
Strong and Elastic Hydrogels from Dual-Crosslinked Composites Composed of Glycol Chitosan and Amino-Functionalized Bioactive Glass Nanoparticles

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
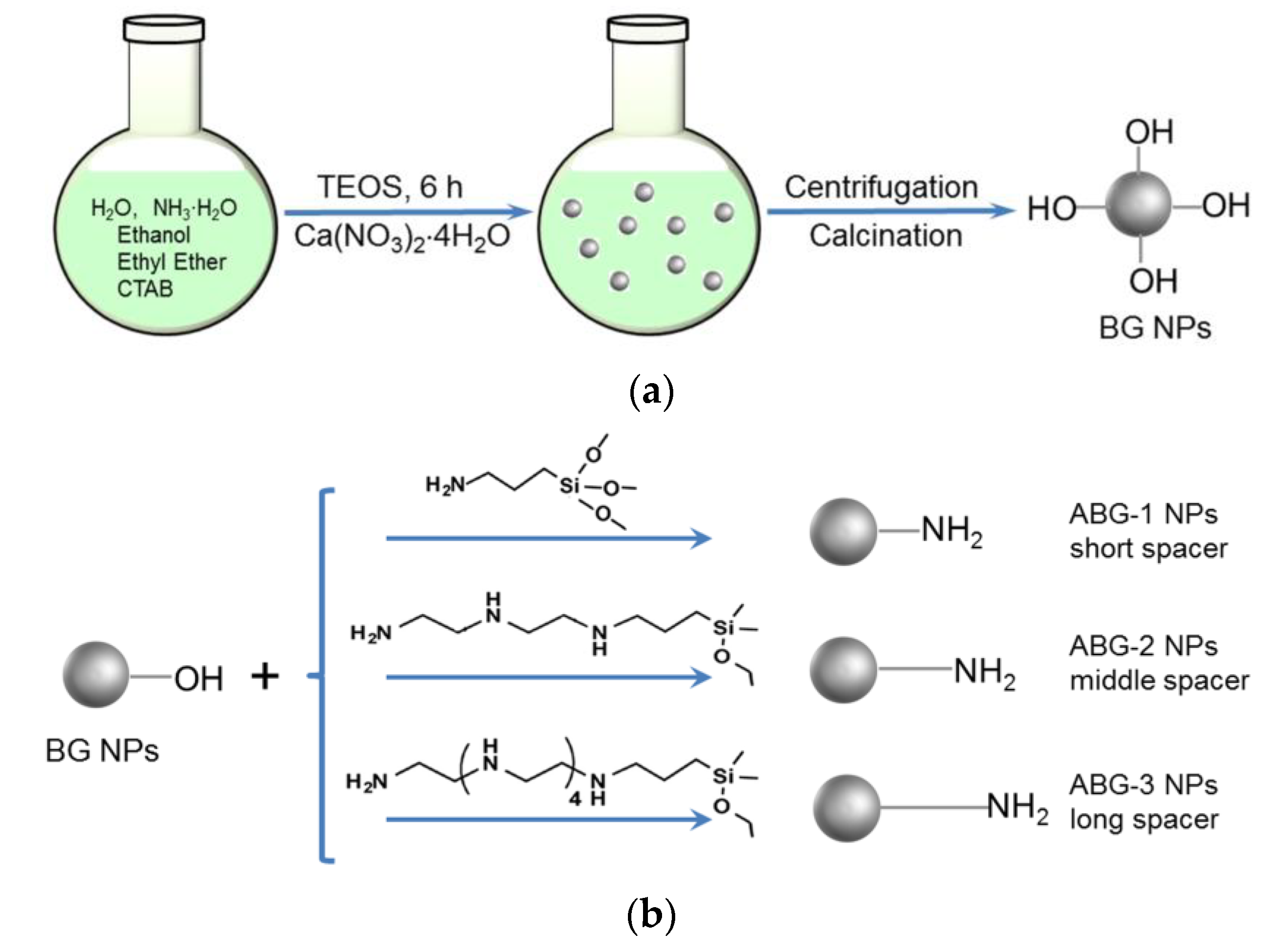
2.2. Synthesis of Bioglass Nanoparticles
2.3. Amino Functionalization of Bioglass Nanoparticles
2.4. Characterization
2.5. Preparation of Hydrogels
2.6. Rheological Measurements
2.7. Cell Culture
2.8. Statistical Analysis
3. Results and Discussion
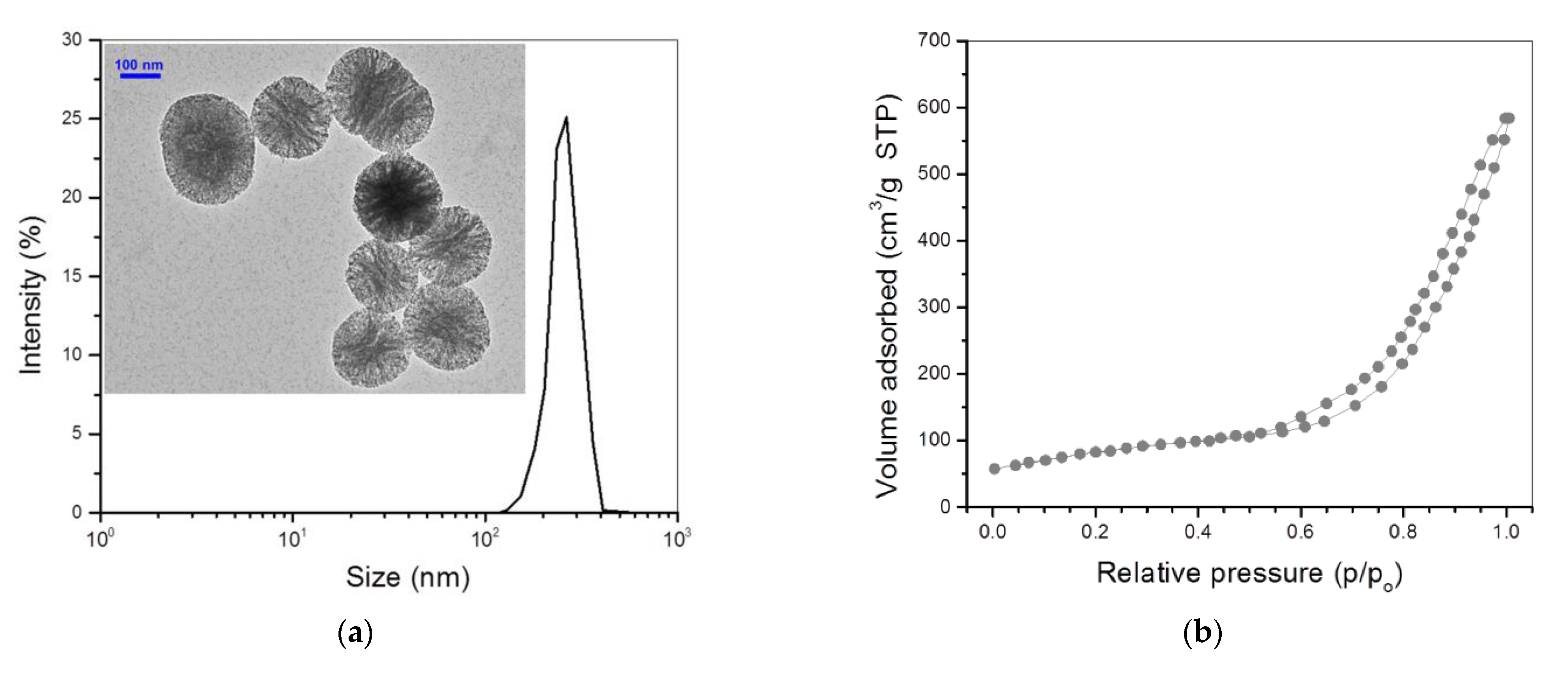
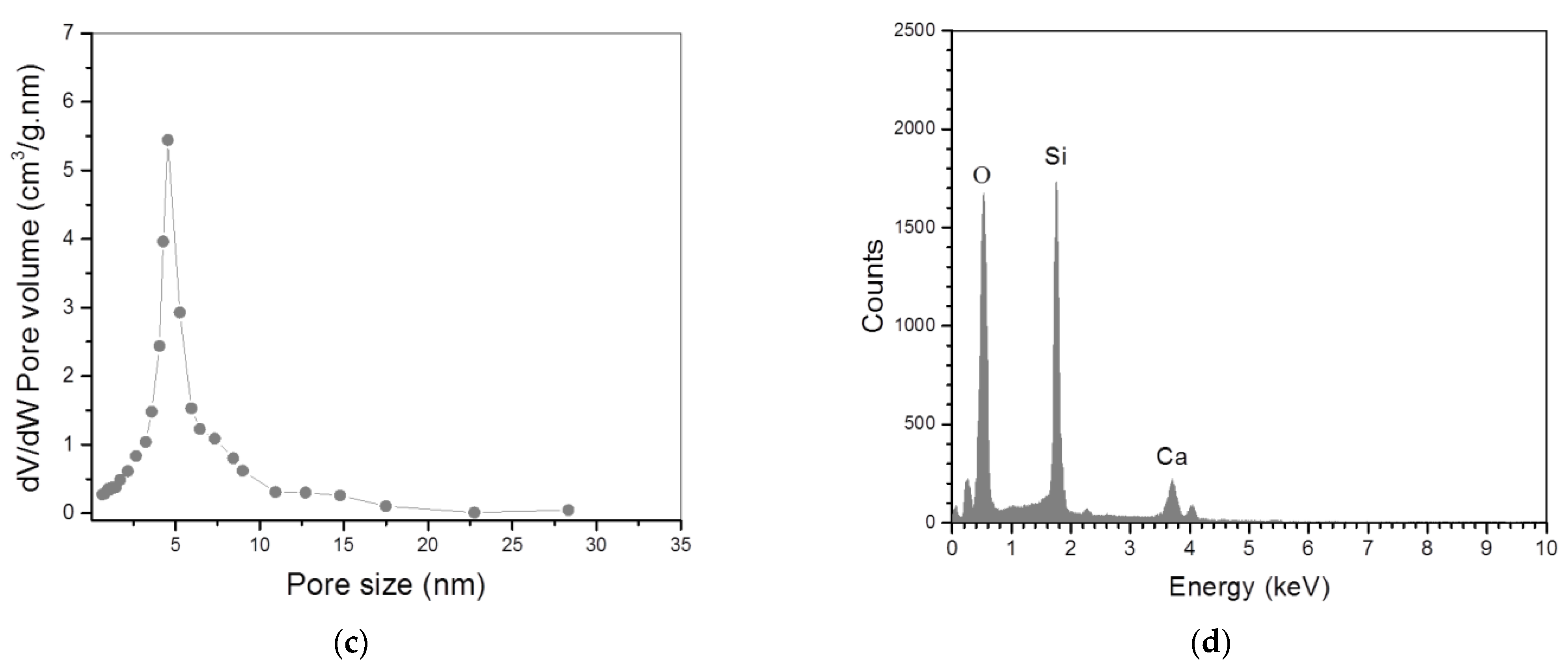
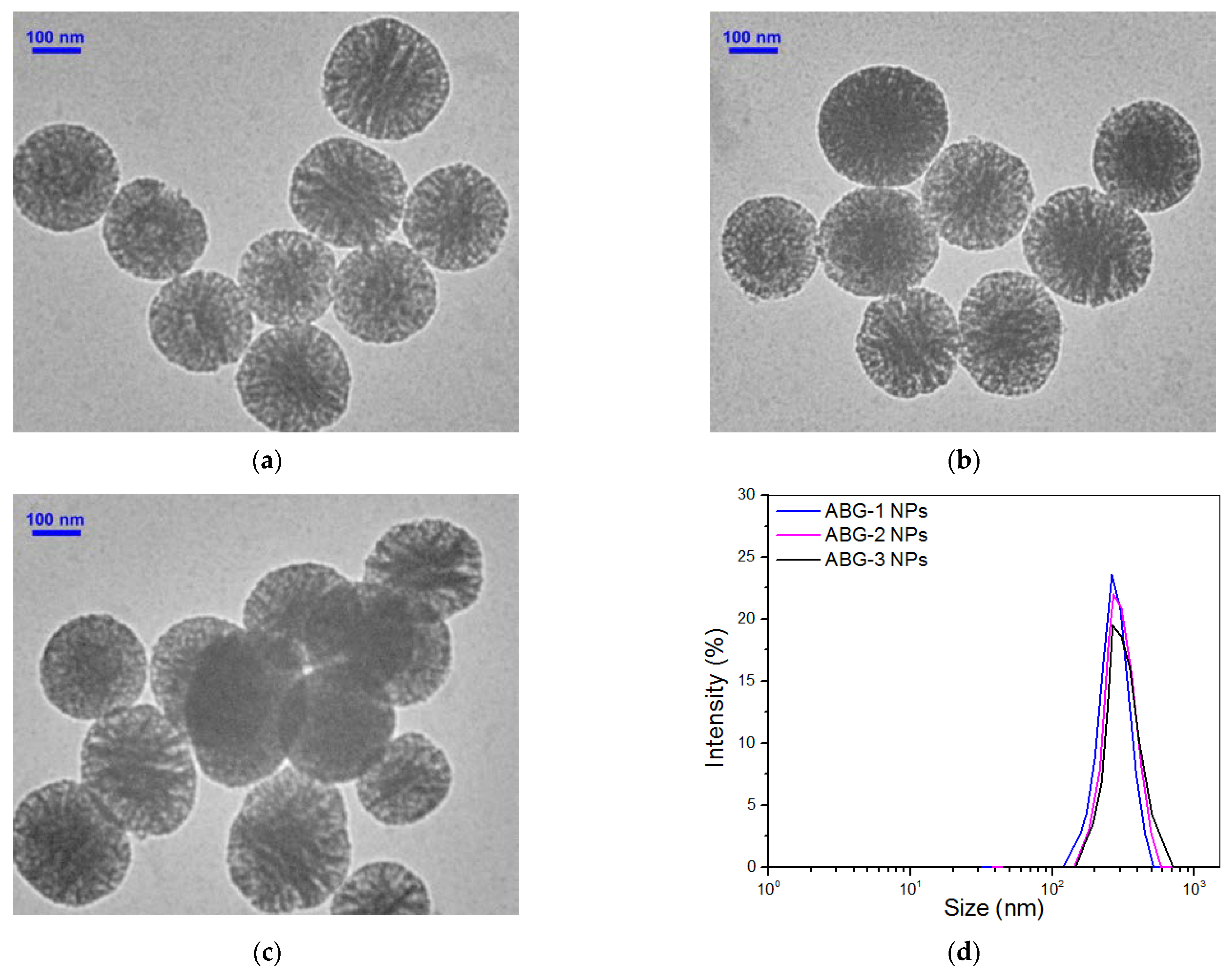
3.1. Characterization of Bioglass Nanoparticles
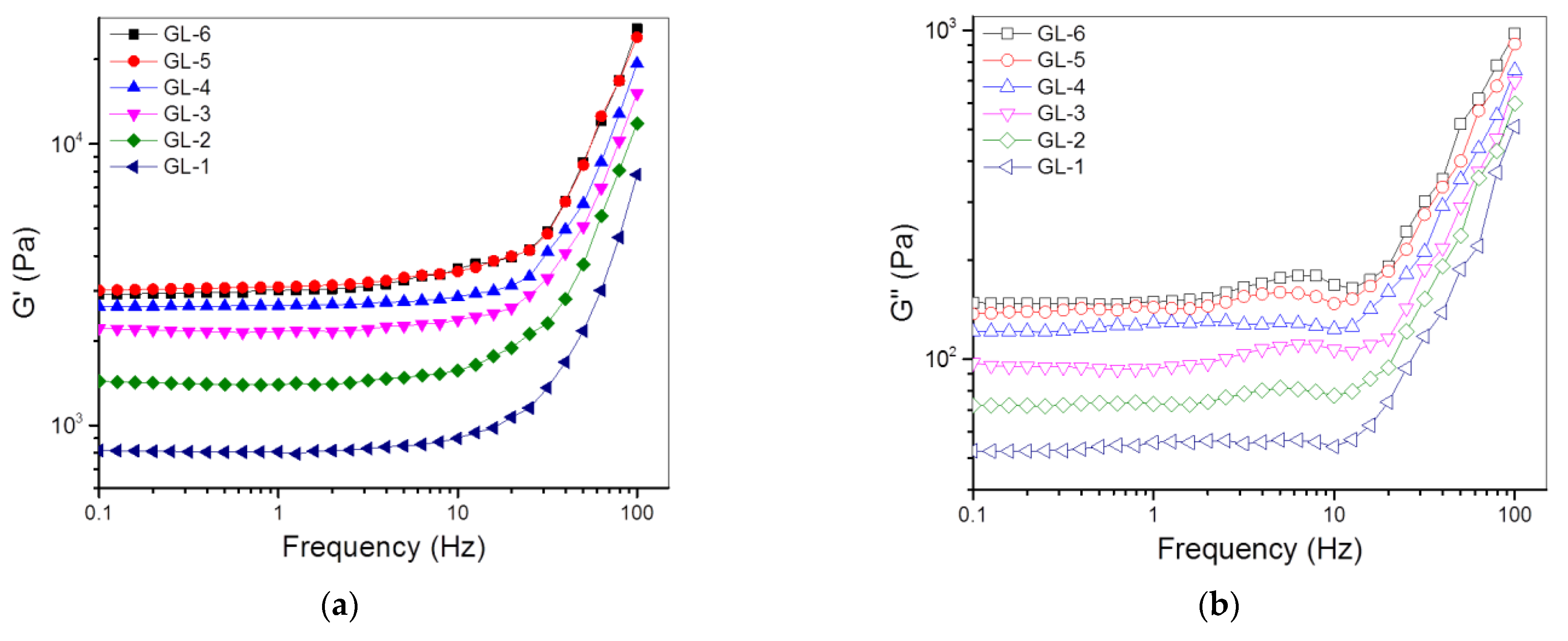
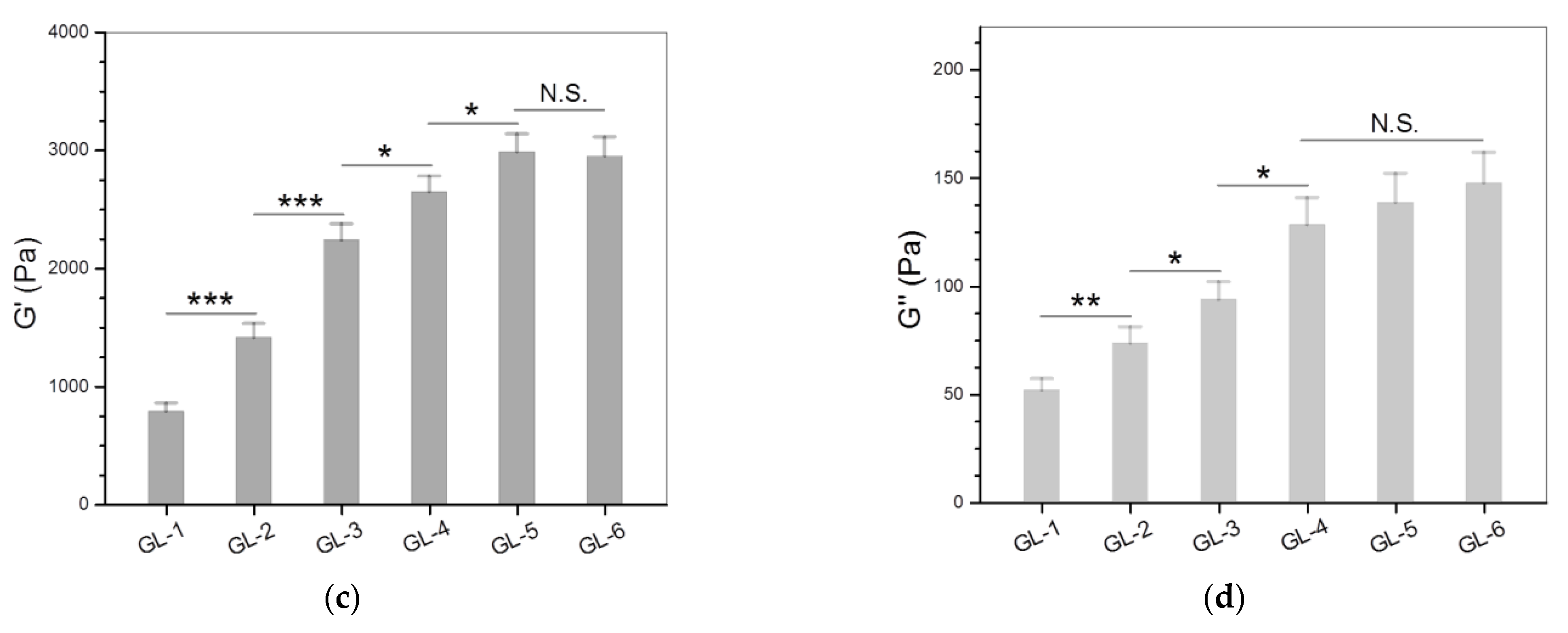
3.2. Single-Crosslinked Hydrogels
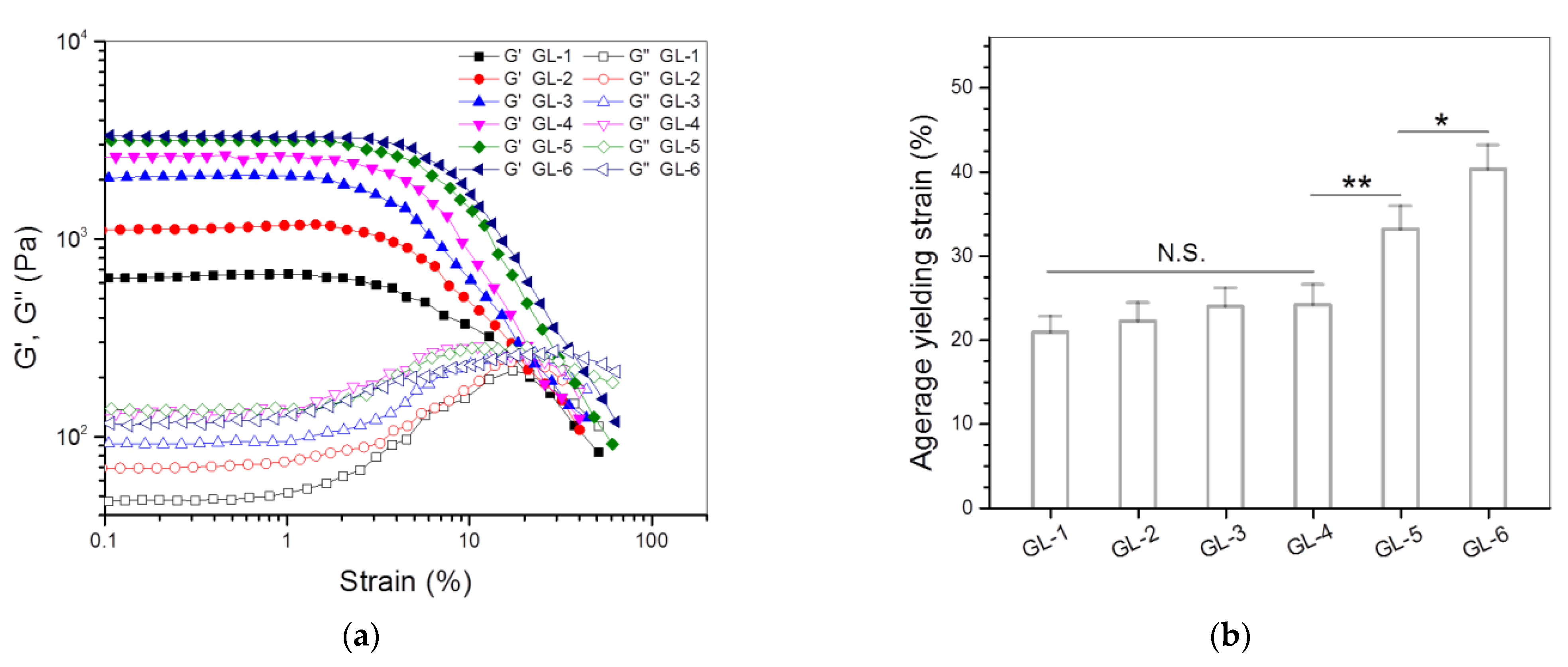
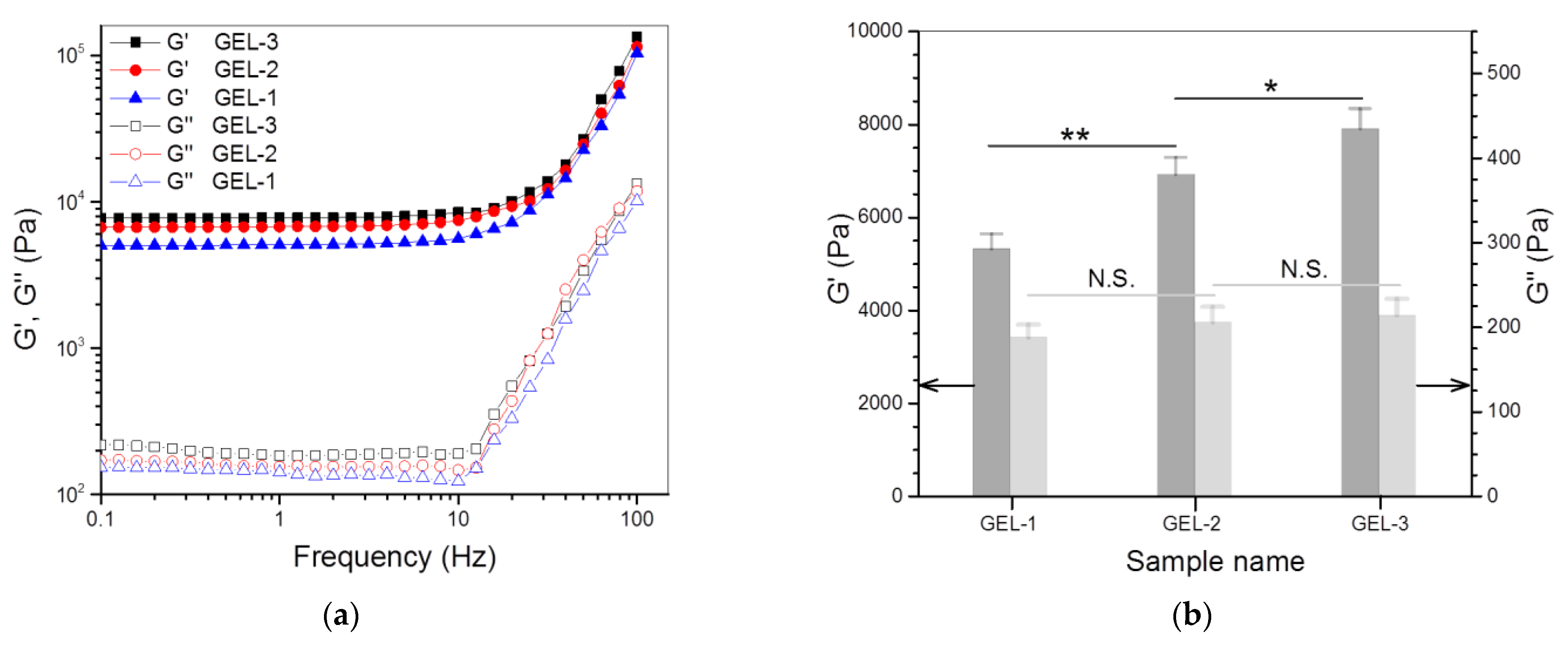
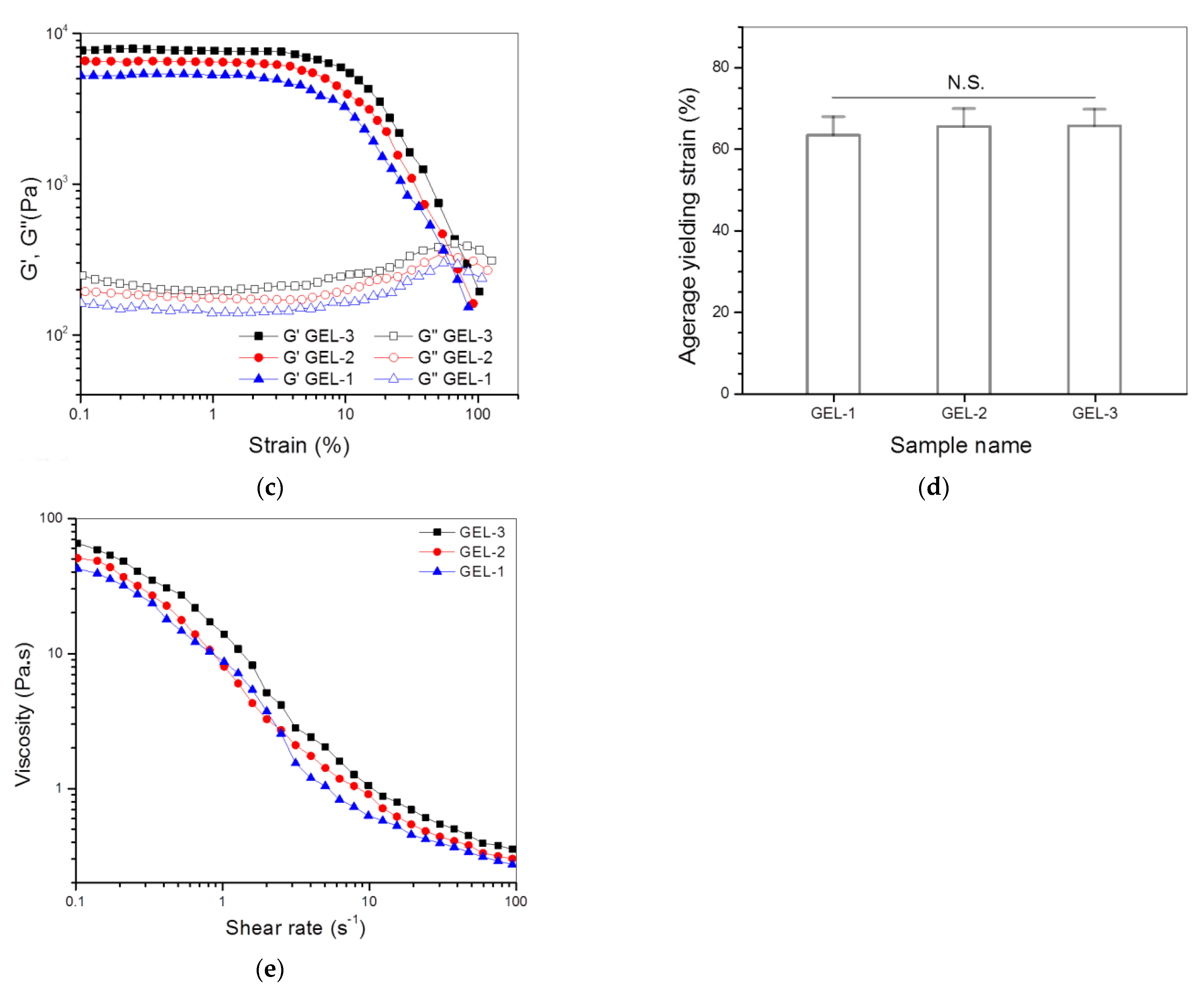
3.3. Dual-Crosslinked Hydrogels
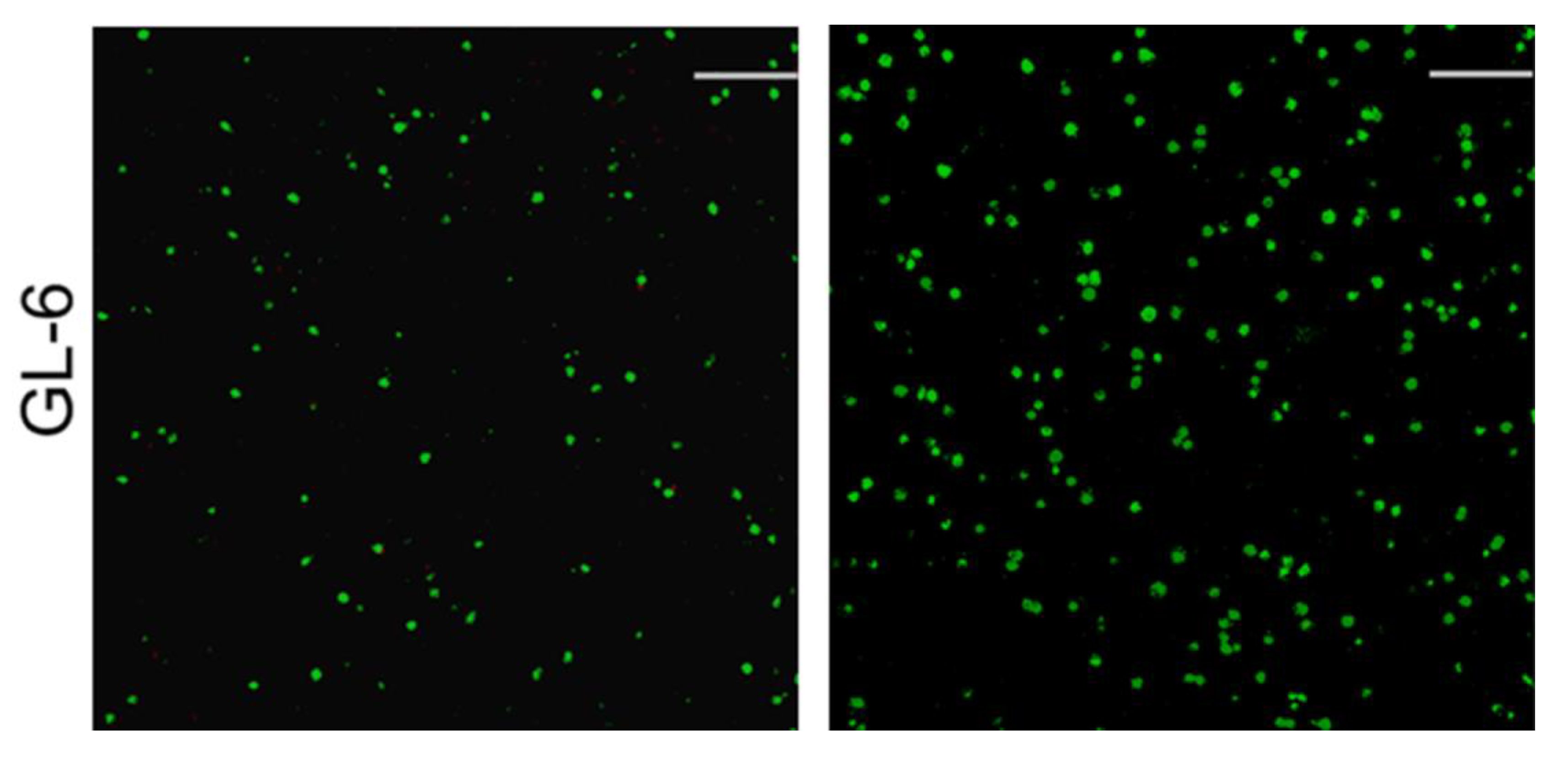
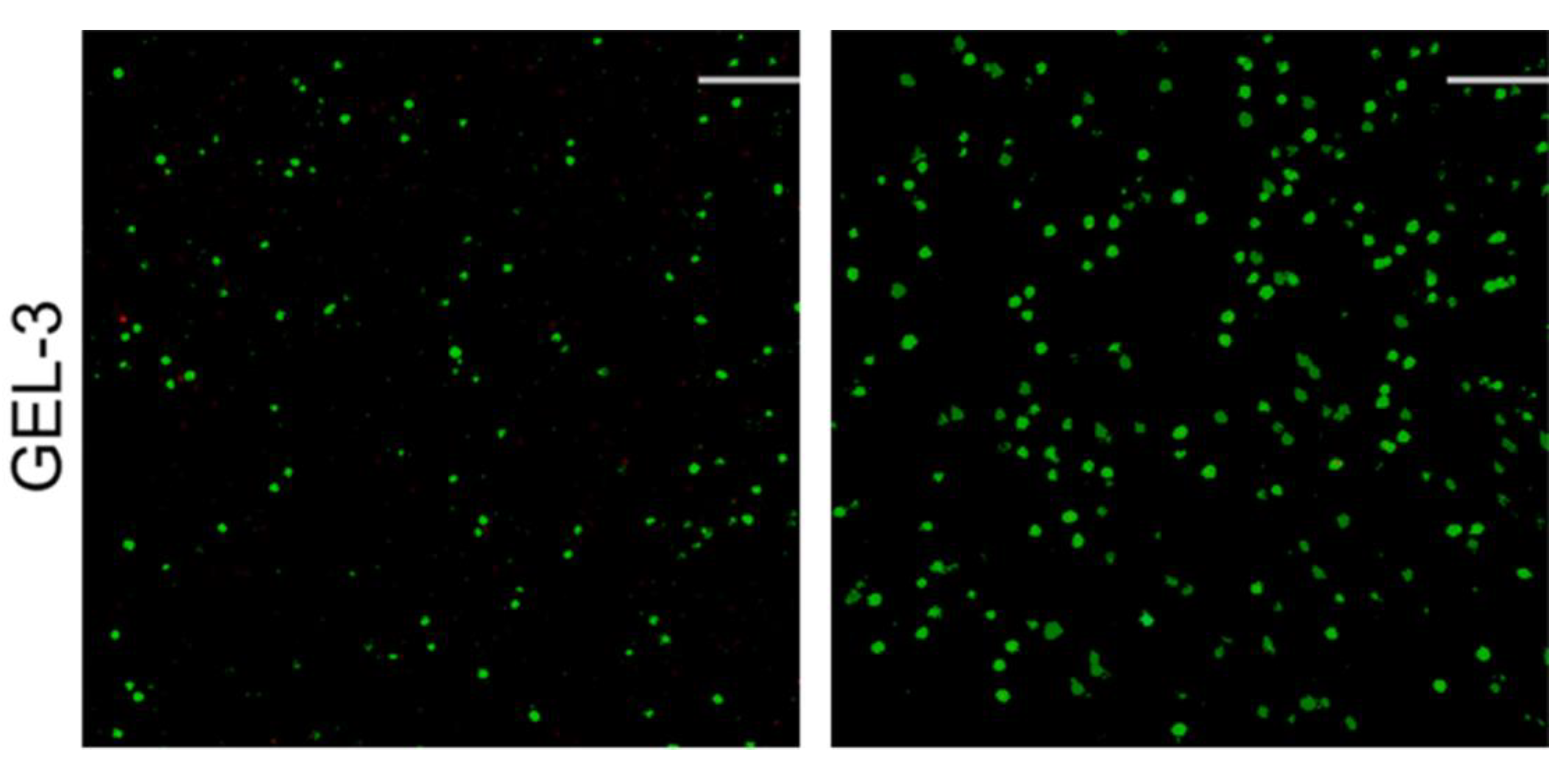
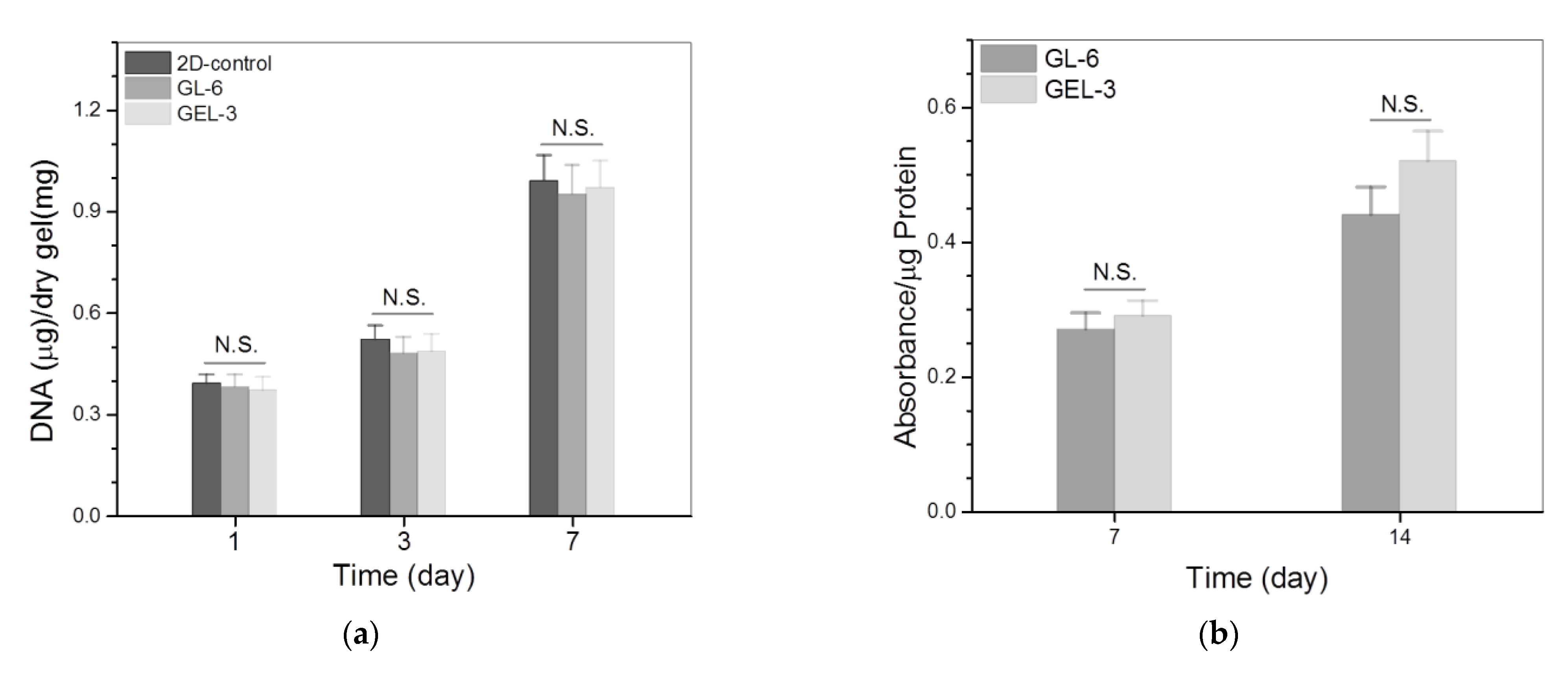
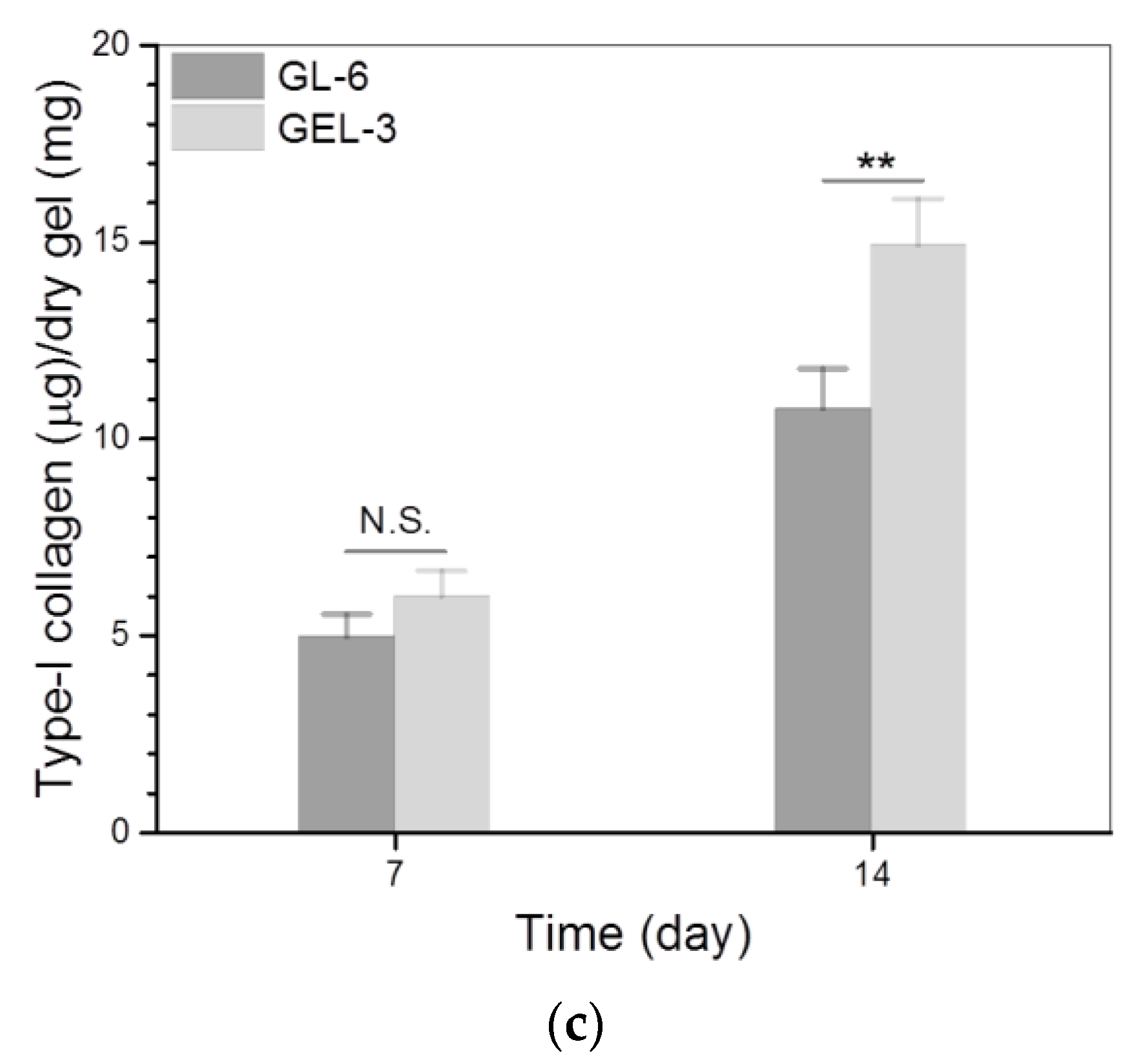
3.4. Cell Growth and Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharma, S.; Tiwari, S. A review on biomacromolecular hydrogel classification and its applications. Int. J. Biol. Macromol. 2020, 163, 737–747. [Google Scholar] [CrossRef]
- Tan, H.; Marra, K.G. Injectable, biodegradable hydrogels for tissue engineering applications. Materials 2010, 3, 1746–1767. [Google Scholar] [CrossRef]
- Couto, D.S.; Hong, Z.; Mano, J.F. Development of bioactive and biodegradable chitosan-based injectable systems containing bioactive glass nanoparticles. Acta Biomater. 2009, 5, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kretlow, J.D.; Klouda, L.; Mikos, A.G. Injectable matrices and scaffolds for drug delivery in tissue engineering. Adv. Drug Deliv. Rev. 2007, 59, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.A.; Chen, R.; Veen, T.; Bryan, N. Hydrogels for tissue engineering and regenerative medicine. J. Mater. Chem. B 2014, 2, 5319–5338. [Google Scholar] [CrossRef]
- Wu, J.; Liu, J.; Shi, Y.; Wan, Y. Rheological, mechanical and degradable properties of injectable chitosan/silk fibroin/hydroxyapatite/glycerophosphate hydrogels. J. Mech. Behav. Biomed. Mater. 2016, 64, 161–172. [Google Scholar] [CrossRef]
- Lee, E.J.; Kang, E.; Kang, S.W.; Huh, K.M. Thermo-irreversible glycol chitosan/hyaluronic acid blend hydrogel for injectable tissue engineering. Carbohydr. Polym. 2020, 244, 116432. [Google Scholar] [CrossRef]
- Muzzarelli, R.A. Genipin-crosslinked chitosan hydrogels as biomedical and pharmaceutical aids. Carbohydr. Polym. 2009, 77, 1–9. [Google Scholar] [CrossRef]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef]
- Majeti, N.V.; Kumar, R. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar]
- Muzzarelli, R.A. Chitins and chitosans for the repair of wounded skin, nerve, cartilage and bone. Carbohydr. Polym. 2009, 77, 167–182. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Discher, D.E.; Janmey, P.; Wang, Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Chen, H.; Zhu, L.; Zheng, J. Fundamentals of double network hydrogels. J. Mater. Chem. B 2015, 3, 3654–3676. [Google Scholar] [CrossRef]
- Xu, C.; Dai, G.; Hong, Y. Recent advances in high-strength and elastic hydrogels for 3D printing in biomedical applications. Acta Biomater. 2019, 95, 50–59. [Google Scholar] [CrossRef]
- Li, J.; Suo, Z.; Vlassak, J.J. Stiff, strong, and tough hydrogels with good chemical stability. J. Mater. Chem. B 2014, 2, 6708–6713. [Google Scholar] [CrossRef]
- El-Rashidy, A.A.; Roether, J.A.; Harhaus, L.; Kneser, U.; Boccaccini, A.R. Regenerating bone with bioactive glass scaffolds: A review of in vivo studies in bone defect models. Acta Biomater. 2017, 62, 1–28. [Google Scholar] [CrossRef]
- Hoppe, A.; Guldal, N.S.; Boccaccini, A.R. A review of the biological response to ionic dissolution products from bioactive glasses and glass-ceramics. Biomaterials 2011, 32, 2757–2774. [Google Scholar] [CrossRef]
- Park, J.E.; Lee, J.Y.; Kim, H.G.; Hahn, T.R.; Paik, Y.S. Isolation and characterization of water-soluble intermediates of blue pigments transformed from geniposide of Gardenia jasminoides. J. Agric. Food Chem. 2002, 50, 6511–6514. [Google Scholar] [CrossRef]
- Sung, H.W.; Huang, R.N.; Huang, L.; Tsai, C.C. In vitro evaluation of cytotoxicity of a naturally occurring crosslinking reagent for biological tissue fixation. J. Biomater. Sci. Polym. Ed. 1999, 10, 63–68. [Google Scholar] [CrossRef]
- Du, X.; He, J. Hierarchically mesoporous silica nanoparticles: Extraction, amino-functionalization, and their multipurpose potentials. Langmuir 2011, 27, 2972–2979. [Google Scholar] [CrossRef]
- Walcarius, A.; Etienne, M.; Lebeau, B. Rate of access to the binding sites in organically modified silicates. 2. ordered mesoporous silicas grafted with amine or thiol groups. Chem. Mater. 2003, 15, 2161–2173. [Google Scholar] [CrossRef]
- Mori, T.; Kubo, T.; Kaya, K.; Hosoya, K. Quantitative evaluations of surface-concentrated amino groups on monolithic-type solid supports prepared by copolymerization method. Colloid Polym. Sci. 2009, 287, 513–523. [Google Scholar] [CrossRef]
- Lu, H.; Ko, Y.G.; Kawazoe, N.; Chen, G. Cartilage tissue engineering using funnel-like collagen sponges prepared with embossing ice particulate templates. Biomaterials 2010, 31, 5825–5835. [Google Scholar] [CrossRef]
- Mahapatra, C.; Singh, R.K.; Kim, J.J.; Patel, K.D.; Perez, R.A.; Jang, J.H.; Kim, H.W. Osteopromoting reservoir of stem cells: Bioactive mesoporous nanocarrier/collagen gel through slow-releasing FGF18 and the activated BMP signaling. ACS Appl. Mater. Interfaces 2016, 8, 27573–27584. [Google Scholar] [CrossRef]
- Clark, A.H.; Ross-Murphy, S.B. Structural and mechanical properties of biopolymer gels. Adv. Polym. Sci. 1987, 83, 57–192. [Google Scholar]
- Lejardi, A.; Hernandez, R.; Criado, M.; Santos, J.I.; Etxeberria, A.; Sarasua, J.R.; Mijangos, C. Novel hydrogels of chitosan and poly(vinyl alcohol)-g-glycolic acid copolymer with enhanced rheological properties. Carbohydr. Polym. 2014, 103, 267–273. [Google Scholar] [CrossRef]
- Kavanagh, G.M.; Ross-Murphy, S.B. Rheological characterization of polymer gels. Prog. Polym. Sci. 1998, 23, 533–562. [Google Scholar] [CrossRef]
- Martinez-Ruvalcaba, A.; Chornet, E.; Rodrigue, D. Viscoelastic properties of dispersed chitosan/xanthan hydrogels. Carbohydr. Polym. 2007, 67, 586–595. [Google Scholar] [CrossRef]
- Yang, J.A.; Yeom, J.; Hwang, B.W.; Hoffman, A.S.; Hahn, S.K. In situ-forming injectable hydrogels for regenerative medicine. Prog. Polym. Sci. 2014, 39, 1973–1986. [Google Scholar] [CrossRef]
- Delmar, K.; Bianco-Peled, H. The dramatic effect of small pH changes on the properties of chitosan hydrogels crosslinked with genipin. Carbohydr. Polym. 2015, 127, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Ramay, H.R.; Gunn, J.; Matsen, F.A.; Zhang, M. PEG-grafted chitosan as an injectable thermosensitive hydrogel for sustained protein release. J. Control. Release 2005, 103, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Jóźwiak, T.; Filipkowska, U.; Szymczyk, P.; Rodziewicz, J.; Mielcarek, A. Effect of ionic and covalent crosslinking agents on properties of chitosan beads and sorption effectiveness of reactive black 5 dye. React. Funct. Polym. 2017, 114, 58–74. [Google Scholar] [CrossRef]
- Tanabe, T.; Okitsu, N.; Yamauchi, K. Fabrication and characterization of chemically crosslinked keratin films. Mater. Sci. Eng. C 2004, 24, 441–446. [Google Scholar] [CrossRef]
- Baker, J.P.; Blanch, H.V.; Prausnits, J.M. Swelling properties of carylamide-based ampholytic hydrogels: Comparison of experiment with theory. Polymer 1995, 36, 1061–1069. [Google Scholar] [CrossRef]
- Senna, A.M.; Novack, K.M.; Botaro, V.R. Synthesis and characterization of hydrogels from cellulose acetate by esterification crosslinking with EDTA dianhydride. Carbohydr. Polym. 2014, 114, 260–268. [Google Scholar] [CrossRef]
- Capitani, D.; Nobile, M.A.D.; Mensitieri, G.; Sannino, A.; Segre, A.L. 13C solid-state NMR determination of cross-linking degree in superabsorbing cellulose-based networks. Macromolecules 2000, 33, 430–437. [Google Scholar] [CrossRef]
- Lenzi, F.; Sannino, A.; Borriello, A.; Porro, F.; Capitani, D.; Mensitieri, G. Probing the degree of crosslinking of a cellulose based superabsorbing hydrogel through traditional and NMR techniques. Polymer 2003, 44, 1577–1588. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornel University Press: Ithica, NY, USA, 1953. [Google Scholar]
- Lionetto, F.; Sannino, A.; Mensitieri, G.; Maffezzoli, A. Evaluation of the degree of cross-linking of cellculose based superabsorbent hydroles: A comparison between different techniques. Macromol. Symp. 2003, 200, 199–207. [Google Scholar] [CrossRef]
- Samavedi, S.; Whittington, A.R.; Goldstein, A.S. Calcium phosphate ceramics in bone tissue engineering: A review of properties and their influence on cell behavior. Acta Biomater. 2013, 9, 8037–8045. [Google Scholar] [CrossRef]
- Koons, G.L.; Diba, M.; Mikos, A.G. Materials design for bone-tissue engineering. Nature Rev. Mater. 2020, 5, 584–603. [Google Scholar] [CrossRef]
- Shi, M.; Zhou, Y.; Shao, J.; Chen, Z.; Song, B.; Chang, J.; Wu, C.; Xiao, Y. Stimulation of osteogenesis and angiogenesis of hBMSCs by delivering Si ions and functional drug from mesoporous silica nanospheres. Acta Biomater. 2015, 21, 178–189. [Google Scholar] [CrossRef]
- LogithKumar, R.; KeshavNarayan, A.; Dhivya, S.; Chawla, A.; Saravanan, S.; Selvamurugan, N. A review of chitosan and its derivatives in bone tissue engineering. Carbohydr. Polym. 2016, 151, 172–188. [Google Scholar] [CrossRef]
- Saravanan, S.; Vimalraj, S.; Thanikaivelan, P.; Banudevi, S.; Manivasagam, G. A review on injectable chitosan/beta glycerophosphate hydrogels for bone tissue regeneration. Int. J. Biol. Macromol. 2019, 121, 38–54. [Google Scholar] [CrossRef]
- Moreira, C.D.F.; Carvalho, S.M.; Sousa, R.G.; Mansur, H.S.; Pereira, M.M. Nanostructured chitosan/gelatin/bioactive glass in situ forming hydrogel composites as a potential injectable matrix for bone tissue engineering. Mater. Chem. Phys. 2018, 218, 304–316. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Surface Area (m2/g) | Pore Volume (mL/g) | Pore Size (nm) | ζ-Potential (mV) | Particle Size (nm) | Content of Amino Groups (mmol/g) |
|---|---|---|---|---|---|---|
| BG | 794.4 ± 63.2 | 1.03 ± 0.09 | 4.94 ± 0.21 | −12.8 ± 0.81 | 251.6 ± 10.8 | − |
| ABG-1 | 462.1 ± 31.7 | 0.81 ± 0.06 | 4.06 ± 0.17 | 27.3 ± 1.27 | 279.9 ± 17.5 | 0.426 ± 0.031 |
| ABG-2 | 445.4 ± 27.4 | 0.74 ± 0.07 | 3.74 ± 0.13 | 26.7 ± 1.32 | 298.1 ± 20.7 | 0.407 ± 0.046 |
| ABG-3 | 417.8 ± 23.1 | 0.68 ± 0.05 | 3.38 ± 0.15 | 26.4 ± 1.49 | 314.4 ± 21.1 | 0.386 ± 0.042 |
| Sample Name | GCH (w/v%) | ABG-1 (w/v%) | ABG-2 (w/v%) | ABG-3 (w/v%) | GN (w/v%) |
|---|---|---|---|---|---|
| GL-1 (a) | 2.5 | − | − | − | 0.2 |
| GL-2 | 2.5 | 1 | − | − | 0.2 |
| GL-3 | 2.5 | 1.5 | − | − | 0.2 |
| GL-4 | 2.5 | 2.0 | − | − | 0.2 |
| GL-5 | 2.5 | − | 2.0 | − | 0.2 |
| GL-6 | 2.5 | − | − | 2.0 | 0.2 |
| Sample Name | GCH (w/v%) | ABG-3 (w/v%) | GN (w/v%) | PEGDE (w/v%) | Gelation Time at 37 °C (s) (a) | Degree of Crosslinking (×10−5 mol/cm3) (b) | ϕ |
|---|---|---|---|---|---|---|---|
| GEL-1 | 2.5 | 2.0 | 0.2 | 0.01 | 630 ± 24.4 | 0.902 (±0.063) | 0.0128 |
| GEL-2 | 2.5 | 2.0 | 0.2 | 0.02 | 285 ± 17.3 | 1.122 (±0.051) * | 0.0147 |
| GEL-3 | 2.5 | 2.0 | 0.2 | 0.03 | 207.5 ± 20.6 | 1.23 (±0.047) # | 0.0161 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, Q.; Wang, C.; Zhang, Y.; Tian, D.; Wan, Y.; Wu, J. Strong and Elastic Hydrogels from Dual-Crosslinked Composites Composed of Glycol Chitosan and Amino-Functionalized Bioactive Glass Nanoparticles. Nanomaterials 2022, 12, 1874. https://doi.org/10.3390/nano12111874
Min Q, Wang C, Zhang Y, Tian D, Wan Y, Wu J. Strong and Elastic Hydrogels from Dual-Crosslinked Composites Composed of Glycol Chitosan and Amino-Functionalized Bioactive Glass Nanoparticles. Nanomaterials. 2022; 12(11):1874. https://doi.org/10.3390/nano12111874
Chicago/Turabian StyleMin, Qing, Congcong Wang, Yuchen Zhang, Danlei Tian, Ying Wan, and Jiliang Wu. 2022. "Strong and Elastic Hydrogels from Dual-Crosslinked Composites Composed of Glycol Chitosan and Amino-Functionalized Bioactive Glass Nanoparticles" Nanomaterials 12, no. 11: 1874. https://doi.org/10.3390/nano12111874