
Comparison of the Morphological and Structural Characteristic of Bioresorbable and Biocompatible Hydroxyapatite-Loaded Biopolymer Composites


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Different Calcium Phosphate Nano-Powders
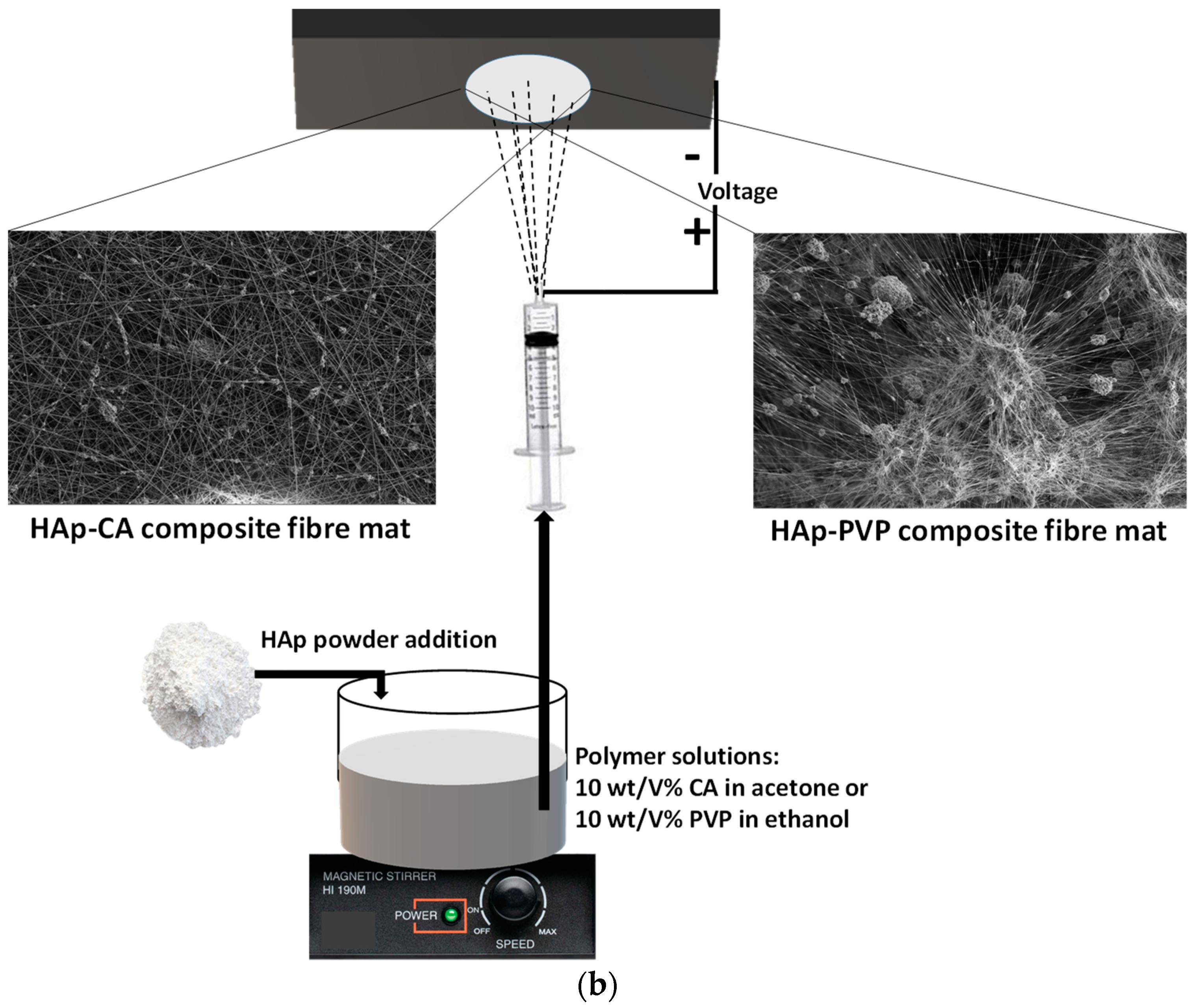
2.2. Preparation of cHAp/PVP and cHAp/CA Composites by Electrospinning
2.3. Characterisation Methods
2.3.1. X-Ray Diffraction Analysis
2.3.2. Scanning Electron Microscopy (SEM)
2.3.3. FT-IR and Raman Analysis
2.3.4. Immersion Tests
3. Results and Discussion
3.1. Morphological and Structural Characterisation of Calcium Phosphate (CaP) Powders Prepared with Different Parameters
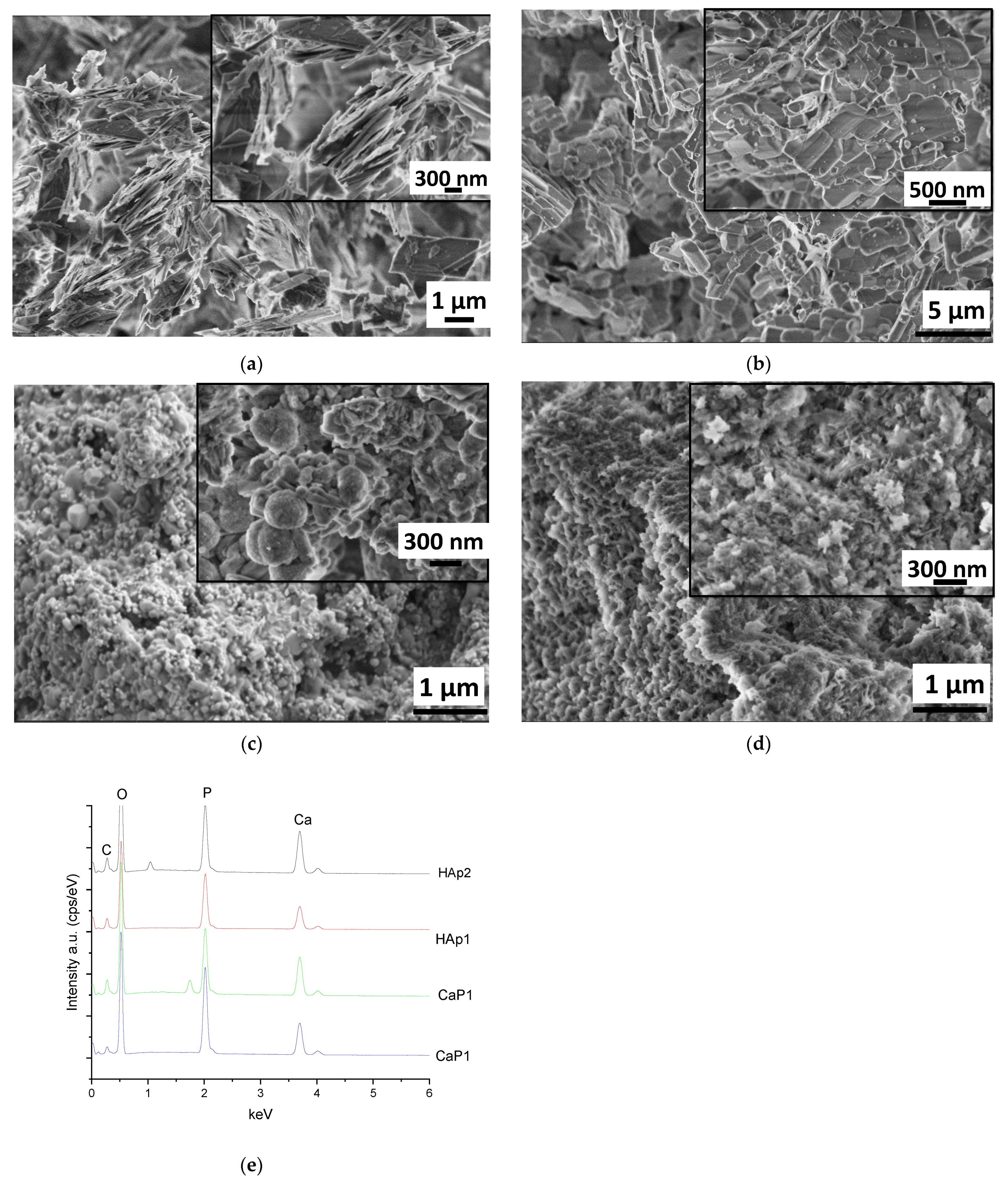
3.1.1. Scanning Electron Microscope Analysis of CaP Powders
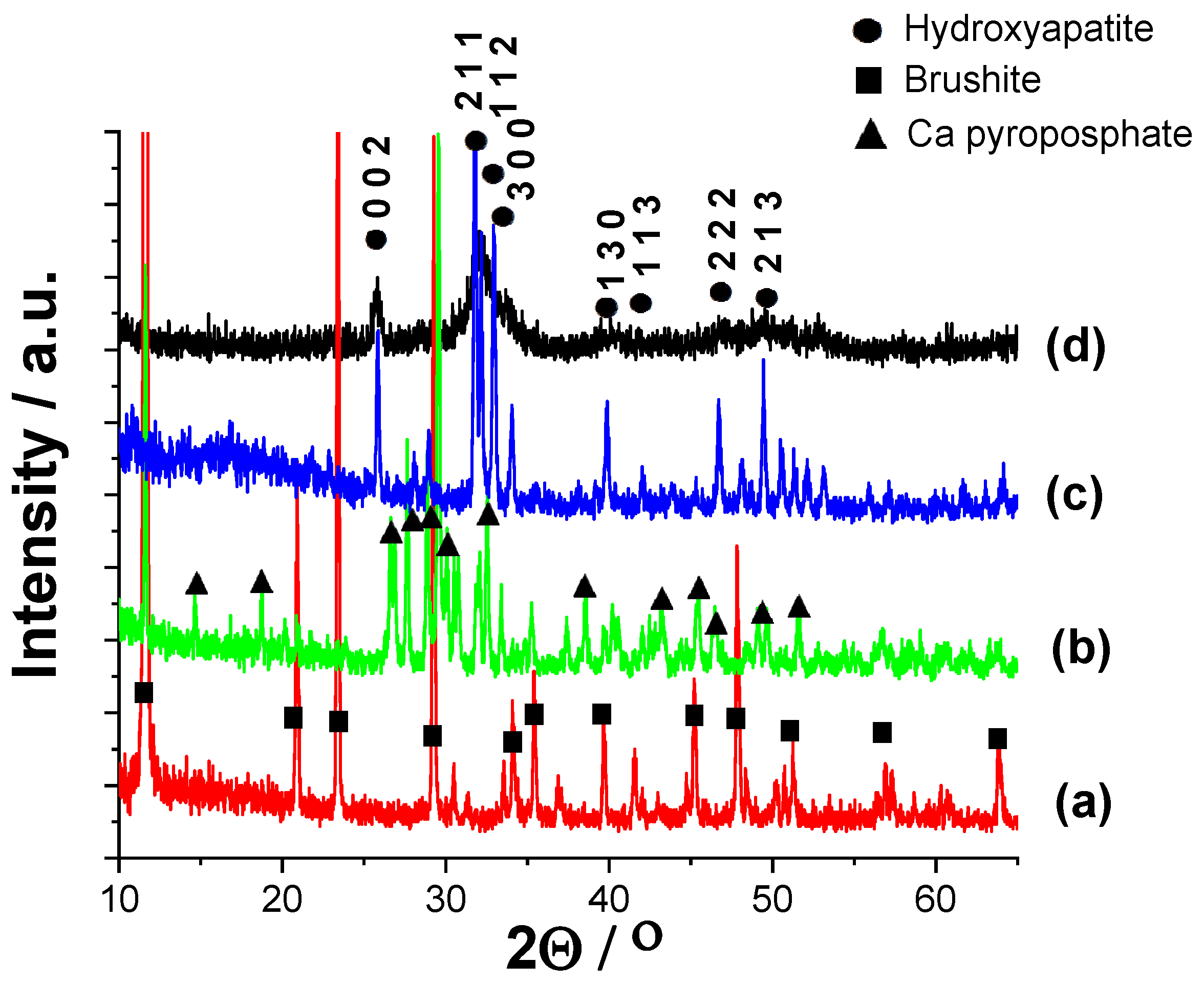
3.1.2. Structural Investigation of Calcium Phosphate Powders Prepared by Wet Precipitation by XRD Measurements
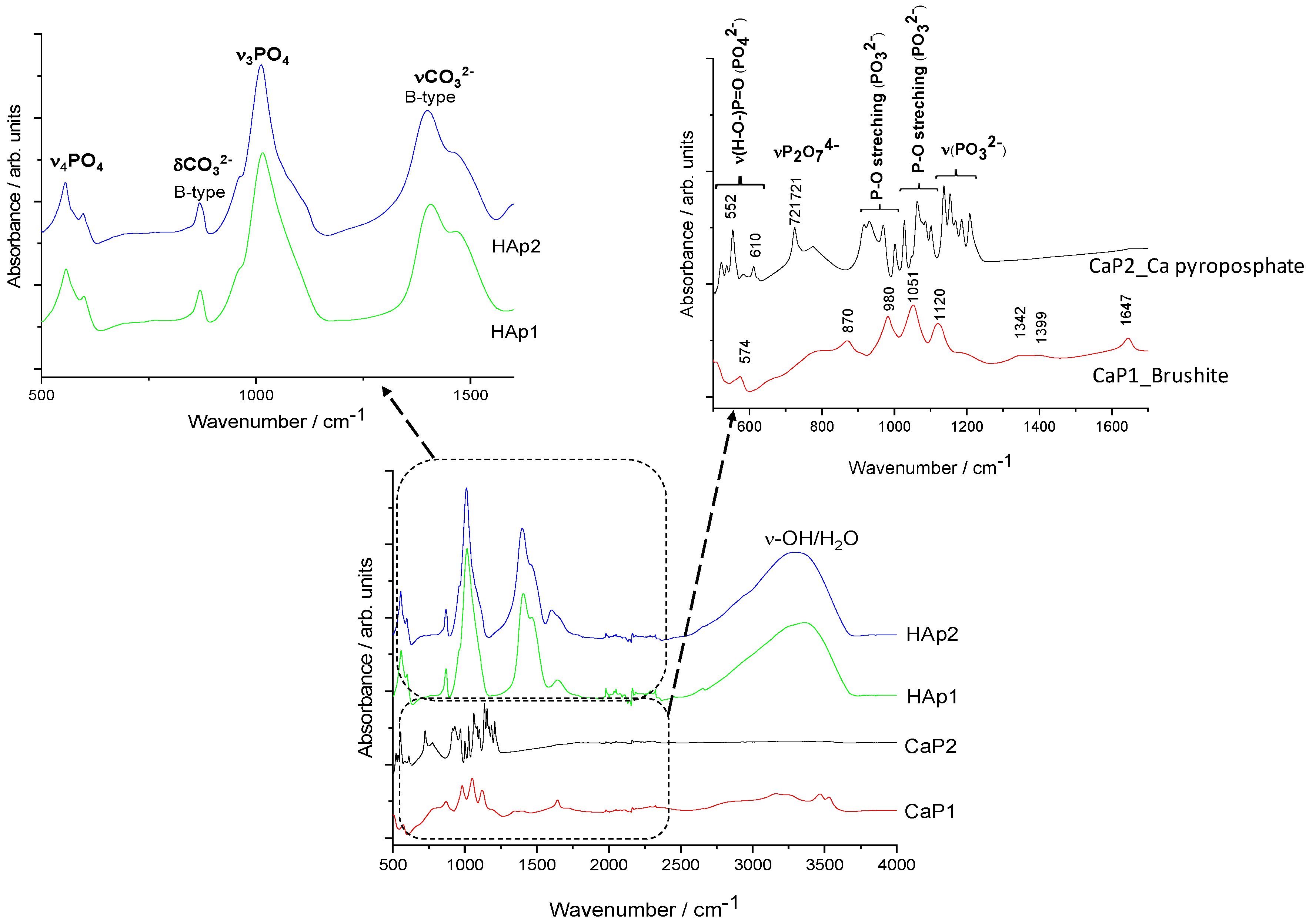
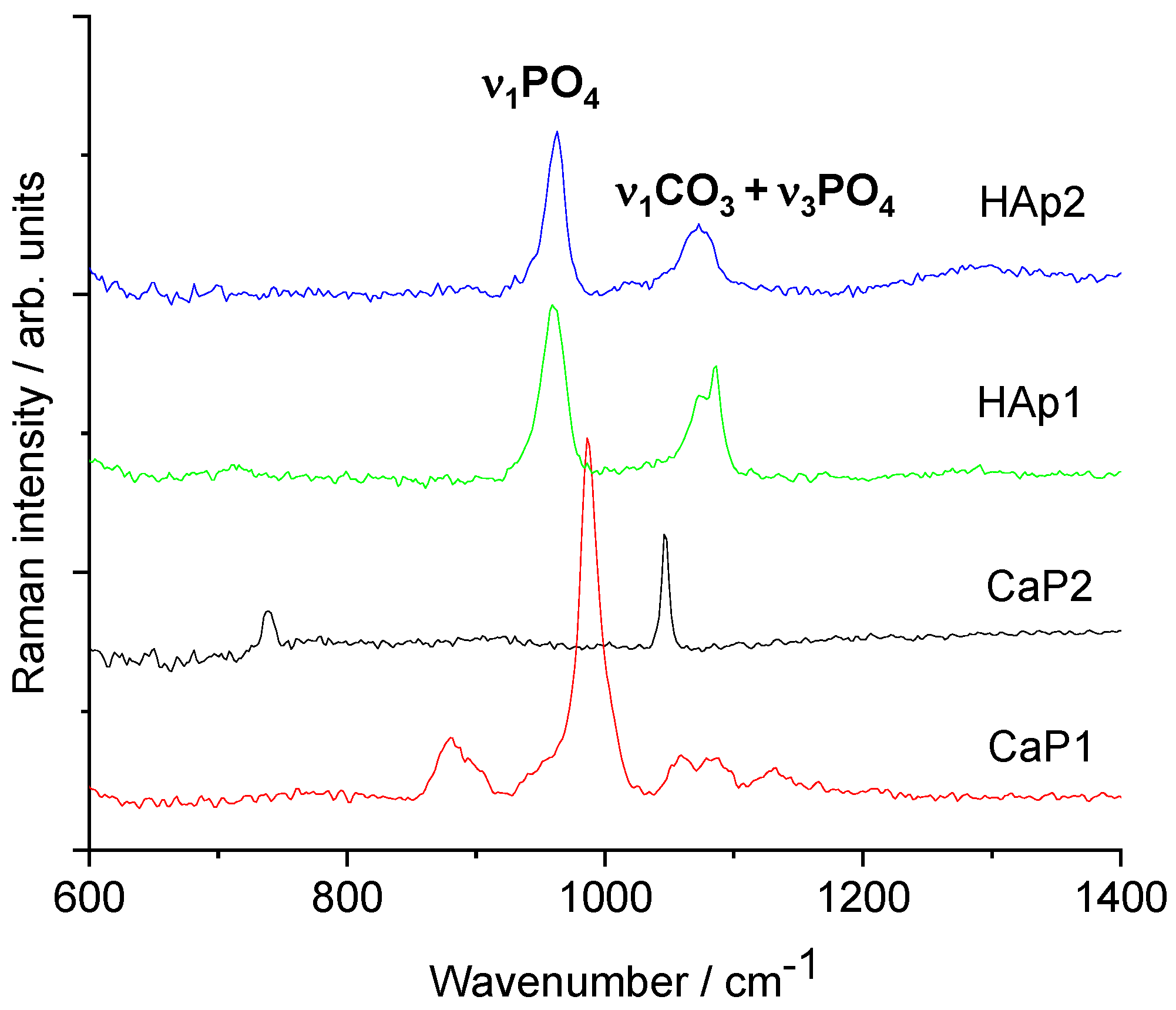
3.1.3. FT-IR and Raman Characterisation of CaP Powders
3.2. Morphological Characterisation of PURE and cHAp-Loaded PVP and CA
3.3. Immersion Test
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodrich, J.T.; Sandler, A.L.; Tepper, O. A review of reconstructive materials for use in craniofacial surgery bone fixation materials, bone substitutes, and distractors. Child. Nerv. Syst. 2012, 28, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- González-Carrasco, J.L.; Cuellar, S.C.C.; Rodríguez, M.L. Chapter 5—Metals. In Bone Repair Biomaterials: Regeneration and Clinical Applications, 2nd ed.; Pawelec, K.M., Planell, J.A., Eds.; Woodhead Publishing Series in Biomaterials; Elsevier: Sawston, UK, 2019; pp. 103–140. [Google Scholar]
- Eliaz, N. Corrosion of Metallic Biomaterials: A Review. Materials 2019, 12, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, K.W.; Wong, K.H. Biodegradable metallic materials for orthopaedic implantations: A review. Technol. Health Care 2012, 20, 345–362. [Google Scholar] [CrossRef]
- LeGeros, R.Z. Biodegradation and bioresorption of calcium phosphate ceramics. Clin. Mater. 1993, 14, 65–88. [Google Scholar] [CrossRef]
- Griffith, L.G.; Naughton, G. Tissue engineering—Current challenges and expanding opportunities. Science 2002, 295, 1009–1014. [Google Scholar] [CrossRef]
- Burg, K.J.; Porter, S.; Kellam, J.F. Biomaterial developments for bone tissue engineering. Biomaterials 2000, 21, 2347–2359. [Google Scholar] [CrossRef]
- Sheikh, Z.; Sima, C.; Glogauer, M. Bone replacement materials and techniques used for achieving vertical alveolar bone augmentation. Materials 2015, 8, 2953–2993. [Google Scholar] [CrossRef]
- Sheikh, Z.; Javaid, M.A.; Hamdan, N.; Hashmi, R. Bone regeneration using bone morphogenetic proteins and various biomaterial carriers. Materials 2015, 8, 1778–1816. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.; Hurzeler, M.B.; Schliephake, H. A review of material properties of biodegradable and bioresorbable polymers and devices for GTR and GBR applications. Int. J. Oral Maxillofac. Implant. 1996, 11, 667–678. [Google Scholar]
- Sheikh, Z.; Najeeb, S.; Khurshid, Z.; Verma, V.; Rashid, H.; Glogauer, M. Biodegradable Materials for Bone Repair and Tissue Engineering Applications. Materials 2015, 8, 5744–5794. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Wu, Z.; Chen, H. Poly(N-vinylpyrrolidone)-Modified surfaces for biomedical applications. Macromol. Biosci. 2013, 13, 147–154. [Google Scholar] [CrossRef]
- Koczkur, K.M.; Mourdikoudis, S.; Polavarapu, L.; Skrabalak, S.E. Polyvinylpyrrolidone (PVP) in nanoparticle synthesis. Dalton Trans. 2015, 44, 17883–17905. [Google Scholar] [CrossRef] [Green Version]
- Sobczak-Kupiec, A.; Pluta, K.; Drabczyk, A.; Włoś, M.; Tyliszczak, B. Synthesis and characterization of ceramic-polymer composites containing bioactive synthetic hydroxyapatite for biomedical applications. Ceram. Int. 2018, 44, 13630–13638. [Google Scholar] [CrossRef]
- Elashmawi, I.S.; Baieth, H.E.A. Spectroscopic studies of hydroxyapatite in PVP/PVA polymeric matrix as biomaterial. Curr. Appl. Phys. 2012, 12, 141–146. [Google Scholar] [CrossRef]
- Kurakula, M.; Rao, G.S.N.K. Moving polyvinyl pyrrolidone electrospun nanofibers and bioprinted scaffolds toward multidisciplinary biomedical applications. Eur. Polym. J. 2020, 136, 109919. [Google Scholar] [CrossRef]
- Fischer, S.; Thümmler, K.; Volkert, B.; Hettrich, K.; Schmidt, I.; Fischer, K. Properties and applications of cellulose acetate. Macromol. Symp. 2008, 262, 89–96. [Google Scholar] [CrossRef]
- Salama, A. Cellulose/calcium phosphate hybrids: New materials for biomedical and environmental applications. Intern. J. Biol. Macromol. 2019, 12, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Angel, N.; Guo, L.; Yan, F.; Wang, H.; Kong, L. Effect of processing parameters on the electrospinning of cellulose acetate studied by response surface methodology. J. Agric. Food Res. 2020, 2, 100015. [Google Scholar] [CrossRef]
- Shahriar, S.M.S.; Mondal, J.; Hasan, M.N.; Revuri, V.; Lee, D.Y.; Lee, Y.K. Electrospinning nanofibers for therapeutics delivery. Nanomaterials 2019, 9, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.I.; Ko, E.K.; Yum, J.; Jung, C.H.; Lee, Y.M.; Shin, H. Nanofibrous poly(lactic acid)/hydroxyapatite composite scaffolds for guided tissue regeneration. Macromol. Biosci. 2008, 8, 328–338. [Google Scholar] [CrossRef]
- Turon, P.; del Valle, L.J.; Alemán, C.; Puiggalí, J. Biodegradable and Biocompatible Systems Based on Hydroxyapatite Nanoparticles. Appl. Sci. 2017, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Cama, G.; Gharibi, B.; Knowles, J.C.; Romeed, S.; DiSilvio, L.; Deb, S. Structural changes and biological responsiveness of an injectable and mouldable monetite bone graft generated by a facile synthetic method. J. R. Soc. Interface 2014, 11, 20140727. [Google Scholar] [CrossRef] [PubMed]
- Idowu, B.; Cama, G.; Deb, S.; Di Silvio, L. In vitro osteoinductive potential of porous monetite for bone tissue engineering. J. Tissue Eng. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Touny, A.H.; Bhaduri, S.B. A reactive electrospinning approach for nanoporous PLA/monetite nanocomposite fibers. Mat. Sci. Eng. C. 2010, 30, 1304–1312. [Google Scholar] [CrossRef]
- Safronova, T.V.; Putlayev, V.I.; Bessonov, K.A.; Ivanov, V.K. Ceramics based on calcium pyrophosphate nanopowders. Proc. Appl. Ceram 2013, 7, 9–14. [Google Scholar] [CrossRef]
- Corrêa, T.H.A.; Holanda, J.N.F. Calcium pyrophosphate powder derived from avian eggshell waste. Cerâmica 2016, 62, 278–280. [Google Scholar] [CrossRef] [Green Version]
- Zyman, Z.Z.; Goncharenko, A.V.; Rokhmistrov, D.V. Phase evolution during heat treatment of amorphous calcium phosphate derived from fast nitrate synthesis. Proc. Appl. Ceram 2017, 11, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.A.; Mansour, S.F.; El-dek, S.I.; Abd-Elwahab, S.M.; Ahmed, M.K. Characterization and annealing performance of calcium phosphate nanoparticles synthesized by co-precipitation method. Ceram Intern. 2014, 40, 12807–12820. [Google Scholar] [CrossRef]
- Londoño-Restrepo, S.M.; Herrera-Lara, M.; Bernal-Alvarez, L.R.; Rivera-Muñoza, E.M.; Rodriguez-García, M.E. In-situ XRD study of the crystal size transition of hydroxyapatite from swine bone. Ceram Int. 2020, 46, 24454–24461. [Google Scholar] [CrossRef]
- Fathi, M.H.; Hanifi, A.; Mortazavi, V. Preparation and bioactivity evaluation of bone-like hydroxyapatite nanopowder. J. Mater. Process. Technol. 2008, 202, 536–542. [Google Scholar] [CrossRef]
- Koerten, H.K.; van der Meulen, J. Degradation of calcium phosphate ceramics. J. Biomed. Mater. Res. 1999, 44, 78–86. [Google Scholar] [CrossRef]
- Al-Qasas, N.S.; Rohani, S. Synthesis of Pure Hydroxyapatite and the Effect of Synthesis Conditions on its Yield, Crystallinity, Morphology and Mean Particle Size. Sep. Sci. Techn. 2007, 40, 3187–3224. [Google Scholar] [CrossRef]
- Anastasiou, A.D.; Thomson, C.L.; Hussain, S.A.; Edwards, T.J.; Strafford, S.; Malinowski, M.; Mathieson, R.; Brown, C.T.A.; Brown, A.P.; Duggal, M.S.; et al. Sintering of calcium phosphates with a femtosecond pulsed laser for hard tissue engineering. Mater. Des. 2016, 101, 346–354. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, A.O.; Jablonski, W.L. X-ray diffraction powder patterns of calcium phosphates. Anal. Chem. 1956, 28, 1424–1427. [Google Scholar] [CrossRef]
- Webb, N. The crystal structure of β-Ca2P2O7. Acta Cryst. 1966, 21, 942–948. [Google Scholar] [CrossRef]
- Smith, D.K.; Johnson, G.G., Jr.; Scheible, A.; Wims, A.M.; Johnson, J.L.; Ullmann, G. Quantitative X-Ray Powder Diffraction Method Using the Full Diffraction Pattern. Powder Diffr. 1987, 2, 73–77. [Google Scholar] [CrossRef]
- Singh, S.; Singh, V.; Aggarwal, S.; Mandal, U.K. Synthesis of brushite nano particles at different temperatures. Chem. Pap. 2010, 64, 491–498. [Google Scholar] [CrossRef]
- Liu, D.M.; Yang, Q.; Troczynski, T.; Tseng, W.J. Structural evolution of sol–gel derived hydroxyapatite. Biomaterials 2002, 23, 1679–1687. [Google Scholar] [CrossRef]
- Zhou, S.; Su, Q.; Li, X.; Weng, J. A novel in situ synthesis of dicalcium phosphate dehydrate nanocrystals in biodegradable polymer matrix. Mater. Sci. Eng. A. 2006, 430, 341–345. [Google Scholar] [CrossRef]
- Salimi, E.; Javadpour, J. Synthesis and Characterization of Nanoporous Monetite Which Can Be Applicable for Drug Carrier. J. Nanomater. 2012, 2012, 5. [Google Scholar] [CrossRef]
- Singh, S.; Bhardwaj, P.; Singh, V.; Aggarwal, S.; Mandal, U.K. Synthesis of nanocrystalline calcium phosphate in microemulsion-effect of nature of surfactants. J. Coll. Interface Sci. 2008, 319, 322–329. [Google Scholar] [CrossRef]
- Sanosh, K.P.; Chu, M.; Balakrishnan, A.; Lee, Y.; Kim, T.N.; Cho, S. Synthesis of nano hydroxyapatite powder that simulate teeth particle morphology and composition. Curr. Appl. Phys. 2009, 9, 1459–1462. [Google Scholar] [CrossRef]
- Antonakosa, A.; Liarokapisa, E.; Leventouri, T. Micro-Raman and FTIR studies of synthetic and natural apatites. Biomaterials 2007, 28, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Kumta, P.N. An alternative chemical route for the synthesis and thermal stability of chemically enriched hydroxyapatite. J. Am. Ceram. Soc. 2006, 89, 444–449. [Google Scholar] [CrossRef]
- Quillard, S.; Mevellec, J.-Y.; Deniard, P.; Bouler, J.-M.; Buisson, J.-P. Polarized Raman spectra of brushite (CaHPO4 2H2O) crystal. Investigation of the phosphate stretching modes, study of the LOTO splitting: Polarized Raman spectra. J. Raman Spectrosc. 2016, 47, 971–977. [Google Scholar] [CrossRef]
- Soulié, J.; Gras, P.; Marsan, O.; Laurencin, D.; Rey, C.; Combes, C. Development of a new family of monolithic calcium (pyro)phosphate glasses by soft chemistry. Acta Biomater. 2016, 41, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Mihály, J.; Gombás, V.; Afishah, A.; Mink, J. FT-Raman investigation of human dental enamel surfaces. J. Raman Spectrosc. 2009, 40, 898–902. [Google Scholar] [CrossRef]
- Awonusi, A.; Morris, M.D.; Tecklenburg, M.M.J. Carbonate Assignment and Calibration in the Raman Spectrum of Apatite. Calcif. Tissue Int. 2007, 81, 46–52. [Google Scholar] [CrossRef]
- Zakaria, F.Z.; Mihály, J.; Sajó, I.; Katona, R.; Hajba, L.; Aziz, F.A.; Mink, J. FT-Raman and FTIR spectroscopic characterization of biogenic carbonates from Philippine venus seashell and Porites sp. coral. J. Raman Spectrosc. 2008, 39, 1204–1209. [Google Scholar] [CrossRef]
- Schmid, T.; Dariz, P. Shedding light onto the spectra of lime: Raman and luminescence bands of CaO, Ca(OH)2 and CaCO3: Raman and luminescence bands of CaO, Ca(OH)2 and CaCO3. J. Raman Spectrosc. 2015, 46, 141–146. [Google Scholar] [CrossRef]
- Von Euw, S.; Wang, Y.; Laurent, G.; Drouet, C.; Babonneau, F.; Nassif, N. Bone mineral: New insights into its chemical composition. Sci. Rep. 2019, 9, 8456. [Google Scholar] [CrossRef] [Green Version]
- Glimcher, M.J. Recent studies of the mineral phase in bone and its possible linkage to the organic matrix by protein-bound phosphate bonds. Phil. Trans. R. Soc. Lond. B. Biol. Sci. 1984, 304, 479–508. [Google Scholar] [CrossRef]
- Vallet-Regi, M.; Navarrete, D.A. Chapter 1—Biological Apatites in Bone and Teeth. In Nanoceramics in Clinical Use: From Materials to Applications, 2nd ed.; O’Brien, P., Ed.; RCS Publishing: London, UK, 2015; pp. 1–29. [Google Scholar] [CrossRef]
- Lee, H.; Nishino, M.; Shon, D.; Lee, J.S.; Kim, I.S. Control of the morphology of cellulose acetate nanofibers via electrospinning. Cellulose 2018, 25, 2829–2837. [Google Scholar] [CrossRef]
- Chuangchote, S.; Sagawa, T.; Yoshikawa, S. Electrospinning of poly(vinyl pyrrolidone): Effects of solvents on electrospinnability for the fabrication of poly(p-phenylene vinylene) and TiO2 nanofibers. J. Appl. Pol. Sci. 2013, 114, 2777–2791. [Google Scholar] [CrossRef]
- Du, Q.; Harding, D.R.; Yang, H. Helical peanut- shaped poly(vinyl pyrrolidone) ribbons generated by electrospinning. Polymer 2013, 54, 6752–6759. [Google Scholar] [CrossRef]
- Amith, V.; Sridhar, R.; Angadi, G.; Prajwal, D.; Mamatha, V.; Murthy, H.N.N. Development of Electrospinning System for Synthesis of Polyvinylpyrrolidone Thin Films for Sensor Applications. Mater Today 2018, 5, 20920–20926. [Google Scholar] [CrossRef]
- Utkarsh; Hegab, H.; Tariq, M.; Syed, N.A.; Rizvi, G.; Pop-Iliev, R. Towards Analysis and Optimization of Electrospun PVP (Polyvinylpyrrolidone) Nanofibers. Hindaw Adv. Pol. Techn. 2020, 9. [Google Scholar] [CrossRef]
- Feng, P.; Peng, S.; Shuai, C.; Gao, C.; Yang, W.; Bin, S.; Min, A. In Situ Generation of Hydroxyapatite on Biopolymer Particles for Fabrication of Bone Scaffolds Owning Bioactivity. ACS Appl. Mater. Interfaces 2020, 12, 46743–46755. [Google Scholar] [CrossRef]
- Fook, A.C.B.M.; Fideles, T.B.; Barbosa, R.C.; Furtado, G.T.F.S.; Sampaio, G.Y.H.; Fook, M.V.L. Hydroxyapatite/Biopolymers Composite Scaffolds for Bone Tissue Engineering. Key Eng. Mater. 2011, 493–494, 826–831. [Google Scholar] [CrossRef]
- Luong, N.D.; Moon, I.-S.; Lee, D.S.; Lee, Y.-K.; Nam, J.-D. Surface modification of poly(L-lactide) electrospun fibers with nanocrystal hydroxyapatite for engineered scaffold applications. Mater. Sci. Eng. C 2008, 28, 1242–1249. [Google Scholar] [CrossRef]
- Nouri-Felekori, M.; Khakbiz, M.; Nezafati, N.; Mohammadi, J.; Eslaminejad, M.B.; Fani, N. Characterization and multiscale modeling of novel calcium phosphate composites containing hydroxyapatite whiskers and gelatin microspheres. J. Alloys Comp. 2020, 832, 154938. [Google Scholar] [CrossRef]
- Gritsch, L.; Perrin, E.; Chenal, J.-M.; Fredholm, Y.; Maçon, A.L.B.; Chevalier, J.; Aldo, R.; Boccaccini, A.R. Combining bioresorbable polyesters and bioactive glasses: Orthopedic applications of composite implants and bone tissue engineering scaffolds. Appl. Mater. Today 2021, 22, 100923. [Google Scholar] [CrossRef]
- Heydari, Z.; Mohebbi-Kalhoria, D.; Afarani, M.S. Engineered electrospun polycaprolactone (PCL)/octacalcium phosphate (OCP) scaffold for bone tissue engineering. Mater. Sci. Eng. C 2017, 81, 127–132. [Google Scholar] [CrossRef] [PubMed]
- El-Habashy, S.E.; Eltaher, H.M.; Gaballah, A.; Zaki, E.I.; Mehanna, R.A.; El-Kamel, A.H. Hybrid bioactive hydroxyapatite/polycaprolactone nanoparticles for enhanced osteogenesis. Mater. Sci. Eng. C 2021, 119, 111599. [Google Scholar] [CrossRef] [PubMed]
- Moeini, S.; Mohammadi, M.R.; Simchi, A. In-situ solvothermal processing of polycaprolactone/hydroxyapatite nanocomposites with enhanced mechanical and biological performance for bone tissue engineering. Bioact. Mater. 2017, 2, 146–155. [Google Scholar] [CrossRef]
- Gouma, P.; Xue, R.; Goldbeck, C.P.; Perrotta, P.; Balázsi, C. Nano-hydroxyapatite—Cellulose acetate composites for growing of bone cells. Mater. Sci. Eng. C 2012, 32, 607–612. [Google Scholar] [CrossRef]
- Sebastian, T.; Preisker, T.R.; Gorjana, L.; Graule, T.; Aneziris, C.G.; Clemens, F.J. Synthesis of hydroxyapatite fibers using electrospinning: A study of phase evolution based on polymer matrix. J. Eur. Ceram. Soc. 2020, 40, 2489–2496. [Google Scholar] [CrossRef]
- Gomes Luz, E.P.C.; Chaves, P.H.S.; Vieira, L.A.P.; Ribeiro, S.F.; Borges, M.F.; Andrade, F.K.; Muniz, C.R.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Rosa, M.F.; et al. In vitro degradability and bioactivity of oxidized bacterial cellulose hydroxyapatite Composites. Carbohydr Polym. 2020, 237, 116174. [Google Scholar] [CrossRef]
- Yamashita, Y.; Endo, T. Deterioration Behavior of Cellulose Acetate Films in Acidic or Basic Aqueous Solutions. J. Appl. Polym. Sci. 2004, 91, 3354–3361. [Google Scholar] [CrossRef]
- Dridi, A.; Riahi, K.Z.; Somrani, S. Mechanism of apatite formation on a poorly crystallized calcium phosphate in a simulated body fluid (SBF) at 37 °C. J. Phys. Chem. Solids 2021, 156, 110122. [Google Scholar] [CrossRef]
- El-Arnaouty, M.B.; Eid, M.; Salah, M.; Hegazy, A.E.-S. Preparation and Characterization of Poly Vinyl Alcohol/Poly Vinyl Pyrrolidone/Clay Based Nanocomposite by Gamma Irradiation. J. Macromol. Sci. A Pure Appl. Chem. 2012, 49, 1041–1051. [Google Scholar] [CrossRef]
- Matsuyama, H.; Maki, T.; Teramoto, M.; Kobayashi, K. Effect of PVP Additive on Porous Polysulfone Membrane Formation by Immersion Precipitation Method. Sep. Sci. Techn. 2003, 38, 3449–3458. [Google Scholar] [CrossRef]
- Naeem, M.A.; Siddiqui, M.Q.; Leroy, L.; Khan, M.R.; Wei, Q. The production and characterization of microbial cellulose—Electrospun membrane hybrid nano-fabrics. J. Ind. Text. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Sheikh, E.; Sheikh, A. Effects of Gelatin and Nano-ZnO on the Performance and Properties of Cellulose Acetate Water Membranes. Chem. Eng. Technol. 2020, 43, 1749–1757. [Google Scholar] [CrossRef]
- Cui, L.-Y.; Zeng, R.-C.; Li, S.-Q.; Zhang, F.; Han, E.-H. Corrosion resistance of layer-by-layer assembled polyvinylpyrrolidone/polyacrylic acid and amorphous silica films on AZ31 magnesium alloys. RSC Adv. 2016, 6, 63107. [Google Scholar] [CrossRef]
- Valbon, A.; Neves, M.A.; Echevarria, A. Anticorrosive Effect of PVP 40000 against AISI 1020 Carbon Steel in HCl. Mater. Res. 2018, 21, e20170847. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements/ At% | O | C | Ca | P | Total | Ca/P Ratio |
|---|---|---|---|---|---|---|
| CaP1 | 35.5 | 4.4 | 31.7 | 28.4 | 100 | 1.11 |
| CaP2 | 34.8 | 1.3 | 37.8 | 26.1 | 100 | 1.45 |
| HAp1 | 37.9 | 3.8 | 37.9 | 20.4 | 100 | 1.85 |
| HAp2 | 31.7 | 5.4 | 39.8 | 23.1 | 100 | 1.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furko, M.; Horváth, Z.E.; Mihály, J.; Balázsi, K.; Balázsi, C. Comparison of the Morphological and Structural Characteristic of Bioresorbable and Biocompatible Hydroxyapatite-Loaded Biopolymer Composites. Nanomaterials 2021, 11, 3194. https://doi.org/10.3390/nano11123194
Furko M, Horváth ZE, Mihály J, Balázsi K, Balázsi C. Comparison of the Morphological and Structural Characteristic of Bioresorbable and Biocompatible Hydroxyapatite-Loaded Biopolymer Composites. Nanomaterials. 2021; 11(12):3194. https://doi.org/10.3390/nano11123194
Chicago/Turabian StyleFurko, Monika, Zsolt E. Horváth, Judith Mihály, Katalin Balázsi, and Csaba Balázsi. 2021. "Comparison of the Morphological and Structural Characteristic of Bioresorbable and Biocompatible Hydroxyapatite-Loaded Biopolymer Composites" Nanomaterials 11, no. 12: 3194. https://doi.org/10.3390/nano11123194