Tailoring Bandgap of Perovskite BaTiO3 by Transition Metals Co-Doping for Visible-Light Photoelectrical Applications: A First-Principles Study

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Calculation Details
3. Results and Discussion
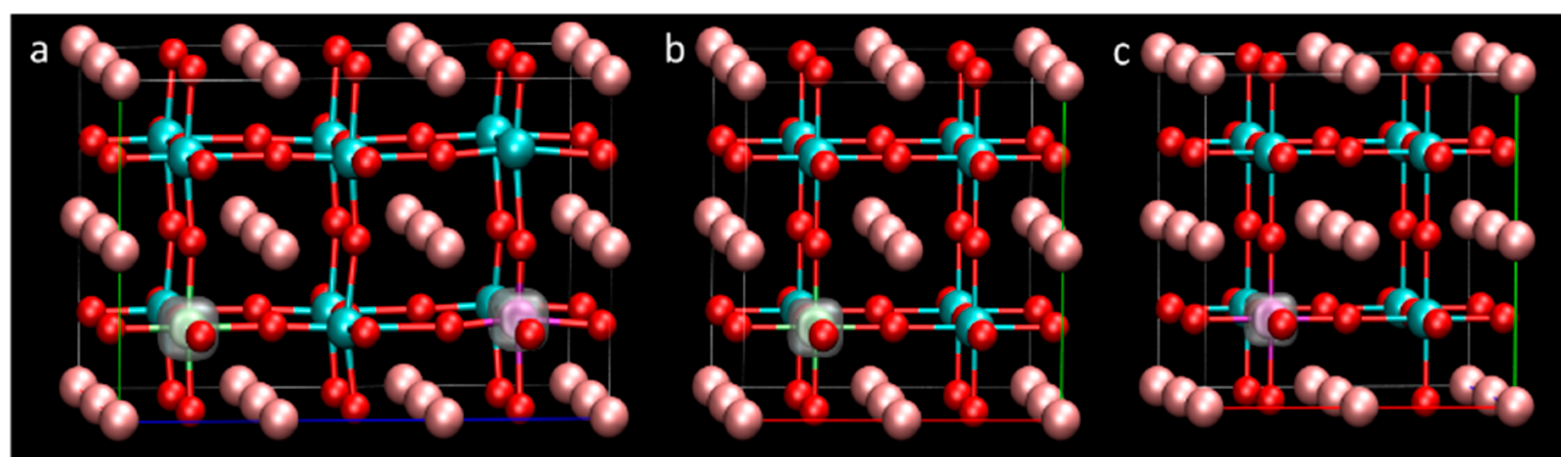
3.1. Structure
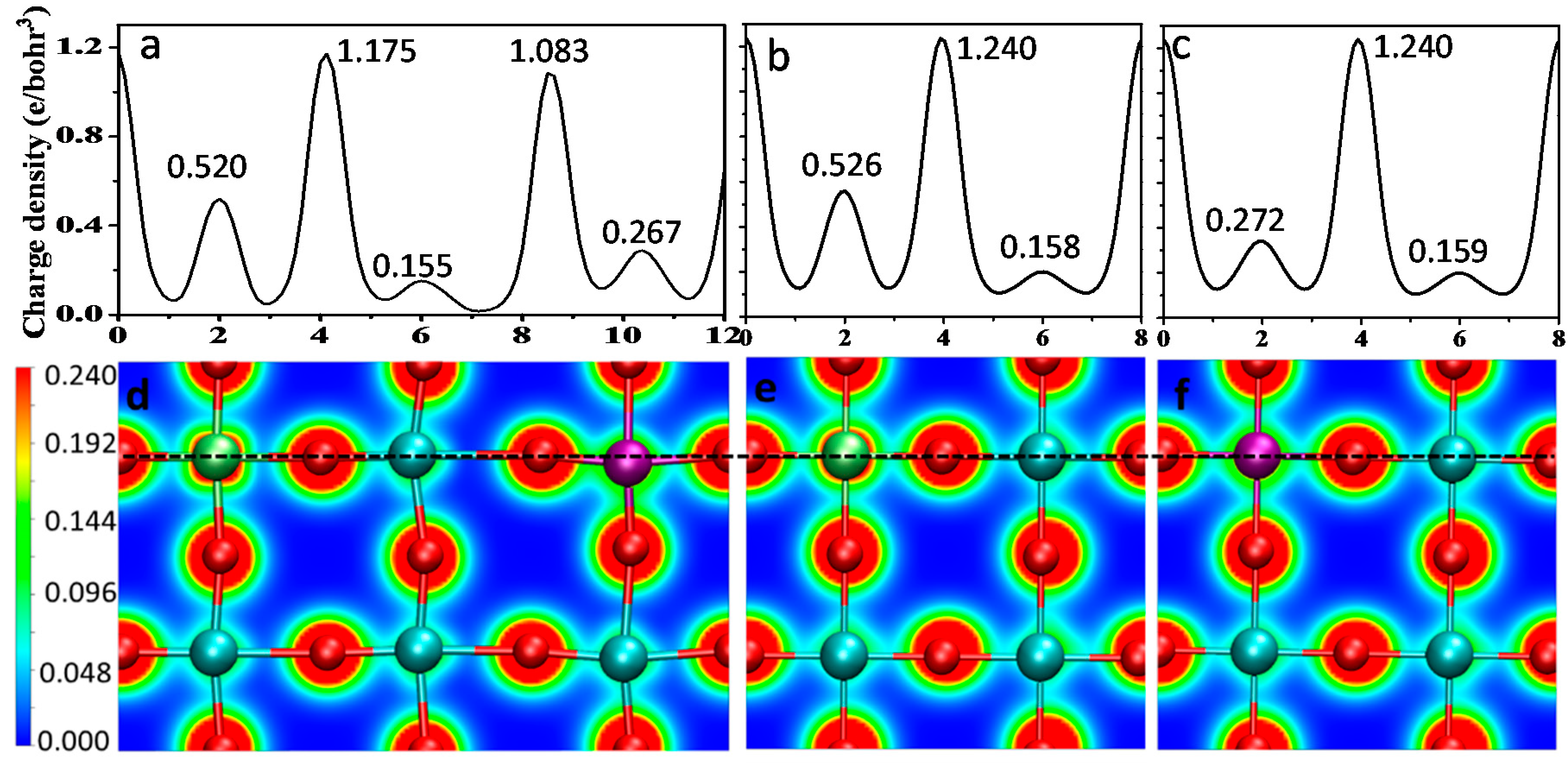
3.2. Defect Formation Energy
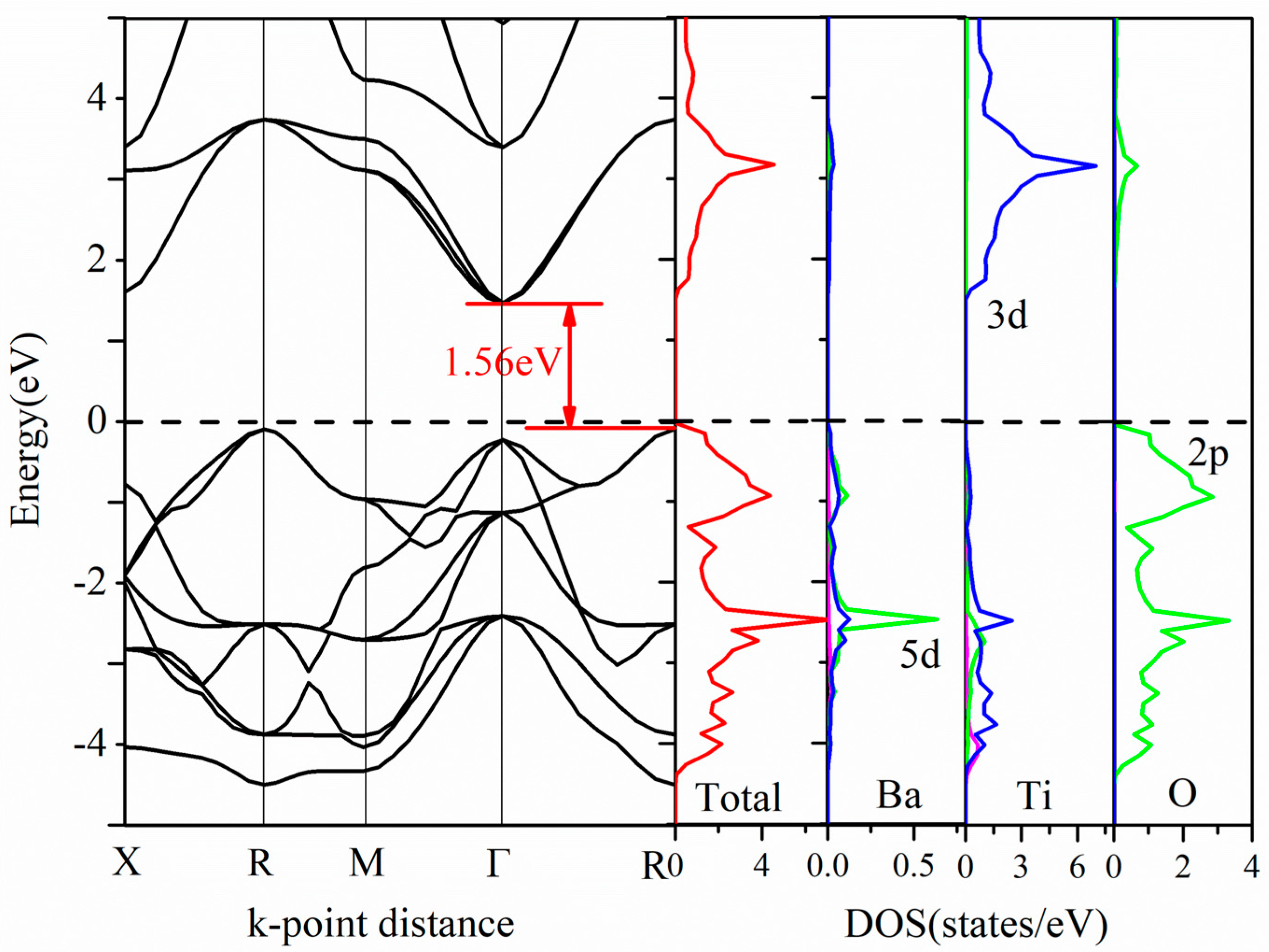
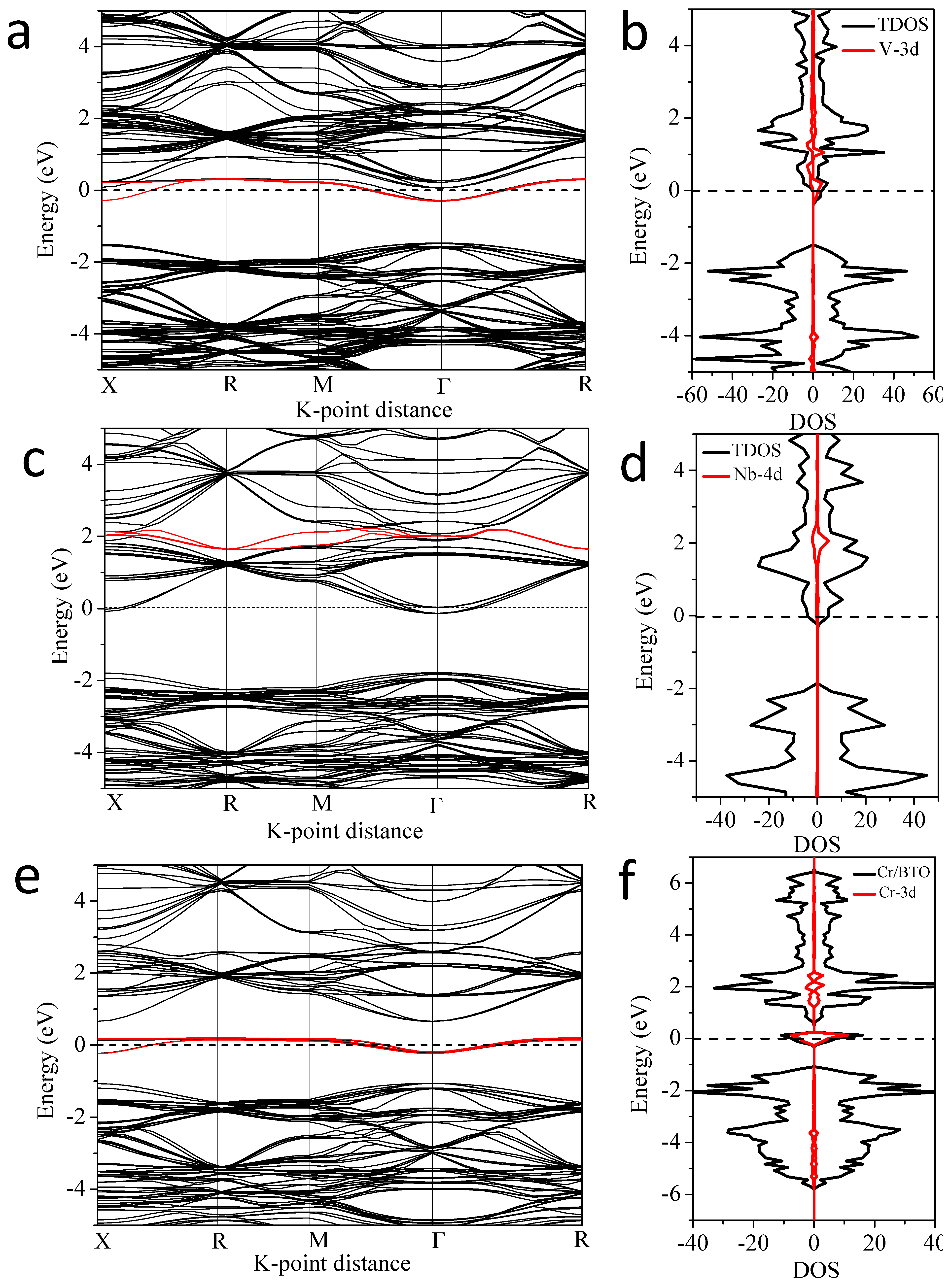
3.3. Electronic Properties
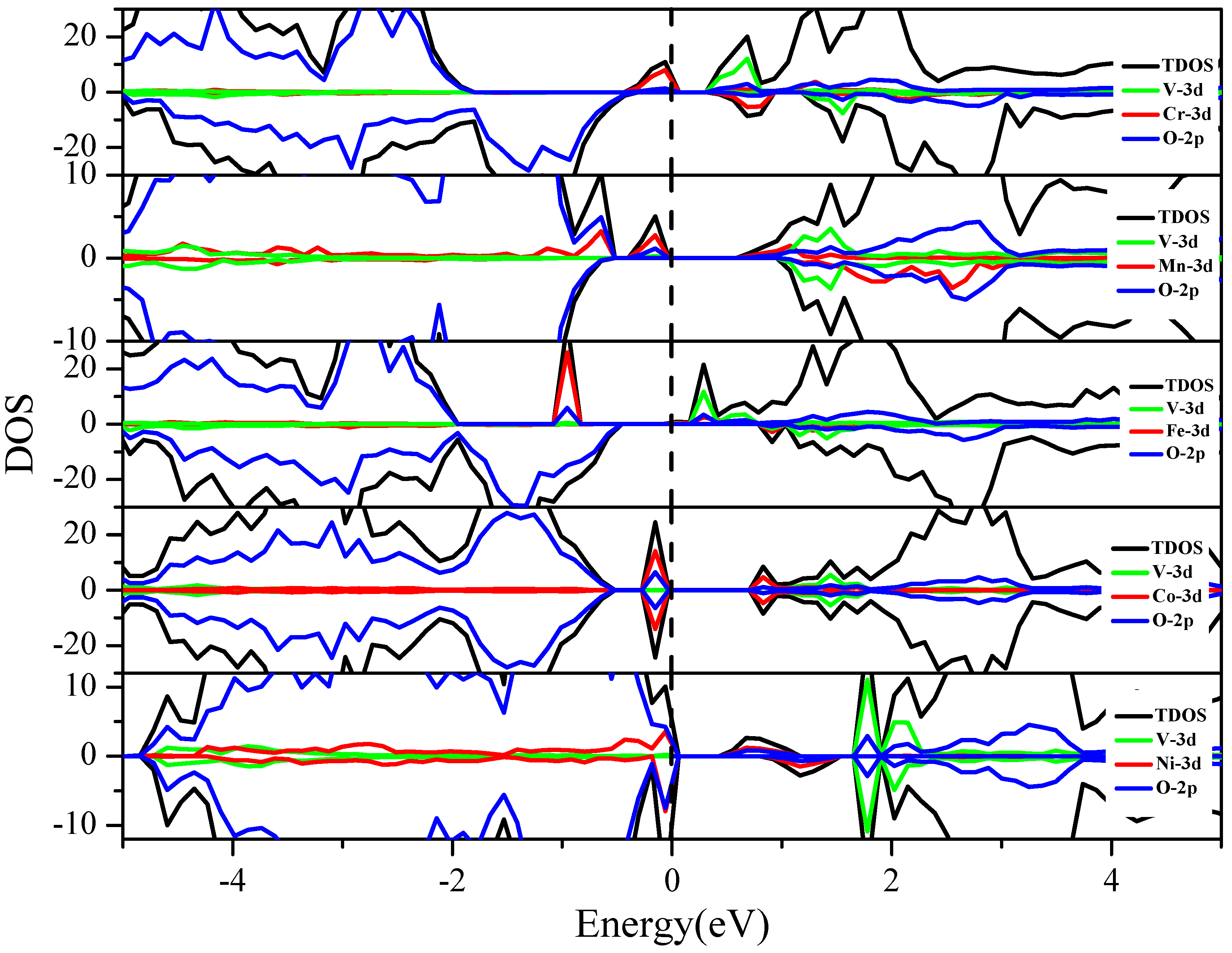
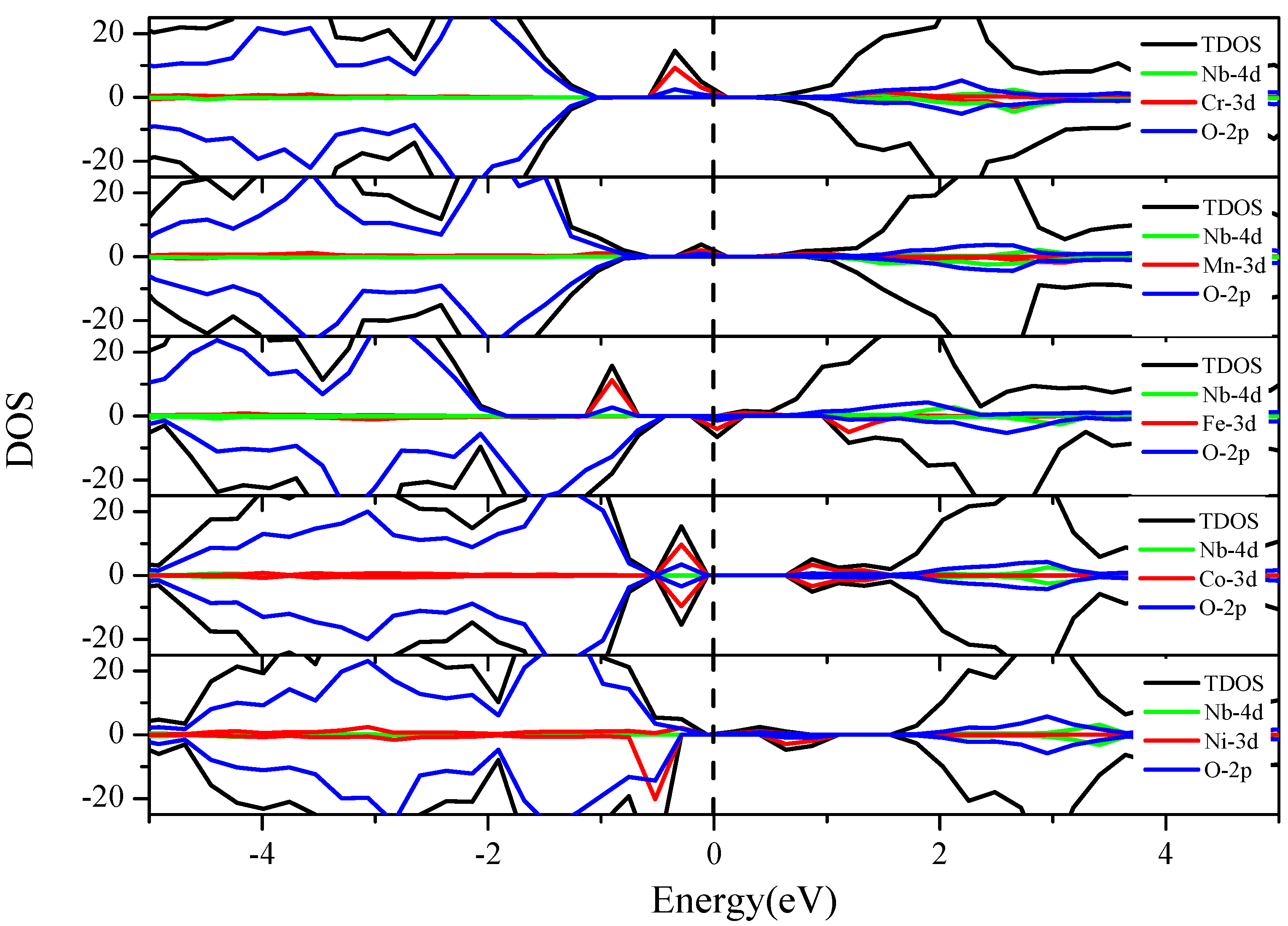
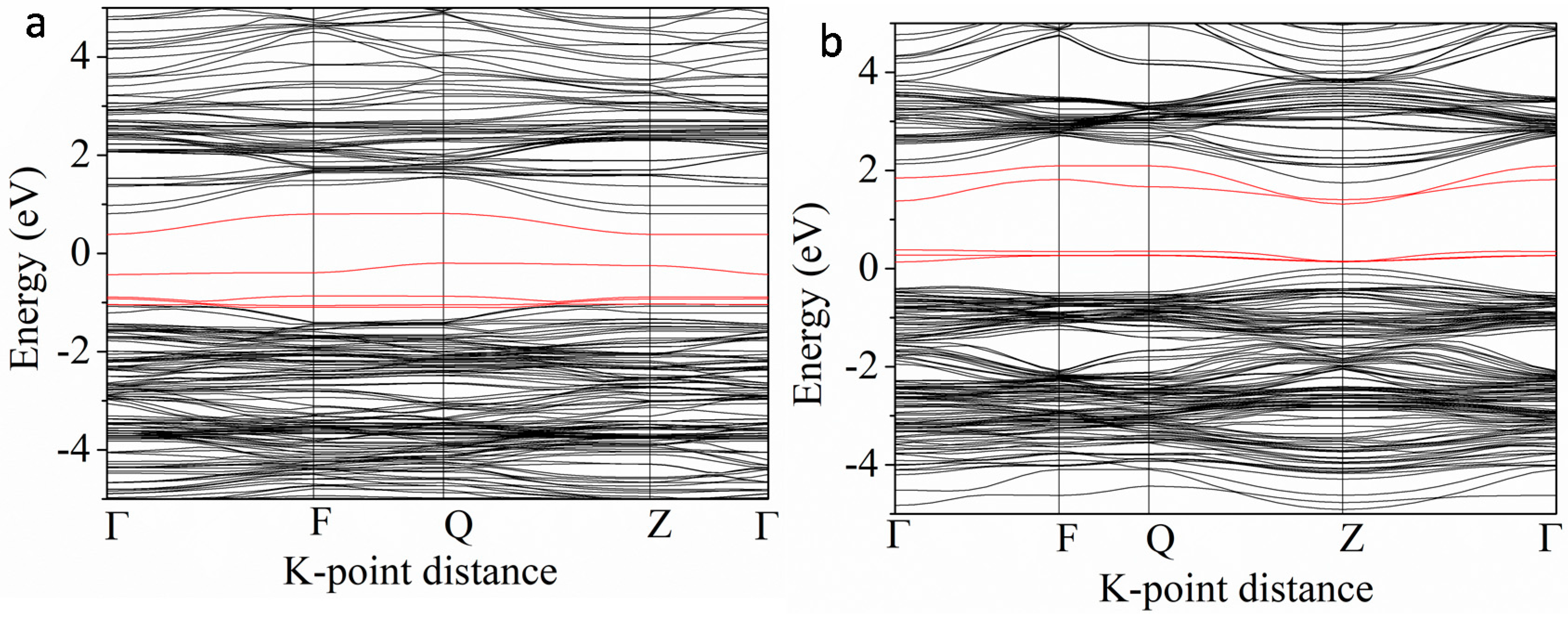
3.3.1. V and 3d M″ Co-Doping (Cr, Mn, Fe, Co and Ni)
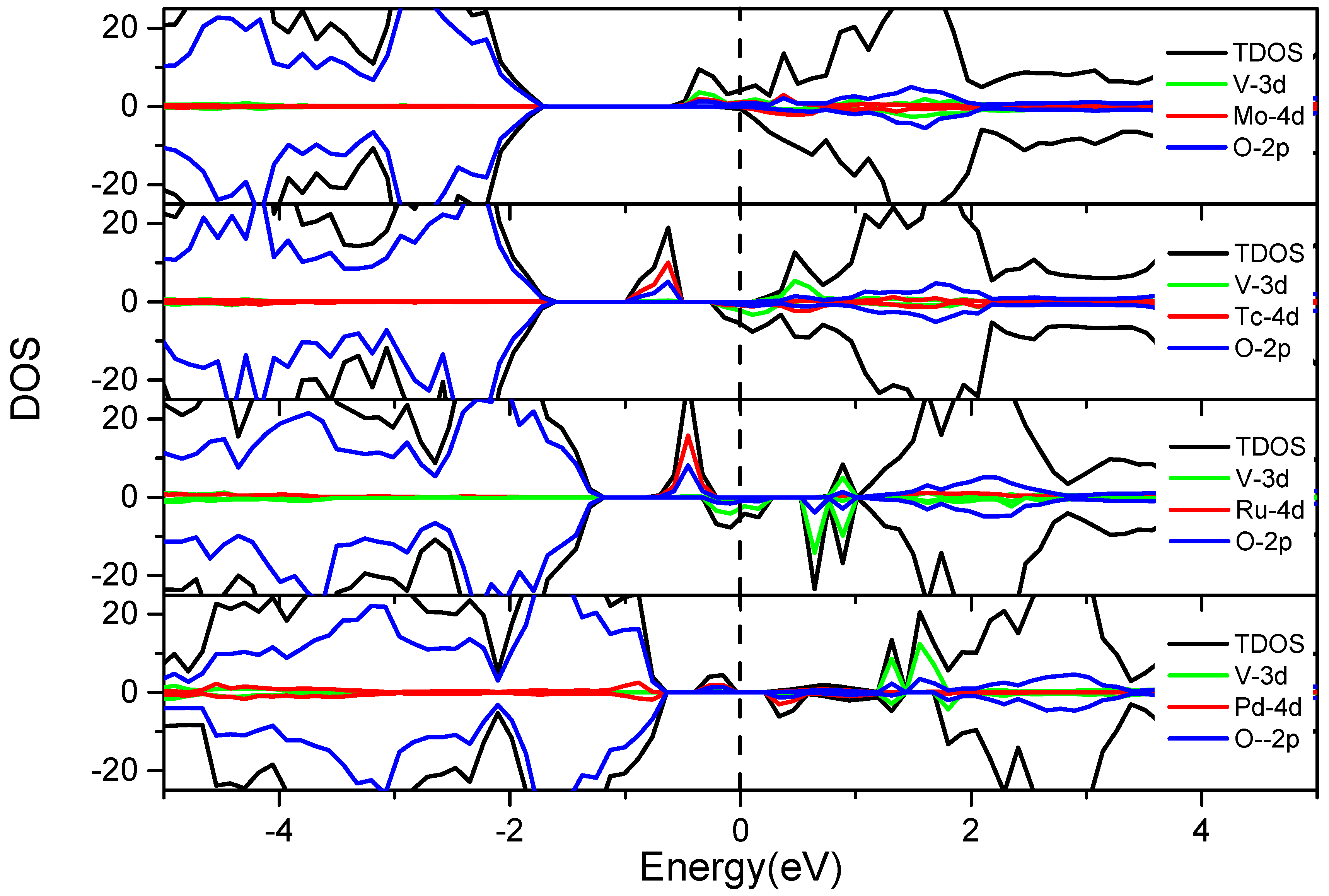
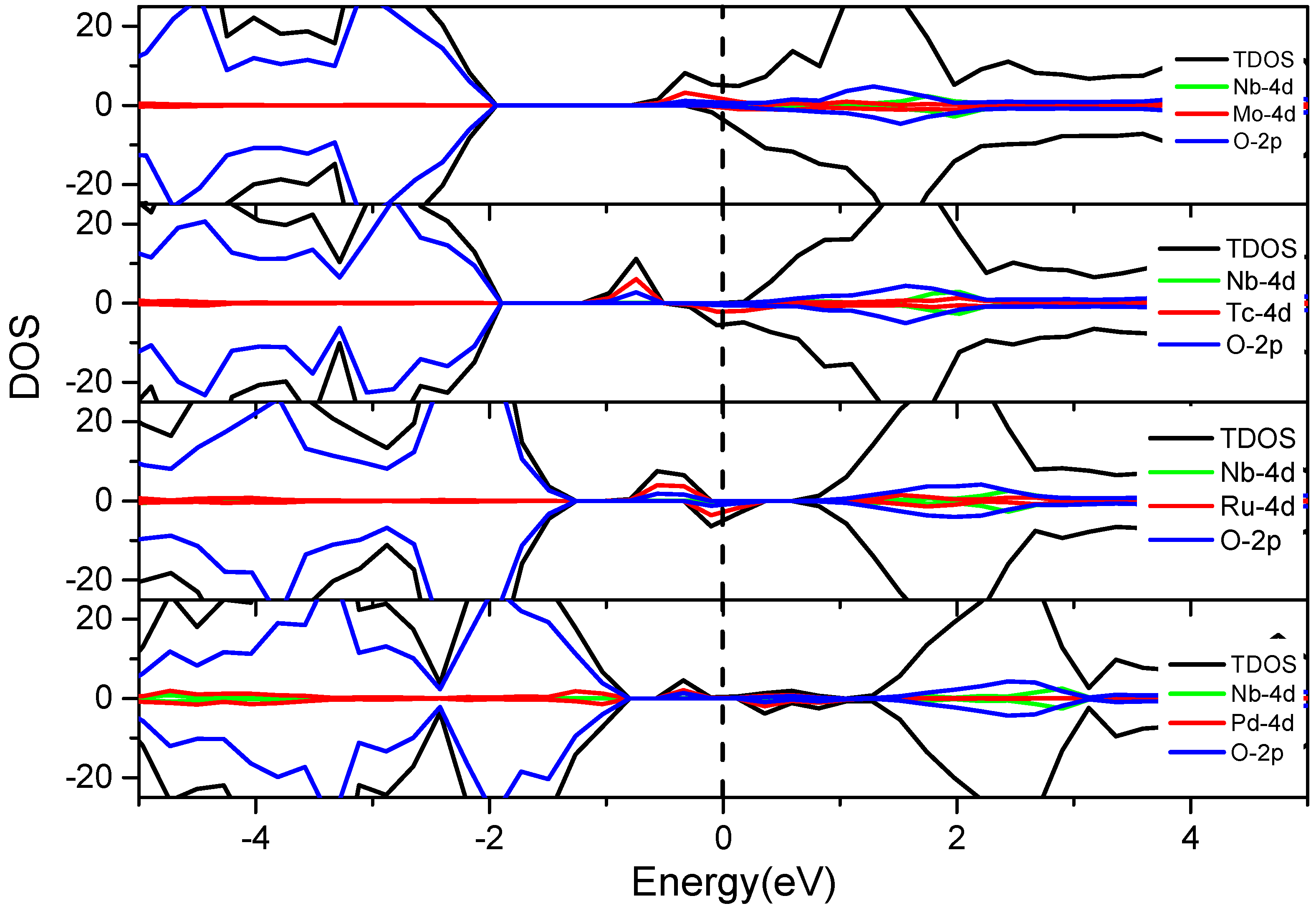
3.3.2. V and 4d M″ Co-Doping (Mo, Tc, Ru and Pd)
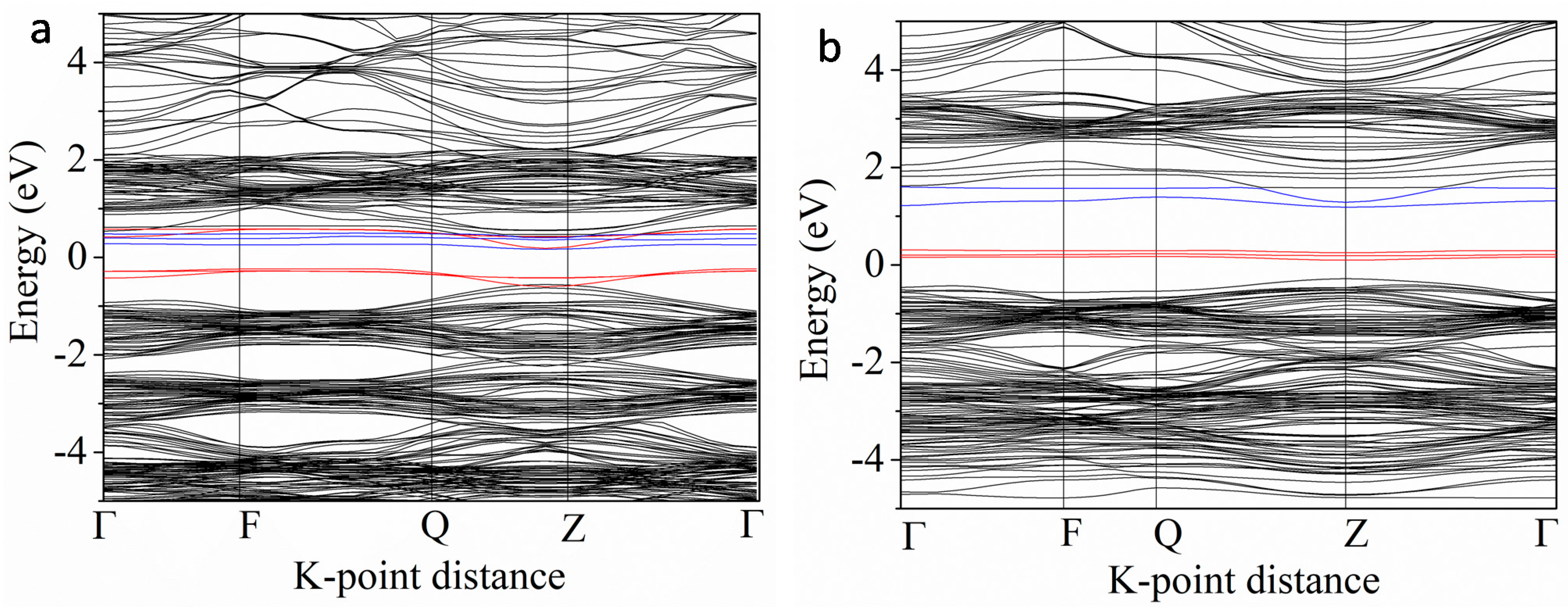
3.3.3. Nb and 3d M″ Co-Doping (Cr, Mn, Fe, Co and Ni)
3.3.4. Nb-4d M″ Co-Doping (Mo, Tc, Ru and Pd)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.H.; Im, S.H.; Heo, J.H.; Mandal, T.N.; Seok, S.I. Chemical management for colorful, efficient, and stable inorganic-organic hybrid nanostructured solar cells. Nano Lett. 2013, 13, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhu, Q.; Qiu, M.; Yang, A.; Wang, Y.; Zhu, D.; Wang, J.; Yang, R. High-performance inverted planar perovskite solar cells without a hole transport layer via a solution process under ambient conditions. J. Mater. Chem. A 2015, 3, 19294–19298. [Google Scholar] [CrossRef]
- Swainson, I.P.; Tucker, M.G.; Wilson, D.J.; Winkler, B.; Milman, V. Pressure response of an organic-inorganic perovskite: Methylammonium lead bromide. Chem. Mater. 2007, 19, 2401–2405. [Google Scholar] [CrossRef]
- Dualeh, A.; Gao, P.; Seok, S., II; Nazeeruddin, M.K.; Grätzel, M. Thermal behavior of methylammonium lead-trihalide perovskite photovoltaic light harvesters. Chem. Mater. 2014, 26, 6160–6164. [Google Scholar] [CrossRef]
- Lopez-Varo, P.; Bertoluzzi, L.; Bisquert, J.; Alexe, M.; Coll, M.; Huang, J.; Jimenez-Tejada, J.A.; Kirchartz, T.; Nechache, R.; Rosei, F.; et al. Physical aspects of ferroelectric semiconductors for photovoltaic solar energy conversion. Phys. Rep. 2016, 653, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Wu, H.; Li, G.; Huang, Q.; Yang, C.; Huang, F.; Liao, F.; Lin, J. New high Tc multiferroics KBiFe2O5 with narrow bandgap and promising photovoltaic effect. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Grinberg, I.; West, D.V.; Torres, M.; Gou, G.; Stein, D.M.; Wu, L.; Chen, G.; Gallo, E.M.; Akbashev, A.R.; Davies, P.K.; et al. Perovskite oxides for visible-light-absorbing ferroelectric and photovoltaic materials. Nature 2013, 503, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schultz, A.M.; Salvador, P.A.; Rohrer, G.S. Spatially selective visible light photocatalytic activity of TiO2/BiFeO3 heterostructures. J. Mater. Chem. 2011, 21, 4168–4174. [Google Scholar] [CrossRef]
- Dharmadhikari, V.S.; Grannemann, W.W. Photovoltaic properties of ferroelectric BaTiO3 thin films rf sputter deposited on silicon. J. Appl. Phys. 1982, 53, 8988–8992. [Google Scholar] [CrossRef]
- Zenkevich, A.; Matveyev, Y.; Maksimova, K.; Gaynutdinov, R.; Tolstikhina, A.; Fridkin, V. Giant bulk photovoltaic effect in thin ferroelectric BaTiO3 films. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 90. [Google Scholar] [CrossRef]
- Choi, T.; Lee, S.; Choi, Y.J.; Kiryukhin, V.; Cheong, S.W. Switchable ferroelectric diode and photovoltaic effect in BiFeO3. Science 2009, 324, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yao, K.; Liang, Y.C. Bulk photovoltaic effect at visible wavelength in epitaxial ferroelectric BiFeO3 thin films. Adv. Mater. 2010, 22, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Seidel, J.; Byrnes, S.J.; Shafer, P.; Yang, C.H.; Rossell, M.D.; Yu, P.; Chu, Y.H.; Scott, J.F.; Ager, J.W.; et al. Above-bandgap voltages from ferroelectric photovoltaic devices. Nat. Nanotechnol. 2010, 5, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechache, R.; Harnagea, C.; Li, S.; Cardenas, L.; Huang, W.; Chakrabartty, J.; Rosei, F. Bandgap tuning of multiferroic oxide solar cells. Nat. Photonics 2014, 9, 61–67. [Google Scholar] [CrossRef]
- Glass, A.M.; Von Der Linde, D.; Negran, T.J. High-voltage bulk photovoltaic effect and the photorefractive process in LiNbO3. Appl. Phys. Lett. 1974, 25, 233–235. [Google Scholar] [CrossRef]
- Peithmann, K.; Hukriede, J.; Buse, K.; Krätzig, E. Photorefractive properties of crystals doped by copper diffusion. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 61, 4615–4620. [Google Scholar] [CrossRef]
- Festl, H.G.; Hertel, P.; Krätzig, E.; von Baltz, R. Investigations of the Photovoltaic Tensor in Doped LiNbO3. Phys. Status Solidi 1982, 113, 157–164. [Google Scholar] [CrossRef]
- Schirmer, O.F.; Imlau, M.; Merschjann, C. Bulk photovoltaic effect of LiNbO3:Fe and its small-polaron-based microscopic interpretation. Phys. Rev. B 2011, 83, 165106. [Google Scholar] [CrossRef]
- Nonaka, K.; Akiyama, M.; Xu, C.N.; Hagio, T.; Komatsu, M.; Takase, A. Enhanced photovoltaic response in lead lanthanum zirconate-titanate ceramics with a-site deficient composition for photostrictor application. Jpn. J. Appl. Phys. Part 1 2000, 39, 5144–5145. [Google Scholar] [CrossRef]
- Qin, M.; Yao, K.; Liang, Y.C. High efficient photovoltaics in nanoscaled ferroelectric thin films. Appl. Phys. Lett. 2008, 93. [Google Scholar] [CrossRef]
- Qin, M.; Yao, K.; Liang, Y.C. Photovoltaic mechanisms in ferroelectric thin films with the effects of the electrodes and interfaces. Appl. Phys. Lett. 2009, 95. [Google Scholar] [CrossRef]
- Fan, Z.; Sun, K.; Wang, J. Perovskites for photovoltaics: A combined review of organic–inorganic halide perovskites and ferroelectric oxide perovskites. J. Mater. Chem. A 2015, 3, 18809. [Google Scholar] [CrossRef]
- Wang, D.; Ye, J.; Kako, T.; Kimura, T. Photophysical and photocatalytic properties of SrTiO3 doped with Cr cations on different sites. J. Phys. Chem. B 2006, 110, 15824–15830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Sun, S.; Jiang, W.; Miao, X.; Zhao, Z.; Zhang, X.; Qu, D.; Zhang, D.; Li, D.; Sun, Z. A Novel Perovskite SrTiO3-Ba2FeNbO6 Solid Solution for Visible Light Photocatalytic Hydrogen Production. Adv. Energy Mater. 2017, 7, 1600932. [Google Scholar] [CrossRef]
- Yang, F.; Lin, S.; Yang, L.; Liao, J.; Chen, Y.; Wang, C.Z. First-principles investigation of metal-doped cubic BaTiO3. Mater. Res. Bull. 2017, 96, 372–378. [Google Scholar] [CrossRef]
- Wei, W.; Dai, Y.; Guo, M.; Yu, L.; Huang, B. Density Functional Characterization of the Electronic Structure and Optical Properties of N-Doped, La-Doped, and N/La-Codoped SrTiO3. J. Phys. Chem. C 2009, 113, 15046–15050. [Google Scholar] [CrossRef]
- Li, W.; Wang, G.; Chen, C.; Liao, J.; Li, Z. Enhanced Visible Light Photocatalytic Activity of ZnO Nanowires Doped with Mn2+ and Co2+ Ions. Nanomaterials 2017, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Kato, H.; Kudo, A. H2 evolution from an aqueous methanol solution on SrTiO3 photocatalysts codoped with chromium and tantalum ions under visible light irradiation. J. Photochem. Photobiol. A Chem. 2004, 163, 181–186. [Google Scholar] [CrossRef]
- Kako, T.; Yao, W.; Ye, J. Preparation and characterization of visible light sensitive Fe- and Ta-codoped TiO2 photocatalyst. J. Mater. Res. 2010, 25, 110–116. [Google Scholar] [CrossRef]
- Miyauchi, M.; Takashio, M.; Tobimatsu, H. Photocatalytic Activity of SrTiO3 Codoped with Nitrogen and Lanthanum under Visible Light Illumination. Langmuir 2004, 20, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented—Wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Perdew, Burke, and Ernzerhof Reply. Phys. Rev. Lett. 1998, 80, 891. [Google Scholar] [CrossRef]
- Skulason, E.; Karlberg, G.S.; Rossmeisl, J.; Bligaard, T.; Greeley, J.; Jonsson, H.; Nørskov, J.K. Density functional theory calculations for the hydrogen evolution reaction in an electrochemical double layer on the Pt(111) electrode. Phys. Chem. Chem. Phys. 2007, 9, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Zhang, F.; Zhang, S.; Wang, X.; Shao, T. Electronic and Magnetic Properties of Ni-Doped Zinc-Blende ZnO: A First-Principles Study. Nanomaterials 2018, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Corà, F.; Catlow, C.R.A. QM investigations on perovskite-structured transition metal oxides: Bulk, surfaces and interfaces. Faraday Discuss. 1999, 114, 421–442. [Google Scholar] [CrossRef]
- Xiao, Z.; Meng, W.; Wang, J.; Yan, Y. Thermodynamic Stability and Defect Chemistry of Bismuth-Based Lead-Free Double Perovskites. ChemSusChem 2016, 9, 2628–2633. [Google Scholar] [CrossRef] [PubMed]
- Petraru, A.I. Optical and Electro-Optical Properties of BaTiO3 Thin Films and Mach-Zehnder Waveguide Modulators. Doctoral Thesis, Forschungszentrum Jülich, Jülich, Germany, 2003. [Google Scholar]
- Weng, B.; Xiao, Z.; Meng, W.; Grice, C.R.; Poudel, T.; Deng, X.; Yan, Y. Bandgap Engineering of Barium Bismuth Niobate Double Perovskite for Photoelectrochemical Water Oxidation. Adv. Energy Mater. 2017, 7. [Google Scholar] [CrossRef]
- Mamouni, N.; El Kenz, A.; Ez-Zahraouy, H.; Loulidi, M.; Benyoussef, A.; Bououdina, M. Stabilization of ferromagnetism in (Cr, V) co-doped ZnO diluted magnetic semiconductors. J. Magn. Magn. Mater. 2013, 340, 86–90. [Google Scholar] [CrossRef]
- Jungwirth, T.; Sinova, J.; Mašek, J.; Kučera, J.; MacDonald, A.H. Theory of ferromagnetic (III,Mn)V semiconductors. Rev. Mod. Phys. 2006, 78, 809–864. [Google Scholar] [CrossRef] [Green Version]
- Olibet, S.; Vallat-Sauvain, E.; Ballif, C. Model for a-Si:H/c-Si interface recombination based on the amphoteric nature of silicon dangling bonds. Phys. Rev. B 2007, 76, 35326. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, Y.; Zhu, S.; Wang, F. Copper doping in titanium oxide catalyst film prepared by dc reactive magnetron sputtering. Catal. Today 2004, 93–95, 589–594. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, F.; Zhang, J.; Chen, H.; Anpo, M. Fe3+-TiO2 photocatalysts prepared by combining sol–gel method with hydrothermal treatment and their characterization. J. Photochem. Photobiol. A Chem. 2006, 180, 196–204. [Google Scholar] [CrossRef]
- Hur, S.G.; Kim, T.W.; Hwang, S.; Park, H.; Choi, W.; Kim, S.J.; Choy, J. Synthesis of New Visible Light Active Photocatalysts of Ba(In1/3Pb1/3M′1/3)O3 (M′ = Nb, Ta): A Bandgap Engineering Strategy Based on Electronegativity of a Metal Component. J. Phys. Chem. B 2005, 109, 17346. [Google Scholar] [CrossRef]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.; Yang, L.; Ai, C.; Xie, P.; Lin, S.; Wang, C.-Z.; Lu, X. Tailoring Bandgap of Perovskite BaTiO3 by Transition Metals Co-Doping for Visible-Light Photoelectrical Applications: A First-Principles Study. Nanomaterials 2018, 8, 455. https://doi.org/10.3390/nano8070455
Yang F, Yang L, Ai C, Xie P, Lin S, Wang C-Z, Lu X. Tailoring Bandgap of Perovskite BaTiO3 by Transition Metals Co-Doping for Visible-Light Photoelectrical Applications: A First-Principles Study. Nanomaterials. 2018; 8(7):455. https://doi.org/10.3390/nano8070455
Chicago/Turabian StyleYang, Fan, Liang Yang, Changzhi Ai, Pengcheng Xie, Shiwei Lin, Cai-Zhuang Wang, and Xihong Lu. 2018. "Tailoring Bandgap of Perovskite BaTiO3 by Transition Metals Co-Doping for Visible-Light Photoelectrical Applications: A First-Principles Study" Nanomaterials 8, no. 7: 455. https://doi.org/10.3390/nano8070455