X.; Hu, W.; Pan, Y.; Hu, Y.; Gong, X. Functionalized Graphene from Electrochemical Exfoliation for Thermoplastic Polyurethane: Thermal Stability, Mechanical Properties, and Flame Retardancy. Ind. Eng. Chem. Res. 2016, 55, 10681–10689. [Google Scholar] [CrossRef]
- Khan, U.; May, P.; O’Neill, A.; Coleman, J.N. Development of stiff, strong, yet tough composites by the addition of solvent exfoliated graphene to polyurethane. Carbon 2010, 48, 4035–4041. [Google Scholar] [CrossRef] [Green Version]
- Pokharel, P.; Pant, B.; Pokhrel, K.; Pant, H.R.; Lim, J.G.; Lee, D.S.; Kim, H.Y.; Choi, S. Effects of functional groups on the graphene sheet for improving the thermomechanical properties of polyurethane nanocomposites. Compos. Part B Eng. 2015, 78, 192–201. [Google Scholar] [CrossRef]
- Appel, A.K.; Thomann, R.; Mülhaupt, R. Polyurethane nanocomposites prepared from solvent-free stable dispersions of functionalized graphene nanosheets in polyols. Polymer 2012, 53, 4931–4939. [Google Scholar] [CrossRef]
- Kim, J.T.; Kim, B.K.; Kim, E.Y.; Park, H.C.; Jeong, H.M. Synthesis and shape memory performance of polyurethane/graphene nanocomposites. React. Funct. Polym. 2014, 74, 16–21. [Google Scholar] [CrossRef]
- Cai, D.; Jin, J.; Yusoh, K.; Rafiq, R.; Song, M. High performance polyurethane/functionalized graphene nanocomposites with improved mechanical and thermal properties. Compos. Sci. Technol. 2012, 72, 702–707. [Google Scholar] [CrossRef]
- Nguyen, D.A.; Lee, Y.R.; Raghu, A.V.; Jeong, H.M.; Shin, C.M.; Kim, B.K. Morphological and physical properties of a thermoplastic polyurethane reinforced with functionalized graphene sheet. Polym. Int. 2009, 58, 412–417. [Google Scholar] [CrossRef]
- Thakur, S.; Karak, N. Bio-based tough hyperbranched polyurethane–graphene oxide nanocomposites as advanced shape memory materials. RSC Adv. 2013, 3, 9476–9482. [Google Scholar] [CrossRef]
- Li, X.; Deng, H.; Li, Z.; Xiu, H.; Qi, X.; Zhang, Q.; Wang, K.; Chen, F.; Fu, Q. Graphene/thermoplastic polyurethane nanocomposites: Surface modification of graphene through oxidation, polyvinyl pyrrolidone coating and reduction. Compos. Part A Appl. Sci. Manuf. 2015, 68, 264–275. [Google Scholar] [CrossRef]
- Han, S.; Chun, B.C. Preparation of polyurethane nanocomposites via covalent incorporation of functionalized graphene and its shape memory effect. Compos. Part A Appl. Sci. Manuf. 2014, 58, 65–72. [Google Scholar] [CrossRef]
- Wu, C.; Huang, X.; Wang, G.; Wu, X.; Yang, K.; Li, S.; Jiang, P. Hyperbranched-polymer functionalization of graphene sheets for enhanced mechanical and dielectric properties of polyurethane composites. J. Mater. Chem. 2012, 22, 7010–7019. [Google Scholar] [CrossRef]
- Wu, S.; Li, J.; Zhang, G.; Sun, R.; Wong, C. High mechanical strength and high dielectric graphene/polyuthane composites healded by near infrared laser. In Proceedings of the 17th International Conference on Electronic Packaging Technology (ICEPT 2016), Wuhan, China, 16–19 August 2016; pp. 157–161. [Google Scholar]
- Kim, J.T.; Jeong, H.J.; Park, H.C.; Jeong, H.M.; Bae, S.Y.; Kim, B.K. Electroactive shape memory performance of polyurethane/graphene nanocomposites. React. Funct. Polym. 2015, 88, 1–7. [Google Scholar] [CrossRef]
- Yadav, S.K.; Cho, J.W. Functionalized graphene nanoplatelets for enhanced mechanical and thermal properties of polyurethane nanocomposites. Appl. Surf. Sci. 2013, 266, 360–367. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Y.; Song, L.; Yang, H.; Xing, W.; Lu, H. In situ polymerization of graphene nanosheets and polyurethane with enhanced mechanical and thermal properties. J. Mater. Chem. 2011, 21, 4222–4227. [Google Scholar] [CrossRef]
- Allen, M.J.; Tung, V.C.; Kaner, R.B. Honeycomb carbon: A Review of graphene. Chem. Rev. 2010, 110, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ruoff, R.S. Chemical methods for the production of graphenes. Nat. Nanotechnol. 2009, 4, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.; Dikin, D.; Park, S.; Cai, W.; Mielke, S.L.; Ruoff, R.S. Effect of Water Vapor on Electrical Properties of Individual Reduced Graphene Oxide Sheets. J. Phys. Chem. C 2008, 112, 20264–20268. [Google Scholar] [CrossRef]
- Hernandez, Y.; Lotya, M.; Nicolosi, V.; Blighe, F.; De, S.; Duesberg, G.; Coleman, J. Liquid phase production of graphene by exfoliation of graphite in surfactant/water solutions. J. Am. Chem. Soc. 2008, 131, 3611–3620. [Google Scholar]
- Guardia, L.; Fernández-Merino, M.J.; Paredes, J.I.; Solís-Fernández, P.; Villar-Rodil, S.; Martínez-Alonso, A.; Tascón, J.M.D. High-throughput production of pristine graphene in an aqueous dispersion assisted by non-ionic surfactants. Carbon 2011, 49, 1653–1662. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron-phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy in graphene. Phys. Rep. 2009, 473, 51–87. [Google Scholar] [CrossRef]
- Ni, Z.; Wang, Y.; Yu, T.; Shen, Z. Raman spectroscopy and imaging of graphene. Nano Res. 2008, 1, 273–291. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.H.; Wang, H.M.; Kasim, J.; Fan, H.M.; Yu, T.; Wu, Y.H.; Feng, Y.P.; Shen, Z.X. Graphene thickness determination using reflection and contrast spectroscopy. Nano Lett. 2007, 7, 2758–2763. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Tong, X.; Wang, Y.; Bao, J.; Wang, Z.M. Graphene oxide liquid crystals: Synthesis, phase transition, rheological property, and applications in optoelectronics and display. Nanoscale Res. Lett. 2015, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gambhir, S.; Jalili, R.; Officer, D.L.; Wallace, G.G. Chemically converted graphene: Scalable chemistries to enable processing and fabrication. NPG Asia Mater. 2015, 7, e186. [Google Scholar] [CrossRef]
- Aboutalebi, S.H.; Gudarzi, M.M.; Zheng, Q.B.; Kim, J.K. Spontaneous formation of liquid crystals in ultralarge graphene oxide dispersions. Adv. Funct. Mater. 2011, 21, 2978–2988. [Google Scholar] [CrossRef]
- Narayan, R.; Kim, J.E.; Kim, J.Y.; Lee, K.E.; Kim, S.O. Graphene Oxide Liquid Crystals: Discovery, Evolution and Applications. Adv. Mater. 2016, 28, 3045–3068. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Guo, C.; Ji, L.; Li, Y.; Tu, Y. Liquid crystalline and shear-induced properties of an aqueous solution of graphene oxide sheets. Langmuir 2013, 29, 8103–8107. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gao, C. Aqueous Liquid Crystals of Graphene Oxide SI. ACS Nano 2011, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.A. Rheology of Fluid, Semisolid, and Solid Foods: Principles and Applications; Springer: New York, NY, USA, 2013; pp. 27–36. [Google Scholar]
- Yousefi, N.; Gudarzi, M.M.; Zheng, Q.; Aboutalebi, S.H.; Sharif, F.; Kim, J.-K. Self-alignment and high electrical conductivity of ultralarge graphene oxide–polyurethane nanocomposites. J. Mater. Chem. 2012, 22, 12709–12717. [Google Scholar] [CrossRef]
- Jung, Y.C.; Kim, J.H.; Hayashi, T.; Kim, Y.A.; Endo, M.; Terrones, M.; Dresselhaus, M.S. Fabrication of transparent, tough, and conductive shape-memory polyurethane films by incorporating a small amount of high-quality graphene. Macromol. Rapid Commun. 2012, 33, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qiu, Z. Crystallization kinetics and morphology of biodegradable poly(L-lactic acid)/graphene oxide nanocomposites: Influences of graphene oxide loading and crystallization temperature. Thermochim. Acta 2012, 527, 40–46. [Google Scholar] [CrossRef]
- Kim, H.M.; Lee, J.K.; Lee, H.S. Transparent and high gas barrier films based on poly(vinyl alcohol)/graphene oxide composites. Thin Solid Films 2011, 519, 7766–7771. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
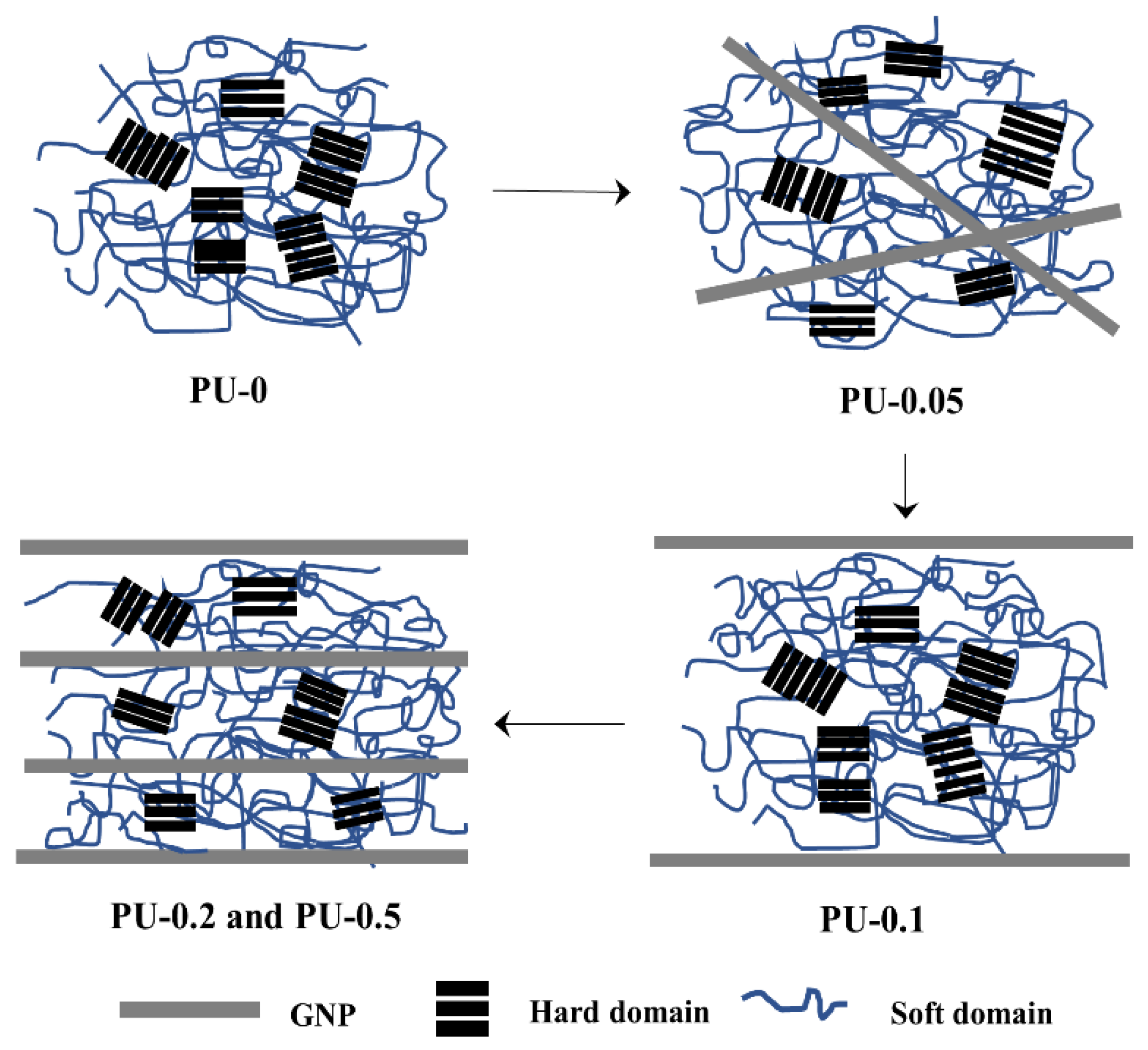{kind=link}
| Sample Code | Composition (wt%) | |||
|---|---|---|---|---|
| MDI | Polyol | BD | GNP | |
| PU-0 | 31.47 | 62.87 | 5.66 | 0 |
| PU-0.05 | 31.45 | 62.84 | 5.66 | 0.05 |
| PU-0.1 | 31.43 | 62.81 | 5.66 | 0.10 |
| PU-0.2 | 31.40 | 62.74 | 5.65 | 0.20 |
| PU-0.5 | 31.31 | 62.56 | 5.64 | 0.50 |
| Sample | Raman Characterization | |
|---|---|---|
| ID/IG | 2D Band (cm−1) | |
| NG | 0.1 | 2677 |
| GO | 1.2 | NA |
| GNP | 0.6 | 2649 |
| Sample Code | FTIR Spectroscopy | DSC | ||||
|---|---|---|---|---|---|---|
| R* | DPS (%) | Tg-s (°C) | Tm-h (°C) | ΔHm-h (J/g) | Tc (°C) | |
| PU-0 | 1.23 | 55.1 | −47 | 169 | 4.05 | 36 |
| PU-0.05 | 1.08 | 51.9 | −52 | 153 | 5.57 | 61 |
| PU-0.1 | 1.25 | 55.5 | −48 | 168 | 4.02 | 38 |
| PU-0.2 | 1.01 | 50.2 | −50 | 158 | 4.77 | 50 |
| PU-0.5 | 1.03 | 50.7 | −50 | 154 | 4.85 | 50 |
| Sample Code | Young’s Modulus (MPa) | Tensile Strength (MPa) | Elongation at Break (%) |
|---|---|---|---|
| U-0 | 4.2 ± 0.25 | 33.1 ± 1.49 | 570 ± 17.82 |
| PU-0.05 | 7.5 ± 0.23 | 35.6 ± 1.23 | 740 ± 12.51 |
| PU-0.1 | 10.6 ± 0.34 | 67.2 ± 1.21 | 470 ± 18.52 |
| PU-0.2 | 7.3 ± 0.28 | 44.8 ± 1.42 | 640 ± 20.24 |
| PU-0.5 | 8.7 ± 0.52 | 36.5 ± 0.82 | 560 ± 25.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-H.; Oh, C.-R.; Lee, D.-S. Large Improvement in the Mechanical Properties of Polyurethane Nanocomposites Based on a Highly Concentrated Graphite Nanoplate/Polyol Masterbatch. Nanomaterials 2019, 9, 389. https://doi.org/10.3390/nano9030389
Lee S-H, Oh C-R, Lee D-S. Large Improvement in the Mechanical Properties of Polyurethane Nanocomposites Based on a Highly Concentrated Graphite Nanoplate/Polyol Masterbatch. Nanomaterials. 2019; 9(3):389. https://doi.org/10.3390/nano9030389
Chicago/Turabian StyleLee, Sang-Hyub, Cho-Rong Oh, and Dai-Soo Lee. 2019. "Large Improvement in the Mechanical Properties of Polyurethane Nanocomposites Based on a Highly Concentrated Graphite Nanoplate/Polyol Masterbatch" Nanomaterials 9, no. 3: 389. https://doi.org/10.3390/nano9030389