
Impacts of the Catalyst Structures on CO2 Activation on Catalyst Surfaces

1
Department of Chemical Engineering, Guangdong Technion-Israel Institute of Technology (GTIIT), Shantou 515063, China
2
Wolfson Faculty of Chemical Engineering, Technion-Israel Institute of Technology (IIT), Haifa 32000, Israel
*
Author to whom correspondence should be addressed.
Nanomaterials 2021, 11(12), 3265; https://doi.org/10.3390/nano11123265
Submission received: 14 October 2021
/
Revised: 14 November 2021
/
Accepted: 23 November 2021
/
Published: 30 November 2021
(This article belongs to the Special Issue Bifunctional Metal Oxides as Heterogeneous Catalysis for CO2 Adsorption and Conversion)
Abstract
:Utilizing CO2 as a sustainable carbon source to form valuable products requires activating it by active sites on catalyst surfaces. These active sites are usually in or below the nanometer scale. Some metals and metal oxides can catalyze the CO2 transformation reactions. On metal oxide-based catalysts, CO2 transformations are promoted significantly in the presence of surface oxygen vacancies or surface defect sites. Electrons transferable to the neutral CO2 molecule can be enriched on oxygen vacancies, which can also act as CO2 adsorption sites. CO2 activation is also possible without necessarily transferring electrons by tailoring catalytic sites that promote interactions at an appropriate energy level alignment of the catalyst and CO2 molecule. This review discusses CO2 activation on various catalysts, particularly the impacts of various structural factors, such as oxygen vacancies, on CO2 activation.
1. Introduction
The presence of CO2 in high concentrations in the atmosphere leads to a myriad of harmful consequences, including climate change and sea-level rise. Therefore, CO2 emission mitigation has become a top priority of various governments globally, and research interest in recent years is being dedicated to deriving the positive impact of the CO2 levels through efficient capture and utilization. An economically viable approach to mitigating the monstrous effect of CO2 and reducing its presence in the environment is to transform it into value-added products. The conversion of CO2 to fuels and chemicals has attracted increasing research attention globally, and great achievements have been recorded in the production of C1 products, such as methanol, methane, and formic acid [1,2,3].
CO2 is a chemically inert molecule due to its kinetically stable nature; thus, its conversion by reduction to economically viable products relies on its activation to kinetically vibrant species [4]. The stable nature of CO2 and high activation barrier (1.9 eV) implies that its transformation over catalyst surfaces should create a unique environment for facilitating activation pathways [5,6,7,8,9]. It is possible to apply either homogeneous or heterogeneous catalysis to transform CO2 into value-added products, the latter through thermo-, electro-, or photocatalysis, to induce the catalytic reactions [10,11]. Each of these processes comes with its shortcomings and benefits. The electro/photocatalytic processes have advantages in tuning the reaction products but are difficult to scale up [12]. Given the intricacy of the accompanying reaction mechanisms and the dynamics of catalysis under reaction conditions, particularly for the heterogeneous system, there is still a lack of very fundamental understanding of the chemistry of CO2 reduction. In the presence of small but reactive molecules such as hydrogen or water, CO2 can be transformed into stable products over heterogeneous catalysts with more favorable thermodynamics. The catalyst must possess activity for activating CO2 in addition to enabling effective conversion reactions. Adsorption and activation are two important steps that occur during the CO2 reduction on catalyst surfaces. The adsorption (physisorption and chemisorption) of CO2 on the surface of heterogeneous catalysts has become a subject of growing research interest in recent years [13].
The activation of CO2 on the heterogeneous catalyst surface yields CO2-derived species that eventually convert to useful products. Several CO2-derived species, including carboxylate and carbonates, have been identified, irrespective of which approach is adopted for CO2 conversion [6]. The generation of specific activated carbon species determines the kind and selectivity of the product(s). Co-reactants such as water contribute to the overall reduction reaction by promoting adsorption and subsequent activation. As with most chemical conversion technologies, the choice of catalyst is also an important factor, which must have suitable selectivity and activity for activating CO2 under relatively mild conditions. Many studies reported in the literature tried to understand how to promote the chemisorption and subsequent activation of CO2 by focusing on catalysts preparation and structures. Specifically engineered or surface-modified catalysts have shown improved performance for CO2 activation. Catalysts (metal oxides) with rich surface defects or high concentrations of oxygen vacancies have been particularly effective for CO2 activation. Oxygen vacancies can greatly influence the interaction of CO2 with the surface and enhance the adsorption of CO2 molecules. Thus, oxygen vacancies play important roles in CO2 conversion. Surface defects can be created in metal oxide catalysts either during reactions or incorporated by external methods. Understanding the nature of CO2 in the activated form and the catalyst characteristics will be important in developing novel strategies for activating CO2. An appreciable understanding of the reaction of CO2 on pure metal surfaces is well documented; however, less is known about the reactivity of CO2 on metal oxide surfaces, e.g., how the surface defects contribute to the mechanism of CO2 activation. The central aim of this work is to discuss the CO2 activation mechanism on metal oxide surfaces, particularly the roles of the surface defects in the activation process.
1.1. CO2 Conversion to Chemicals: Challenges, Thermodynamics, and Kinetics
The development of environmentally viable technologies for the utilization of CO2 as a chemical feedstock remains a great challenge because of its thermodynamic stability (CO2 possesses a highly stable π-conjugated structure) and kinetic inertness. Regarding the energy requirement, reactions involved in CO2 conversion can be classified into two major categories: The first category involves adding CO2 to a reaction where the other reactants with higher Gibbs free energy supply energy; such reactions produce the likes of carboxylates and lactones, carbamates, urea, isocyanates, and carbonates. These reactions are energetically favorable and often can be run without any catalyst. The second category is the CO2 reduction reaction, through which important industrial chemicals such as formate, oxalates, carbon monoxide, formaldehyde, methanol, and methane can be obtained. Table 1 presents an overview of the standard enthalpies (ΔH°298K) and Gibbs free energies (ΔG°298K) for different products formed from CO2 conversion reactions. These reactions require an external energy source to activate the strong C=O bond in CO2 and a catalyst to lower the energy barrier. It will be energy-demanding if CO2 is used as a single reactant (Table 1, Equation (1)). However, it becomes thermodynamically easier if another substrate molecule, such as H2, with higher Gibbs free energy, is introduced as a co-reactant (Table 1, Equations (2)–(10)). Another difficulty is that the final products derived from CO2 are mostly in the liquid phase (e.g., formic acid, methanol, and higher hydrocarbons). As these reactions involve a phase change from gaseous reactants into liquid products, they are thus entropically unfavorable.
The unfavorable thermodynamic character of CO2 stems from its highly stable chemical nature. Both ∆S and ∆H (−393.5 kJ·mol−1), which govern the thermodynamics, are unfavorable for CO2 conversion as a result of the very strong C=O bond with a higher dissociation energy (~750 kJ·mol−1) [14,15]. Thus, highly active catalysts are necessary for the activation of CO2. Good catalysts should be able to break the kinetic barriers and enable the chemical conversion of CO2 under relatively mild reaction conditions [16]. While efforts have been demonstrated in this regard, leading to fair knowledge of the reaction mechanisms, more research inputs are still needed to comprehend the principles governing the catalytic transformation of CO2 into value-added chemicals that can drive the industrial scale utilization.
1.2. General Properties of CO2 Molecule: Molecular Structure and Bonding Properties
CO2 is a linear non-polar molecule in its ground state possessing two equivalent C–O double bonds, and its main properties are summarized in Table 2 [18,19,20,21]. Its high oxidation state of carbon (+4) makes it a thermodynamically stable molecule, requiring a high energy input for activation and reaction for producing value-added products. The activation of CO2 typically involves altering the molecular properties, such as the C–O bond length and O–C–O angle. According to the molecular orbital diagram of CO2 (Figure 1a), there are a total of 16 valence electrons distributed among the three atoms (one carbon and two oxygen atoms) in the molecule. The carbon atom exhibits an electrophilic character, conveying an exception to the overall energetics of the CO2 molecule, and thus the nucleophilic and electrophilic reactions are preferred on the C- and O-atoms, respectively. In other words, activation of CO2 can be achieved both nucleophilically and/or electrophilically through the carbon or oxygen atom, respectively [22]. For example, nucleophilic compounds such as water and hydroxides prefer to react at the carbon center; the carbon atom could also gain electrons from a catalyst surface. On the other hand, oxygen atoms in the CO2 molecule with lone pairs of electrons can donate them; thus, CO2 molecules can simultaneously donate or accept electrons, leading to mixed coordination (i.e., binding on both C and O atoms). In the presence of a substrate such as water, the charge on the carbon atom can increase according to a DFT study using a polarizable continuum model with a linear geometry [7], which can boost the carbon atom reactivity of CO2. Non-bonding orbitals are located on the 1s shells of the C and O atoms and do not participate in CO2 bonding [23,24]. The introduction of an electron into the antibonding orbital 2πu, as shown in Figure 1b, results in the formation of the CO2 radical anion (CO2−) or CO2δ−, which can be stabilized by changing the structure from the linear to the bent form [23]. For the anionic CO2 molecule, the carbon atom is nucleophilic, and the molecule has a bent geometry (Figure 1c).
2. CO2 Activation and Different Modes
CO2 is an inert molecule, but its reactivity can be increased by activation, which can be measured by the difference in molecular properties (geometry and electronic properties) of the charged species relative to those in the stable ground state [5]. The activation of a stable CO2 molecule can take one of the following modes:
- bending of the O–C–O angle from 180 degrees,
- elongation of at least one of the two C–O bonds,
- polarization of the charges on C and O, leading to the transfer of charge/electron to CO2,
- hydride transfer, or
- redistribution of charges.
Generally, over heterogeneous catalysts, the activation of CO2 molecule involves a charge transfer (effect iii) from the catalyst to the molecule, which results in elongation of the C–O bond length and reduction in/bending of O–C–O bond angle (effects i and ii). The activation can proceed in the presence or absence of reducing species such as hydrogen [26,27]. Mondal et al. [27], via ab initio DFT calculations, observed the C–O bond in CO2 to increase from 1.16 Å to between 1.27 and 1.42 Å on Zr clusters. Furthermore, the O–C–O bond angle changed from 180 to between 115 and 136°. These occurred through chemisorption and subsequent activation of the free CO2 on the surface of the metal cluster as a result of charge migration from the Zr cluster to the CO2, forming partially charged CO2δ− species.
At the molecular level, the CO2 activation results from partial electron transfer into the LUMO [28]. Based on the molecular orbitals analysis, the reverse charge donation to LUMO of the activated CO2 is responsible for the CO2 binding, which determines the degree of CO2 activation. Consequently, the binding energy originating from the backward donation can be explored to explain the degree of CO2 activation [29]. However, the binding energy is affected by the coordination number and the deformation of the ligand. Definitive evidence for CO2 activation is the bending of the linear OCO configuration. The more bent the geometry, the lower the energy level of the in-plane (i.e., to the plane of bending) contribution of the 2πu LUMO will be.
CO2 bending lowers the LUMO’s energy to a significant degree, particularly that of the in-plane 2πu orbital (Figure 1b), and increases the carbon electron density associated with it, facilitating the transfer of an electron to the molecule. As a result, bending leads to the C–O bond weakening by stretching up to 0.11 Å in comparison with the linear state. This may lead to the dissociation of CO2 on the catalyst surface into CO and O species. Unlike the HOMO that possess the characteristics of the O p-orbital, the LUMOs exhibit mainly the C p-orbital character. These features enhance the CO2 reduction (electron accepting) ability.
However, some of the above indicators or differences in other properties may not sufficiently explain the activation of the CO2 molecule. It has been shown that bending CO2 molecules to smaller O–C–O angles (less than 180 degrees) and an electron transfer to the π*-antibonding orbital correlate more with strong adsorption, leading to the formation of undesired carbon species such as carbonates [30]. CO2 activation on metal oxide surface as studied using an Artificial Intelligence (AI) model revealed the binding of an O atom to a surface cation, resulting in an increase in the C–O bond length and weakening of the bond strength. This confirms only the C–O bond elongation as a valid indicator of CO2 activation [30].
The transfer of electrons to CO2 always precedes a key step—adsorption (chemisorption or physisorption), and the structural properties of the activated CO2 species are evident after adsorption of the CO2 molecule. In many examples, the interaction that results upon chemisorption leads to the formation of CO2δ−. This carbon species present different structures depending on the mode of adsorption [31,32], including oxygen coordination, carbon coordination, and mixed coordination (Figure 2). Coordination through the oxygen atom results in two possible structures of bidentate species as shown in Figure 2a. The formation of a carbonate-like species is evident in the carbon binding mode. Structures of both species can be observed in the combined coordination mode. These different binding modes play a crucial part in determining the reaction pathways as a result of the different possible intermediates that can form [32].
The other chemisorbed CO2 species is the CO32−, which is direct evidence of the transfer of charge from surface oxygen to the CO2 and a molecule bending to form a [O–CO2]2− complexes [33].
The chemisorption of CO2 is a critical step in CO2 conversion reactions [34,35,36]. For example, the dissociation into CO and O or hydrogenation to form different products such as methane or methanol follows the adsorption of CO2. However, the adsorption process must avoid producing inactive surface species (e.g., carbonates) that poison catalysts [26]. From both experimental and theoretical investigations, the activation of CO2 over a heterogeneous catalyst surface involves its adsorption followed by charge transfer (electron) from the catalyst active site to the CO2 molecule [35,37,38]. Upon accepting an extra electron, the molecular CO2 forms the radical anion, CO2− or CO2δ− [31]. This perturbation of the molecular state results in weakening of the C–O bond and reduction in O–C–O bond angle (bending) [34,39]. CO2 activation was observed from the infrared vibrational spectra of the CO2 molecule adsorbed onto Ti8C12 [35]. A peak that was obviously lacking in the gaseous state CO2 molecule was observed. CO2 is adsorbed and activated by dissociating into CO and O. In some CO2 transformation reaction mechanisms, CO has been identified as an important intermediate in the reaction pathway.
The adsorption and activation of CO2 molecules can be enhanced by adopting the strategies presented in Scheme 1.
2.1. Photocatalytic Activation of CO2
Solar-driven catalytic (photo)reduction of CO2 with H2O at low temperature (room temperature) and pressure (atmospheric pressure) is an approach for economically utilizing large amounts of CO2 emitted [40]. CO2 photoreduction mainly involves the following elementary steps:
- Photon absorption and excited carrier generation.
- Activation of CO2 to form an anion radical, CO2•−, or other intermediates by the photoexcited electrons.
- Dissociation of the C–O bond, involving the participation of protons and electron transfer, generating different products.
During the photoreduction process in the presence of a photocatalyst, an electron–hole pair is generated upon photo-illumination of the catalyst surface. The generated electrons transfer to the stable CO2 molecule and activate it to intermediates species that can be much easily converted to a variety of chemical and fuel products, including CO, CH4, CH3OH, and HCOOH. However, water, which is typically used as the reducing agent in CO2 photoreduction reaction, results in low photocatalytic efficiency [40,43]. Some of the reasons for the observed low efficiency on TiO2 photocatalyst include:
- the one-electron transfer to form CO2− is thermodynamically unfavorable as it requires a very negative reduction potential of −1.9 V NHE;
- the photoexcited holes (or OH radicals) easily oxidize water to oxygen, or oxidize the intermediates and products converted from CO2 to undesirable products (reverse reactions); and
- the electron–hole pairs recombination rate is much faster.
Almost no photocatalyst can provide sufficiently high energy to transfer a single photoexcited electron to a neutral state CO2. This remains the most important obstacle to the photoreduction of CO2. Consequently, the multielectron process is often adopted; however, this leads to slow reaction kinetics. The above limitations can be checked by stabilizing the LUMO of CO2 close to or lower than the conduction band minimum (CBM) of the semiconductor; this especially can decrease the strongly negative potential [44]. In the photoreduction process, it is possible to regulate light irradiation to tune the CO2 conversion as desired in comparison with the conventional thermal reaction. Thus, the photo-assisted heterogeneous catalytic reaction can proceed with quite low energy consumption for product formation and a much faster response for the changes in the reaction condition.
The photoreduction of CO2 also involves important steps, namely, adsorption, activation of CO2, and dissociation of the C=O bond. Although the adsorption precedes the activation step followed by the dissociation step, they all work synchronously to realize the overall process, and these steps are often paid less attention in the literature. The transfer of charge (photogenerated electrons) to the CO2 molecule from the photocatalyst surface occurs during the activation step. At the metal-oxide interface, the ease of electron transfer depends on the electron affinity of CO2 and its interaction with the catalyst surface [45]. In addition, the adsorption and activation properties of CO2 on the surface of photocatalyst greatly affect the activity and product selectivity. Generally, the activation and subsequent reduction of CO2 involves the participation of protons and electrons transfer (single or multiple electrons). This generates CO2− by one-electron transfer to CO2, which is an important activated intermediate in CO2 reduction reactions [40]. A more feasible pathway involves multiple electrons and a corresponding number of protons. Therefore, controlling the transfer of electrons and protons is crucial for the activation of CO2 [46]. Understanding the CO2 activation mechanism is essential to guide the improvement and development of effective photocatalysts to promote CO2 reduction efficiency [47]. We herein briefly discuss the activation of CO2 through the photocatalytic reduction mechanism. The adsorption of CO2 on photocatalysts through interactions with surface atoms can activate CO2 by transferring an electron from excited photocatalysts to the CO2, although adsorption and the interactions between CO2 and the photocatalyst surface sometimes are not strong enough to sufficiently lower the barrier and enable the transfer of conduction band electrons. In this case, efforts are directed at improving the photocatalytic efficiency by fabricating photocatalysts with various features to enhance the chemisorption of CO2 [46].
Researchers have studied many strategies to promote adsorption and activation of CO2 on metal oxides. Both theoretical and experimental studies have revealed that these processes are significantly influenced by the crystal phase of TiO2 photocatalysts and their surface defects [48,49,50,51]. Graphene-laden catalysts have been observed with high photocatalytic activity. In addition to increasing CO2 adsorption, there are reports about the increasing separation efficiency of photogenerated electrons and holes of graphene containing photocatalysts. Reduced graphene (rGO)-adorned Pt/TiO2 photocatalysts with enhanced photogenerated charges separation and CO2 adsorption was reported by Zhao et al. [52]. The surface hydroxyl and abundant p electron of rGO provide adsorption and activation sites for CO2. Doping photocatalysts with alkaline-earth or transition metal oxides can provide more surface basic sites for CO2 adsorption and activation, thus improving reaction kinetics and product selectivity [53,54]. For example, 1.0 wt% MgO–Pt–TiO2 gave a superior photocatalyst performance in reducing CO2 into CH4 in the presence of H2O in comparison with Pt–TiO2 [55]. MgO presence provided more surface basic sites for chemisorption of CO2. Insertion of defects or vacancies in photocatalysts can provide more sites for CO2 adsorption and activation. Zhao et al. [56] constructed O vacancies in ZnAl-LDH nanosheets for photocatalytic reduction of CO2. The resulting Zn+–VO complexes served as trapping sites for adsorption of CO2 and H2O that boosted the photocatalytic CO2 reduction activity. Incorporating an appropriate co-catalyst can enhance CO2 activation. Pt/Cu2O-modified TiO2 exhibited high activity for the selective photocatalytic reduction of CO2 into CH4. Deposited Pt can capture photogenerated electrons and generate H2 and CH4, while the Cu2O can enhance the CO2 chemisorption and inhibit H2O [57]. In general, on the surface of photocatalysts, these strategies can be adopted to activate CO2 molecules essential for CO2 conversion. Photocorrosion of oxide photocatalysts can generate in situ photoinduced oxygen vacancies to activate a defect reaction for splitting CO2 under mild reaction conditions that eliminate the need for high temperature and water presence. Wang et al. [58] used an amorphous semiconductor photocatalyst with weak lattice constraint via the photocorrosion mechanism to facilitate the generation of oxygen vacancies, which can react with oxygen atoms of CO2, splitting it into C and O2. Such a defect reaction can be sustained by continuous photogenerated hole oxidation of surface oxygen atoms of the photocatalyst to form an oxygen vacancy and O2. On the other hand, the photogenerated electrons would reduce the carbon species of CO2 to solid carbon. A major challenge is how to generate a high concentration of oxygen vacancies on the photocatalyst surface [58].
Recently, photoinduced electrons have been shown to benefit the activation of stable CO2 molecules for the photoreduction process [44,59]. Hot electrons generated upon light irradiation enhanced the CO2 conversion. The enhancement was related to the change in the HOMO–LUMO energy gap of the CO2 adsorbed on the metal surface. According to DFT calculations, CO2 can strongly bind to Ru surface, and the energy bandgap is reduced from 8.5 eV to 2.4 eV in free CO2 and CO2 adsorbed to the Ru surface, respectively. The CO2 dissociation into CO is promoted with light irradiation because it lowers the reaction energy for CO2 reduction. Total energy consumption of 37% was required for the light-irradiated process compared to the case without light irradiation at a CO2 conversion rate of 15% [59]. On the rutile TiO2 (110) surface investigated theoretically, photo-excited electron induced CO2 reduction through a process that involves the formation of a transient CO2•− with bent geometry through the photoexcitation of one electron to the LUMO of CO2 as shown in Figure 3, and summarized as follows: (1) photoexcitation generates CO2•−, (2) transient CO2•− excites the bending and antisymmetric stretching vibrations that stabilize the CO2 LUMO, (3) CO2 traps hot electrons and forms a new CO2•−, and (4) CO2•− dissociates on oxygen vacancy [44]. The excitation of the bending and antisymmetric stretching vibrations can sufficiently decrease the energy of the CO2 LUMO to below that of the CBM of TiO2. Although this was demonstrated with TiO2, it is believed that conclusion can be widely applied to other metal oxides as well as to other semiconductors.
2.2. Electrocatalytic Activation of CO2
Electroreduction of CO2 is another appealing approach for CO2 valorization. The principles of operation, striking features, and the challenges of this process have been discussed in the literature [10,12,60,61,62]. The electrochemical CO2 reduction is a multielectron and multiproton process involving several transfer pathways that result in different products [63], and this is of immense advantage over the single electron process in terms of electrochemical potential requirements [62]. As in the thermo- and photo-catalytic processes, the electrocatalytic reduction involves the adsorption and activation of CO2 on the catalyst surface to product precursors. Following adsorption of CO2 on the electrocatalytic surface is the electron transfer and/or proton migration that initiates the C=O bond cleavage and subsequent formation of C–O and/or C–H bonds [64]. The adsorbed product species then desorb and diffuse from the catalyst surface. From both theoretical and experimental studies, the adsorption and activation steps are the most critical of the CO2 electroreduction process not only due to the high thermodynamic stability of the neutral CO2 molecule, but also the requirement of a large amount of energy for breaking the kinetic barrier [62], considering the strongly negative redox potential (−1.90 V) compared with SHE [65]. As the rate-determining step, the chemisorption and subsequent activation of CO2 on the electrocatalyst surface would form CO2•− intermediate that can further be reduced to the *CO, *COH, *CH2 product precursors, or effect C–C coupling to form multicarbon products [65,66,67,68]. Consequently, understanding the CO2 activation mechanisms over electrocatalysts is necessary for a proper comprehension of the electroreduction transformation to different products, and for improving its performance. To this end, researchers have focused on how to achieve efficient CO2 activation on electrocatalysts. This includes electrocatalyst engineering and surface modification [69]. Such catalysts need to be tailored with electron-donating centers for the efficient chemisorption of CO2 molecules.
The proper design of the electrocatalyst active phase can improve the activation of the neutral CO2 molecule. Xiao et al. [69] conducted a detailed study of the CO2 adsorption on Cu electrocatalyst employing different catalyst models. It was found that only the catalyst with a combination of surface Cu0/Cu+ sites promoted CO2 activation, favoring kinetics and thermodynamics. The Cu0 sites bind to activated CO2, whereas Cu+ sites dilute the negative charge by binding to water molecules in the electrolyte solution. This translated into the observed increase in the onset potential and peak Faradaic efficiency for CO production [69].
The combination of experiments and theoretical calculations on the surface and electronic structure of Cu electrocatalyst revealed that the presence of a thin suboxide species below the Cu surface could bind the CO2 in the physisorbed configuration at 298 K; in the presence of H2O, this suboxide is essential for further converting to the chemisorbed CO2 and subsequently toward CO2 reduction products such as formate and CO [70]. Indeed, it has been suggested that the CO2 might dissociate more easily on pre-oxidized Cu surfaces [71], but evidence to support this is scarce. The results of the CO2 reduction to CO with annealed Cu2O revealed a linear Tafel plot over the range of overpotentials from 0.05 to 0.3 V with a slope of 116 mV/decade. The slope of this plot was in agreement with the rate-determining initial electron transfer to CO2 to form a surface adsorbed CO2•− intermediate. The difference in Faradic Efficiency (FE) between the reduced Cu electrode and that of the polycrystalline Cu suggested that the Cu surface formed by reducing thick Cu2O layers could form the CO2•− intermediate [71]. This presents the need for understanding the surface structures of electrocatalytic Cu particles for the CO2 activation and subsequent reduction to different products.
Engineering oxygen vacancy/surface defects in CO2-reduction catalysts has proven an indispensable method for generating and transferring surface electrons to CO2 molecules [72,73,74]. Gu et al. [73] reduced copper oxide nanodendrites with rich surface oxygen vacancies and obtained partially reduced Cu oxide catalyst with Lewis base sites for enhanced CO2 adsorption and subsequent electroreduction. Furthermore, strong binding of *CO and *COH intermediates to CuOxsurface was observed. Theoretical calculations and catalytic test results showed that oxygen vacancy can provide a surface for binding, and as a result, the partially reduced CuOxnanodendrites had a high electrocatalytic activity for C2H4 production. ZnO nanosheets rich in oxygen vacancies exhibited a current density of −16.1 mA cm−2 with a FE of 83% for CO production at −1.1 V versus RHE in the CO2 electrochemical reduction [74]. According to DFT calculations, introducing oxygen vacancies increased the charge density of ZnO around the valence band maximum, resulting in the enhanced activation of CO2. In addition, mechanistic studies revealed that introducing oxygen vacancies into ZnO nanosheets increased the binding strength of CO2 and facilitated CO2 activation [74]. The electrochemical properties of InOx electrocatalysts (P-InOx NRs, O-InOx NRs, and H-InOx NRs) in CO2-saturated NaHCO3 solution were investigated. It was found that H-InOx NRs exhibited the best performance, generating a current density of 26.6 mA·cm−2 at −1.05 V versus RHE. This catalyst had the highest oxygen vacancy concentration compared with others. The current densities for HCOO− at various overpotentials for the different InOx catalysts are presented in Figure 4a. The Tafel plot of O-InOx NRs, P-InOx NRs, and H-InOx NRs gave slopes of 150.2, 117.0, and 57.9 mV/decade, respectively, pointing to the different adsorption strength of the catalysts for CO2 molecules. H-InOx yielding the least slope value indicates the pre-equilibrium transfer step (CO2 + H+ + e− + * → HCOO*) rather than a chemical H+ transfer step as the rate-determining step [72]. The availability of hydroxyl groups on the surface of the electrocatalyst can enhance its adsorption properties. Huang et al. [64] functionalized the MOF surface with OH− moieties and used it for the electrochemical reduction of CO2. They observed single-crystal to single-crystal structural transformation between the MOF and MOF-CO2, activating it to HCO3−, an important intermediate in the electrocatalytic reduction process [75]. Theoretical calculations revealed the formation of the HCO3− species from the geometries of CO2 adsorption at both the O- and C-atoms. In addition, the OH-incorporated MOF sample exhibited high FE for CO, achieving approximately 100% at −0.6 V versus RHE, surpassing the 96% (−0.6 to −0.9 V) in most MOF-based catalysts [64].
2.3. Activation of CO2 by Homogeneous Catalysts
In an aqueous solution of transition metal salts, a CO2 molecule can be activated by forming a transition metal–CO2 complex via direct coordination [4,22,76,77,78]. This can be considered a combination of π electrons transfer to the metal and the oxidative addition of the C–O bond to the metal (Scheme 2) [22]. For a multi-metal complex, CO2 can also be activated by bridging between metals in the complex. The successful formation of the metal–CO2 complexes is key to CO2 activation. Upon forming the metal–CO2 bond, the activation energy for further reactions is sufficiently lowered, thus accelerating the reactions rates in various CO2 transformation processes. A number of factors, including the central metal itself, ligand set, and metal oxidation state, have been identified to affect the activation of CO2 on metal complexes [22].
Infrared spectroscopy (IR) has emerged as an excellent tool for the structural investigation of metal–CO2 interactions in complexes, such as M+(CO2)n or M−(CO2)n [79,80,81,82,83]. Review articles discussing the spectroscopic studies of the various types of CO2 interactions with metal complexes are available in the literature, which readers are directed to. A recent review on the subject was authored by Paparo and Okuda in 2017 [84].
We briefly discuss recent studies on the conversion of CO2 to chemicals and fuel via the formation of CO2-activated complexes. Catalytic reduction of CO2 to methanol and other products has been observed with transition metal complexes [22]; it is possible to develop highly active catalysts based on early transition metal complexes. It was found that anionic metal clusters (Mn−) with 1, 2, and 6 atoms formed the activated complex (Mn–CO2)− for Cu, Ag, and Au [4]. The activation was a result of a strong interaction between the CO2 moiety and Mn− via the formation of a partial covalent M–C bond with a full delocalization of the electronic charge due to electron transfer from the HOMO of Mn− to the LUMO of CO2 as in metal–CO2 π-backbonding. This can easily be understood from the perspective of the orbital geometry and energetics following orbital mixing (orbital interaction between HOMO of Mn− and LUMO of CO2) [4,29].
3. Activation of CO2 on Heterogeneous Catalyst Surface
The interaction of CO2 molecules with metal-based surfaces has attracted intense research attention for a long time. Reduction of CO2 by H2 has been studied on some transition metals (e.g., Cu, Co, and Ni) and metal oxide catalysts (e.g., TiO2, CeO2, and In2O3). The catalytic surfaces for the activation of CO2 include metal sites, metal-oxide interfaces, and oxygen vacancies. For facile CO2 activation and conversion, efforts should be made to know, design, and optimize functional sites in heterogeneous catalysts, which involves constructing surface sites with a charge density gradient capable of reorganizing the electronic structures of CO2 and polarizing the adsorbed species [85].
Metal-based catalysts are capable of effectively activating the CO2 even in the absence of hydrogen [27]. These include pure metal surfaces, doped or promoted metal surfaces, and supported metal nanoparticles. On supported metal surfaces, CO2 activation occurs mainly through acceptance of charge from the metal; the oxide support plays a big part in the activation process through different mechanisms such as acid sites interactions and provision of oxygen vacant sites [86]. The metal surface is also important in the dissociation of H2 in CO2 hydrogenation reactions [6].
3.1. CO2 Activation on Representative Pure Metals
Metal nanoparticles sit on a metal oxide serve as active sites for the electron transfer. Transition metals such as Cu, Ni, and Fe are particularly energetic for activating CO2. Their surfaces possess high binding abilities to CO2. DFT calculations evidenced that on Pt, Rh, Ni, Cu, Ag, and Pd (111) surfaces, the affinity toward oxygen is different for the metals, which selects their reaction pathways for the CO2 activation in the RWGS) reaction. Pt, Ag, and Pd tend to favor the COOH-mediated mechanism, whereas Rh, Ni, and Cu dissociate CO2 into CO and O. This difference underlines the variation in the CO2 dissociation barrier of the different metal groups. Thus, the nature of interaction between the adsorbed O and the surface is critical for determining the CO2 dissociation barrier [87]. In the activation of CO2 via the charge transfer mode, which is the prevalent on metal surfaces, the partial and full charge transfer leads to the formation of CO2δ− and CO2−, respectively. The degree of charge transfer can be analyzed on metal surfaces after adsorption. Physisorption and chemisorption of CO2 on single crystal surfaces of various metals have been studied by means of DFT calculations [88]. Physisorption imposes difficulty in transferring charge due to the weak interaction of adsorbed CO2 with the surface. It was found that, approximately −0.06 electron charge transferred from the surface of metals listed in Table 3 (little or no dependence on the kinds of metals) as a result of the location of adsorbed CO2 (~3 Å) above the surface [88]. On the contrary, chemisorption states are highly dependent on both the metal itself and adsorption sites. Chemisorbed CO2 molecules often have a bent structure with O–C–O angle varying between 121 and 140° compared with the nearly linear coordination of the physisorbed CO2 molecules, and their extent depends on the kind of metal surface. In other words, the degree of CO2 activation varies with metal surfaces. A similar behavior can be observed for the amount of charge transferred (Table 3). According to Wang et al. [89], who investigated CO2 chemisorption on nine transition metal surfaces (Fe, Co, Ni, Cu, Rh, Pd, Ag, Pt, and Au), the adsorption strength is affected by both the d-band center of the metal surface and the charge transfer, which control the activation of C=O bond. It is possible to promote chemisorption by adjusting the properties of the catalysts, such as the catalyst surface area, surface defects, basic sites and the addition of promoters [31].
Generally, the DFT study of the CO2 activation on transition metal surfaces suggests different barriers for CO2 dissociation on different monometallic or bimetallic surfaces [88]. The coordination of CO2 with metals can take different modes, e.g., with the electron-deficient C atom as the electron acceptor and the C=O bonds or O atoms as the electron donor as shown in Scheme 3 [85]. Electron-rich metal surfaces such as Ni (110) and Cu (100) generally activates CO2 by attaching to the carbon atom [90,91]. In the process, electrons in the dz2 orbital of the metal transfer to the unpopulated antibonding π* orbital of the CO2 molecule. Consequently, negatively charged surfaces are efficient for CO2 activation via the charge transfer mode. Such charged surfaces can be generated through inserting strong metal-support interaction, bimetallic and ligand effects, etc. [85]. Electron-deficient metal surfaces, such as Ti, Cr, V, and Mn, favor the end-on coordination with CO2 [92]. In this mode, the bending or distortion of the linear CO2 molecule is difficult [85]. For metal centers with combined binding behavior, i.e., possessing both an electron acceptor site and an electron donor site, CO2 adsorption generally prefers the bridge sites. More efficient activation can result through this mechanism and lead to bending of the linear CO2 molecule from different sites [85].
Investigations by DFT calculations revealed the characteristic adsorption and activation of CO2 on Rh, Pd, Pt, Ni, Fe, Cu, Re, Al, Mg, and Ag metals [87,93]. Strong evidence has been provided for the formation of CO2− according to spectroscopic results. Depending on the type of metal, CO2 can also dissociate into CO and O or be transformed into CO32− and CO. The activation of CO2 on Pt, Rh, Ni, Cu, Ag, and Pd followed different elementary steps as a result of the different levels of interaction of the metals with adsorbed oxygen in the RWGS reaction. Metals with high affinities toward oxygen presented lower activation barriers, leading to facile hydrogenation reactions [87]. The presence of preadsorbed oxygen was responsible for forming carbonates of different structures [87,93]. On most metal surfaces, CO2 activation is highly surface orientated, pressure- and particle size-dependent [89,94,95,96]. Yu et al. [96] demonstrated through spin-polarized DFT calculations that adsorption and dissociation of CO2 was dependent on the Co particle size. They showed that Co55 nanoclusters had the highest CO2 dissociation activity in comparison to Co13 and Co38. However, Co13 activated CO2 with the smallest O–C–O angle (123°) against 137° for both Co38 and Co55 nanoclusters.
Cu-based materials have gained much attention in the CO2 conversion process due to their wide applicability in the different conversion processes and low cost [12,97]. Despite these and other massive studies, the activation of CO2 on Cu catalysts is still an issue due to the poor understanding of its mechanism. Studies have shown that CO2 interacts weakly on low-index Cu surfaces under UHV conditions [98,99]. However, a recent study found Cu (100) surface to be more active in dissociating CO2 than Cu (111), producing oxygen [91]. Ambient pressure X-ray photoelectron spectroscopy (APXPS) and DFT calculations revealed the activation of CO2 on Cu surfaces. APXPS showed that CO2 adsorbed as CO2δ− on Cu (111) surface under a pressure of 0.01 mbar at 300 K (Figure 5). With an increase in pressure to 1 mbar, adsorbed CO2δ− partially transformed into carbonate as a result of the disproportionation reaction between CO2 molecules. Subsequent annealing at 400 K or higher temperatures led to the dissociation of CO2δ− and carbonate and the formation of a chemisorbed oxygen-covered surface. On Cu (110) surface, the CO2δ− gradually dissociated into CO and chemisorbed oxygen under the same CO2 pressure at room temperature. On both surfaces, atomic oxygen was generated that catalyzed the self-deactivation of CO2 adsorption. The DFT results, which collaborated the experimental findings, further indicated that the Cu (110) surface was more active than the Cu (111) surface in breaking C–O bonds [95]. Comparing the adsorption of CO2 on Cu (111), (100), and (110) surfaces, it was found that CO2 molecules aligned parallel to Cu (111) and (100), whereas a vertical configuration was more stable for the adsorbed CO2 on Cu (110) with one of two oxygen atoms towards the surface. The degree of CO2 activation followed the order: Cu (110) > Cu (100) > Cu (111) [89]. A decrease in the activation energies for CO2 dissociation has been observed when Cu surfaces have step or kink defects in comparison with the flat surface. However, chemisorption of CO2 has been reported on Cu stepped surfaces [98,99,100,101].
Ni-based catalysts can dissociate and convert CO2 into value-added products, such as methane; thus, a fundamental understanding of the interaction between CO2 and Ni surface at the atomic level is crucial to design even more efficient Ni-based catalysts. According to theoretical studies, the surface orientation of Ni influences the activation of CO2 by altering the energetics for subsequent C–O bond breakage [102]. Ab initio calculations using slab models have shown that CO2 reactions on model Ni are surface sensitive, with reactivity in the following trend: Ni (110) > Ni (100) > Ni (111) [102,103]. Experimental investigations revealed the capability of Ni (110) surface to molecularly adsorb and subsequently dissociate CO2 at room temperature [104]. By using in situ APXPS, carbonate was identified as the dominating surface intermediate at room temperature upon CO2 adsorption on Ni (111) and Ni (110) surfaces [105,106,107]. However, where there are multiple dissociation products, their distribution depends on the surfaces. Carbonate, CO, and graphitic carbon were all observed on both Ni (111) and Ni (100) surfaces under a CO2 pressure of 0.2 Torr. Ni (111) was predominantly covered with carbonate, whereas adsorbed CO* and graphitic carbon were prevalent on the Ni (100) surface as indicated in Figure 6a–d [107]. The CO2 adsorption and dissociation on ideal Ni (111) and stepped Ni (211) surfaces are shown in Figure 6e. It can be seen that CO2 adsorption is endothermic by 20 kJ·mol−1 on the Ni (111) surface, whereas it is exothermic by 40 kJ·mol−1 on the Ni (211) surface [108].
Based on the experimental results of the ultrahigh vacuum (UHV), Auger electron spectroscopy (AES), temperature-programmed desorption (TPD), and high-resolution electron energy loss spectroscopy (HREELS), it is difficult for CO2 to adsorb on a clean Pt (111) surface between 110 and 300 K, but the dissociation capability can be improved by doping alkali metals such as potassium [109,110]. Conversely, it was reported that Pt foil treated with CO2 in the gas phase during in situ UHVXPS experiments at 77 K formed chemisorbed CO species [111]. In collaboration with the latter assertion, on the clean Pt (111) surface, CO2 dissociated into adsorbed CO and O at both room and elevated temperatures [112]. The production of adsorbed CO increased upon the introduction of H2 (hydrogenation reaction). This observation was obvious for the CO2 adsorption at all temperatures and led to the deoxygenation (consumption of oxygen) of the surface, resulting in cleaning the sites for further CO production and desorption from the surface at elevated temperatures. The Pt surface was active in the RWGS reaction. At a low pressure, the RWGS was initiated at 300 °C; on the contrary, at a high pressure (H2:CO2 of 150 mtorr: 15 mtorr), a low temperature (200 °C) favored the initiation of the RWGS reaction, and the conversion of CO2 continued to increase with increasing temperatures [112]. Moreover, under a pressure of 40 mtorr of pure CO2 and at temperatures below 150 °C, graphitic carbon has been also observed as a product of the Boudouard reaction. IR spectroscopy revealed the size-dependent nature of CO2 adsorption on Pt clusters. On small Pt clusters anions (Ptn−, n = 4–7), CO2 was highly activated but remained molecularly adsorbed on Pt4−. On large clusters, dissociative adsorption was observed [13].
The CO2δ− state of adsorbed CO2 molecule has been observed on Co (100) and Co (110) surfaces upon CO2 dissociation based on the energetics, geometries, vibrational frequencies, and charge transfer analysis [113].
3.2. CO2 Activation on Bimetallic/Alloyed Catalyst Surfaces
Bimetallic surfaces are highly unique and active for a wide range of CO2 transformation reactions due to the electronic and geometric alterations within the structure. These alterations could be observed as a change in the morphology of metal, adsorption mode and configuration, and chemical ordering with varying composition and particle size [8,34]. This can help to control the adsorption properties to attain desired adsorption coverages. Numerous studies have appeared in the open literature discussing both activation of CO2 and the subsequent conversion of CO2 to fuels and chemicals on bimetallic catalysts. Ko et al. [88] studied the CO2 activation and adsorption by performing DFT calculations on a range of bimetallic alloy surfaces. The dissociation energy barriers of CO2 were screened by combining Bronsted–Evans–Polanyi (BEP) relation, scaling relation, and surface mixing rule. It was found that CO2 dissociated into CO and O, which sum of their adsorption energies was linearly related to both the energy for CO2 dissociation and that for CO2δ− adsorption. The activation of CO2 proceeded through a direct dissociation (CO2 → CO + O) mechanism in three successive elementary steps: physisorption of CO2 from the gas phase on the metal surface, chemisorption of CO2δ− from the physisorbed CO2, and direct dissociation of CO2δ− into CO and O.
The predicted activation energies for the CO2 dissociation on bimetallic alloy surfaces are shown in Figure 7, with the row and column indicating the host and solute metals of the bimetallic alloys, respectively. The alloying effect would lead to reduction for surface reactions when the solute metals are placed in the bulk region. In the figure, the activation energy (Eaact) decreases from right to left and from bottom to top, thus alloys with relatively low activation energies (~0.75 eV) are Fe-, Ru-, Co-, Ni-, Rh-, and Ir-based alloys, whereas Pd-, Pt-, and Cu-based alloys possess high activation energies (~1.51 eV). Still, some metals, including Ru-, Co-, Ni-, Rh-, Ir-, and Cu-based alloys, have activation energies in between the extremes (0.76–1.50 eV). This picture makes sense for fabricating bimetallic catalysts for CO2 conversion reactions by manipulating the activation energies [88].
The mechanisms of CO2 dissociation on bimetallic clusters (Pt3Ni1, Pt2Ni2, and Pt1Ni3) were investigated and characterized by the activation barrier [21]. Compared with the single metallic clusters (Pt4 and Ni4), results showed the activation barrier to be between those of the monometallic clusters. Increasing Ni atoms in the bimetallic clusters moderately raised the activation barrier, which means that such a bimetal combination could have a significant negative impact on the activity of Pt for CO2 reaction. In the presence of H2, CO2 dissociation became less difficult for all metallic clusters [21].
Results from an ab initio study of chemisorption and activation of CO2 on Pt-based transition-metal nanoalloys on 55-atom nanoclusters (PtnTM55−n), where n = 0, 13, 42, 55, and TM = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Au, indicated a linear correlation between the interaction energy, charge transfer from the nanoclusters toward CO2 and the bent CO2 angle [8]. It was further realized that the interaction energy was enhanced for larger angles and molecular charge. With 55 atoms for Cu, Ag and Au in the Pt alloy, a change from physisorption to chemisorption was observed, whereas the strong interaction energy of CO2 with Os55, Ru55, and Fe55 can be decreased by alloying with Pt [8]. It was concluded that certain metals (Fe, Co and Ni) activate CO2 more strongly as monometals than in an alloyed form due to weaker adsorption energy in the latter.
3.3. CO2 Adsorption and Activation on Metal Oxide Surfaces
3.3.1. Metal Oxide
Various metal oxides (MOx) or (MxOy) are investigated as supports or directly as catalysts for CO2 conversion, including In2O3, CeO2, ZnO, ZrO2, TiO2, and CeO2. Metal oxide surfaces consist of both metal (Mn+) and oxygen (O2−) ions, which are effective sites for CO2 activation. The activation can occur by coordination to one or two adjacent metal sites through the terminal oxygen atoms or C atom of the CO2 molecule, forming monodentate or bidentate carbonate species, as illustrated in Scheme 4a,b [114]. Interaction of the C atom is on the surface oxygen sites of the metal oxide. The CO2 activation can also occur via the σ-bond and π-bonds activation on metal ions and oxygen ions, respectively, as observed upon chemisorption on metal oxides applied as catalyst supports (as shown in Scheme 4c) [115]. Due to the large surface areas, switchable redox properties, and rich oxygen vacancies, metal oxides can act as adsorption and activation sites for small molecules, including O2, H2, and CO2 [116]. The surface oxygen vacancies interact with the carbon and/or oxygen atoms of CO2 through which electron transfer from the oxide defective site to adsorbed CO2 becomes feasible. One example is In2O3, which is rich in oxygen vacancies and has shown a high activity for CO2 activation and methanol synthesis by hydrogenation [116]. On oxide surfaces, generally, CO2 interactions can vary from physisorption to chemisorption, the extent that affects the structure and reactivity of the adsorbed CO2 and the kinetics and mechanisms of surface catalytic reactions [114]. The surface structure is important for CO2 adsorption and activation [117]; thus, the interaction of CO2 with metal oxides can be structure-dependent. It was found that CO2 adsorption on Zn2GeO4 (001) was higher than that on Zn2GeO4 (010) surface [117]. The interaction with (010) surface led to bidentate carbonate species, whereas on the (001) surface, stronger interaction with CO2 resulted in a bridged carbonate-like species. The strongest adsorption based on calculated CO2 adsorption energies were around the surface oxygen vacancy site on both surfaces. Analysis of the LDOS and Mulliken charge for adsorbed CO2 on perfect Zn2GeO4 surfaces revealed that CO2 formed a CO2δ− species upon receiving electrons from the surface. CO2 molecule was found to be activated on the CuO surfaces ((011), (111), and (−111)), with strong adsorption only on the (011) surface. The CuO (111) and CuO (−111) surfaces showed relatively weak adsorption. CO2 activation was characterized by structural transformations and charge transfer that resulted in the formation of bent CO2δ− species with an elongation of the C–O bonds [94].
The ability of metal oxides to bind and activate CO2 depends greatly on several factors, including their preparation methods, physiochemical properties, redox properties, and electronic and geometric structures [108,114,118]. The preparation methods were found to impact the properties of CeO2 nanostructures for the photocatalytic reduction of CO2 [119]. A high surface area was obtained for catalysts synthesized through the sunlight-assisted combustion process, in addition to possessing a small particle size, high concentration of oxygen vacancies, and a narrow bandgap. Compared to that prepared from the conventional combustion process with a spongy-like structure, a porous network consisting of small and uniformed pores was also obtained for the sunlight-assisted process. The superior catalytic properties could be attributed to the combustion process aided by solar irradiation during the synthesis process. As demonstrated with the CeOx/Cu catalyst, the important roles of metal oxide ions were revealed on the catalytic cycle of H2O and CO2 activation. The Cu phase reduced into Cu0 that promoted Ce4+ reduction into Ce3+. H2O and CO2 activation occurred on the Ce3+ sites. Without the presence of Cu, Ce3+ would lead to oxidation into Ce4+. However, in contact with Ce4+, Cu0 reacted to form Cu+ and Ce3+, sustaining ceria in the more active state. The cycle is closed when Cu+ reduced to Cu0 [120]. This synergistic effect afforded the catalysts with high reactivity in the RWGS reactions. The chemisorption of CO2 molecules on CeO2 at RT as studied using in situ DRIFTS indicated adsorption at both the Ce3+ and Ce4+ sites, although adsorption was also found at the oxygen sites that resulted in carbonates and bicarbonates species [121]. In the same study, the CO2 chemisorption on TiO2 under similar reaction conditions and instrumentation was observed at both Ti3+ and Ti4+ sites, exhibiting O–C–O vibrations at 1667 and 1248 cm−1 and 2339–2345 cm−1, respectively [121]. The CO2 molecules adsorbed at Ti3+ sites formed CO2− species, which concentration increased with the amount of oxygen vacancies present. Like with CeO2, CO2 chemisorption at the oxygen sites formed carbonates and bicarbonates species. It was observed that the interaction between TiO2 and CO2 molecules is somewhat weak compared with that of CeO2. Such weak interactions can be improved by doping TiO2 with CeO2. CeO2 doping can improve the interaction of TiO2 with CO2 as a result of the introduction of Ce3+, which strengthens the bonding of CO2 with catalyst surfaces and enhances the production of bidentate carbon species that can readily be transformed to surface CO2− in the presence of H2O under sunlight irradiation. The formation of adsorbed species of CO2 over CeO2/TiO2 could be attributed to the binding of CO2 species to Ti/Ce atoms that have reductive capabilities under photo-irradiation. Furthermore, the Ce3+ availability from CeO2 facilitates photogenerated charge separation; thus, the CO2 adsorption and enhanced charge separation can be tuned for increased activity of CeO2/TiO2 catalyst [122]. The surface area of a material positively correlates with its adsorption capacity. It was found that the Bi12O17Cl2 nanotubes had higher adsorption capacity for CO2 (~4.3 times) than bulk Bi12O17Cl2 due to the higher BET specific surface area of the former. As a result, the effective adsorption of CO2 on Bi12O17Cl2 nanotubes than bulk Bi12O17Cl2 favored the photocatalytic process [123]. In addition, the high surface area correlated with strong adsorption. Weak chemisorption of CO2 has been reported for CeO2 nanostructures with low exposed surface area [121]. Mesoporous structured photocatalyst displayed improved activity for CO2 reduction into CH4 due to the presence of high specific surface area and mesostructure that enhanced adsorption of CO2 [124]. Highly mesoporous In(OH)3 synthesized via a sol–gel hydrothermal treatment exhibited ~20-fold higher efficiency for CO2 reduction in comparison with that lacking mesopores [125]. It is reported that the methanol activity of the In2O3 catalyst could also be improved by increasing the (111) surface area [126].
3.3.2. Characteristic Adsorption of Representative Metal Oxides
Ceria (CeO2) has shown catalytic activity in the reduction of CO2 to liquid fuels and chemicals. It has rich oxygen vacancies and a high oxygen storage/release capacity. Several studies demonstrating the interaction of CO2 with high-surface-area ceria catalysts have been reported. As noted in [127], CO2 dissociates into CO and an oxygen-containing surface species on the surface Ce3+ ions, which are considered active sites for CO2 activation due to formation of carbonates or inorganic carboxylates. Graciani et al. reported a highly active CeOx/Cu nanoparticles catalyst for methanol synthesis from CO2 [128]. The catalyst activated CO2 as CO2δ− and exhibited a faster methanol production rate than Cu/ZnO, on which CO2 was chemisorbed as CO32−. A study on the CO2 adsorption sites of CeO2 (110) surface using DFT was carried out by Cheng et al. [129]. Reduced and stoichiometric ceria (110) surfaces were compared. Results revealed that CO2 adsorption on the reduced ceria (110) surface was thermodynamically favored than on the stoichiometric ceria (110) surface. Furthermore, the most stable adsorption configuration consisted of CO2 adsorbed parallel to the reduced ceria (110) surface at the oxygen vacancy. Upon adsorption, the CO2 molecule distorted out of the plane and formed carbonates with the remaining oxygen anion at the surface [129]. It was suggested that the structural changes in the catalyst after CO2 adsorption were due to charge transfer between the surface and adsorbate molecule. The formation of two different adsorbate species: a carbonate and a weakly bound and linear physisorbed species, were observed upon exposure of reduced CeO2−x (110) substrates to CO2 at low temperatures. There was no evidence for the formation of CO2δ−. Furthermore, based on angle-dependent C K-edge NEXAFS spectra, the most preferred orientation of the adsorbate was lacking. The physisorbed CO2 species and carbonate species were completely desorbed at 250 and 500 K, respectively. The authors remarked that it is most unlikely that the activation of CO2 on the reduced CeO2−x (110) surface was via breaking the C=O bond to form CO and O. However, on fully oxidized CeO2 (110), CO2 adsorbed as a carbonate which was completely decomposed and desorbed as CO2 at 400 K [130]. CO2 adsorbed on the CeO2 (111) surface formed a monodentate carbonate species found to be most stable on CeO2 at low coverages [131]. Increasing the CO2 coverage destabilized the formed species, indicating a mixed adsorption mechanism with both carbonate and linear adsorbed CO2 species. Although CeO2 has been studied for CO2 reduction reactions, the insights into CO2 adsorption, activation, and reaction on ceria surfaces are not yet fully understood.
TiO2 possesses good photocatalytic properties for many chemical reactions, including CO2 reduction. Since its first demonstration in the photoelectrochemical CO2 reduction to formic acid and formaldehyde by Inoue et al. [132], TiO2-based materials have attracted great research interests in CO2 photoreduction reactions. The adsorption properties of CO2 on both the rutile and anatase phases of TiO2 have been widely studied using various surface science techniques [133,134,135,136]. Sorescu et al. [137] investigated the adsorption and dissociation of CO2 on an oxidized anatase (101) surface using dispersion-corrected DFT and found CO2 to adsorbed at a fivefold coordinated Ti site in a tilted configuration. Based on in situ FTIR experiments, the CO2 adsorption formed CO32− and CO2 bonded to Ti, with absorption bands at 1319, 1376, 1462, 1532, 1579, and 2361 cm−1. The band at 2361 cm−1 was assigned to adsorbed CO2 with Ti–O–C–O adsorption configuration [138]. The 1319 and 1579 cm−1 bands were assigned to bidentate carbonate, while the band at 1461 cm−1 was due to monodentate or free carbonate. Under the vacuum condition, the intensities of all of the bands were reduced at 35 °C. The bidentate carbonate was the predominant species for CO2 on TiO2. The scanning tunneling microscopy (STM) enabled the study of the dissociation of CO2 adsorbed at the oxygen vacancy of TiO2 (110) at the single-molecule level [139]. It was found that the electrons injected from the STM tip into the adsorbed CO2 caused its dissociation into CO and O, and the released O was observed to heal the oxygen vacancy. According to experimental analysis, ~1.4 eV above the conduction band minimum of TiO2 is needed for the electron induction process to dissociate CO2. The formation of a transient negative ion by the injected electron is an important step in the CO2 dissociation, and this can only be possible above the threshold voltage. TiO2 modified with metal oxide nanoclusters possess enhanced activity to adsorb and convert CO2 [26,140]. The Bi2O3-TiO2 heterostructures obtained by modifying TiO2 with Bi exhibited low coordinated Bi sites in the nanoclusters and a valence band edge consisting mainly of Bi–O states due to the presence of the Bi lone pair. Upon interaction of CO2 with the reduced heterostructures, CO or CO2− were observed mainly through electron transfer to CO2, and the Bi2O3–TiO2 heterostructures became oxidized in the process with adsorbed CO2 in carbonate form [140]. In a related study, clean or hydroxylated extended rutile and anatase TiO2 surfaces modified with Cr nanoclusters presented an upshift valence band edge related to the existence of Cr 3d–O 2p interactions, which promoted the CO2 activation. [26]. The activated CO2 molecule reduced its O–C–O angle to 127–132° and increased the C–O bond length 1.30 Å. It was concluded that the strong CO2–Cr–O interaction induced the structural distortions.
Iron oxides (FeOx) are an important component of catalysts for the conversion of CO2 to hydrocarbons (liquid fuels). The adsorption and activation of CO2 on FeOx have been investigated by researchers [141,142,143]. It is suggested that Fe2+ and Fe3+ cations are crucial for CO2 adsorption. Using TPD, Pavelec et al. [141] observed a weak interaction between CO2 and Fe3O4 (001) surface. On this surface, CO2 molecules existed in the physisorbed state as they desorbed at a low temperature (115 K). However, strong CO2 adsorption was observed on the defects and surface Fe3+ sites. Weak CO2 adsorption has also been observed on Fe3O4 (111) as investigated by various experimental techniques [144]. At different CO2 dosages and temperatures (between 120 and 140 K), TPD experiments suggested CO2 adsorb very weakly on a regular Fe3O4 (111) surface. However, CO2 chemisorption was also observed but at relatively long CO2 exposure times [144,145] via binding to under-coordinated oxygen sites [145]. The formation of chemisorbed species such as carboxylates and carbonates was facilitated by surface imperfections. Conclusively, FeOx exhibit weak interaction with CO2 molecules, and studies are recommended in this direction to adjust its CO2 adsorption strength.
ZrO2 has been demonstrated as an active catalyst or catalyst support for the CO2 hydrogenation reactions to a variety of products. In a study on CO2 hydrogenation on Cu/ZrO2 interface using the first-principles kinetic Monte Carlo simulations by Tang et al. [146], the authors showed that CO2 prefers to adsorb on the bare ZrO2 nanoparticles surface rather than at the Cu/ZrO2 interface. This led to the bending of the CO2 molecule with a calculated adsorption energy of 0.69 eV. The stretching of the C–O bonds and charge transfer from the ZrO2 surface to the antibonding 2πμ orbital of CO2 were also observed. On the bare ZrO2 surface, bidentate bicarbonate (HCO3) was formed upon CO2 adsorption based on observable IR frequencies at ~1225, ~1620, and ~3615 cm−1 [147].
3.3.3. Oxygen Vacancies and Their Roles in CO2 Activation
Many metal oxides, particularly the reducible ones (e.g., TiO2, CeO2, and In2O3), contain certain number oxygen vacancies, depending on the preparation and treatment conditions. The formation of oxygen vacancies can alter the physicochemical and electronic properties, improve surface adsorption, and generate additional active centers of metal oxides [148]. As reported in the literature, the generation of oxygen vacancies in catalytic materials is linked with enhanced light harvesting, charge separation and transfer in photochemical reactions, charge (electron) production and transfer in thermochemical reactions, and charge density increase in electrochemical reactions. During CO2 conversion by the mentioned approaches, surface oxygen defects on metal oxides can significantly influence the interaction of CO2 with the surface and enhance the adsorption compared to the perfect surface due to modification of adsorption and binding properties [117]. In theory, removing O atoms in metal oxides leads to the formation of oxygen defects/voids that can activate CO2 due to the unsaturated chemical bond in the metal oxide and the unshared pair electrons of CO2 [72]. This reconfigures the coordination environment and delocalizes charge, facilitating the conversion of reaction intermediates [149]. The backward transfer of electrons from the defect to the adsorbed molecule alters the d-band center of the oxide catalyst, causing further interaction with the adsorbed CO2 and resulting in the reduction of activation energy barrier and enhanced CO2 reduction [72]. In summary, oxygen vacancies have valuable input to the overall reaction mechanism, thermodynamics, and kinetics.
How to Generate Oxygen Vacancies
Theoretical studies predict that reducible metal oxides with oxygen vacancies possess high CO2 adsorption and activation properties and can improve selectivity toward a characteristic reaction product [58,73,74,139,150,151,152]. Thus, the presence of oxygen vacancies in proper concentration can improve the catalytic performance of oxide-based catalysts. While oxygen vacancies in a catalyst sample can be detected by various techniques [153], X-ray photoelectron spectroscopy (XPS) is a good technique for confirming and measuring the relative oxygen vacancy concentration [119,154,155]. The ratio of the peak areas of lattice oxygen (Olat) to adsorbed oxygen (Oads), i.e., Olat/Oads area ratio, obtained from the XPS can be used to estimate the relative concentrations of oxygen vacancy. A low oxygen vacancy concentration would exhibit a high Olat/Oads ratio [154,155]. Oxygen vacancies can be formed during catalytic reactions [85] or be deliberately introduced in metal oxides during synthesis [153].
The formation of oxygen vacancies during reactions can be enabled by reaction conditions, via thermal desorption of lattice oxygen or reduction by reactive molecules (H2 or CO) [152,156,157,158]. Their generation through the former process is highly endothermic, while it is exothermic when initiated in the presence of H2 or CO; it is more exothermic using CO than H2 [152]. As an example, the surface reduction of In2O3 with H2 at above 500 K reordered the surface and formed defective sites [157]. During CO2 hydrogenation on oxide supported metal catalysts, H2 that dissociates on the metal site spills over and attacks the oxide at the metal-oxide interface, generating oxygen vacancies [156]. It was revealed in CO2 methanation over Ru/CeO2 catalyst, using operando XAENS, IR, and Raman that Ru species reduced to metallic Ru, which provided the ability to dissociate H2. The dissociated H atom attacked the Ce–O bond on the CeO2 surface, generating surface OH, Ce3+, and oxygen vacancies [156]. The hydrogen spillover effect of metallic Ru would facilitate the attacking of Ce–O bond by H atoms. Pd/In2O3 catalyst with highly dispersed Pd NPs (~3.6 nm in size) and predominately exposed (111) facets showed a better ability to dissociatively adsorb hydrogen and supply it for creating oxygen vacancy. The interfacial sites were also active for enhanced CO2 adsorption and hydrogenation [158].
Catalytic activities of irreducible metal oxides such as Al2O3, ZnO, and MgO can be improved by introducing oxygen vacancies in them. Methods including doping, plasma-assisted techniques, photo-irradiation of synthesis reaction, high-temperature annealing under inert environment, and wet and solid-state chemical reactions have been reported to create oxygen vacancies in metal oxides [74,119,153]. Doping suitable elements, both metallic (Cu, Fe, La, etc.) and non-metallic (C, N, S, P, etc.), is a proven method for introducing oxygen vacancies in metal oxides, which can be carried out either during synthesis or by post-synthesis treatment. For example, doping of non-metallic elements in TiO2 has been reported as an effective approach to producing oxygen vacancies [153]. Wang et al. [159] increased the oxygen vacancy concentration in CeO2−x by doping Cu to it. Cu doping stabilized the pre-existed vacancies in CeO2−x and resulted in improved and sustained photocatalytic activity in CO2 reduction. Co-doping or self-doping has also been reported. In co-doping, any two or more combinations of metals, metal/non-metal or non-metal/non-metal, can effectively dope and create oxygen vacancies [160,161]. Self-doping, on the other hand, occurs intrinsically within the metal oxide structure. Such structural doping can be achieved by other oxygen vacancies creation methods such as treatment with plasma, H2 or via redox reactions [162].
Solid-state and wet chemical redox reactions can produce metal oxides with sufficient oxygen vacancies, applying various treatments or reactions [153,163]. In the solid-state process, the reaction uses gaseous or solid reductants such as graphene, H2, NH3, S, CaH2, NaH, and LiH at high temperatures. The wet reaction is carried out in the liquid phase in the presence of a suitable reducing agent such as NaBH4 at room temperature or by using the hydrothermal process. The mechanism involves decreasing the oxidation states of the metal cations during the redox reactions, leading to the formation of oxygen vacancies. In addition, oxygen vacancies can be created in metal oxides by decreasing the particle size of the bulk material [56,164]. Guo et al. [164] introduced oxygen vacancies in Bi2Sn2O7 nanoparticles by decreasing the nanoparticle size to ~4 nm via the solution chemistry synthesis technique. The formation of oxygen vacancies was confirmed by an obvious increase in the visible light absorption according to the UV–visible spectroscopy results. The obtained oxidic nanoparticles with abundant oxygen vacancies exhibited an 8-fold enhanced photocatalytic performance compared with the bulk Bi2Sn2O7 in CO production from CO2 in pure water. Layered double hydroxide (LDH) nanosheets were synthesized via the templated hydrothermal methods (inverse microemulsion technique or controlled hydrolysis) [56]. These methods decreased the thickness of the Zn-containing LDH nanosheets from 5 μm to 40 nm, with platelet thicknesses of ≈2.7 nm. A combination of several characterization results (EXAFS, positron annihilation spectrometry, ESR, and DFT calculations) concluded that oxygen vacancies were created with decreasing LDH thickness. The created oxygen vacancies formed coordinatively unsaturated Zn with the nanosheets represented as Zn+–Vo complexes that serve as trapping sites for the efficient adsorption of CO2 and H2O molecules, promoting photoinduced charge separation, thus significantly improving the catalytic activity for CO2 photoreduction. These illustrations demonstrate the possibility of engineering metal oxide catalysts with abundant oxygen vacancies for enhanced activity for CO2 reduction.
Thermal treatment of metal oxides in reducing gases (H2, Ar or vacuum) at a high temperature is a proven strategy to generate or enrich oxygen vacancies in metal oxides. The process ejects some lattice oxygen from the crystal and effects a change in the bulk phase. This method is very easy and efficient because the oxygen vacancy concentration can be easily tuned by simply controlling either the annealing temperature or reducing gas flow rate during treatment [163,165]. Ye et al. [154] applied a zeolite imidazolate framework-8 (ZIF-8)-derived ZnO as the carrier and synthesized carbon-modified CuO/ZnO catalyst with high oxygen vacancy content for CO2 hydrogenation to methanol. They revealed that the pyrolysis temperature of ZIF-8 affected the surface oxygen defects of ZnO, and the CuO/ZnO catalyst (pyrolyzed at 400 °C) achieved the best CO2 conversion and methanol selectivity due to the presence of more oxygen vacancies and carbon modification. Oxygen vacancies were engineered in bulk Bi24O31Cl10 by thermal annealing at 250 °C under an inert atmosphere consisting of H2/Ar (10/90 v/v) [148]. In the process, H2 interacted with the lattice oxygen, forming oxygen vacancies, which created a new level near the conduction band minimum, enabling a fast charge transfer and high carrier density (Figure 8a,b). As shown in Figure 8e, the existence of oxygen vacancies led to a reduction in the energy barrier for the *COOH intermediates formation during CO2 photochemical reduction. The photocatalytic activity of Bi24O31Cl10 for photoreduction of CO2 to CO was enhanced. CO was generated at a rate of 0.9 mmol h−1 g−1, which is 4 times higher compared with that of bulk Bi24O31Cl10 [148]. A controlled thermal process was adopted to prepare different indium oxide InOx nanoribbons (NRs) with tunable O-vacancy: P-InOx NRs, O-InOx NRs, and H-InOx NRs. The H-InOx NRs rich in O-vacancy showed enhanced performance for the electrocatalytic CO2 conversion to HCOOH with the selectivity up to 91.7% [72].
In the plasma method, the material is subjected to high-energy ion (such as Ar+, N2+, H2O+) bombardment [166]. Metal oxides bombardment with these high-energy ions generates oxygen vacancies on the surface. An advantage is that the plasma treatment is fast and can effectively etch the oxide to expose more surface sites and selectively remove oxygen from the surface to produce oxygen vacancies. Geng et al. [74] introduced oxygen vacancies in ZnO nanosheets via H2 plasma treatment for the electrochemical reduction of CO2. This induced the formation of a new defect level around the valence band maximum, where abundant localized electrons accumulation caused increasing charge density. The new electronic structure promoted the activation of CO2, and the ZnO nanosheets with rich oxygen vacant sites (Vo-rich ZnO nanosheets) exhibited a current density for CO production of −16.1 mA cm−2 with a Faradaic efficiency of 83% at −1.1 V versus RHE. The oxygen vacancies concentration can be controlled by the plasma treatment time.
The photochemical reactions for generating oxygen vacancies involve directing a photo source to the synthesis reaction system, as shown in Figure 9 [119]. Hezam et al. [119] used sunlight as the energy source to effect exothermic combustion reaction between Ce-(NO3)3·6H2O and C2H5NO2. The presence of the light source improved the oxygen vacancies generation in CeO2 compared with CeO2 synthesized in the conventional way, without sunlight.
The Roles of Oxygen Vacancies
- (1)
- CO2 adsorption and dissociation, creation of binding and active sites
In the adsorption and dissociation of CO2 on the anatase TiO2 (101) surface, by dispersion-corrected DFT calculations, Sorescu et al. [137] found that CO2 adsorbed at a fivefold coordinated Ti site in a skewed configuration on an oxidized surface. The presence of a bridging oxygen defect allowed for the creation of strong binding configurations. Lee et al. [139] found that CO2 was preferentially adsorbed at the oxygen vacancy defects on the TiO2 (110) surface. During the CO2 reduction process, one oxygen atom of CO2 filled an oxygen vacancy, decreasing the surface concentration. Oxygen vacancies provided active sites and increased the CO2 adsorption energy, promoting CO2 adsorption and activation on the photocatalyst surface. It was also found that subsurface oxygen vacancy enhanced the binding of CO2 molecules to the surface, and the CO2 dissociation from the defect sites was exothermic with a barrier of less than 21 kcal/mol [70]. CO2 adsorbed directly at the oxygen vacant sites of Zn2GeO4 dissociated into CO and O [117]. As the oxygen vacancies can be healed by oxygen atoms released during the dissociation process, the dissociative adsorption of CO2 on the oxide followed a stepwise mechanism that could be described as: CO2 + Vo → CO2δ−/Vo → COad + Osur, where Vo is the oxygen vacancy.
Huygh et al. [150] studied the adsorption and activation of CO2 on the fully oxidized and reduce anatase TiO2 (001) surfaces using DFT calculations with long-range dispersion energy corrections (Figure 10a,b). They found the monodentated carbonate-like structure to be the most stable adsorption configuration for the fully oxidized surface. CO2 dissociation was not observed on the stoichiometric anatase TiO2 (001) surface. The introduction of oxygen-vacant defects gave rise to new highly stable adsorption configurations with stronger C–O bond activation. These reactions caused the formation of a CO molecule, which will easily desorbed, and the reduced surface became oxidized.
By DFT slab calculations, Pan et al. [151] studied the effects of hydration and oxygen vacancy on CO2 adsorption on the β-Ga2O3 (100) surface. Adsorption of CO2 on the perfect dry β-Ga2O3 (100) surface was slightly endothermic, with an adsorption energy of 0.07 eV, which resulted in a carbonated species. The presence of oxygen vacancies promoted the adsorption and activation of CO2. On the most stable CO2 adsorption configuration, one of the oxygen atoms of the adsorbed CO2 occupied the oxygen vacancy site with low adsorption energy (−0.31 eV). Feng et al. [167] synthesized a series of GaZrOx catalysts by the evaporation-induced self-assembly (EISA) method and proposed the Zr3+–OV–Ga–O species (OV represents the oxygen vacancy) on the GaZrOx surface as the active site (Figure 10c). The synergistic effect stems from neighboring Ga–O and Zr3+–OV sites. H2 dissociated on the Ga–O sites to produce Ga–H and OH species, while CO2 was trapped by the oxygen vacancies and activated by electron transfer from the Zr3+ ions. Liu et al. [47] studied the activation of CO2 on the defective surface of Cu(I)/TiO2−x using the in situ diffuse reflectance infrared Fourier transformed spectroscopy (DRIFTS). They observed the formation of CO2− species upon the transfer of an electron to CO2. The anionic CO2− species further dissociated into CO on a partially oxygen-depleted Cu(I)/TiO2−x surface obtained by thermal annealing in a non-reactive atmosphere. The dissociation process was enhanced by the surface Cu+ species on the catalyst. The defective surface also delivered the electronic charge for the formation of Ti3+. It was observed that the dissociation of CO2− in the absence of light related to the surface oxygen vacancies that provided electronic charge and sites for the adsorption of oxygen atoms from CO2. DFT studies also confirmed that the adsorption of CO2 at the oxygen vacancy site over the Zn-terminated ZnO (0001) surface occurs by inserting one of the O atoms of CO2 into the vacancy, resulting in the formation of a bent CO2 species [168]. Singh et al. [169] investigated the role of oxygen vacancies and basic site density in tuning methanol selectivity over Cu/CeO2, Cu/ZnO, and Cu/ZrO2 catalysts during CO2 hydrogenation. The strong basic sites for CO2 adsorption over catalyst surface were attributed to unsaturated, low coordination oxygen atoms (derived from oxygen vacancies), the oxygen vacancy concentrations of Cu/CeO2 catalyst was higher than those of Cu/ZnO and Cu/ZrO2 catalysts, so the Cu/CeO2 catalyst exhibited more than 90% methanol selectivity at 220 °C and 30 bar. The adsorption capacity of m-ZrO2 was increased due to the high concentration of oxygen vacancy on its surface, which led to the higher catalytic activity of Cu/m-ZrO2 compared with that of Cu/t-ZrO2 [170]. This benefited the activation of CO2, thus improving the catalytic activity for methanol synthesis. Martin et al. [171] synthesized In2O3/ZrO2 catalyst and observed the catalyst had high activity, high stability, and high selectivity due to the possession of a large number of oxygen vacancies. It was difficult to form oxygen vacancy on the Pt/SiO2 interface, and also weak CO2 adsorption was observed due to an obvious lack of the oxygen vacancies. However, CO2 adsorbed strongly on TiO2 due to the presence of oxygen vacancies on the TiO2 surface [172].
- (2)
- Selection of reaction pathway, stabilization of key intermediates and electronic structure modification
CO2 hydrogenation to methanol on In2O3 surface with an oxygen vacancy was investigated by Ye et al. [152] using periodic DFT calculations. Different kinds of oxygen vacancies were identified with dissimilar energy states. On the surface with the highest energy, one of the O atoms of the CO2 molecule filled in the site upon adsorption. The hydrogenation process that proceeded passed through the HCOO* intermediate. Further hydrogenation of HCOO* formed the C–H bond following the C–O bond scission, resulting in H2CO* and hydroxyl. This step was slightly endothermic with a barrier of 0.57 eV but both thermodynamically and kinetically favorable. These observations confirm that the oxygen vacancy on the In2O3 (110) surface assists CO2 activation and hydrogenation and also stabilizes the key intermediates (HCOO*, H2COO*, and H2CO*) involved in methanol formation. The methanol molecules healed the oxygen vacant sites by replenishing them (Figure 11) with the assistance of H2, sustaining the catalytic cycle. Oxygen vacancy catalyzed the formate dissociation to methanol, which was the rate-determining step on Ru/CeO2 catalyst. Moreover, the activation temperature for the formate route was much lower than that for the CO route (150 °C vs. 250 °C), which shows the advantage of oxygen vacancy in promoting the rate-determining step [173].
The introduction of oxygen vacancies into ZnO nanosheets created a new defect level around the valence band maximum where abundant localized electrons accumulated. This formed a new electronic structure that benefited the activation of CO2 as revealed by the lower Gibbs free energy and lower energy barrier of the rate-determining step than that of the ZnO slab for the formation and activation of *COOH intermediates [74]. The Bi2Sn2O7 NPs with oxygen vacancies possess the ability to back donate an electron and optimize the electronic structure for effective CO2 activation as proven by DFT calculations, revealing through the catalytic performance efficient stabilization of the *COOH intermediates, and energy barrier reduction for CO desorption which was the determining step [164]. Abundant oxygen vacancies in Bi24O31Cl10 improved separation of electron–hole pairs, enhanced charge density, and lowered activation energy for the formation of intermediates during the photocatalytic CO2 reduction that led to an optimized CO2 conversion efficiency [148]. The photocatalytic performance (enhanced light absorption and improved charge carrier separation) of CeO2 nanoparticles (12–18 nm) was improved in the presence of higher concentration of oxygen vacancies, which suppressed the electron–hole recombination as studied using photoluminescence, and confirmed with photocurrent measurement results. The surface oxygen vacancies modified the surface state above the valance band, as seen in the upward shift relative to the vacuum (Figure 12). In the photoelectrochemical CO2 reduction system, introducing oxygen vacancies into the lattice of a semiconductor catalyst can alter its intrinsic electronic properties and band gap, promote the transfer of electrons and facilitate adsorption and activation of CO2 [139].
3.3.4. The Roles of Solvent in CO2 Transformation
There have been interesting attempts to replace water with other solvents as reductants in photocatalytic CO2 reduction. For example, as described in a study by Srinivas et al. [174], CO2 reduction with CdS in various solvents (water, methanol, ethanol, and 1-propanol of different dielectric constants of 80, 33, 24.3, and 20.1, respectively) was experimentally investigated. Formate and CO were observed as the major products in various solvents deployed as reductants in the photocatalytic reduction of CO2 using TiO2 nanocrystals embedded in SiO2 matrices as the photocatalyst [175]. The characteristics of the solvent (defined with polarity/dielectric constant) and CO2 adsorption/dissolution will determine the selectivity toward the product. Based on the polarity and dielectric constant, solvents with low dielectric constant or low polarity could in situ generate CO2− anionic radicals that were strongly adsorbed on the surface of the catalyst through the carbon atom because the CO2− anion radicals were poorly dissolved in the solutions [174,175]. This will lead to the formation of CO as the major product. In solutions with high dielectric constants, such as water, the CO2− anion radicals are well stabilized in the solvent, resulting in a weak interaction with the photocatalyst surface. In this case, the carbon atom tends to be attracted to a proton, exhibiting a high propensity toward formate formation [175]. However, water is often deployed for the photocatalytic (artificial photosynthesis) and, in some cases, electrocatalytic CO2 reduction reactions.
The change in the adsorbed surface species and surface chemical state of catalysts has been correlated with the moisture or humidity change during chemical reactions [107]. Adsorbed water takes part in the reaction chemistry of CO2 on metal oxide surfaces. Water molecules involve in the CO2 reduction process by providing hydroxyl ions and protons that facilitate the electron transfer [176]; thus, H2O is beneficial for hydroxylation of the catalyst surface. In addition, the adsorption and dissociation of H2O to protons on the catalyst surface can activate deep reduction, leading to highly reduced state hydrocarbon products [32]. On the hydroxylated metal/metal oxide surfaces, CO2 can bind as bicarbonates and carbonates in monodentate and bidentate modes [114]. Co-adsorbed water was essential for activating CO2 by forming surface adsorbed intermediates during methanol synthesis on metal/oxide catalysts [177]. Most photocatalytic CO2 reduction reactions proceed in the presence of H2O. Water participates in the reaction by capturing photogenerated holes to generate O2, thus reducing the recombination rate of photogenerated holes and electrons. Under this condition, the utilization efficiency of photogenerated electrons can be improved [32]. Although water is often used as the reducing agent in photocatalytic process, pure water is hardly used. The use of NaOH-containing water solution is practiced due to undisputed benefits that include: (i) facilitating the dissolution of adsorbed CO2 due to its high affinity with caustic NaOH solution, and (ii) slowing the electron-hole pairs recombination, leading to more utilization of surface electrons that facilitates CO2 reduction. The OH ions in aqueous basic solutions act as strong hole scavengers and readily form •OH radicals [174].
In the absence of co-adsorbed H2O, the in situ H2O formation is possible via the following mechanism. In the presence of H2 as a reducing agent, over the surface of supported metals/metal oxides, H2 dissociates at the metal sites or metal-oxide interface to adsorbed H* atoms, which then migrate to the active sites and react with the surface oxygen atoms of metal oxide or interact with the adsorbed CO2 species in a stepwise fashion to form water [178]. Therefore, the hydroxylation of the catalyst surface is still possible when H2 is deployed as a reductant, and the facile and efficient activation of hydrogen is necessary. Using DFT, CO2 adsorption was investigated on the In2O3 (110) surface for methanol synthesis to understand the benefits of H2 and H2O presence. Results revealed the hydroxylation of the catalyst surface. H2 dissociated homolytically on the oxide surface with a high surface coverage, leading to the formation of water. This induced the formation of surface OH-groups, which reduced under-coordinated In atoms at the surface, giving rise to In ion sites and enhanced the formation of oxygen vacancies observed at high hydrogen coverages [179]. However, the oxygen vacancies formed on In2O3 (110) surfaces did not facilitate CO2 adsorption during the reaction. CO2 adsorption was facilitated on hydroxylated surface sites, leading to activation by accepting one electron from the In2O3 (110) surface [179]. Su et al. [112] observed the promotion of a reaction with adsorbed oxygen which formed H2O after the introduction of H2 during the RWGS, although the adsorbed H2O desorbed from the surface at high temperatures. It was also found that adsorbed CO produced more in the presence of H2.
The effect of H2O on Ni surfaces pre-exposed to CO2 was investigated by Cai et al. [107]. As observed using ambient air XPS measurements (Figure 13c,d), a new peak evolved in the O 1s spectra at 535.3 eV due to the gas-phase H2O, that transformed to surface hydroxyl and adsorbed water with increased concentrations as revealed by the enhanced peak intensities at 530.9 and 532.6 eV, respectively. It obviously indicates that adsorption and dissociation of H2O occurred on the Ni surfaces, producing H atom and OH, which further reacted with CO2 and other surface species to produce CO2 hydrogenation intermediates and product species (formate, methoxy/CO*, and carbonate species) as shown in Figure 13a,b.
The interaction of CO2 with a ZrO2 model surface (a O–Zr–O trilayer grown on Pt3Zr (0001)) with and without the presence of H2O was measured by in situ near ambient (atmospheric) pressure XPS (NAP-XPS) and infrared reflection absorption spectroscopy (IRAS). No interaction was observed with the ZrO2 model surface at room temperature upon exposure to pure CO2 up to 3 × 10−2 mbar. Co-adsorption of CO2 with H2O led to the formation of various carbonaceous surface species. This means that activation of CO2 occurred near ambient pressures under the humid condition due to surface hydroxylation that led to the formation of surface species identified by the combined NAP-XPS and IRAS measurements [177]. On reduced CeOx-TiO2 composite surfaces, the interaction of CO2 and H2O was thermodynamically favorable at multiple sites. H2O molecules were observed to spontaneously dissociate at the oxygen vacant sites, generating surface hydroxyl groups that reacted with CO2 to form carbonaceous species—carboxylate or bicarbonate species—according to the NEXAFS results [130]. The apparent correlation between CO2 and water was investigated via infrared experiments performed using regular and deuterated water to pre-cover the surface prior to the CO2 adsorption. It was found that the degree of hydroxylation of the surface plays a crucial role in CO2 adsorption on regular (Fetet1) tetrahedrally terminated magnetite (Fe3O4) surface, leading to the formation of surface bicarbonates [144]. Surface hydroxide sites in hydroxylated indium oxide (In2O3−x(OH)y) acted as Lewis base for CO2 activation [180]. In the presence of co-adsorbed water, metal oxides (FeOx, Al2O3, and TiO2) exhibited high reaction rates for CO2 adsorption under ambient conditions and, thus, can improve the CO2 adsorption kinetics [176].
Although the positive effects of moisture have been discussed, an experimental report revealed a 20% decrease in methanol yield under standard methanol synthesis conditions when water was co-fed into an H2/CO2 mixture over In2O3 surface [181]. The co-adsorption of H2O suppressed the interfacial interaction between CO2 and Ni surface [107]. On defective β-Ga2O3 (100) with rich oxygen vacancy, water spontaneously enhanced dissociative adsorption of CO2 at the oxygen vacancy site, with reaction energy of −0.62 eV. These results indicate that the co-existence of water and CO2 in the adsorption system imposes competition with CO2 for the oxygen vacancy sites, which negatively impacts CO2 chemisorption and conversion [151].
3.3.5. General Methods for Modifying Metal Oxide Surface Structure for CO2 Transformation
Several approaches can be applied to modify the structure of metal oxides for the facile adsorption and dissociation of CO2 molecules, mainly, through tailoring their physicochemical (e.g., improving surface basicity) [182,183] and electronic properties (e.g., doping and creating defect sites) [182].
- (i)
- Insertion of acidic and basic surface sites. The basic sites on the catalyst surface can facilitate CO2 adsorption because CO2 is an acidic compound whose interaction with the catalyst surface will be enhanced with a basic surface. The basic sites of metal oxide catalysts can be generated by pre-treatment with basic promoters such as La2O3, K2O, Na2O, and MgO [183]. Tian et al. [182] reported that introducing potassium into iron oxide catalyst increased the surface basicity. Thus, alkali metals can be incorporated into the metal oxides to tune the concentration of surface basic sites. The basic sites enhance the activation of acidic CO2 and also help to limit carbon deposition. The overall benefit is improvement in catalytic activity and stability. For example, in the CO2 dry reforming of methane, the catalytic reaction was initiated by an acid–base interaction [183]. On the In2O3−x(OH)y surface, comprising frustrated Lewis pairs (FLP) (A frustrated Lewis pairs consists of both Lewis acid and Lewis base that cannot combine to form an adduct due to steric hindrance.), the surface hydroxide site acts as Lewis base and the coordinately unsaturated In surface site acts as a Lewis acid to activate CO2 [180]. The Lewis acid and Lewis base synergistically interact to heterolytically dissociate H2 adsorbed on the In2O3−x(OH)y surface, forming protonic surface FLP sites that can capture and convert CO2 to CO and H2O. The surface Lewis basicity can be tuned by the nature of the metal site, which can be controlled by the size, charge, coordination number, geometry, and electronegativity of the metal in a particular oxidation state [114]. Surface basic sites can be characterized using CO2-TPD. Typically, desorption at high temperatures signifies the presence of strong basic sites. The CO2 adsorption strength on metal oxides has been related to the basicity, improving as the concentration increases. However, too strong adsorption may result in the formation of undesirable intermediate species such as surface carbonate and bicarbonate species.
- (ii)
- Metal oxide doping. Doping metal oxides with metallic elements can optimize their electronic and geometric structures and enhance catalytic performance. Alkali metals doping can modify the electronic structure by increasing the concentration of electron-withdrawing groups such as surface hydroxyl groups or reducing the kinetic barrier for CO2 dissociation [184]. Moreover, oxides with electron-rich defect centers may interact readily with CO2 by donating an electron. These, in turn, will facilitate surface reaction activity, boosting catalytic efficiency for CO2 activation. The list is not exclusive to alkali metals, as some transition metals have the potentials to tune the electronic characteristics of the doped oxide. Doping transition metal oxide to the based oxide can as well tune the concentration of oxygen vacancies
- (iii)
- Defect engineering. It involves creating or adjusting surface oxygen vacancies or hydroxyl groups in an oxide. The presence of oxygen defective sites can similarly impact the surface electronic property. Evidence has linked the activity of an oxide catalyst with surface defects. For example, it was demonstrated that the activity of MnO2 in HCHO oxidation correlated with the concentration of surface defects [185]. CeO2 nanoparticles with a high concentration of oxygen vacancies had high photocatalytic performance in converting CO2 to methanol (0.702 μmol h−1 g−1) [119]. Furthermore, oxygen vacancies are sites for adsorption and anchoring of CO2 molecules. Consequently, surface adsorption and reactivity of adsorbates, including H2 and H2O, would be facilitated in the presence of extra electrons in the vicinity of oxygen vacancies. The amount of defective sites in a metal oxide can be controlled by oxidative treatment during the catalyst synthesis and catalytic reactions.
- (iv)
- Generation of surface hydroxyls. The surface hydroxyl groups participate in CO2 hydrogenation by incorporating hydrogen atom into CO2, facilitating CO2 activation and accelerating reaction rate. Mechanistic studies evidenced the surface hydroxyl species on SiC quantum dots (QDs) to directly participate in CO2 hydrogenation through the addition of H atoms of hydroxyl groups into CO2 to form HCOO* [186]. The surface hydroxide site on the In2O3−x(OH)y surface acted as a Lewis base that interacted with CO2 [180].
- (v)
- Mixed metal oxides or composites catalysts can yield good catalytic performances. Introducing a second oxide component can improve adsorption, increase the effective oxygen vacancies, and present an interfacial surface for reactions (adsorption/activation and desorption). When preparing a mixed-metal oxide catalyst, consideration should be given to factors that can improve catalytic performance, such as elemental composition since catalytic activity can be a function of how elements in the composite are combined. The deposition of sub-monolayer amounts of a second oxide over a host oxide can create nanostructures that can enhance the overall catalytic properties of the composite system [114,187].
- (vi)
- Forming metallic–non-metallic hybrid catalysts. These kinds of catalysts have high surface areas and improved adsorption properties. Carbon materials, such as carbon nanotubes (CNTs) and graphene oxides (GOs), and metal oxides (e.g., TiO2, CeO2, Al2O3, ZrO2, In2O3, Ga2O3, and NiO) can be grafted to form hybrid nanostructured materials [154,188,189,190]. Carbon materials possess very high surface areas, whereas metal oxides are endowed with rich oxygen vacancies. Combining materials with these features can result in a unique hybrid material with superior catalytic performance toward CO2 reduction reactions. The high surface area will enhance adsorption complemented by oxygen vacancies, which are also sites for CO2 activation on oxide catalysts. Modification of CuO–ZnO–ZrO2 with GO increased the adsorption capacities of both CO2 and H2, resulting in an improved catalytic activity (methanol selectivity) in methanol synthesis than the GO-free catalyst [188]. The electronic structure of the traditional methanol synthesis catalyst (Cu–ZnO–Al2O3) was altered by adding the N-doped graphene, which provided a synergistic effect for methanol synthesis [189]. Incorporating metal oxides to single-wall CNT electrode can form a hybrid structure with an increase in specific surface area, which also improved electrical conductivity and charge transfer [191,192]. Moreover, doping carbon into photocatalysts can enhance light absorption and improve the photothermal conversion efficiency by reducing the energy for oxygen vacancy formation, thus generating a high concentration of active sites [190]. Therefore, hybrid nanostructured materials with good electronic and charge transfer properties can be explored for both thermochemical and electrochemical reduction of CO2.
- (vii)
- Stabilization of metal oxide surface. Reduced oxides are more susceptible to react with CO2 [118,193]. Stabilizing the reduced surface can be achieved by creating a conducive environment for reactions such as forming oxide–metal or oxide–oxide interfaces [128,187,194]. The synergistic properties associated with these interfaces can improve the surface chemistry of metal oxides and could be beneficial for synthesizing efficient catalysts for CO2 transformation.
4. Conclusions
Controlling the emission of CO2 by converting it to products of economic value is highly attractive. However, CO2 is a kinetically stable molecule that requires high energy input for the C–O bond breaking. Its proper activation can reduce the high energy barrier substantially, easing conversion by various processes. The CO2 activation is an important step that precedes the conversion of CO2 to chemicals and fuels. It can be effected in the presence of a catalyst by decreasing the O–C–O angle from 180 degrees (bending), weakening and elongation of the C–O bonds, polarizing the charges on C and O by charge/electron transfer, and redistribution of charges on the CO2 molecule. Upon accepting an extra electron from a substrate, the neutral CO2 molecule forms an anion with a full charge (CO2−) or partial charge (CO2δ−). However, a recent study has proposed that just a charge transfer is insufficient to activate CO2 molecules on metal oxide catalysts, but the binding of an O atom to a surface metal cation weakens and elongates the molecular C–O bond.
Some metals and metal oxides are efficient catalysts for CO2 conversion reactions; thus, they should be good for CO2 activation. In general, metal nanoparticles serve as active sites for electron transfer, with certain factors such as change in morphology of metal particles, nanoparticle size, adsorption mode and configuration, and chemical ordering as the CO2 activation marker. The interaction of CO2 with pure metals is rather weak but can be improved by incorporating promoters (e.g., alkali metals) with low electronegativity. Metal oxide nanoparticles are utilized as supports or directly as catalysts for the CO2 conversion. Their surfaces comprise both metal (Mn+) and oxygen (O2−) ions, which can act as active sites for CO2 activation. They can activate CO2 by coordinating to one or two adjacent metal sites through the terminal oxygen atoms of the CO2 or interaction of the carbon atom of CO2 with surface oxygen sites. A particularly interesting feature in metal oxides is the oxygen vacancies.
Oxygen vacancies are of great benefits to the overall reaction thermodynamics and kinetics and can greatly enhance the adsorption energy of CO2 molecules, promoting interaction of CO2 with the oxide surface. Indeed, oxygen vacancies play vital roles in the overall CO2 conversion process, ranging from electronic structure modification, selection of reaction pathway, and stabilization of key intermediates to the creation of binding surface, sites for CO2 adsorption, and dissociation. Although being an integral part of reducible oxides, the concentration of oxygen vacancies can be increased or created in metal oxide catalysts either in situ during reactions or incorporated by external methods, including thermal annealing at high temperatures, doping, plasma-assisted techniques, photo-irradiation, and solid-state chemical reactions during synthesis or by post-synthesis techniques.
The surface hydroxyl groups, which can be introduced in metal oxide catalysts in the presence of moisture, also present significant CO2 activation enhancement by facilitating interaction and providing additional binding sites.
Several strategies are applicable to modify metal oxide catalysts for CO2 activation and conversion. Creating more basic surface sites can stimulate surface reaction with acidic CO2. Other strategies include elemental doping and defect engineering. Surface defect sites are good adsorption sites for CO2 on metal oxides. Doping with low electronegative elements can enrich the surface with electrons to easily transfer to CO2 upon adsorption. Like the case of basic sites, generating more hydroxyl groups to make the surface hydrophilic can boost the catalytic performance of metal oxides, similar to those of metal hydroxides or layered double hydroxides. Hybrid catalysts, such as metal oxide/carbon material and composite oxide catalysts, tend to improve the performance of the resulting system through the synergism of the individual components. In these processes, it is possible to stabilize the metal surface by forming oxide–metal or oxide–oxide interfaces. The synergetic properties associated with such interfaces can improve the surface chemistry for reactions. In view of the catalysts discussed in this work, metal oxides with large numbers of oxygen vacancies exhibit excellent performance for CO2 adsorption and activation. In the present, researchers have demonstrated the ability of pure (e.g., In2O3) or doped (ZnO-ZrO2) metal oxides and metal–organic framework to reduce CO2 to methanol, and of course, some other products. However, challenges remain due to the relatively low catalytic activity and selectivity of these materials. Perhaps, their bulk forms are presented with these limitations. Furthermore, other factors, such as the strong metal-support interactions (SMSI) for supported metal catalysts on the reducible oxide supports, should be considered, as the reduced support oxide can migrate and cover the active metal particles, significantly impacting the electronic and interface structure of the catalysts [195,196].
Specially designing and engineering surface functionalities in these materials, including inserting a controllable amount of oxygen vacancies in the right proportion and surface hydroxyls, is capable of tuning the catalytic properties, in which activation of CO2 molecule should be a topmost consideration. The future offers great opportunities to develop even more active heterogeneous catalysts for the various CO2 reduction reactions. The possible structural transformations of the catalysts, which remain elusive, can be unveiled to address the complexities involved in these reactions fully. Identifying the catalyst structural defects, their formation or transformation during the reaction, and their contribution to the overall activity is still a challenging task, yet of significant importance. Theoretical models can provide predictions on the catalyst system, and such methods should be deployed to model new metal oxide catalysts with incorporated functionalities.
Author Contributions
U.J.E. and C.Z. wrote the manuscript; U.J.E. organized and revised the manuscript; Z.Z. provided the original thoughts about the content and structure of the manuscript and technical inputs; Z.Z. has structured, modified, and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
2021 Guangdong Province Key discipline fund, Guangdong Province key Laboratory of Energy Conversion Materials and Technology and 2020 Li Ka Shing Foundation Cross-Disciplinary Research Grant. This work is financially supported by the Guangdong Province Key discipline fund (GTIIT) in 2021, Guangdong Province key Laboratory of Energy Conversion Materials and Technology, by the GTIIT Project KD2000079, and by the 2020 Li Ka Shing Foundation Cross-Disciplinary Research Grant (2020LKSFG09A).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sun, R.; Liao, Y.; Bai, S.-T.; Zheng, M.; Zhou, C.; Zhang, T.; Sels, B.F. Heterogeneous catalysts for CO2 hydrogenation to formic acid/formate: From nanoscale to single atom. Energy Environ. Sci. 2021, 14, 1247–1285. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Li, Z.; Tang, C.; Feng, Z.; An, H.; Liu, H.; Liu, T.; Li, C. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 2017, 3, e1701290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, R.-P.; Li, Q.; Gong, W.; Wang, T.; Razink, J.J.; Lin, L.; Qin, Y.-Y.; Zhou, Z.; Adidharma, H.; Tang, J. High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl. Catal. B Environ. 2020, 268, 118474. [Google Scholar] [CrossRef]
- Lim, E.; Heo, J.; Zhang, X.; Bowen, K.H.; Lee, S.H.; Kim, S.K. Anionic Activation of CO2 via (Mn–CO2)–Complex on Magic-Numbered Anionic Coinage Metal Clusters Mn–(M= Cu, Ag, Au). J. Phy. Chem. A 2021, 125, 2243–2248. [Google Scholar] [CrossRef]
- Alvarez-Garcia, A.; Flórez, E.; Moreno, A.; Jimenez-Orozco, C. CO2 activation on small Cu-Ni and Cu-Pd bimetallic clusters. Mol. Catal. 2020, 484, 110733. [Google Scholar] [CrossRef]
- Álvarez, A.; Borges, M.; Corral-Pérez, J.J.; Olcina, J.G.; Hu, L.; Cornu, D.; Huang, R.; Stoian, D.; Urakawa, A. CO2 activation over catalytic surfaces. ChemPhysChem 2017, 18, 3135–3141. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Hatakeyama, M.; Wang, Y.; Ogata, K.; Fujii, K. A basic quantum chemical review on the activation of CO2. In Advances in CO2 Capture, Sequestration, and Conversion; ACS Publications: Washington, DC, USA, 2015; pp. 123–134. [Google Scholar]
- Mendes, P.C.; Verga, L.G.; Da Silva, J.L. Ab initio screening of Pt-based transition-metal nanoalloys using descriptors derived from the adsorption and activation of CO2. Phys. Chem. Chem. Phys. 2021, 23, 6029–6041. [Google Scholar] [CrossRef]
- Koppenol, W.; Rush, J. Reduction potential of the carbon dioxide/carbon dioxide radical anion: A comparison with other C1 radicals. J. Phys. Chem. 1987, 91, 4429–4430. [Google Scholar] [CrossRef]
- Das, S.; Pérez-Ramírez, J.; Gong, J.; Dewangan, N.; Hidajat, K.; Gates, B.C.; Kawi, S. Core–shell structured catalysts for thermocatalytic, photocatalytic, and electrocatalytic conversion of CO2. Chem. Soc. Rev. 2020, 49, 2937–3004. [Google Scholar] [CrossRef]
- Modak, A.; Bhanja, P.; Dutta, S.; Chowdhury, B.; Bhaumik, A. Catalytic reduction of CO2 into fuels and fine chemicals. Green Chem. 2020, 22, 4002–4033. [Google Scholar] [CrossRef]
- Etim, U.J.; Semiat, R.; Zhong, Z. CO2 Valorization Reactions over Cu-Based Catalysts: Characterization and the Nature of Active Sites. Am. J. Chem. Eng. 2021, 9, 53–78. [Google Scholar] [CrossRef]
- Green, A.E.; Justen, J.; Schöllkopf, W.; Gentleman, A.S.; Fielicke, A.; Mackenzie, S.R. IR Signature of Size-Selective CO2 Activation on Small Platinum Cluster Anions, Ptn−(n= 4–7). Angew. Chem. 2018, 130, 15038–15042. [Google Scholar] [CrossRef]
- Xie, S.; Zhang, Q.; Liu, G.; Wang, Y. Photocatalytic and photoelectrocatalytic reduction of CO2 using heterogeneous catalysts with controlled nanostructures. Chem. Commun. 2016, 52, 35–59. [Google Scholar] [CrossRef]
- Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.; Kubiak, C.P. Photochemical and photoelectrochemical reduction of CO2. Annu. Rev. Phys. Chem. 2012, 63, 541–569. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Alvarez, F.J.; Oro, L.A. Homogeneous Catalytic Reduction of CO2 with Silicon-Hydrides, State of the Art. ChemCatChem 2018, 10, 4783–4796. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Dai, Y.; Zhang, J.; Yang, Y. The thermodynamics analysis and experimental validation for complicated systems in CO2 hydrogenation process. J. Energy Chem. 2016, 25, 1027–1037. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. State of the art and perspectives in catalytic processes for CO2 conversion into chemicals and fuels: The distinctive contribution of chemical catalysis and biotechnology. J. Catal. 2016, 343, 2–45. [Google Scholar] [CrossRef]
- Ronda-Lloret, M.; Rothenberg, G.; Shiju, N.R. A critical look at direct catalytic hydrogenation of carbon dioxide to olefins. ChemSusChem 2019, 12, 3896–3914. [Google Scholar] [CrossRef]
- Radhakrishnan, S.G.; Roduner, E. Carbon dioxide activation. In Carbon Dioxide Utilization; Michael, N., Peter, S., Eds.; De Gruyter: Berlin, Germany, 2019; pp. 227–248. [Google Scholar]
- Niu, J.; Ran, J.; Ou, Z.; Du, X.; Wang, R.; Qi, W.; Zhang, P. CO2 dissociation over PtxNi4− x bimetallic clusters with and without hydrogen sources: A density functional theory study. J. CO2 Util. 2016, 16, 431–441. [Google Scholar] [CrossRef]
- Grice, K.A. Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 2017, 336, 78–95. [Google Scholar] [CrossRef]
- Taifan, W.; Boily, J.-F.; Baltrusaitis, J. Surface chemistry of carbon dioxide revisited. Surf. Sci. Rep. 2016, 71, 595–671. [Google Scholar] [CrossRef] [Green Version]
- Freund, H.-J.; Roberts, M.W. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef] [Green Version]
- Mondal, B.; Song, J.; Neese, F.; Ye, S. Bio-inspired mechanistic insights into CO2 reduction. Curr. Opin. Chem. Biol. 2015, 25, 103–109. [Google Scholar] [CrossRef]
- Nolan, M.; Fronzi, M. Activation of CO2 at chromia-nanocluster-modified rutile and anatase TiO2. Catal. Today 2019, 326, 68–74. [Google Scholar] [CrossRef]
- Mondal, K.; Banerjee, A.; Ghanty, T.K. Adsorption and activation of C on Zr n (n= 2–7) clusters. Phys. Chem. Chem. Phys. 2020, 22, 16877–16886. [Google Scholar]
- Weber, J.M. The interaction of negative charge with carbon dioxide–insight into solvation, speciation and reductive activation from cluster studies. Int. Rev. Phys. Chem. 2014, 33, 489–519. [Google Scholar] [CrossRef]
- Park, J.; Cho, M.; Rhee, Y.M.; Jung, Y. Theoretical Study on the Degree of CO2 Activation in CO2-Coordinated Ni (0) Complexes. ACS Omega 2021, 6, 7646–7654. [Google Scholar] [CrossRef]
- Mazheika, A.; Wang, Y.; Valero, R.; Ghiringhelli, L.M.; Vines, F.; Illas, F.; Levchenko, S.V.; Scheffler, M. Ab initio data-analytics study of carbon-dioxide activation on semiconductor oxide surfaces. arXiv 2019, arXiv:1912.06515. [Google Scholar]
- Chang, X.; Wang, T.; Gong, J. CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Fu, J.; Jiang, K.; Qiu, X.; Yu, J.; Liu, M. Product selectivity of photocatalytic CO2 reduction reactions. Mater. Today 2020, 32, 222–243. [Google Scholar] [CrossRef]
- Yang, B.; Wang, L.; Hua, Z.; Guo, L. How CO2 Chemisorption States Affect Hydrogenation Activity. Ind. Eng. Chem. Res. 2019, 58, 9838–9843. [Google Scholar] [CrossRef]
- Megha; Mondal, K.; Ghanty, T.K.; Banerjee, A. Adsorption and Activation of CO2 on Small-Sized Cu–Zr Bimetallic Clusters. J. Phys. Chem. A 2021, 125, 2558–2572. [Google Scholar] [CrossRef]
- Megha; Banerjee, A.; Ghanty, T.K. Role of metcar on the adsorption and activation of carbon dioxide: A DFT study. Phys. Chem. Chem. Phys. 2021, 23, 5559–5570. [Google Scholar] [CrossRef]
- Liu, X.; Kunkel, C.; Ramirez de la Piscina, P.; Homs, N.; Vines, F.; Illas, F. Effective and highly selective CO generation from CO2 using a polycrystalline α-Mo2C catalyst. ACS Catal. 2017, 7, 4323–4335. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Porosoff, M.D. Development of tandem catalysts for CO2 hydrogenation to olefins. ACS Catal. 2019, 9, 2639–2656. [Google Scholar] [CrossRef]
- Zhao, B.; Liu, Y.; Zhu, Z.; Guo, H.; Ma, X. Highly selective conversion of CO2 into ethanol on Cu/ZnO/Al2O3 catalyst with the assistance of plasma. J. CO2 Util. 2018, 24, 34–39. [Google Scholar] [CrossRef]
- Li, Y.; Hui, D.; Sun, Y.; Wang, Y.; Wu, Z.; Wang, C.; Zhao, J. Boosting thermo-photocatalytic CO2 conversion activity by using photosynthesis-inspired electron-proton-transfer mediators. Nat. Commun. 2021, 12, 123. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y. Understanding the reaction mechanism of photocatalytic reduction of CO2 with H2O on TiO2-based photocatalysts: A review. Aerosol Air Qual. Res. 2014, 14, 453–469. [Google Scholar] [CrossRef]
- Montoya, J.H.; Seitz, L.C.; Chakthranont, P.; Vojvodic, A.; Jaramillo, T.F.; Nørskov, J.K. Materials for solar fuels and chemicals. Nat. Mater. 2017, 16, 70–81. [Google Scholar] [CrossRef]
- Chen, S.; Wang, H.; Kang, Z.; Jin, S.; Zhang, X.; Zheng, X.; Qi, Z.; Zhu, J.; Pan, B.; Xie, Y. Oxygen vacancy associated single-electron transfer for photofixation of CO2 to long-chain chemicals. Nat. Commun. 2019, 10, 788. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Das, T.; Kumar, S.; Bhosale, R.; Kabir, M.; Ogale, S. Photocatalytic activation and reduction of CO2 to CH4 over single phase nano Cu3SnS4: A combined experimental and theoretical study. ACS Appl. Energy Mater. 2019, 2, 5677–5685. [Google Scholar] [CrossRef]
- Chu, W.; Zheng, Q.; Prezhdo, O.V.; Zhao, J. CO2 photoreduction on metal oxide surface is driven by transient capture of hot electrons: Ab initio quantum dynamics simulation. J. Am. Chem. Soc. 2020, 142, 3214–3221. [Google Scholar] [CrossRef]
- Indrakanti, V.P.; Kubicki, J.D.; Schobert, H.H. Photoinduced activation of CO2 on Ti-based heterogeneous catalysts: Current state, chemical physics-based insights and outlook. Energy Environ. Sci. 2009, 2, 745–758. [Google Scholar] [CrossRef]
- Yuan, L.; Xu, Y.-J. Photocatalytic conversion of CO2 into value-added and renewable fuels. Appl. Surf. Sci. 2015, 342, 154–167. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, C.; Li, Y. Spontaneous Dissociation of CO2 to CO on Defective Surface of Cu(I)/TiO2–x Nanoparticles at Room Temperature. J. Phys. Chem. C 2012, 116, 7904–7912. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, H.; Andino, J.M.; Li, Y. Photocatalytic CO2 reduction with H2O on TiO2 nanocrystals: Comparison of anatase, rutile, and brookite polymorphs and exploration of surface chemistry. ACS Catal. 2012, 2, 1817–1828. [Google Scholar] [CrossRef]
- Rodriguez, M.M.; Peng, X.; Liu, L.; Li, Y.; Andino, J.M. A density functional theory and experimental study of CO2 interaction with brookite TiO2. J. Phys. Chem. C 2012, 116, 19755–19764. [Google Scholar] [CrossRef]
- He, H.; Zapol, P.; Curtiss, L.A. A theoretical study of CO2 anions on anatase (101) surface. J. Phys. Chem. C 2010, 114, 21474–21481. [Google Scholar] [CrossRef]
- Pan, H.; Gu, B.; Zhang, Z. Phase-dependent photocatalytic ability of TiO2: A first-principles study. J. Chem. Theory Comput. 2009, 5, 3074–3078. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, Y.; Wu, X.; Zheng, H.; Zhao, Z.; Liu, J.; Li, J. Graphene-wrapped Pt/TiO2 photocatalysts with enhanced photogenerated charges separation and reactant adsorption for high selective photoreduction of CO2 to CH4. Appl. Catal. B Environ. 2018, 226, 360–372. [Google Scholar] [CrossRef]
- Lu, L.; Wang, B.; Wang, S.; Shi, Z.; Yan, S.; Zou, Z. La2O3-Modified LaTiO2N Photocatalyst with Spatially Separated Active Sites Achieving Enhanced CO2 Reduction. Adv. Funct. Mater. 2017, 27, 1702447. [Google Scholar] [CrossRef]
- Xu, F.; Meng, K.; Cheng, B.; Yu, J.; Ho, W. Enhanced Photocatalytic Activity and Selectivity for CO2 Reduction over a TiO2 Nanofibre Mat Using Ag and MgO as Bi-Cocatalyst. ChemCatChem 2019, 11, 465–472. [Google Scholar] [CrossRef]
- Xie, S.; Wang, Y.; Zhang, Q.; Deng, W.; Wang, Y. MgO-and Pt-promoted TiO2 as an efficient photocatalyst for the preferential reduction of carbon dioxide in the presence of water. ACS Catal. 2014, 4, 3644–3653. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, G.; Bian, T.; Zhou, C.; Waterhouse, G.I.; Wu, L.Z.; Tung, C.H.; Smith, L.J.; O’Hare, D.; Zhang, T. Defect-rich ultrathin ZnAl-layered double hydroxide nanosheets for efficient photoreduction of CO2 to CO with water. Adv. Mater. 2015, 27, 7824–7831. [Google Scholar] [CrossRef]
- Xiong, Z.; Lei, Z.; Kuang, C.-C.; Chen, X.; Gong, B.; Zhao, Y.; Zhang, J.; Zheng, C.; Wu, J.C. Selective photocatalytic reduction of CO2 into CH4 over Pt-Cu2O TiO2 nanocrystals: The interaction between Pt and Cu2O cocatalysts. Appl. Catal. B Environ. 2017, 202, 695–703. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.; Lu, L.; Zhou, C.; Xin, Z.; Wang, J.; Ke, X.-K.; Sheng, G.; Yan, S.; Zou, Z. Oxygen-vacancy-activated CO2 splitting over amorphous oxide semiconductor photocatalyst. ACS Catal. 2018, 8, 516–525. [Google Scholar] [CrossRef]
- Kim, C.; Hyeon, S.; Lee, J.; Kim, W.D.; Lee, D.C.; Kim, J.; Lee, H. Energy-efficient CO2 hydrogenation with fast response using photoexcitation of CO2 adsorbed on metal catalysts. Nat. Commun. 2018, 9, 3027. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Huang, Y.; Wu, M.; Wang, Y. Electrochemical carbon dioxide splitting. ChemElectroChem 2019, 6, 1587–1604. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.; Chan, K.; Hahn, C. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.J.; McGibbon, R.T.; Bocarsly, A.B. Electrocatalytic Carbon Dioxide Activation: The Rate-Determining Step of Pyridinium-Catalyzed CO2 Reduction. ChemSusChem 2011, 4, 191–196. [Google Scholar] [CrossRef]
- Yang, P.; Zhao, Z.J.; Chang, X.; Mu, R.; Zha, S.; Zhang, G.; Gong, J. The functionality of surface hydroxy groups on the selectivity and activity of carbon dioxide reduction over cuprous oxide in aqueous solutions. Angew. Chem. 2018, 130, 7850–7854. [Google Scholar] [CrossRef]
- Huang, Q.; Li, Q.; Liu, J.; Wang, R.; Lan, Y.-Q. Disclosing CO2 activation mechanism by hydroxyl-induced crystalline structure transformation in electrocatalytic process. Matter 2019, 1, 1656–1668. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO 2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Zhu, D.D.; Liu, J.L.; Qiao, S.Z. Recent advances in inorganic heterogeneous electrocatalysts for reduction of carbon dioxide. Adv. Mater. 2016, 28, 3423–3452. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Xiao, H.; Goddard, W.A.; Cheng, T.; Liu, Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl. Acad. Sci. USA 2017, 114, 6685–6688. [Google Scholar] [PubMed] [Green Version]
- Favaro, M.; Xiao, H.; Cheng, T.; Goddard, W.A.; Yano, J.; Crumlin, E.J. Subsurface oxide plays a critical role in CO2 activation by Cu (111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl. Acad. Sci. USA 2017, 114, 6706–6711. [Google Scholar] [PubMed] [Green Version]
- Li, C.W.; Kanan, M.W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yin, R.; Shao, Q.; Zhu, T.; Huang, X. Oxygen vacancies in amorphous InOx nanoribbons enhance CO2 adsorption and activation for CO2 electroreduction. Angew. Chem. Int. Ed. 2019, 58, 5609–5613. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Yang, N.; Han, P.; Kuang, M.; Mei, B.; Jiang, Z.; Zhong, J.; Li, L.; Zheng, G. Oxygen vacancy tuning toward efficient electrocatalytic CO2 reduction to C2H4. Small Methods 2019, 3, 1800449. [Google Scholar]
- Geng, Z.; Kong, X.; Chen, W.; Su, H.; Liu, Y.; Cai, F.; Wang, G.; Zeng, J. Oxygen vacancies in ZnO nanosheets enhance CO2 electrochemical reduction to CO. Angew. Chem. 2018, 130, 6162–6167. [Google Scholar] [CrossRef]
- Baruch, M.F.; Pander III, J.E.; White, J.L.; Bocarsly, A.B. Mechanistic insights into the reduction of CO2 on tin electrodes using in situ ATR-IR spectroscopy. ACS Catal. 2015, 5, 3148–3156. [Google Scholar] [CrossRef]
- Barwa, E.; Pascher, T.F.; Ončák, M.; van der Linde, C.; Beyer, M.K. Carbon Dioxide Activation at Metal Centers: Evolution of Charge Transfer from Mg.+ to CO2 in [MgCO2 (H2O) n].+, n= 0–8. Angew. Chem. Int. Ed. 2020, 59, 7467–7471. [Google Scholar] [CrossRef] [Green Version]
- Iskra, A.; Gentleman, A.S.; Cunningham, E.M.; Mackenzie, S.R. Carbon dioxide binding to metal oxides: Infrared spectroscopy of NbO2+ (CO2) n and TaO2+ (CO2) n complexes. Int. J. Mass Spectrom. 2019, 435, 93–100. [Google Scholar] [CrossRef]
- Yin, X.; Moss, J.R. Recent developments in the activation of carbon dioxide by metal complexes. Coord. Chem. Rev. 1999, 181, 27–59. [Google Scholar] [CrossRef]
- Iskra, A.; Gentleman, A.S.; Kartouzian, A.; Kent, M.J.; Sharp, A.P.; Mackenzie, S.R. Infrared spectroscopy of gas-phase M+ (CO2) n (M = Co, Rh, Ir) ion–molecule complexes. J. Phys. Chem. A 2017, 121, 133–140. [Google Scholar] [CrossRef]
- Thompson, M.C.; Ramsay, J.; Weber, J.M. Solvent-driven reductive activation of CO2 by bismuth: Switching from metalloformate complexes to oxalate products. Angew. Chem. Int. Ed. 2016, 55, 15171–15174. [Google Scholar] [CrossRef]
- Knurr, B.J.; Weber, J.M. Solvent-driven reductive activation of carbon dioxide by gold anions. J. Am. Chem. Soc. 2012, 134, 18804–18808. [Google Scholar] [CrossRef]
- Zheng, H.; Kong, X.; Wang, C.; Wang, T.; Yang, D.; Li, G.; Xie, H.; Zhao, Z.; Shi, R.; Han, H. Spectroscopic Identification of Transition-Metal M [η2-(O, O) C] Species for Highly-Efficient CO2 Activation. J. Phys. Chem. Lett. 2020, 12, 472–477. [Google Scholar] [CrossRef]
- Zimmermann, N.; Bernhardt, T.M.; Bakker, J.M.; Barnett, R.N.; Landman, U.; Lang, S.M. Infrared Spectroscopy of Gas-Phase Mn x O y (CO2) z+ Complexes. J. Phys. Chem. A 2020, 124, 1561–1566. [Google Scholar] [CrossRef]
- Paparo, A.; Okuda, J. Carbon dioxide complexes: Bonding modes and synthetic methods. Coord. Chem. Rev. 2017, 334, 136–149. [Google Scholar] [CrossRef]
- Li, H.; Zhao, J.; Luo, L.; Du, J.; Zeng, J. Symmetry-Breaking Sites for Activating Linear Carbon Dioxide Molecules. Acc. Chem. Res. 2021, 54, 1454–1464. [Google Scholar] [CrossRef]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef] [Green Version]
- Dietz, L.; Piccinin, S.; Maestri, M. Mechanistic Insights into CO2 activation via reverse water–gas shift on metal surfaces. J. Phys. Chem. C 2015, 119, 4959–4966. [Google Scholar] [CrossRef]
- Ko, J.; Kim, B.-K.; Han, J.W. Density functional theory study for catalytic activation and dissociation of CO2 on bimetallic alloy surfaces. J. Phys. Chem. C 2016, 120, 3438–3447. [Google Scholar] [CrossRef]
- Wang, S.-G.; Liao, X.-Y.; Cao, D.-B.; Huo, C.-F.; Li, Y.-W.; Wang, J.; Jiao, H. Factors controlling the interaction of CO2 with transition metal surfaces. J. Phys. Chem. C 2007, 111, 16934–16940. [Google Scholar] [CrossRef]
- Ding, X.; De Rogatis, L.; Vesselli, E.; Baraldi, A.; Comelli, G.; Rosei, R.; Savio, L.; Vattuone, L.; Rocca, M.; Fornasiero, P. Interaction of carbon dioxide with Ni (110): A combined experimental and theoretical study. Phys. Rev. B 2007, 76, 195425. [Google Scholar] [CrossRef]
- Eren, B.; Weatherup, R.S.; Liakakos, N.; Somorjai, G.A.; Salmeron, M. Dissociative carbon dioxide adsorption and morphological changes on Cu (100) and Cu (111) at ambient pressures. J. Am. Chem. Soc. 2016, 138, 8207–8211. [Google Scholar] [CrossRef] [PubMed]
- Hammami, R.; Dhouib, A.; Fernandez, S.; Minot, C. CO2 adsorption on (0 0 1) surfaces of metal monoxides with rock-salt structure. Catal. Today 2008, 139, 227–233. [Google Scholar] [CrossRef]
- Solymosi, F. The bonding, structure and reactions of CO2 adsorbed on clean and promoted metal surfaces. J. Mol. Catal. 1991, 65, 337–358. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Roldan, A.; de Leeuw, N.H. CuO surfaces and CO2 activation: A dispersion-corrected DFT+ U study. J. Phys. Chem. C 2016, 120, 2198–2214. [Google Scholar] [CrossRef]
- Yang, T.; Gu, T.; Han, Y.; Wang, W.; Yu, Y.; Zang, Y.; Zhang, H.; Mao, B.; Li, Y.; Yang, B. Surface orientation and pressure dependence of CO2 activation on Cu surfaces. J. Phys. Chem. C 2020, 124, 27511–27518. [Google Scholar] [CrossRef]
- Yu, H.; Cao, D.; Fisher, A.; Johnston, R.L.; Cheng, D. Size effect on the adsorption and dissociation of CO2 on Co nanoclusters. Appl. Surf. Sci. 2017, 396, 539–546. [Google Scholar] [CrossRef]
- Etim, U.J.; Song, Y.; Zhong, Z. Improving the Cu/ZnO-Based Catalysts for Carbon Dioxide Hydrogenation to Methanol, and the Use of Methanol As a Renewable Energy Storage Media. Front. Energy Res. 2020, 8, 545431. [Google Scholar] [CrossRef]
- Fu, S.S.; Somorjai, G.A. Interactions of O2, CO, CO2, and D2 with the stepped Cu (311) crystal face: Comparison to Cu (110). Surf. Sci. 1992, 262, 68–76. [Google Scholar] [CrossRef]
- Rasmussen, P.; Taylor, P.; Chorkendorff, I. The interaction of carbon dioxide with Cu (100). Surf. Sci. 1992, 269, 352–359. [Google Scholar] [CrossRef]
- Muttaqien, F.; Hamamoto, Y.; Inagaki, K.; Morikawa, Y. Dissociative adsorption of CO2 on flat, stepped, and kinked Cu surfaces. J. Chem. Phys. 2014, 141, 034702. [Google Scholar] [CrossRef]
- Pohl, M.; Otto, A. Adsorption and reaction of carbon dioxide on pure and alkali-metal promoted cold-deposited copper films. Surf. Sci. 1998, 406, 125–137. [Google Scholar] [CrossRef]
- Wang, S.-G.; Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. Chemisorption of CO2 on nickel surfaces. J. Phys. Chem. B 2005, 109, 18956–18963. [Google Scholar] [CrossRef]
- Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. CO2 dissociation on Ni (2 1 1). Surf. Sci. 2009, 603, 2991–2998. [Google Scholar] [CrossRef]
- Illing, G.; Heskett, D.; Plummer, E.; Freund, H.-J.; Somers, J.; Lindner, T.; Bradshaw, A.; Buskotte, U.; Neumann, M.; Starke, U. Adsorption and reaction of CO2 on Ni {110}: X-ray photoemission, near-edge X-ray absorption fine-structure and diffuse leed studies. Surf. Sci. 1988, 206, 1–19. [Google Scholar] [CrossRef]
- Heine, C.; Lechner, B.A.; Bluhm, H.; Salmeron, M. Recycling of CO2: Probing the chemical state of the Ni (111) surface during the methanation reaction with ambient-pressure X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 2016, 138, 13246–13252. [Google Scholar] [CrossRef] [PubMed]
- Roiaz, M.; Monachino, E.; Dri, C.; Greiner, M.; Knop-Gericke, A.; Schlögl, R.; Comelli, G.; Vesselli, E. Reverse water–gas shift or Sabatier methanation on Ni (110)? Stable surface species at near-ambient pressure. J. Am. Chem. Soc. 2016, 138, 4146–4154. [Google Scholar] [CrossRef]
- Cai, J.; Han, Y.; Chen, S.; Crumlin, E.J.; Yang, B.; Li, Y.; Liu, Z. CO2 activation on Ni (111) and Ni (100) surfaces in the presence of H2O: An ambient-pressure X-ray photoelectron spectroscopy study. J. Phys. Chem. C 2019, 123, 12176–12182. [Google Scholar] [CrossRef]
- Silaghi, M.-C.; Comas-Vives, A.; Coperet, C. CO2 Activation on Ni/γ–Al2O3 catalysts by first-principles calculations: From ideal surfaces to supported nanoparticles. ACS Catal. 2016, 6, 4501–4505. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, Y.; Solymosi, F.; White, J. Spectroscopic study of K-induced activation of CO2 on Pt (111). Surf. Sci. 1991, 245, 289–304. [Google Scholar] [CrossRef]
- Ricart, J.M.; Habas, M.P.; Clotet, A.; Curulla, D.; Illas, F. Theoretical study of CO2 activation on Pt (111) induced by coadsorbed K atoms. Surf. Sci. 2000, 460, 170–181. [Google Scholar] [CrossRef]
- Huang, M.; Adnot, A.; Suppiah, S.; Kaliaguine, S. XPS observation of surface interaction between H2 and CO2 on platinum foil. J. Mol. Catal. A: Chem. 1995, 104, L131–L137. [Google Scholar] [CrossRef]
- Su, H.; Ye, Y.; Lee, K.-J.; Zeng, J.; Mun, B.S.; Crumlin, E.J. Probing the surface chemistry for reverse water gas shift reaction on Pt (1 1 1) using ambient pressure X-ray photoelectron spectroscopy. J. Catal. 2020, 391, 123–131. [Google Scholar] [CrossRef]
- Víctor, A.; González, S.; Illas, F.; Fierro, J.L. Evidence for spontaneous CO2 activation on cobalt surfaces. Chem. Phys. Lett. 2008, 454, 262–268. [Google Scholar]
- Jia, J.; Qian, C.; Dong, Y.; Li, Y.F.; Wang, H.; Ghoussoub, M.; Butler, K.T.; Walsh, A.; Ozin, G.A. Heterogeneous catalytic hydrogenation of CO2 by metal oxides: Defect engineering–perfecting imperfection. Chem. Soc. Rev. 2017, 46, 4631–4644. [Google Scholar] [CrossRef]
- Seiferth, O.; Wolter, K.; Dillmann, B.; Klivenyi, G.; Freund, H.-J.; Scarano, D.; Zecchina, A. IR investigations of CO2 adsorption on chromia surfaces: Cr2O3 (0001)/Cr (110) versus polycrystalline α-Cr2O3. Surf. Sci. 1999, 421, 176–190. [Google Scholar] [CrossRef]
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024. [Google Scholar] [CrossRef]
- Liu, L.; Fan, W.; Zhao, X.; Sun, H.; Li, P.; Sun, L. Surface dependence of CO2 adsorption on Zn2GeO4. Langmuir 2012, 28, 10415–10424. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Liu, P.; Stacchiola, D.J.; Senanayake, S.D.; White, M.G.; Chen, J.G. Hydrogenation of CO2 to methanol: Importance of metal–oxide and metal–carbide interfaces in the activation of CO2. ACS Catal. 2015, 5, 6696–6706. [Google Scholar] [CrossRef]
- Hezam, A.; Namratha, K.; Drmosh, Q.A.; Ponnamma, D.; Wang, J.; Prasad, S.; Ahamed, M.; Cheng, C.; Byrappa, K. CeO2 nanostructures enriched with oxygen vacancies for photocatalytic CO2 reduction. ACS Appl. Nano Mater. 2019, 3, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Bu, Y.; Weststrate, C.; Niemantsverdriet, J.; Fredriksson, H.O. Role of ZnO and CeO x in Cu-Based Model Catalysts in Activation of H2O and CO2 Dynamics Studied by in Situ Ultraviolet–Visible and X-ray Photoelectron Spectroscopy. ACS Catal. 2016, 6, 7994–8003. [Google Scholar] [CrossRef]
- Chen, S.; Cao, T.; Gao, Y.; Li, D.; Xiong, F.; Huang, W. Probing surface structures of CeO2, TiO2, and Cu2O nanocrystals with CO and CO2 chemisorption. J. Phys. Chem. C 2016, 120, 21472–21485. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, J.; Wang, T.; Li, Y.; Li, X.; Yin, J.; Wang, C. CO2 photoreduction with H2O vapor on highly dispersed CeO2/TiO2 catalysts: Surface species and their reactivity. J. Catal. 2016, 337, 293–302. [Google Scholar] [CrossRef]
- Di, J.; Zhu, C.; Ji, M.; Duan, M.; Long, R.; Yan, C.; Gu, K.; Xiong, J.; She, Y.; Xia, J. Defect-rich Bi12O17Cl2 nanotubes self-accelerating charge separation for boosting photocatalytic CO2 reduction. Angew. Chem. Int. Ed. 2018, 57, 14847–14851. [Google Scholar] [CrossRef]
- Yan, S.C.; Ouyang, S.X.; Gao, J.; Yang, M.; Feng, J.Y.; Fan, X.X.; Wan, L.J.; Li, Z.S.; Ye, J.H.; Zhou, Y. A room-temperature reactive-template route to mesoporous ZnGa2O4 with improved photocatalytic activity in reduction of CO2. Angew. Chem. 2010, 122, 6544–6548. [Google Scholar] [CrossRef]
- Guo, J.; Ouyang, S.; Kako, T.; Ye, J. Mesoporous In(OH)3 for photoreduction of CO2 into renewable hydrocarbon fuels. Appl. Surf. Sci. 2013, 280, 418–423. [Google Scholar] [CrossRef]
- Cao, A.; Wang, Z.; Li, H.; Nørskov, J.K. Relations between Surface Oxygen Vacancies and Activity of Methanol Formation from CO2 Hydrogenation over In2O3 Surfaces. ACS Catal. 2021, 11, 1780–1786. [Google Scholar] [CrossRef]
- Staudt, T.; Lykhach, Y.; Tsud, N.; Skála, T.S.; Prince, K.C.; Matolín, V.R.; Libuda, J.R. Electronic structure of magnesia–ceria model catalysts, CO2 adsorption, and CO2 activation: A synchrotron radiation photoelectron spectroscopy study. J. Phys. Chem. C 2011, 115, 8716–8724. [Google Scholar] [CrossRef]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Sanz, J.F. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Sherman, B.J.; Lo, C.S. Carbon dioxide activation and dissociation on ceria (110): A density functional theory study. J. Chem. Phys. 2013, 138, 014702. [Google Scholar] [CrossRef]
- Yang, C.; Bebensee, F.; Chen, J.; Yu, X.; Nefedov, A.; Wöll, C. Carbon dioxide adsorption on CeO2 (110): An XPS and NEXAFS study. ChemPhysChem 2017, 18, 1874–1880. [Google Scholar] [CrossRef]
- Hahn, K.R.; Iannuzzi, M.; Seitsonen, A.P.; Hutter, J.R. Coverage effect of the CO2 adsorption mechanisms on CeO2 (111) by first principles analysis. J. Phys. Chem. C 2013, 117, 1701–1711. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Thompson, T.L.; Diwald, O.; Yates, J.T. CO2 as a probe for monitoring the surface defects on TiO2 (110) temperature-programmed desorption. J. Phys. Chem. B 2003, 107, 11700–11704. [Google Scholar] [CrossRef]
- Funk, S.; Burghaus, U. Adsorption of CO2 on oxidized, defected, hydrogen and oxygen covered rutile (1 × 1)-TiO2 (110). Phys. Chem. Chem. Phys. 2006, 8, 4805–4813. [Google Scholar] [CrossRef]
- Suriye, K.; Praserthdam, P.; Jongsomjit, B. Control of Ti3+ surface defect on TiO2 nanocrystal using various calcination atmospheres as the first step for surface defect creation and its application in photocatalysis. Appl. Surf. Sci. 2007, 253, 3849–3855. [Google Scholar] [CrossRef]
- Suriye, K.; Jongsomjit, B.; Satayaprasert, C.; Praserthdam, P. Surface defect (Ti3+) controlling in the first step on the anatase TiO2 nanocrystal by using sol–gel technique. Appl. Surf. Sci. 2008, 255, 2759–2766. [Google Scholar] [CrossRef]
- Sorescu, D.C.; Al-Saidi, W.A.; Jordan, K.D. CO2 adsorption on TiO2 (101) anatase: A dispersion-corrected density functional theory study. J. Chem. Phys. 2011, 135, 124701. [Google Scholar] [CrossRef]
- Liao, L.-F.; Lien, C.-F.; Shieh, D.-L.; Chen, M.-T.; Lin, J.-L. FTIR study of adsorption and photoassisted oxygen isotopic exchange of carbon monoxide, carbon dioxide, carbonate, and formate on TiO2. J. Phys. Chem. B 2002, 106, 11240–11245. [Google Scholar] [CrossRef]
- Lee, J.; Sorescu, D.C.; Deng, X. Electron-induced dissociation of CO2 on TiO2 (110). J. Am. Chem. Soc. 2011, 133, 10066–10069. [Google Scholar] [CrossRef]
- Nolan, M. Adsorption of CO2 on heterostructures of Bi2O3 nanocluster-modified TiO2 and the role of reduction in promoting CO2 activation. ACS Omega 2018, 3, 13117–13128. [Google Scholar] [CrossRef] [Green Version]
- Pavelec, J.; Hulva, J.; Halwidl, D.; Bliem, R.; Gamba, O.; Jakub, Z.; Brunbauer, F.; Schmid, M.; Diebold, U.; Parkinson, G.S. A multi-technique study of CO2 adsorption on Fe3O4 magnetite. J. Chem. Phys. 2017, 146, 014701. [Google Scholar] [CrossRef] [Green Version]
- Hakim, A.; Marliza, T.S.; Abu Tahari, N.M.; Wan Isahak, R.W.; Yusop, R.M.; Mohamed Hisham, W.M.; Yarmo, A.M. Studies on CO2 adsorption and desorption properties from various types of iron oxides (FeO, Fe2O3, and Fe3O4). Ind. Eng. Chem. Res. 2016, 55, 7888–7897. [Google Scholar] [CrossRef]
- Li, X.; Paier, J. Vibrational properties of CO2 adsorbed on the Fe3O4 (111) surface: Insights gained from DFT. J. Chem. Phy 2020, 152, 104702. [Google Scholar] [CrossRef]
- Mirabella, F.; Zaki, E.; Ivars-Barcelo, F.; Schauermann, S.; Shaikhutdinov, S.; Freund, H.-J. CO2 adsorption on magnetite Fe3O4 (111). J. Phys. Chem. C 2018, 122, 27433–27441. [Google Scholar] [CrossRef]
- Su, T.; Qin, Z.; Huang, G.; Ji, H.; Jiang, Y.; Chen, J. Density functional theory study on the interaction of CO2 with Fe3O4 (111) surface. Appl. Surf. Sci. 2016, 378, 270–276. [Google Scholar] [CrossRef]
- Tang, Q.-L.; Hong, Q.-J.; Liu, Z.-P. CO2 fixation into methanol at Cu/ZrO2 interface from first principles kinetic Monte Carlo. J. Catal. 2009, 263, 114–122. [Google Scholar] [CrossRef]
- Fisher, I.A.; Bell, A.T. In-situinfrared study of methanol synthesis from H2/CO2 over Cu/SiO2 and Cu/ZrO2/SiO2. J. Catal. 1997, 172, 222–237. [Google Scholar] [CrossRef]
- Jin, X.; Lv, C.; Zhou, X.; Ye, L.; Xie, H.; Liu, Y.; Su, H.; Zhang, B.; Chen, G. Oxygen vacancy engineering of Bi24O31Cl10 for boosted photocatalytic CO2 conversion. ChemSusChem 2019, 12, 2740–2747. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Shi, W.; Ling, P.; Sun, Y.; Jiao, X.; Gao, S.; Liang, L.; Xu, J.; Yan, W. Efficient visible-light-driven CO2 reduction mediated by defect-engineered BiOBr atomic layers. Angew. Chem. 2018, 130, 8855–8859. [Google Scholar] [CrossRef]
- Huygh, S.; Bogaerts, A.; Neyts, E.C. How oxygen vacancies activate CO2 dissociation on TiO2 anatase (001). J. Phys. Chem. C 2016, 120, 21659–21669. [Google Scholar] [CrossRef]
- Pan, Y.-x.; Liu, C.-J.; Mei, D.; Ge, Q. Effects of Hydration and Oxygen Vacancy on CO2 Adsorption and Activation on β-Ga2O3(100). Langmuir 2010, 26, 5551–5558. [Google Scholar] [CrossRef]
- Ye, J.; Liu, C.; Mei, D.; Ge, Q. Active oxygen vacancy site for methanol synthesis from CO2 hydrogenation on In2O3 (110): A DFT study. ACS Catal. 2013, 3, 1296–1306. [Google Scholar] [CrossRef]
- Sarkar, A.; Khan, G.G. The formation and detection techniques of oxygen vacancies in titanium oxide-based nanostructures. Nanoscale 2019, 11, 3414–3444. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Na, W.; Gao, W.; Wang, H. Carbon-Modified CuO/ZnO Catalyst with High Oxygen Vacancy for CO2 Hydrogenation to Methanol. Energy Technol. 2020, 8, 2000194. [Google Scholar] [CrossRef]
- Jiang, D.; Wang, W.; Zhang, L.; Zheng, Y.; Wang, Z. Insights into the surface-defect dependence of photoreactivity over CeO2 nanocrystals with well-defined crystal facets. ACS Catal. 2015, 5, 4851–4858. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Zhang, X.; Wei, M.; Evans, D.G.; Duan, X. Catalytic behavior of supported Ru nanoparticles on the {1 0 0},{1 1 0}, and {1 1 1} facet of CeO2. J. Catal. 2015, 329, 177–186. [Google Scholar] [CrossRef]
- Bielz, T.; Lorenz, H.; Jochum, W.; Kaindl, R.; Klauser, F.; Klötzer, B.; Penner, S. Hydrogen on In2O3: Reducibility, Bonding, Defect Formation, and Reactivity. J. Phys. Chem. C 2010, 114, 9022–9029. [Google Scholar] [CrossRef]
- Rui, N.; Wang, Z.; Sun, K.; Ye, J.; Ge, Q.; Liu, C.-J. CO2 hydrogenation to methanol over Pd/In2O3: Effects of Pd and oxygen vacancy. Appl. Catal. B: Environ. 2017, 218, 488–497. [Google Scholar] [CrossRef]
- Wang, M.; Shen, M.; Jin, X.; Tian, J.; Li, M.; Zhou, Y.; Zhang, L.; Li, Y.; Shi, J. Oxygen vacancy generation and stabilization in CeO2–x by Cu introduction with improved CO2 photocatalytic reduction activity. ACS Catal. 2019, 9, 4573–4581. [Google Scholar] [CrossRef]
- Li, X.; Liu, Q.; Jiang, X.; Huang, J. Enhanced photocatalytic activity of Ga-N Co-doped anatase TiO2 for water decomposition to hydrogen. Int. J. Electrochem. Sci 2012, 7, 11519–11527. [Google Scholar]
- Wang, P.; Qi, C.; Wen, P.; Hao, L.; Xu, X.; Agathopoulos, S. Synthesis of Si, N co-doped nano-sized TiO2 with high thermal stability and photocatalytic activity by mechanochemical method. Nanomaterials 2018, 8, 294. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.; Tian, Y.; Chen, Z.; Yang, Z.; Li, W.; Wang, K.; Wang, L.; Wang, K.; Zhang, W. Synthesis of Ti3+ self-doped TiO2 nanocrystals based on Le Chatelier’s principle and their application in solar light photocatalysis. RSC Adv. 2016, 6, 74376–74383. [Google Scholar] [CrossRef]
- Zhu, K.; Shi, F.; Zhu, X.; Yang, W. The roles of oxygen vacancies in electrocatalytic oxygen evolution reaction. Nano Energy 2020, 73, 104761. [Google Scholar] [CrossRef]
- Guo, S.; Di, J.; Chen, C.; Zhu, C.; Duan, M.; Lian, C.; Ji, M.; Zhou, W.; Xu, M.; Song, P. Oxygen vacancy mediated bismuth stannate ultra-small nanoparticle towards photocatalytic CO2-to-CO conversion. Appl. Catal. B Environ. 2020, 276, 119156. [Google Scholar] [CrossRef]
- Santara, B.; Giri, P.; Imakita, K.; Fujii, M. Evidence of oxygen vacancy induced room temperature ferromagnetism in solvothermally synthesized undoped TiO2 nanoribbons. Nanoscale 2013, 5, 5476–5488. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Neyts, E.C.; Cao, X.; Zhang, X.; Jang, B.W.-L.; Liu, C.-J. Catalyst preparation with plasmas: How does it work? ACS Catal. 2018, 8, 2093–2110. [Google Scholar] [CrossRef]
- Feng, W.-H.; Yu, M.-M.; Wang, L.-J.; Miao, Y.-T.; Shakouri, M.; Ran, J.; Hu, Y.; Li, Z.; Huang, R.; Lu, Y.-L. Insights into Bimetallic Oxide Synergy during Carbon Dioxide Hydrogenation to Methanol and Dimethyl Ether over GaZrO x Oxide Catalysts. ACS Catal. 2021, 11, 4704–4711. [Google Scholar] [CrossRef]
- Tabatabaei, J.; Sakakini, B.; Waugh, K. On the mechanism of methanol synthesis and the water-gas shift reaction on ZnO. Catal. Lett. 2006, 110, 77–84. [Google Scholar] [CrossRef]
- Singh, R.; Tripathi, K.; Pant, K.K. Investigating the role of oxygen vacancies and basic site density in tuning methanol selectivity over Cu/CeO2 catalyst during CO2 hydrogenation. Fuel 2021, 303, 121289. [Google Scholar] [CrossRef]
- Rhodes, M.D.; Bell, A.T. The effects of zirconia morphology on methanol synthesis from CO and H2 over Cu/ZrO2 catalysts: Part I. Steady-state studies. J. Catal. 2005, 233, 198–209. [Google Scholar] [CrossRef]
- Martin, O.; Mondelli, C.; Cervellino, A.; Ferri, D.; Curulla-Ferré, D.; Pérez-Ramírez, J. Operando synchrotron X-ray powder diffraction and modulated-excitation infrared spectroscopy elucidate the CO2 promotion on a commercial methanol synthesis catalyst. Angew. Chem. Int. Ed. 2016, 55, 11031–11036. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; He, S.; Chen, H.; Wang, B.; Zheng, L.; Wei, M.; Evans, D.G.; Duan, X. Active site dependent reaction mechanism over Ru/CeO2 catalyst toward CO2 methanation. J. Am. Chem. Soc. 2016, 138, 6298–6305. [Google Scholar] [CrossRef]
- Srinivas, B.; Shubhamangala, B.; Lalitha, K.; Anil Kumar Reddy, P.; Durga Kumari, V.; Subrahmanyam, M.; De, B.R. Photocatalytic Reduction of CO2 over Cu-TiO2/Molecular Sieve 5A Composite. Photochem. Photobiol. 2011, 87, 995–1001. [Google Scholar] [CrossRef]
- Liu, B.-J.; Torimoto, T.; Matsumoto, H.; Yoneyama, H. Effect of solvents on photocatalytic reduction of carbon dioxide using TiO2 nanocrystal photocatalyst embedded in SiO2 matrices. J. Photochem. Photobiol. A Chem. 1997, 108, 187–192. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Schuttlefield, J.; Zeitler, E.; Grassian, V.H. Carbon dioxide adsorption on oxide nanoparticle surfaces. Chem. Eng. J. 2011, 170, 471–481. [Google Scholar] [CrossRef]
- Li, H.; Rameshan, C.; Bukhtiyarov, A.V.; Prosvirin, I.P.; Bukhtiyarov, V.I.; Rupprechter, G. CO2 activation on ultrathin ZrO2 film by H2O co-adsorption: In situ NAP-XPS and IRAS studies. Surf. Sci. 2019, 679, 139–146. [Google Scholar] [CrossRef]
- Zhu, M.; Chen, J.; Shen, L.; Ford, M.E.; Gao, J.; Xu, J.; Wachs, I.E.; Han, Y.-F. Probing the surface of promoted CuO-Cr2O3-Fe2O3 catalysts during CO2 activation. Appl. Catal. B: Environ. 2020, 271, 118943. [Google Scholar] [CrossRef]
- Posada-Borbón, A.; Grönbeck, H. CO2 adsorption on hydroxylated In2O3 (110). Phys. Chem. Chem. Phys. 2019, 21, 21698–21708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghuman, K.K.; Hoch, L.B.; Wood, T.E.; Mims, C.; Singh, C.V.; Ozin, G.A. Surface analogues of molecular frustrated Lewis pairs in heterogeneous CO2 hydrogenation catalysis. ACS Catal. 2016, 6, 5764–5770. [Google Scholar] [CrossRef]
- Martin, O.; Martin, A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferre, D.; Perez-Ramirez, J. Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO2 Hydrogenation. Angew. Chem. Int. Ed. 2016, 55, 6261–6265. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Gu, M.; Qiu, R.; Yang, Z.; Xuan, F.; Zhu, M. Tunable Carbon Dioxide Activation Pathway over Iron Oxide Catalysts: Effects of Potassium. Ind. Eng. Chem. Res. 2021, 60, 8705–8713. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Wongsakulphasatch, S.; Vo, D.-V.N. Understanding the role of surface basic sites of catalysts in CO2 activation in dry reforming of methane: A short review. Catal. Sci. Technol. 2020, 10, 35–45. [Google Scholar] [CrossRef]
- Nie, X.; Meng, L.; Wang, H.; Chen, Y.; Guo, X.; Song, C. DFT insight into the effect of potassium on the adsorption, activation and dissociation of CO2 over Fe-based catalysts. Phys. Chem. Chem. Phys. 2018, 20, 14694–14707. [Google Scholar] [CrossRef]
- Miao, L.; Wang, J.; Zhang, P. Review on manganese dioxide for catalytic oxidation of airborne formaldehyde. Appl. Surf. Sci. 2019, 466, 441–453. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, L.; Luo, Q.; Cao, Y.; Dai, Y.; Li, Z.; Li, H.; Zheng, X.; Yan, W.; Yang, J. Molecular-level insight into how hydroxyl groups boost catalytic activity in CO2 hydrogenation into methanol. Chem 2018, 4, 613–625. [Google Scholar] [CrossRef] [Green Version]
- Stacchiola, D.J.; Senanayake, S.D.; Liu, P.; Rodriguez, J.A. Fundamental studies of well-defined surfaces of mixed-metal oxides: Special properties of MOx/TiO2 (110){M= V, Ru, Ce, or W}. Chem. Rev. 2013, 113, 4373–4390. [Google Scholar] [CrossRef]
- Witoon, T.; Numpilai, T.; Phongamwong, T.; Donphai, W.; Boonyuen, C.; Warakulwit, C.; Chareonpanich, M.; Limtrakul, J. Enhanced activity, selectivity and stability of a CuO-ZnO-ZrO2 catalyst by adding graphene oxide for CO2 hydrogenation to methanol. Chem. Eng. J. 2018, 334, 1781–1791. [Google Scholar] [CrossRef]
- Ma, Q.; Geng, M.; Zhang, J.; Zhang, X.; Zhao, T.S. Enhanced Catalytic Performance for CO2 Hydrogenation to Methanol over N-doped Graphene Incorporated Cu-ZnO-Al2O3 Catalysts. ChemistrySelect 2019, 4, 78–83. [Google Scholar] [CrossRef]
- Qi, Y.; Jiang, J.; Liang, X.; Ouyang, S.; Mi, W.; Ning, S.; Zhao, L.; Ye, J. Fabrication of black In2O3 with dense oxygen vacancy through dual functional carbon doping for enhancing photothermal CO2 hydrogenation. Adv. Funct. Mater. 2021, 31, 2100908. [Google Scholar] [CrossRef]
- Tomboc, G.M.; Tesfaye Gadisa, B.; Jun, M.; Chaudhari, N.K.; Kim, H.; Lee, K. Carbon Transition-metal Oxide Electrodes: Understanding the Role of Surface Engineering for High Energy Density Supercapacitors. Chem.—Asian J. 2020, 15, 1628–1647. [Google Scholar] [CrossRef]
- Chatterjee, D.P.; Nandi, A.K. A review on the recent advances in hybrid supercapacitors. J. Mater. Chem. A 2021, 9, 15880–15918. [Google Scholar] [CrossRef]
- Yang, Y.; White, M.G.; Liu, P. Theoretical study of methanol synthesis from CO2 hydrogenation on metal-doped Cu (111) surfaces. J. Phys. Chem. C 2012, 116, 248–256. [Google Scholar] [CrossRef]
- Surnev, S.; Fortunelli, A.; Netzer, F.P. Structure–property relationship and chemical aspects of oxide–metal hybrid nanostructures. Chem. Rev. 2013, 113, 4314–4372. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Lin, D.; Feng, X.; Niu, Y.; Zhang, B.; Xiao, F.-S. Strong metal–support interactions on gold nanoparticle catalysts achieved through Le Chatelier’s principle. Nat. Catal. 2021, 4, 418–424. [Google Scholar] [CrossRef]
- Meng, C.; Zhao, G.; Shi, X.-R.; Chen, P.; Liu, Y.; Lu, Y. Oxygen-deficient metal oxides supported nano-intermetallic InNi3C0. 5 toward efficient CO2 hydrogenation to methanol. Sci. Adv. 2021, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) Molecular orbital of CO2 at the ground state. Reproduced with permission from the authors of [23]. Copyright 2016 Elsevier B.V. (b) Walsh diagram for the orbital energies upon CO2 bending, and (c) molecular orbitals of CO2 at different OCO angles (carbon and oxygen weights are given in %). Reproduced with permission from the authors of [25]. Copyright 2015 Elsevier B.V.
Figure 1.
(a) Molecular orbital of CO2 at the ground state. Reproduced with permission from the authors of [23]. Copyright 2016 Elsevier B.V. (b) Walsh diagram for the orbital energies upon CO2 bending, and (c) molecular orbitals of CO2 at different OCO angles (carbon and oxygen weights are given in %). Reproduced with permission from the authors of [25]. Copyright 2015 Elsevier B.V.

Figure 2.
Possible structures of CO2δ− on heterogeneous catalyst surface: (a) oxygen coordination, (b) carbon coordination and (c) mixed coordination. Reproduced with permission from the authors of [31]. Copyright 2016 The Royal Society of Chemistry.
Figure 2.
Possible structures of CO2δ− on heterogeneous catalyst surface: (a) oxygen coordination, (b) carbon coordination and (c) mixed coordination. Reproduced with permission from the authors of [31]. Copyright 2016 The Royal Society of Chemistry.

Scheme 1.
Strategies for enhancing CO2 activation of oxide-based catalyst surface.

Figure 3.
CO2 photoactivation on the TiO2 surface (a–c). CB: conduction band; CBM: conduction band minimum. Schematic of the CO2 molecule adsorbed in the oxygen vacant site and Ti5C sites on the TiO2 (110) surface (d). Reproduced with permission from the authors of [44]. Copyright 2020 American Chemical Society.
Figure 3.
CO2 photoactivation on the TiO2 surface (a–c). CB: conduction band; CBM: conduction band minimum. Schematic of the CO2 molecule adsorbed in the oxygen vacant site and Ti5C sites on the TiO2 (110) surface (d). Reproduced with permission from the authors of [44]. Copyright 2020 American Chemical Society.

Figure 4.
(a) Tafel plots and (b) Nyquist plots of HCOO− for the electrocatalytic reduction of CO2 on InOx catalysts. Reproduced with permission from the authors of [72]. Copyright 2019 John Wiley & Sons, Inc.
Figure 4.
(a) Tafel plots and (b) Nyquist plots of HCOO− for the electrocatalytic reduction of CO2 on InOx catalysts. Reproduced with permission from the authors of [72]. Copyright 2019 John Wiley & Sons, Inc.

Scheme 2.
Activation of CO2 by transition metal complexes.

Scheme 3.
CO2 coordination modes at the transition metal center.

Figure 5.
Ambient pressure XPS spectra of Cu (111) obtained in under 1 mbar CO2 at 300 K and at 350, 400, 450, and 500 K; (a) O 1s and (b) C 1s. Reproduced with permission from the authors of [95]. Copyright 2020 American Chemical Society.
Figure 5.
Ambient pressure XPS spectra of Cu (111) obtained in under 1 mbar CO2 at 300 K and at 350, 400, 450, and 500 K; (a) O 1s and (b) C 1s. Reproduced with permission from the authors of [95]. Copyright 2020 American Chemical Society.

Figure 6.
Ambient pressure XPS spectra of (a,b) C 1s, and (c,d) O 1s Ni (111) and Ni (100), respectively. CO2 adsorption was performed under 0.2 Torr CO2 at room temperature. Reproduced with permission from [107]. Copyright 2019 American Chemical Society; (e) Energy profile diagram for the CO2 activation on Ni (111) and Ni (211) surfaces, edges of Ni13 particle, and edges and terraces of the Ni55 particle and the activation geometries on the Ni13. Reproduced with permission from [108]. Copyright 2016 American Chemical Society.
Figure 6.
Ambient pressure XPS spectra of (a,b) C 1s, and (c,d) O 1s Ni (111) and Ni (100), respectively. CO2 adsorption was performed under 0.2 Torr CO2 at room temperature. Reproduced with permission from [107]. Copyright 2019 American Chemical Society; (e) Energy profile diagram for the CO2 activation on Ni (111) and Ni (211) surfaces, edges of Ni13 particle, and edges and terraces of the Ni55 particle and the activation geometries on the Ni13. Reproduced with permission from [108]. Copyright 2016 American Chemical Society.

Figure 7.
Screening for Eaact for CO2 dissociation on pure metals and bimetallic alloys. Gray cells indicate the bimetallic alloys which are not preferred to the surface segregation of solute atoms. Eaact were estimated by combining BEP relation, scaling relation, and surface mixing rule. Reproduced with permission from the authors of [88]. Copyright 2016 American Chemical Society.
Figure 7.
Screening for Eaact for CO2 dissociation on pure metals and bimetallic alloys. Gray cells indicate the bimetallic alloys which are not preferred to the surface segregation of solute atoms. Eaact were estimated by combining BEP relation, scaling relation, and surface mixing rule. Reproduced with permission from the authors of [88]. Copyright 2016 American Chemical Society.

Scheme 4.
Possible modes of CO2 adsorption on metal oxide surfaces: (a) monodentate, (b) bidentate (c) π-coordination. Reproduced with permission from the authors of [114]. Copyright 2017 The Royal Society of Chemistry.
Scheme 4.
Possible modes of CO2 adsorption on metal oxide surfaces: (a) monodentate, (b) bidentate (c) π-coordination. Reproduced with permission from the authors of [114]. Copyright 2017 The Royal Society of Chemistry.

Figure 8.
Calculated electronic band structures of (a) Bi24O31Cl10, (b) Bi24O31Cl10-OV, and (c) the corresponding DOS plots. (d) CO evolution rate of Bi24O31Cl10 and Bi24O31Cl10-OV. (e) Free-energy diagram for CO2 reduction to CO on Bi24O31Cl10-OV. (f) Surface photovoltage spectroscopy showing carrier separation behavior. Reproduced with permission from the authors of [148]. Copyright 2019 John Wiley & Sons, Inc.
Figure 8.
Calculated electronic band structures of (a) Bi24O31Cl10, (b) Bi24O31Cl10-OV, and (c) the corresponding DOS plots. (d) CO evolution rate of Bi24O31Cl10 and Bi24O31Cl10-OV. (e) Free-energy diagram for CO2 reduction to CO on Bi24O31Cl10-OV. (f) Surface photovoltage spectroscopy showing carrier separation behavior. Reproduced with permission from the authors of [148]. Copyright 2019 John Wiley & Sons, Inc.

Figure 9.
Photo-assisted solution combustion synthesis of CeO2 with rich oxygen vacancies. Redrawn with permission from the authors of [119]. Copyright 2019 American Chemical Society.
Figure 9.
Photo-assisted solution combustion synthesis of CeO2 with rich oxygen vacancies. Redrawn with permission from the authors of [119]. Copyright 2019 American Chemical Society.

Figure 10.
CO2 dissociation minimal energy pathway on (a) stoichiometric anatase (001) surface and (b) reduced anatase (001). Reproduced with permission from the authors of [150]. Copyright 2016 American Chemical Society (c) Proposed mechanism for the hydrogenation of CO2 to methanol and DME on the GaZrOx catalyst. Reproduced with permission from the authors of [167]. Copyright 2021 American Chemical Society.
Figure 10.
CO2 dissociation minimal energy pathway on (a) stoichiometric anatase (001) surface and (b) reduced anatase (001). Reproduced with permission from the authors of [150]. Copyright 2016 American Chemical Society (c) Proposed mechanism for the hydrogenation of CO2 to methanol and DME on the GaZrOx catalyst. Reproduced with permission from the authors of [167]. Copyright 2021 American Chemical Society.

Figure 11.
Possible oxygen vacancies formation mechanism and their healing in CO2 hydrogenation to methanol. Reproduced with permission from the authors of [152]. Copyright 2013 American Chemical Society.
Figure 11.
Possible oxygen vacancies formation mechanism and their healing in CO2 hydrogenation to methanol. Reproduced with permission from the authors of [152]. Copyright 2013 American Chemical Society.

Figure 12.
Valence band top and conduction band bottom energies of CeO2 surface without (CeO2) and with (CeO2−x) oxygen vacancies. Modified surface state and defect peak are shown with the shaded and stripped black blocks, respectively. Reproduced with permission from the authors of [119]. Copyright 2019 American Chemical Society.
Figure 12.
Valence band top and conduction band bottom energies of CeO2 surface without (CeO2) and with (CeO2−x) oxygen vacancies. Modified surface state and defect peak are shown with the shaded and stripped black blocks, respectively. Reproduced with permission from the authors of [119]. Copyright 2019 American Chemical Society.

Figure 13.
Ambient pressure XPS spectra of (a) C 1s, (c) O 1s of Ni (111) and (b) C 1s, (d) O 1s of Ni (100) in 0.2 Torr CO2 + 0.2 Torr H2O at room temperature. Reproduced with permission from the authors of [107]. Copyright 2019 American Chemical Society.
Figure 13.
Ambient pressure XPS spectra of (a) C 1s, (c) O 1s of Ni (111) and (b) C 1s, (d) O 1s of Ni (100) in 0.2 Torr CO2 + 0.2 Torr H2O at room temperature. Reproduced with permission from the authors of [107]. Copyright 2019 American Chemical Society.


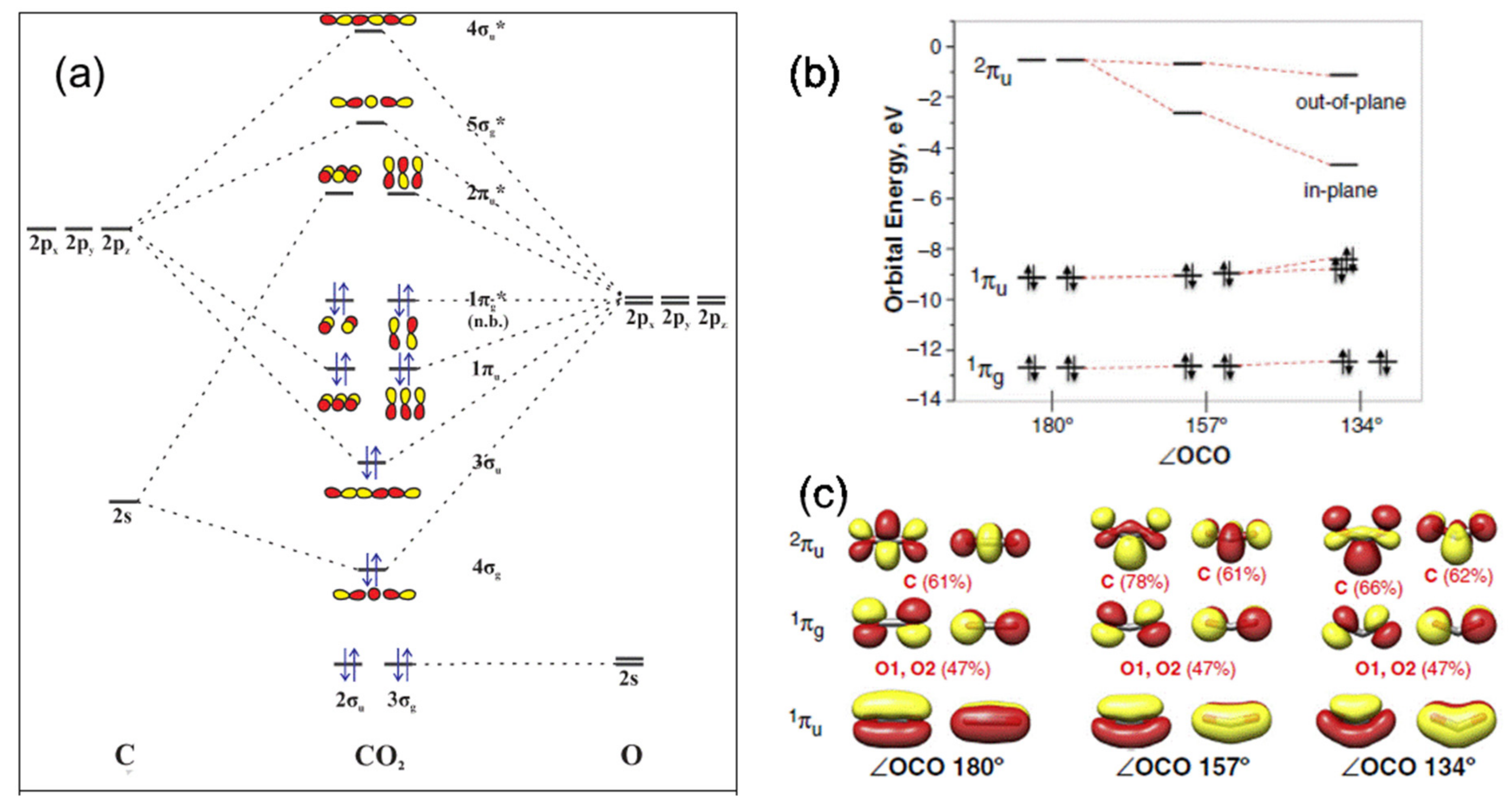{kind=link}
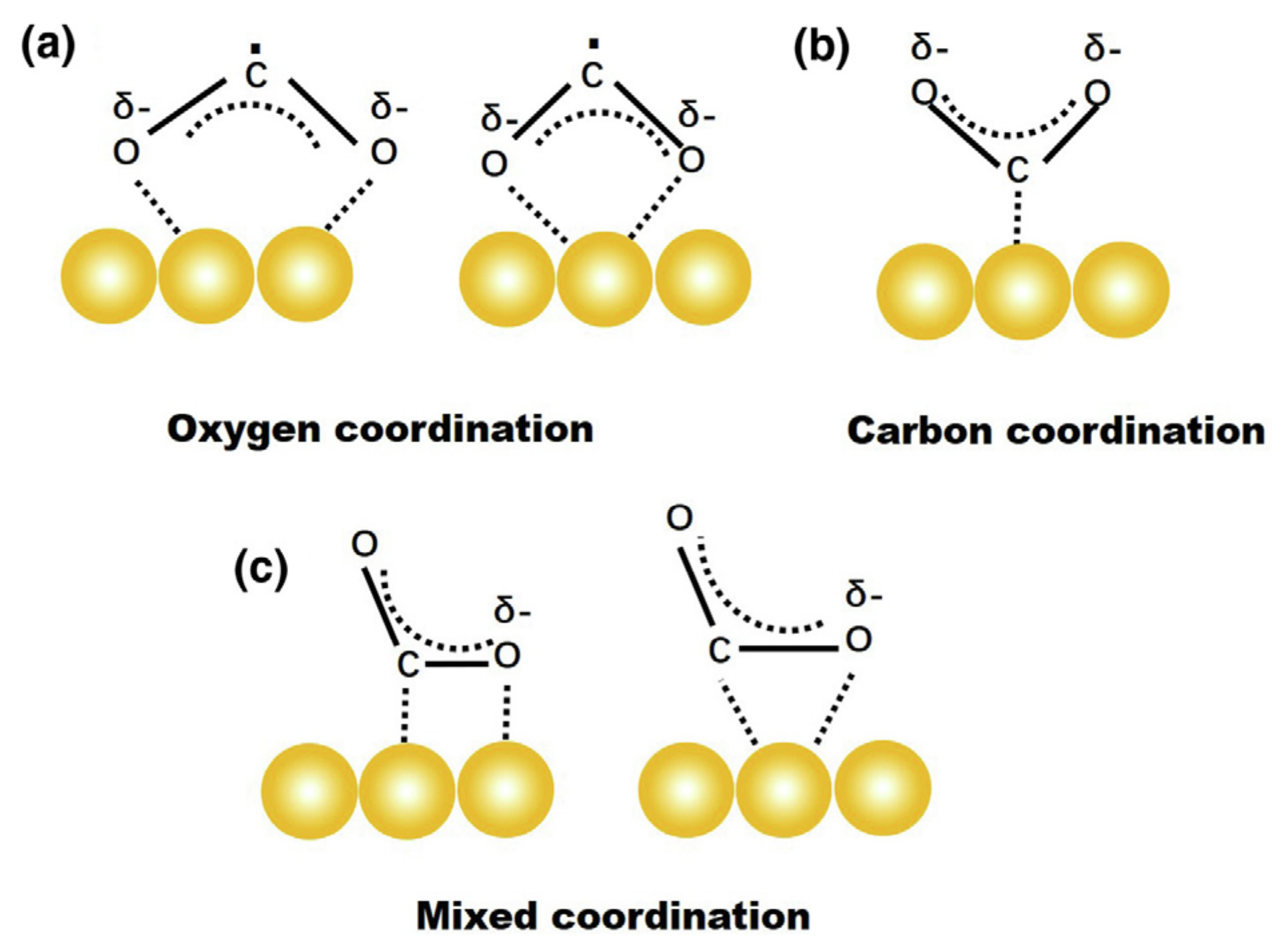{kind=link}
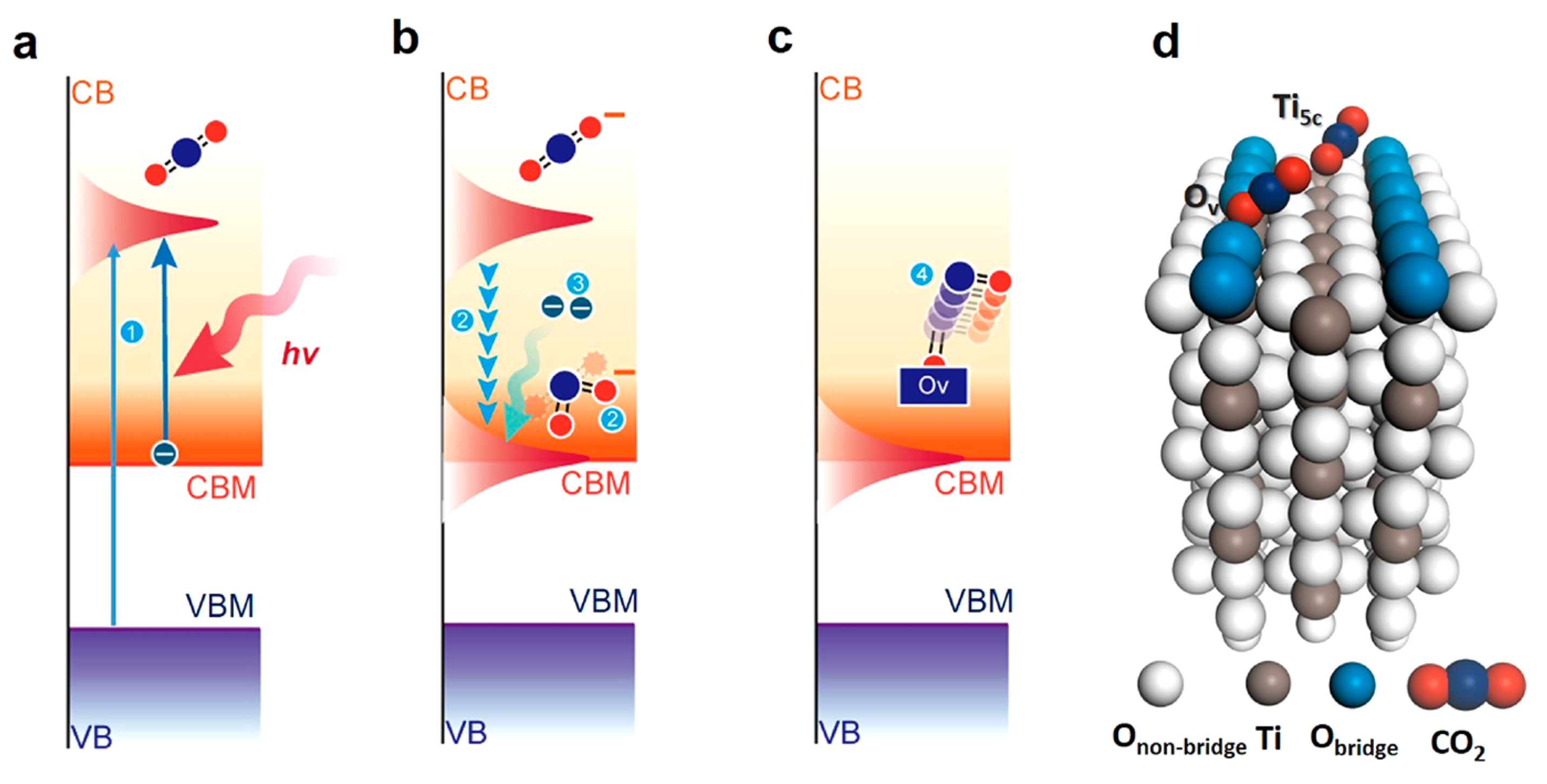{kind=link}
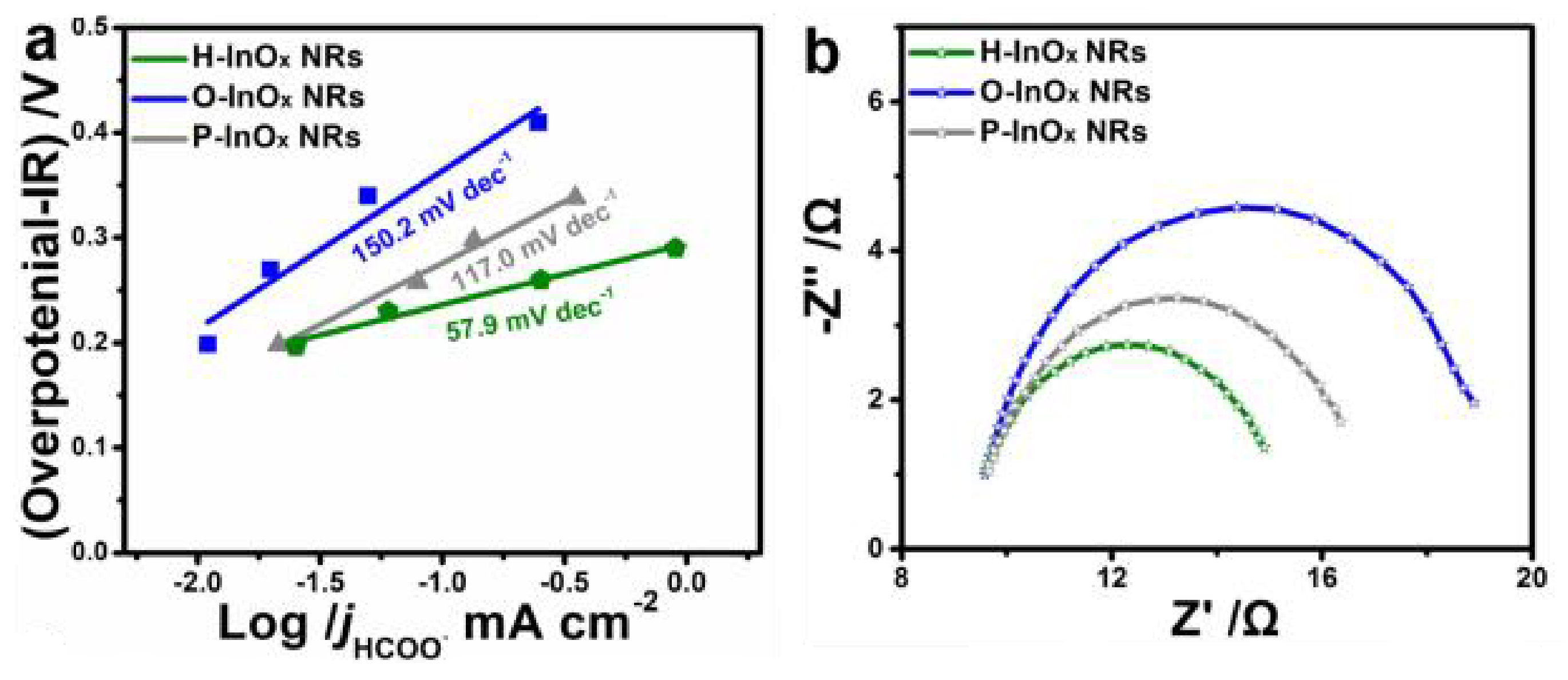{kind=link}
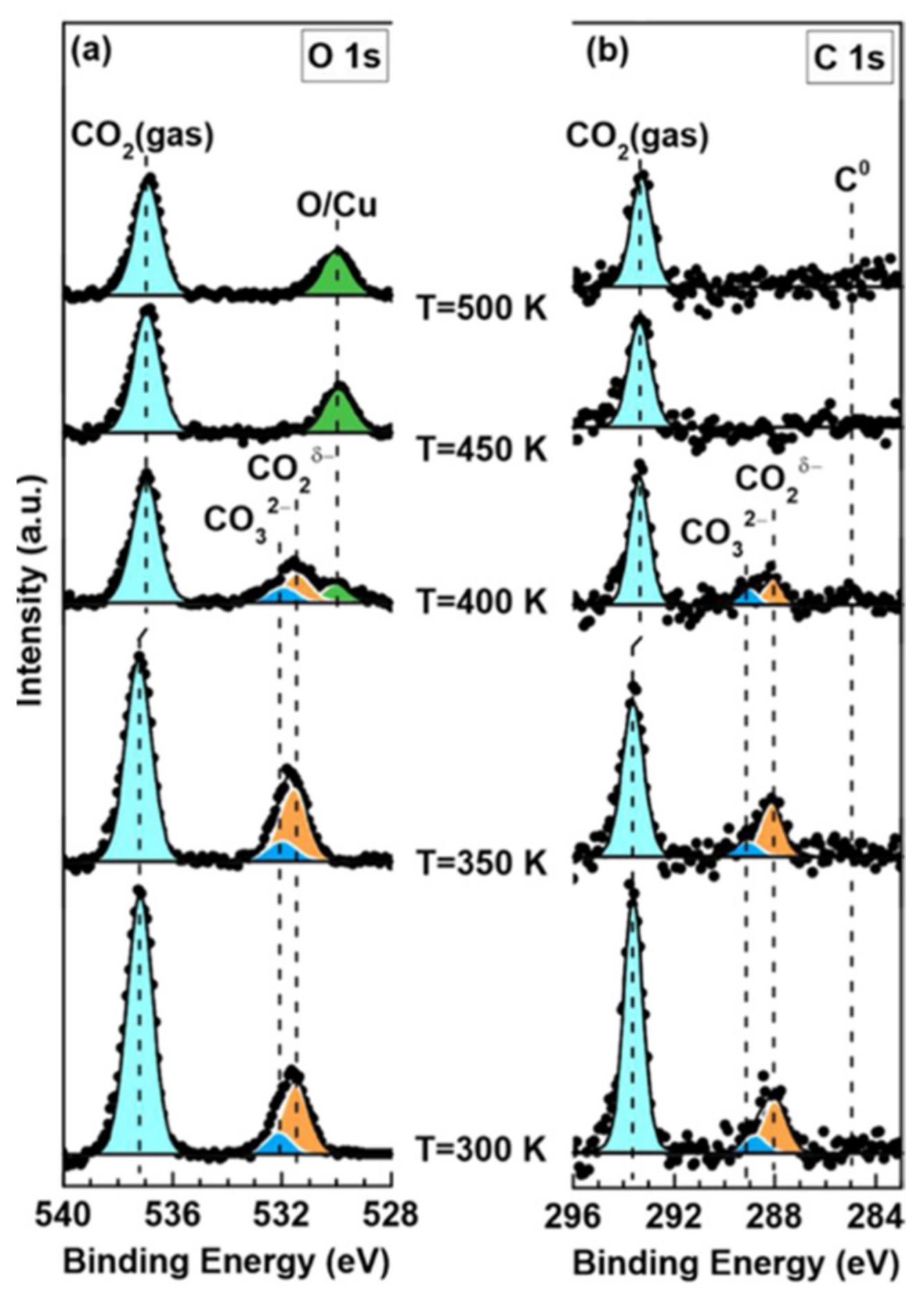{kind=link}
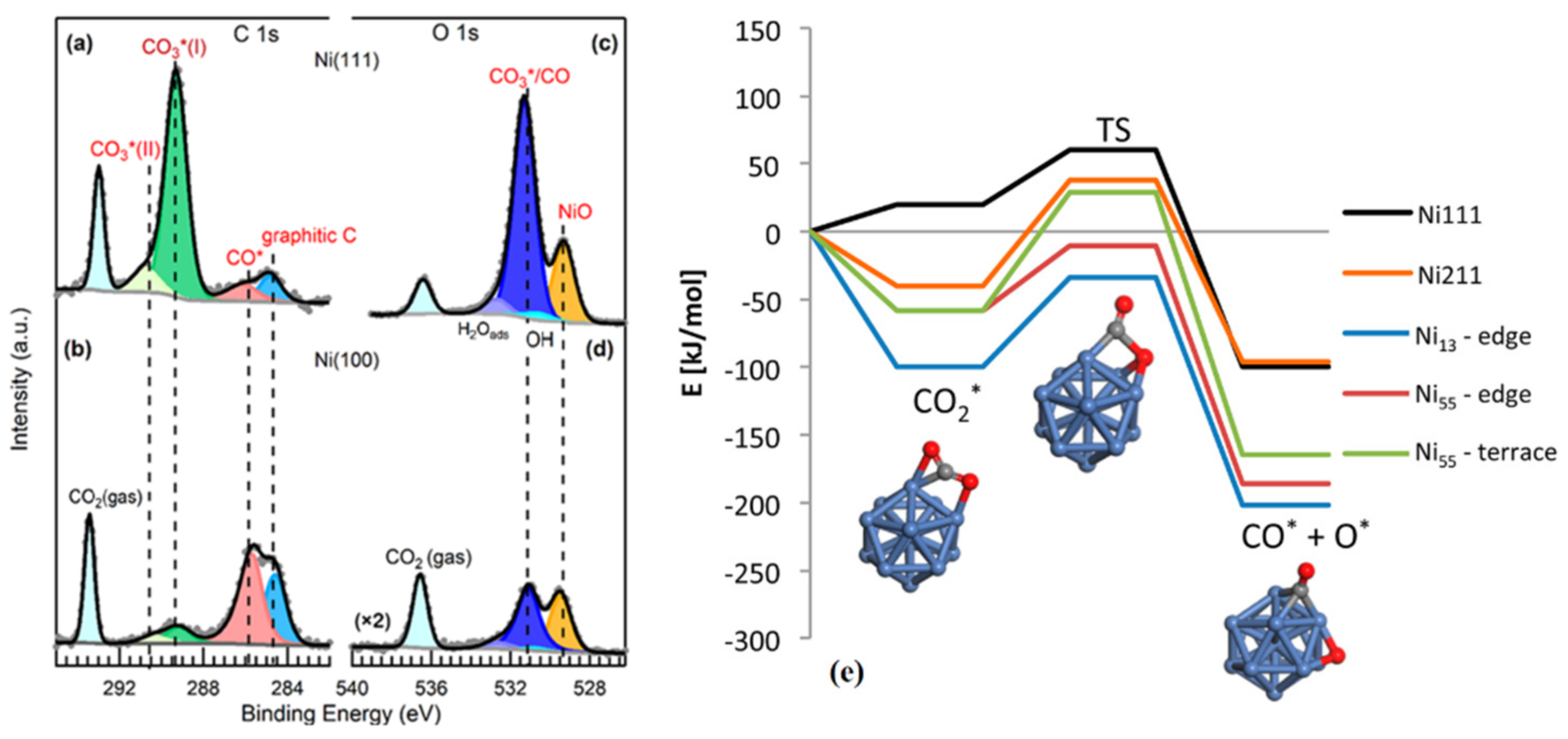{kind=link}
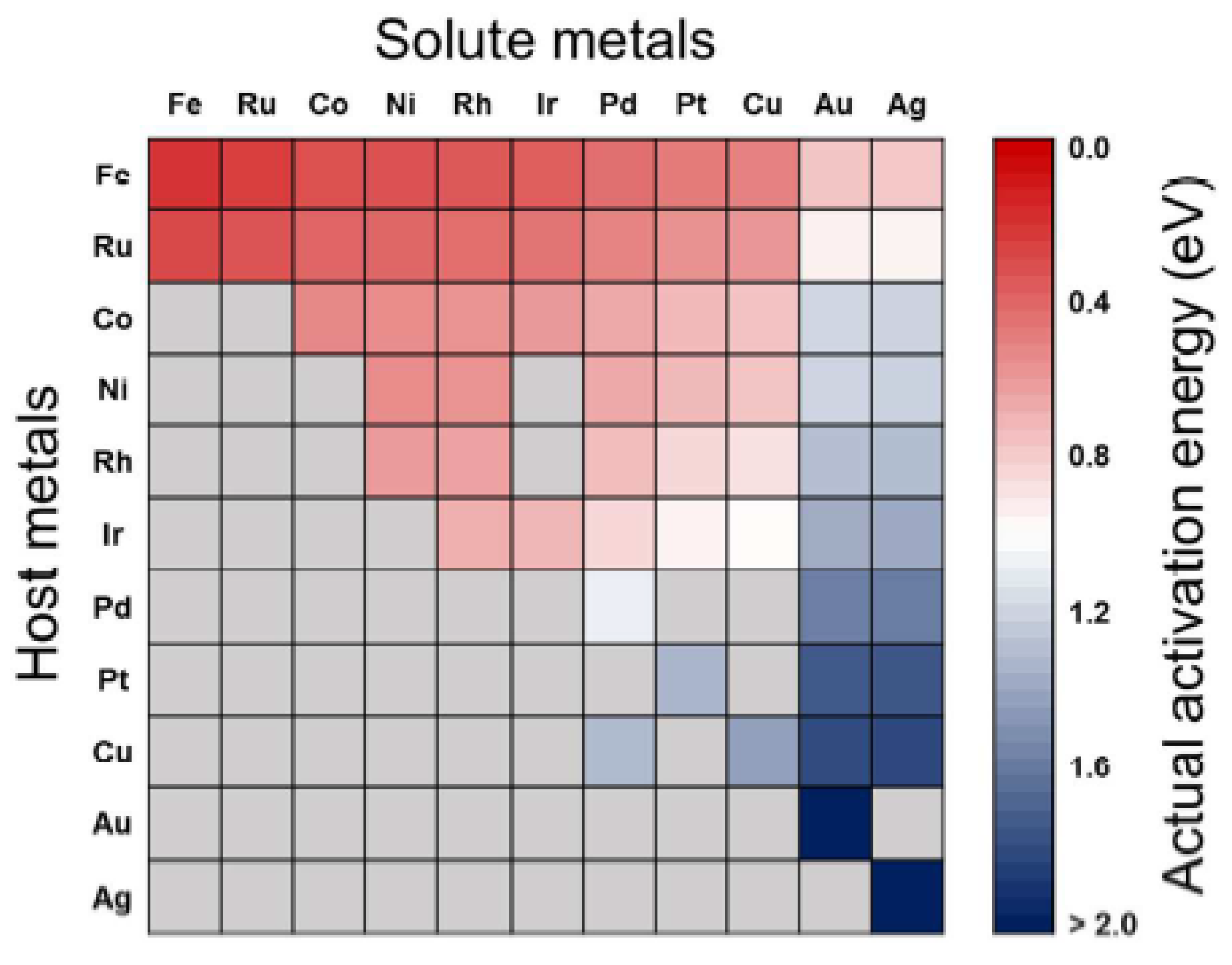{kind=link}
{kind=link}
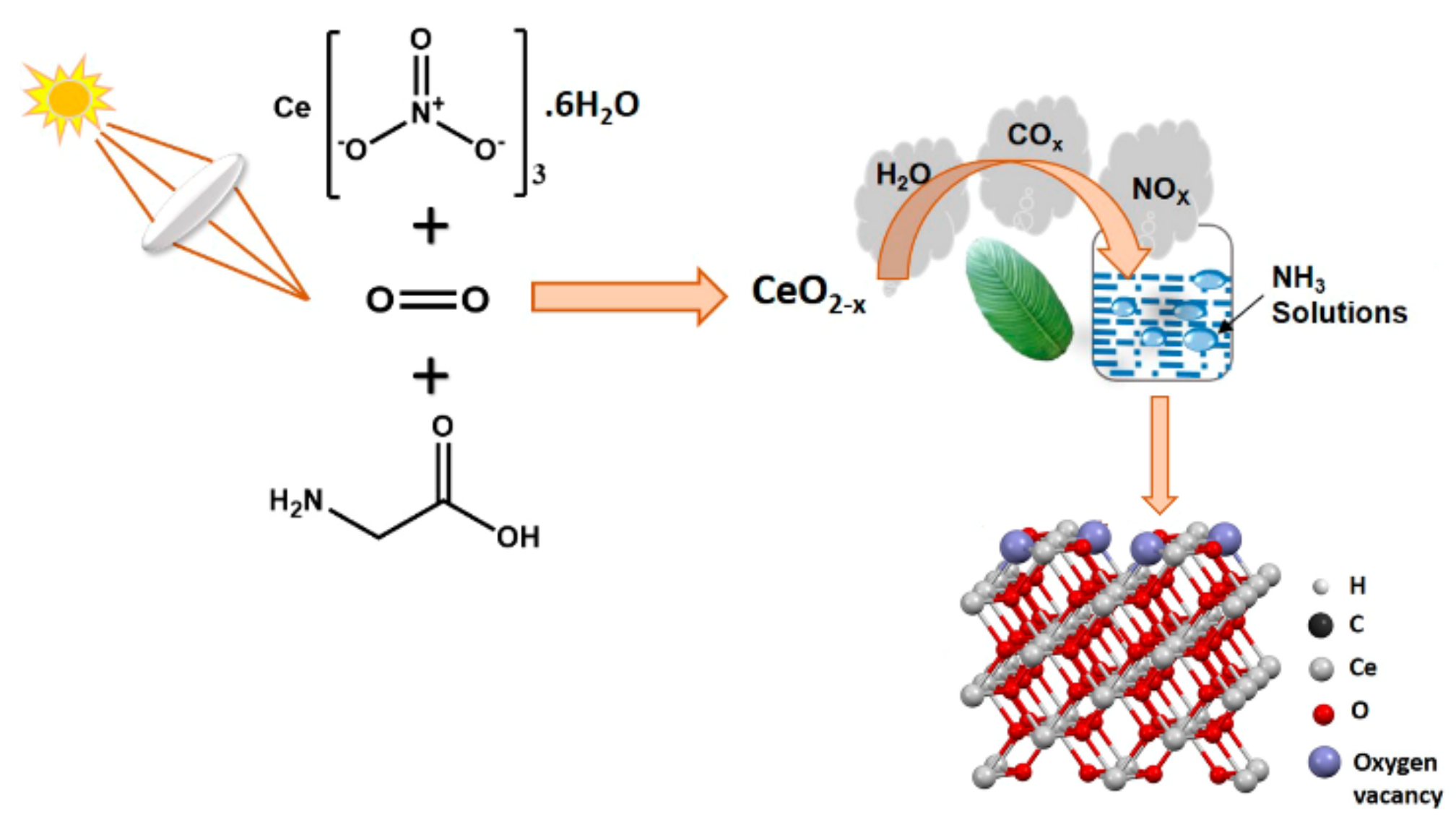{kind=link}
{kind=link}
{kind=link}
{kind=link}
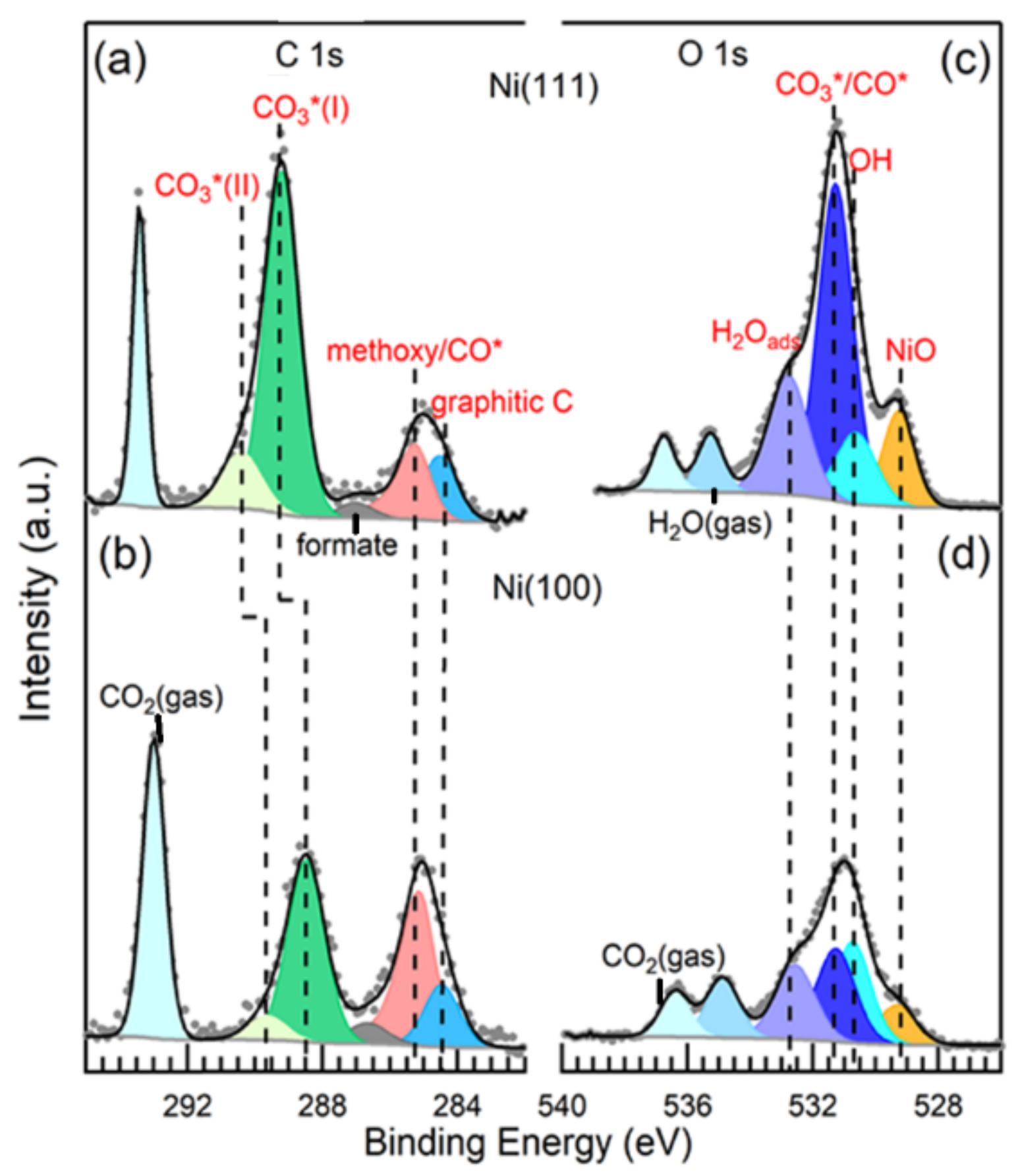{kind=link}
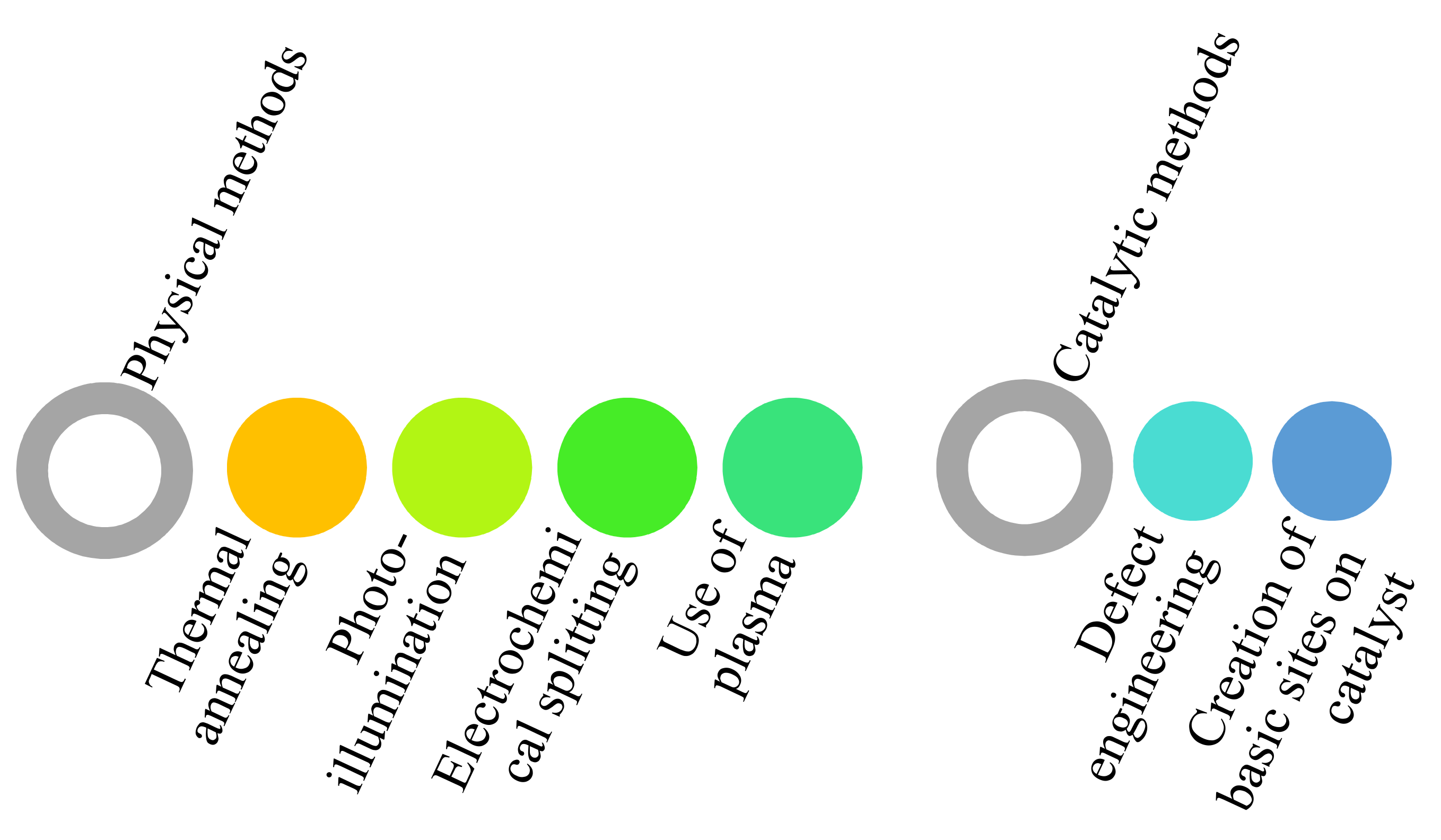{kind=link}
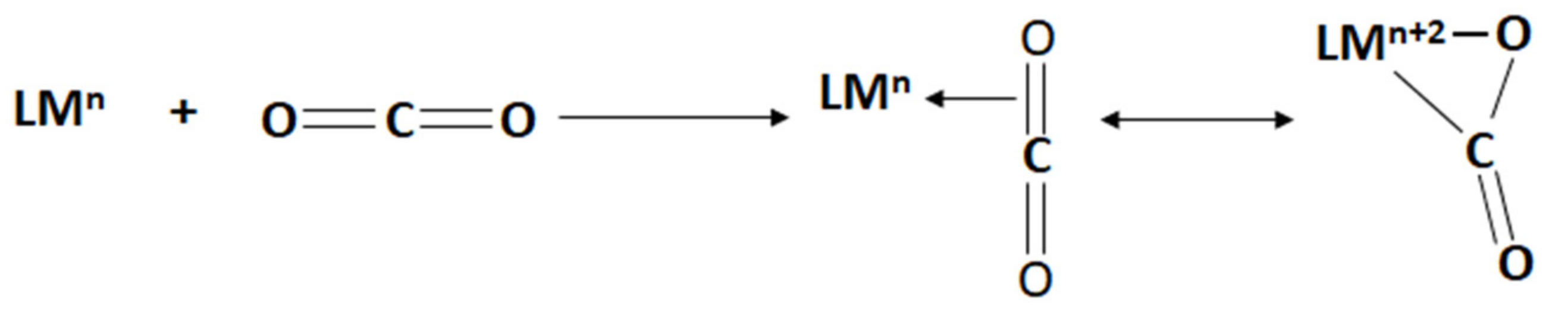{kind=link}
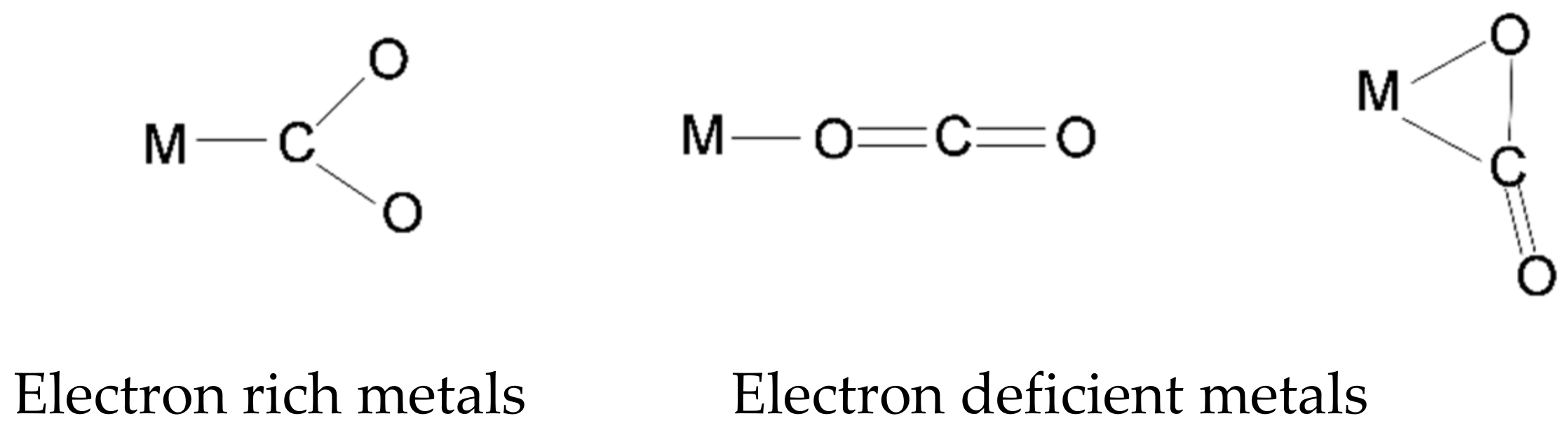{kind=link}
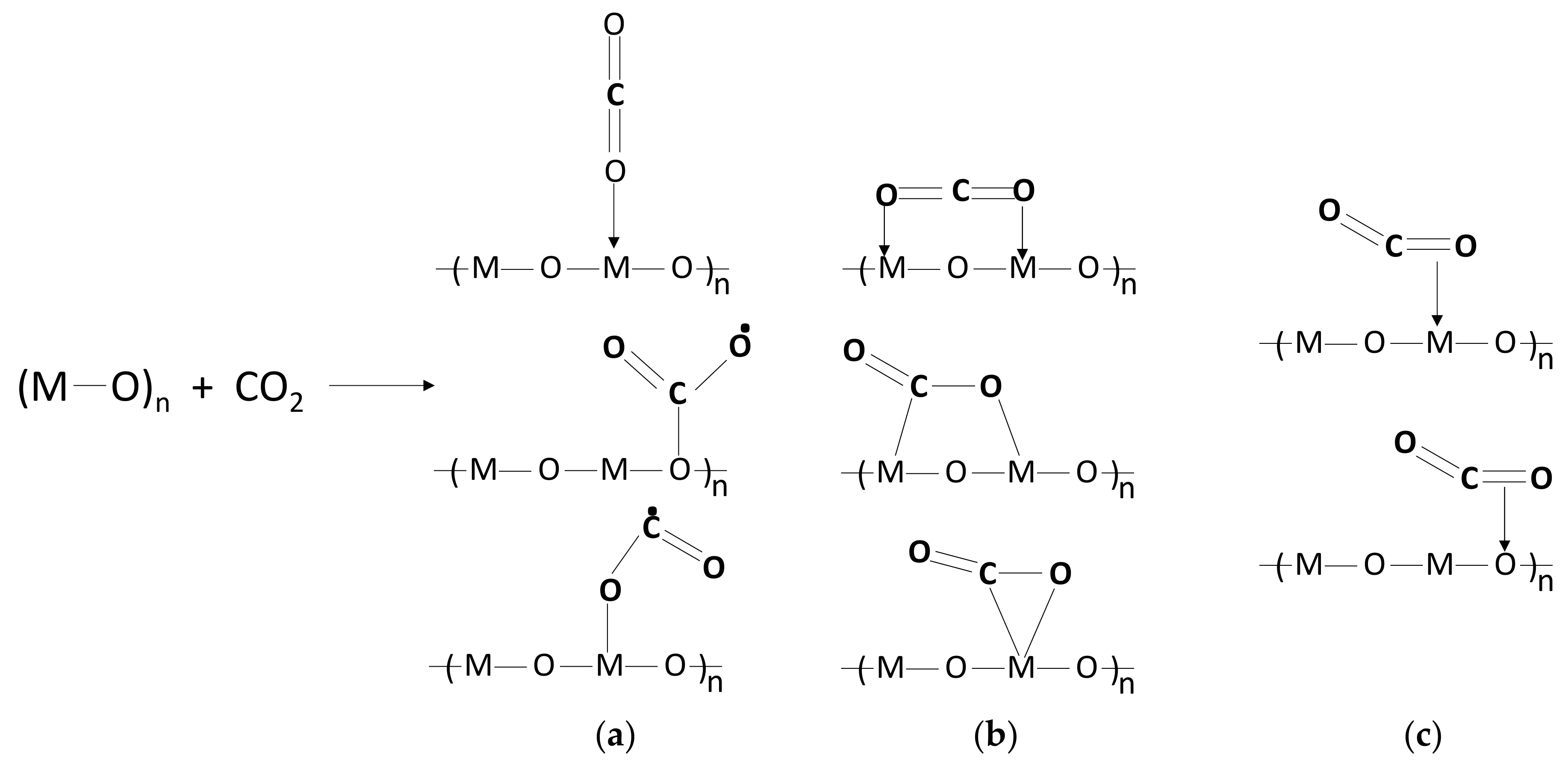{kind=link}
Table 1.
Standard enthalpies (ΔH°298K) and Gibbs free energies (ΔG°298K) for different products formed via CO2.
Table 1.
Standard enthalpies (ΔH°298K) and Gibbs free energies (ΔG°298K) for different products formed via CO2.
| Equation | Reaction | ΔH° 298 K (kJ·mol−1) | ΔG° 298 K (kJ·mol−1) |
|---|---|---|---|
| (1) | 293.0 | 257.2 | |
| (2) | 41.2 | 28.6 | |
| (3) | −49.5 | 3.5 | |
| (4) | −86.7 | −32.4 | |
| (5) | −165.0 | −113.5 | |
| (6) | −132.1 | −78.7 | |
| (7) | −125.0 | −70.9 | |
| (8) | −121.6 | −66.9 | |
| (9) | −64.0 | −28.7 | |
| (10) | −83.6 | −42.1 | |
| (11) | −90.3 | −45.2 |
Thermodynamics data adapted with permission from the authors of [17]. Copyright 2016 Elsevier B.V.
Table 2.
Properties of CO2 molecule.
| Property | Boiling Point (° C) | Density Gas (g/L) | Dipole Moment | Bond Polarity | ∆Hf (298 K) (kJ·mol−1) | Bond Energy (kJ·mol−1) | C–O Bond Distance (Å) | Bond Angle (degree) | Band Gap | Charge | a HOMO Structure | b LUMO Structure |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CO2 | −78.5 (sublimes) | 1.98 | 0 | 1.0 | −393.5 | 806 | 1.16 | 180 | ca. 8 e | C: −0.360 O: +719 |  |  |
a Highest occupied molecular orbital, b Lowest unoccupied molecular orbital (high energy for ground state CO2 molecule).
Table 3.
DFT-calculated adsorption energies, geometries, and charge transferred to CO2 on single crystal surfaces of metals. Data extracted with permission from the authors of [88]. Copyright 2016 American Chemical Society.
Table 3.
DFT-calculated adsorption energies, geometries, and charge transferred to CO2 on single crystal surfaces of metals. Data extracted with permission from the authors of [88]. Copyright 2016 American Chemical Society.
| Metal | Physisorption | Chemisorption | ||
|---|---|---|---|---|
| Binding Energy (kJ·mol−1) | OCO Angle/° | Binding Energy (kJ·mol−1) | Net Charge/e | |
| Fe (110) | −23 | 121 | −90 | −1.11 |
| Ir (111) | −33 | 128 | −34 | −0.47 |
| Pd (111) | −32 | 140 | −17 | −0.35 |
| Ru (0001) | −31 | 123 | −61 | −0.83 |
| Rh (111) | −32 | 135 | −35 | −0.46 |
| Ni (111) | −26 | 136 | −20 | −0.50 |
| Co (0001) | −25 | 139 | −30 | −0.64 |
| Pt (111) | −21 | 131 | −3 | −0.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Etim, U.J.; Zhang, C.; Zhong, Z. Impacts of the Catalyst Structures on CO2 Activation on Catalyst Surfaces. Nanomaterials 2021, 11, 3265. https://doi.org/10.3390/nano11123265
AMA Style
Etim UJ, Zhang C, Zhong Z. Impacts of the Catalyst Structures on CO2 Activation on Catalyst Surfaces. Nanomaterials. 2021; 11(12):3265. https://doi.org/10.3390/nano11123265
Chicago/Turabian StyleEtim, Ubong J., Chenchen Zhang, and Ziyi Zhong. 2021. "Impacts of the Catalyst Structures on CO2 Activation on Catalyst Surfaces" Nanomaterials 11, no. 12: 3265. https://doi.org/10.3390/nano11123265
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.