Turning Waste into Useful Products by Photocatalysis with Nanocrystalline TiO2 Thin Films: Reductive Cleavage of Azo Bond in the Presence of Aqueous Formate


Abstract
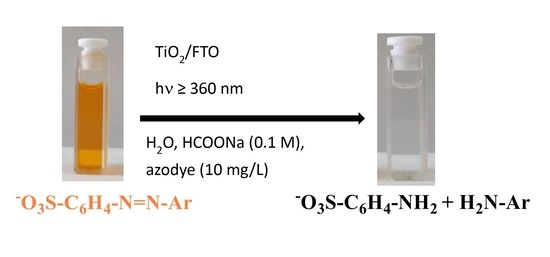
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of TiO2 on Glass Substrate
2.3. Structural Characterization
2.4. Electrochemistry and Photoelectrochemistry
2.5. Quantum Chemical Computation
2.6. Laser Spectroscopy
2.7. ESR Spin Trapping
2.8. Photoluminescence Experiments
2.9. Prolonged Irradiations
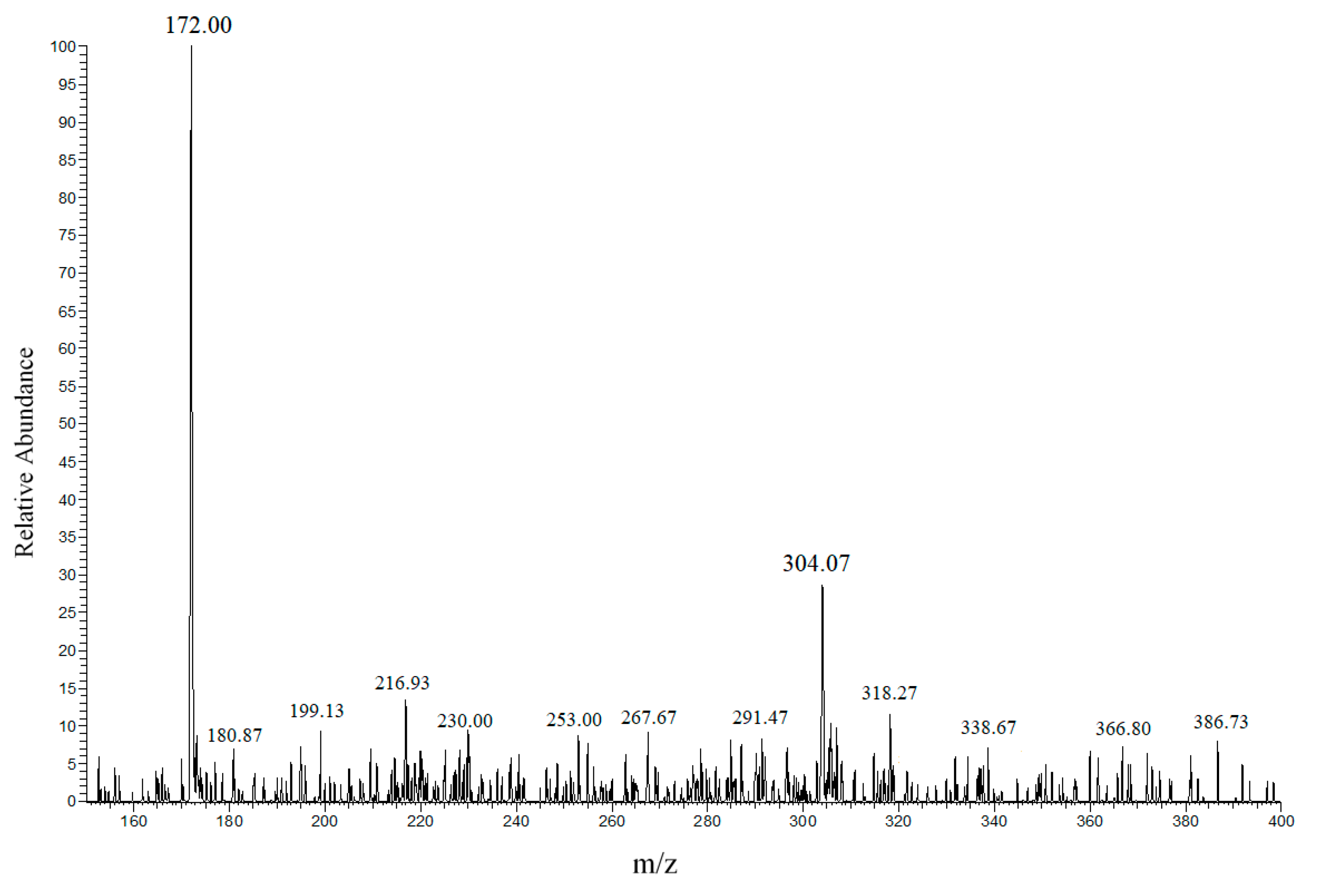
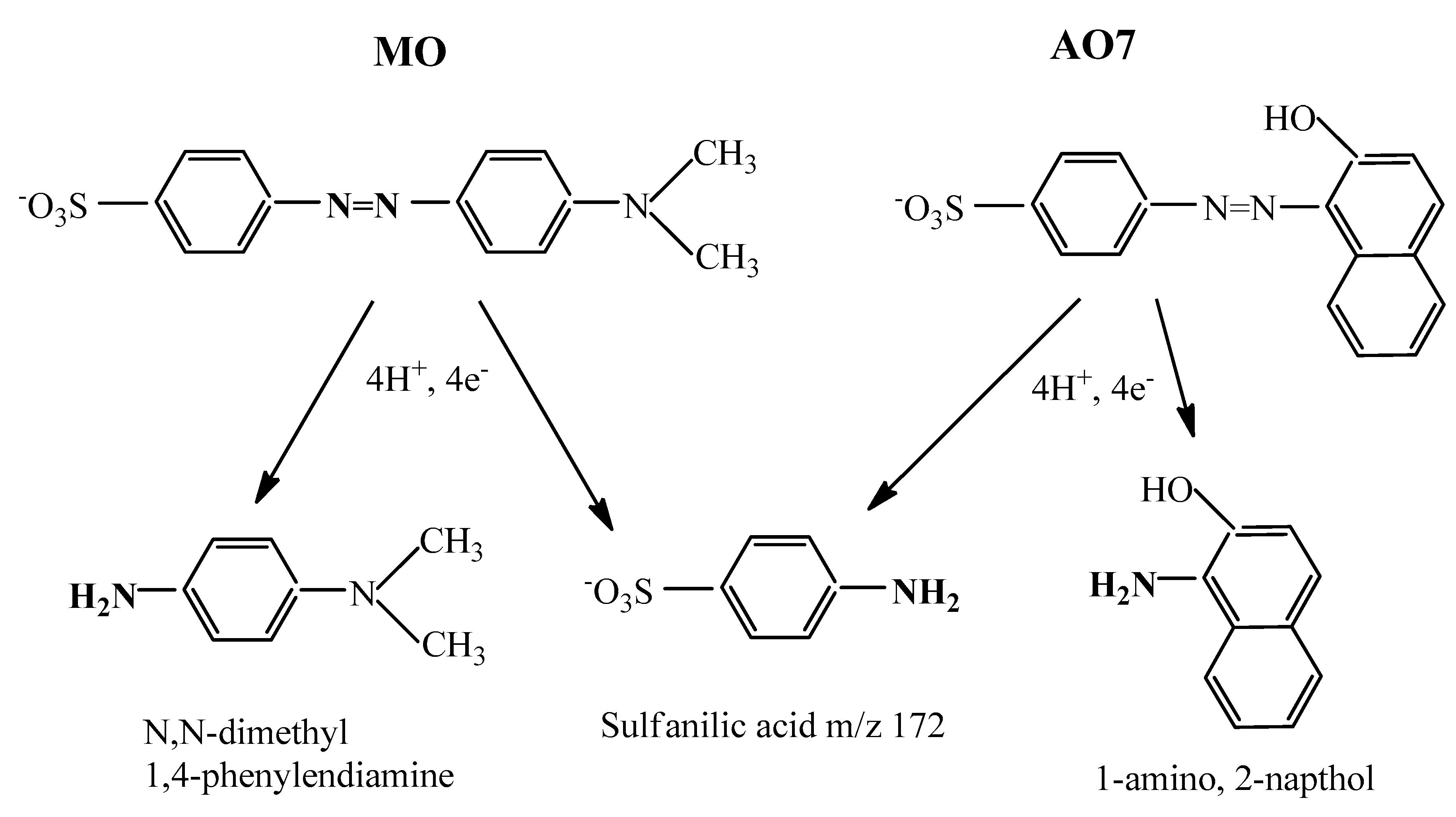
2.10. ESI–MS Investigation
3. Results and Discussion
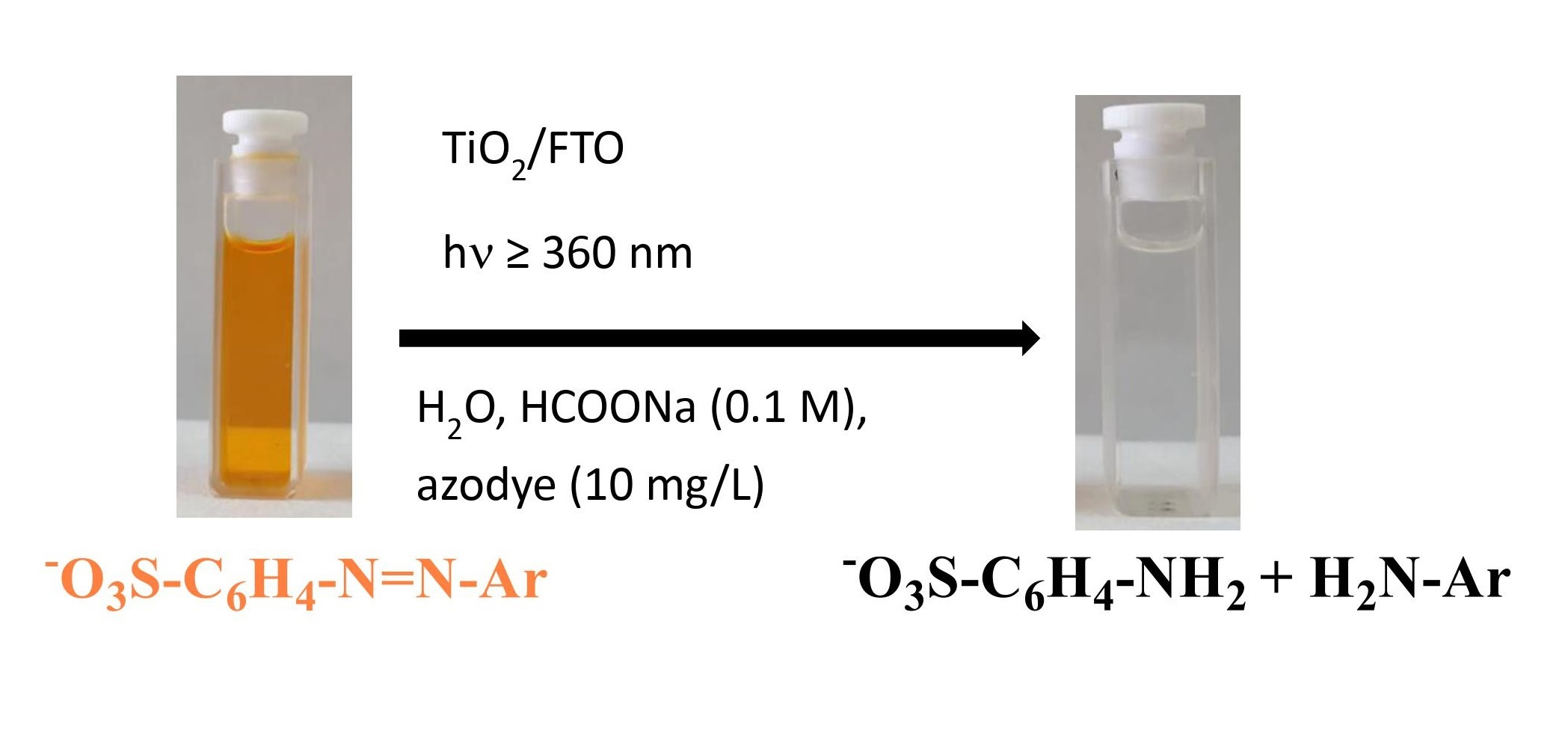
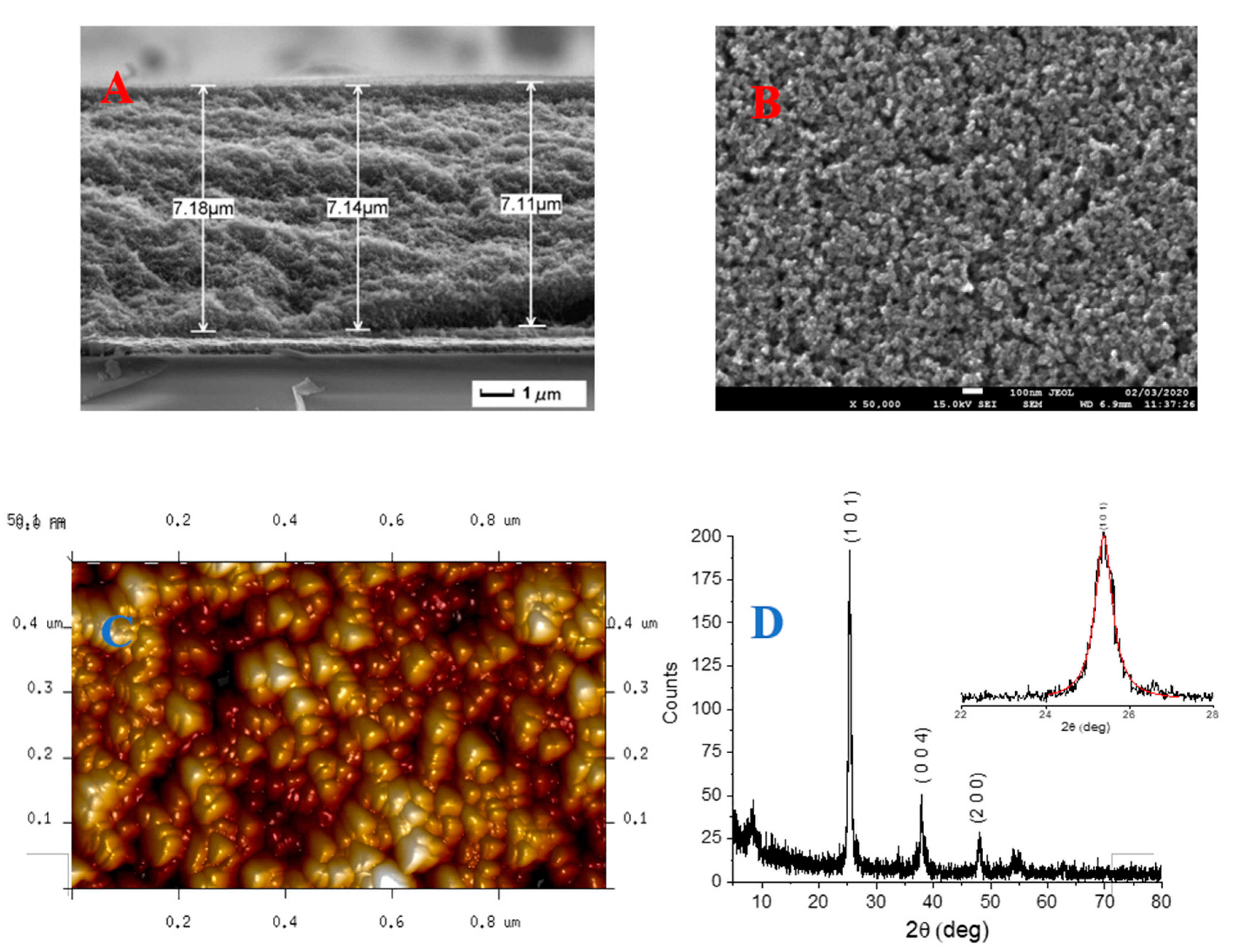
3.1. Structural Properties
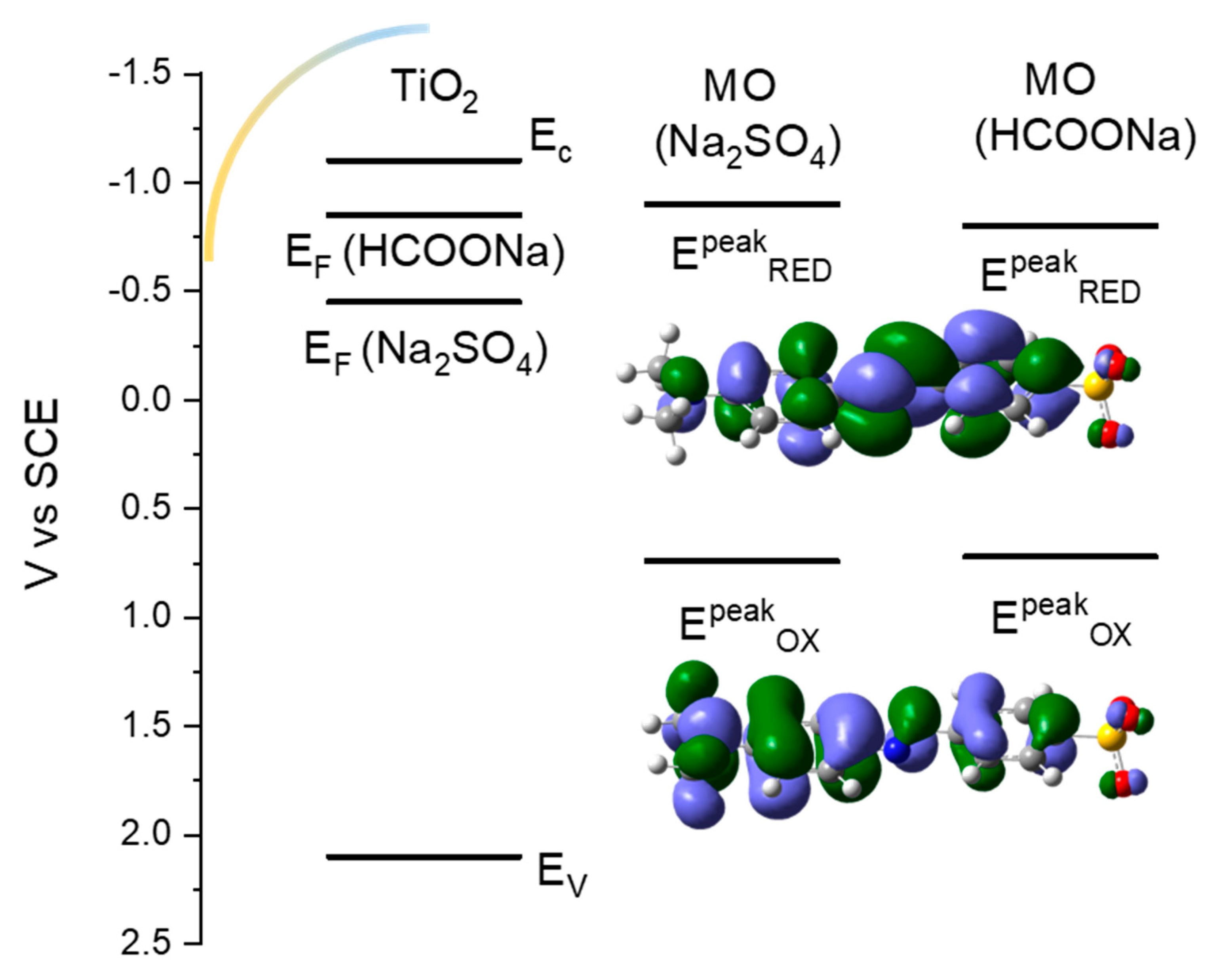
3.2. Energetics
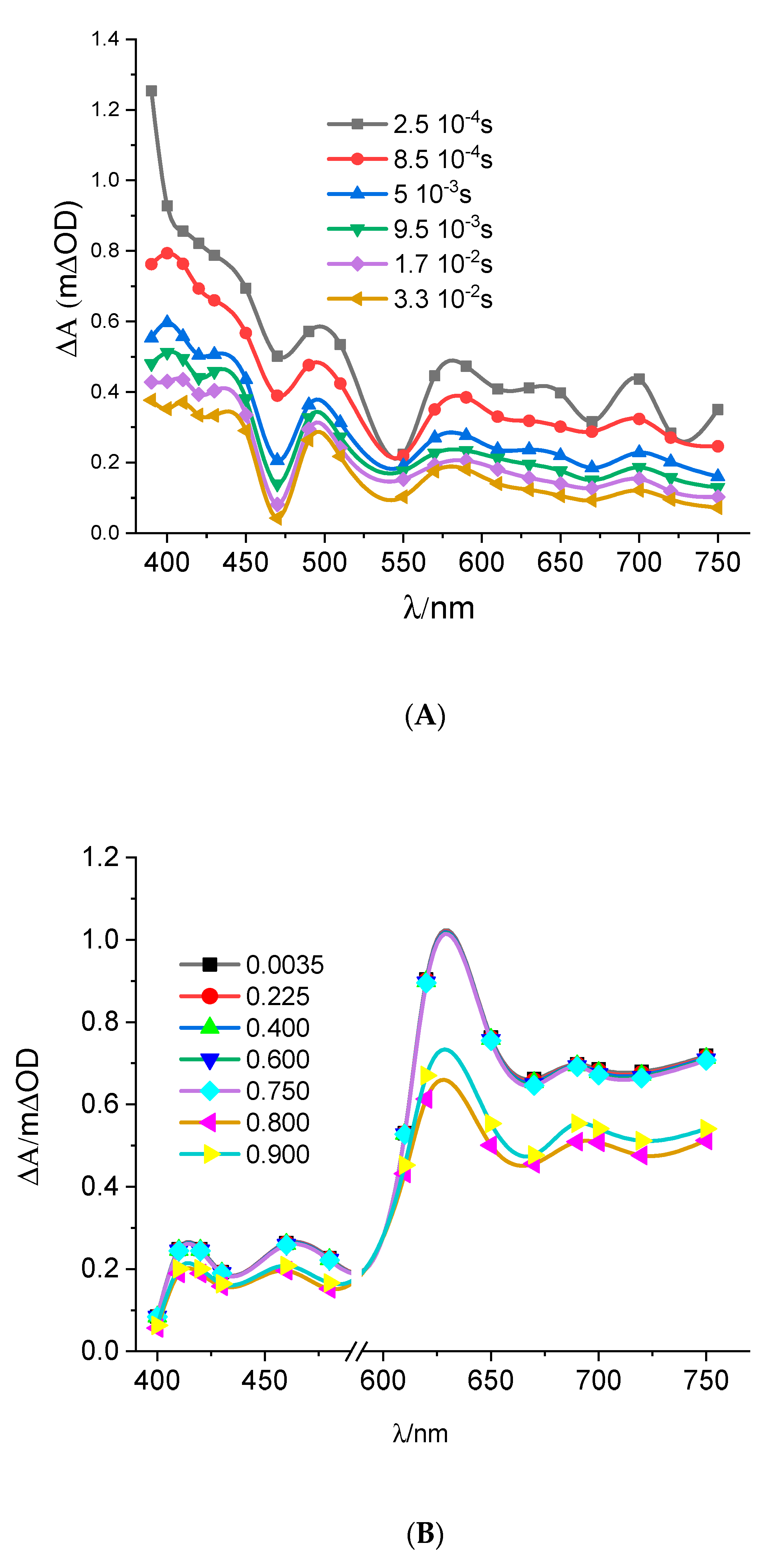
3.3. Transient Spectra of TiO2 Thin Films
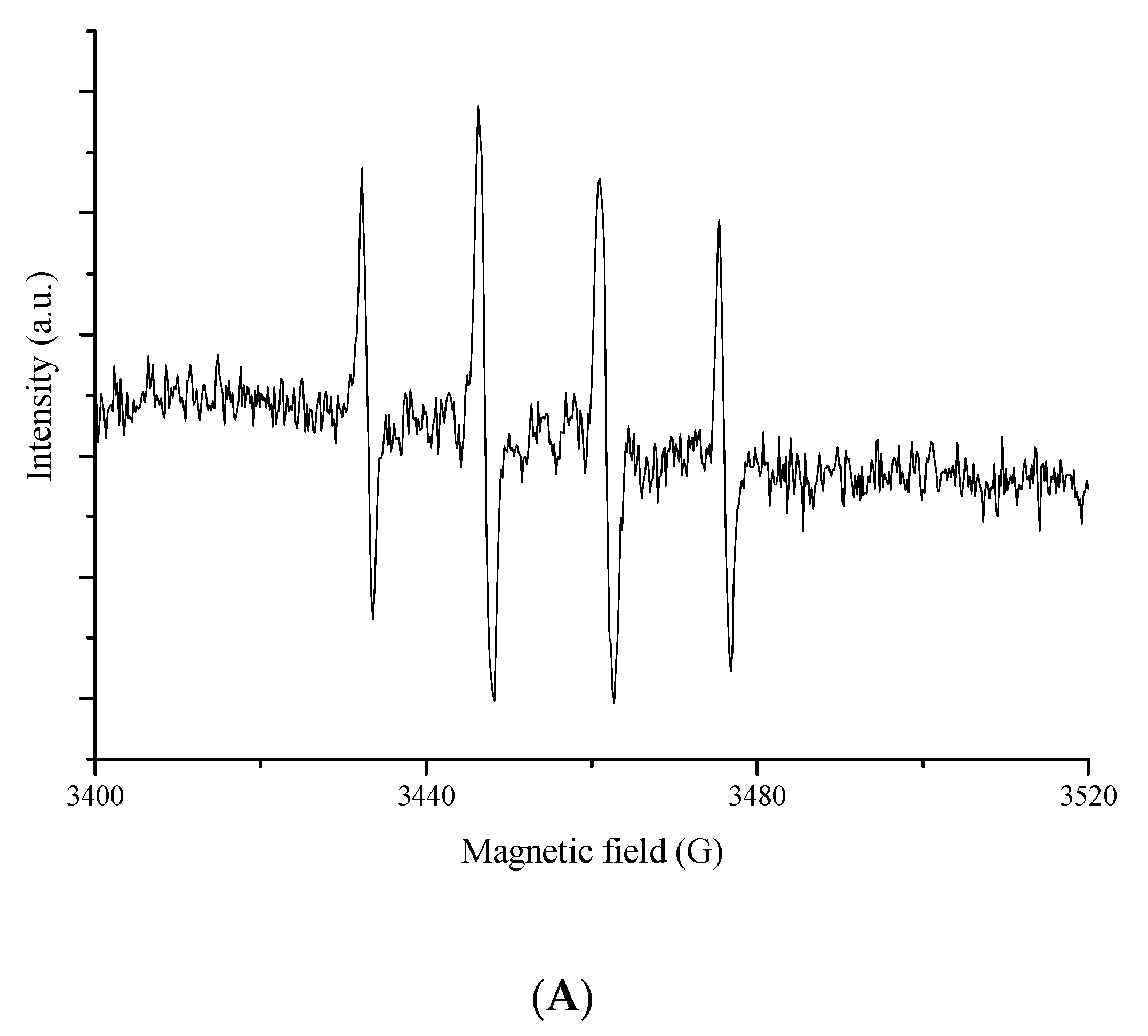
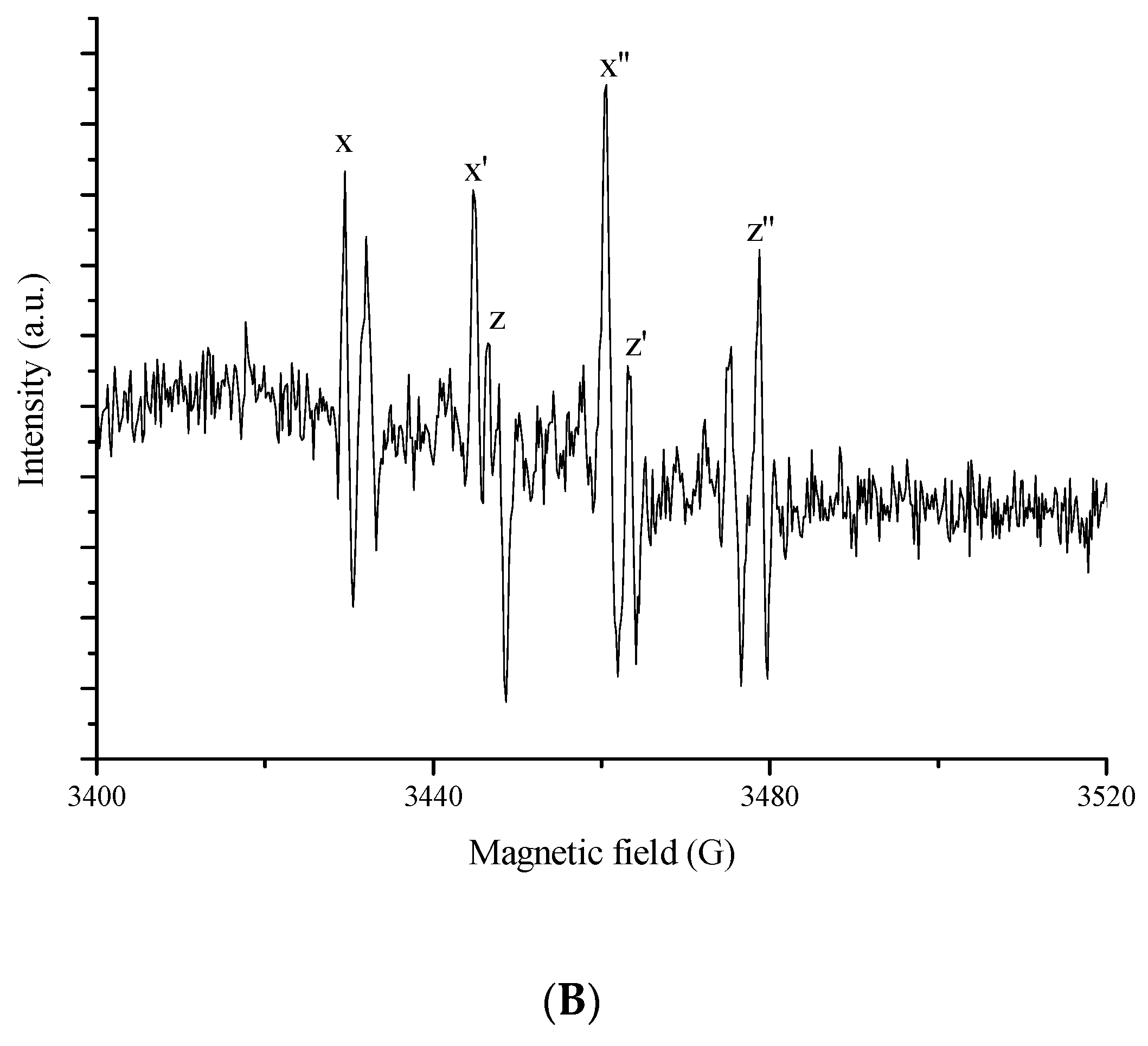
3.4. ESR Spin Trapping Experiments
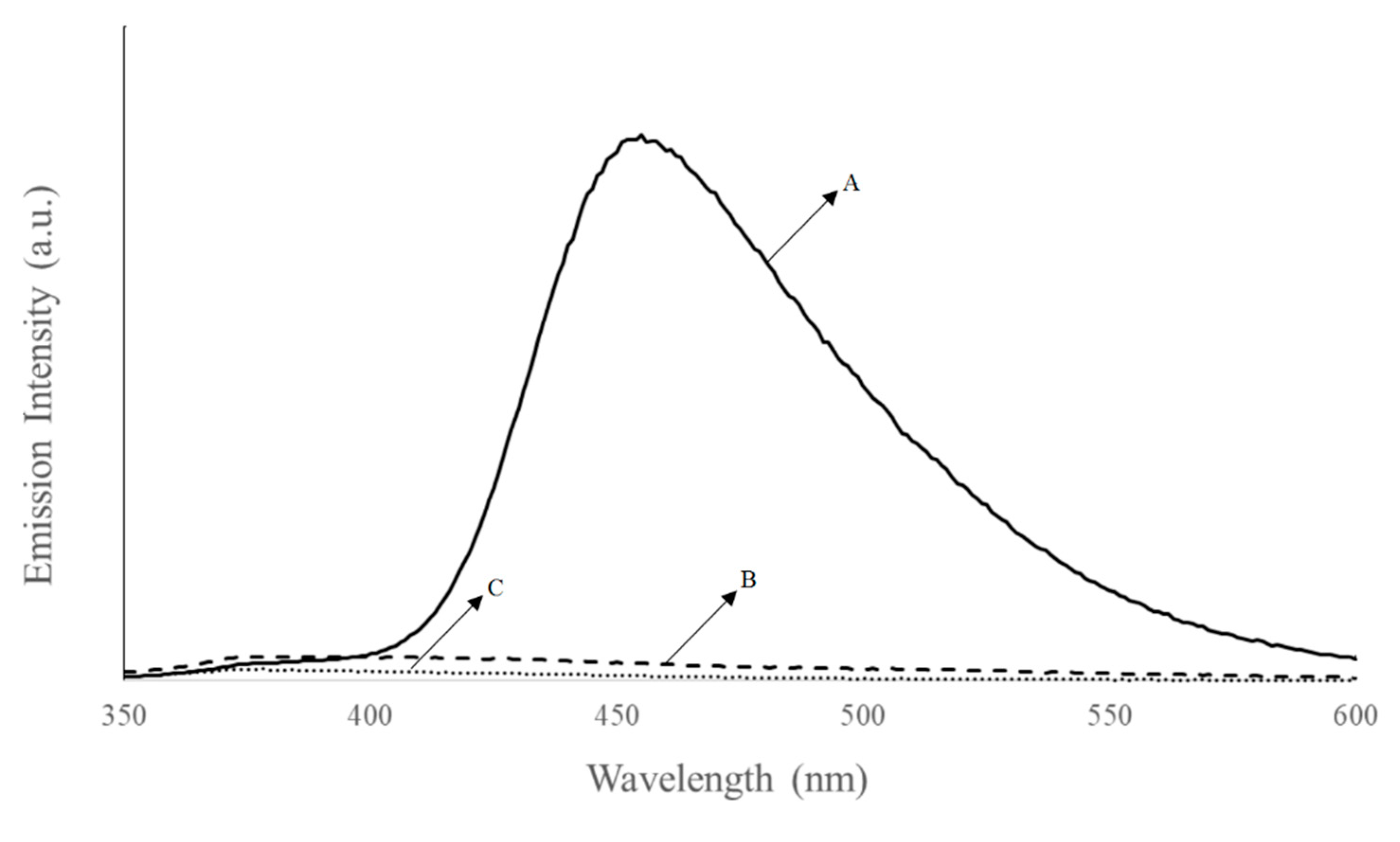
3.5. Photoluminescence Experiments

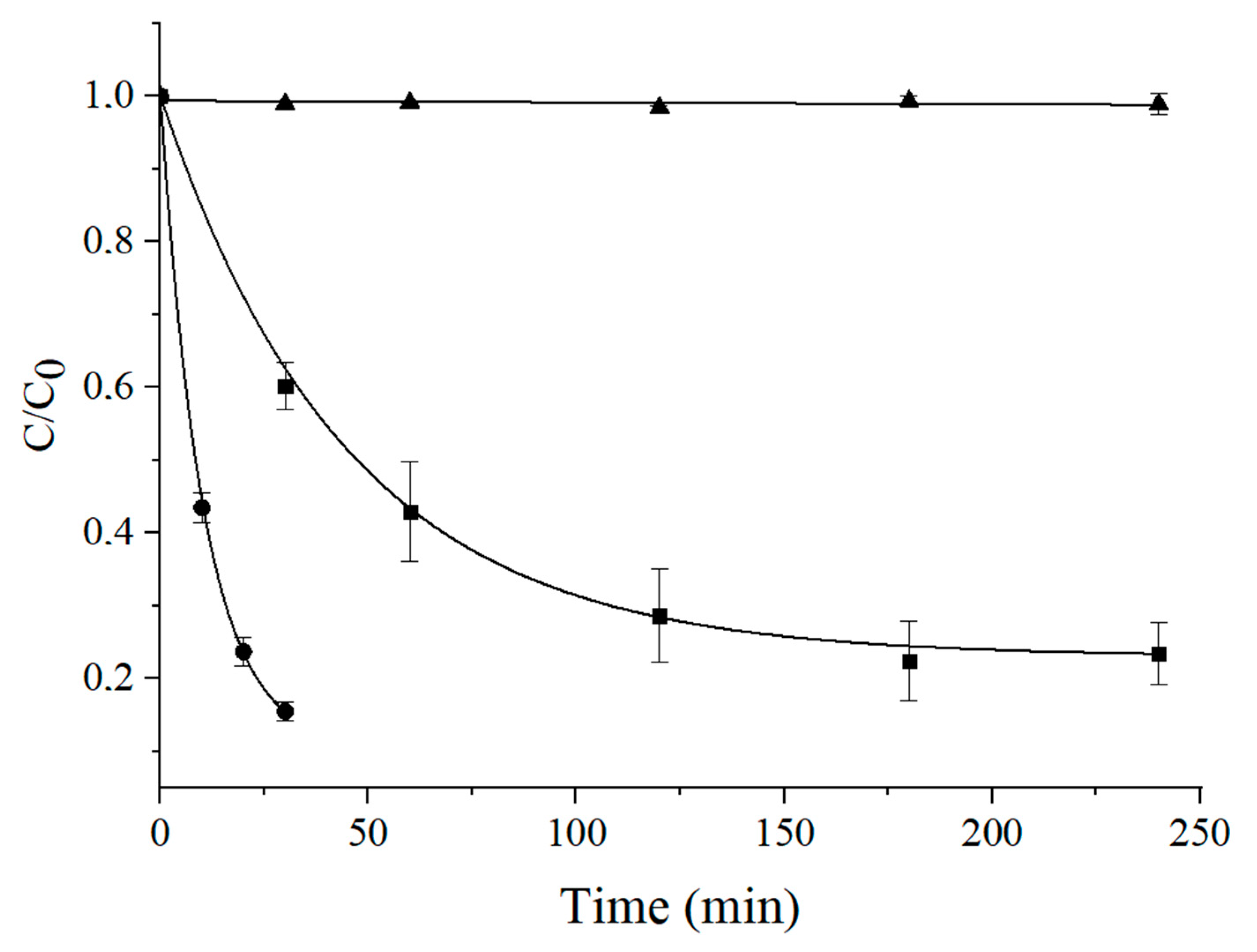
3.6. Prolonged Irradiation Experiments
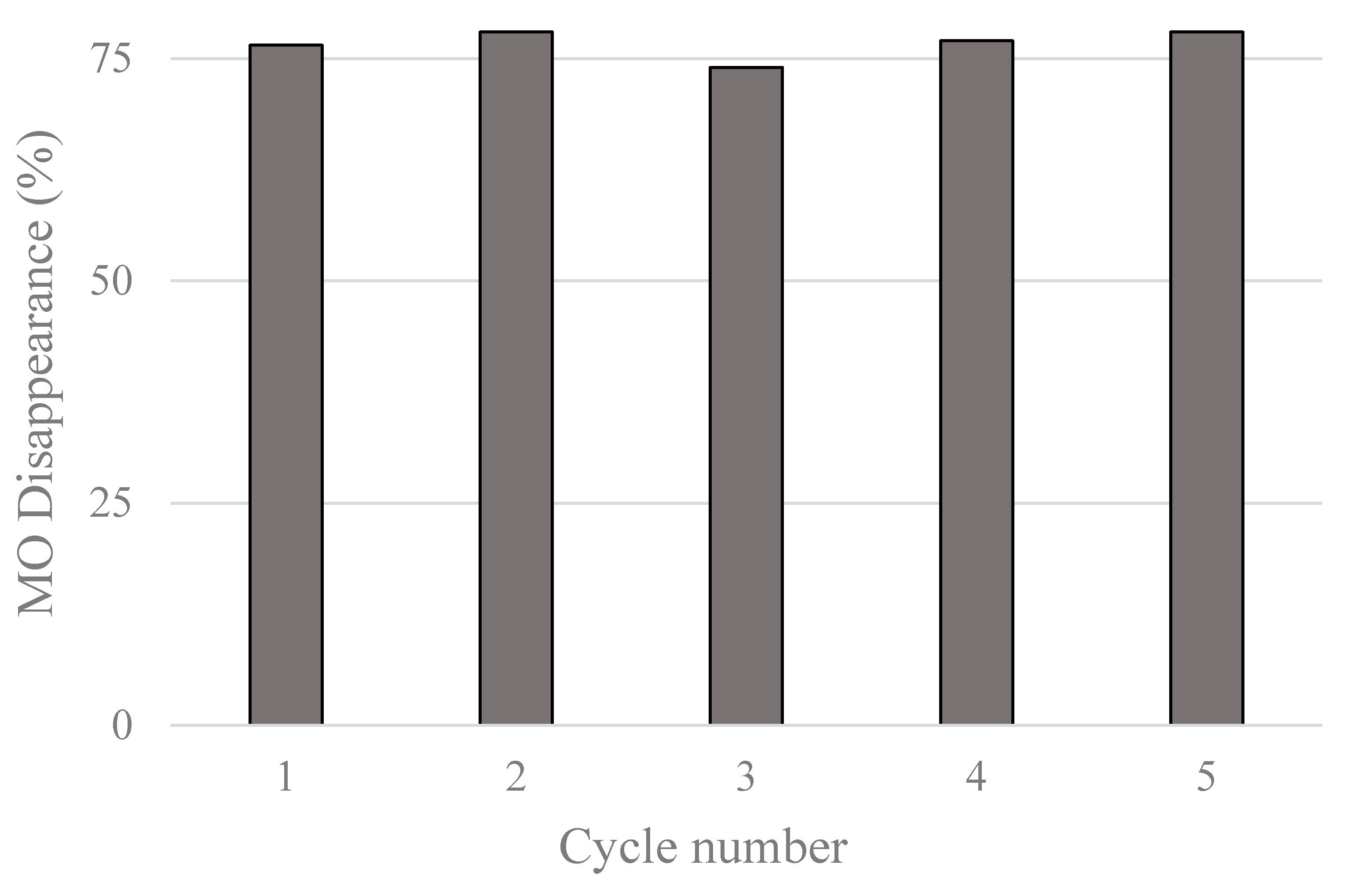
3.7. Recycle
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zollinger, H. Color Chemistry: Synthesis, Properties and Applications of Organic Dyes and Pigments, 2nd ed.; VCH: Vancouver, BC, Canada, 1991. [Google Scholar]
- Gomes da Silva, C.; Faria, J.L. Photochemical and photocatalytic degradation of an azo dye in aqueous solution by UV irradiation. J. Photochem. Photobiol. A Chem. 2003, 155, 133–143. [Google Scholar] [CrossRef]
- Brown, M.A.; De Vito, S.C. Predicting azo dye toxicity. Crit. Rev. Environ. Sci. Technol. 1993, 23, 249–324. [Google Scholar] [CrossRef]
- Tang, W.Z.; An, H. UV/TiO2 photocatalytic oxidation of commercial dyes in aqueous solutions. Chemosphere 1995, 31, 4157–4170. [Google Scholar] [CrossRef]
- Meshko, V.; Markovska, L.; Mincheva, M.; Rodrigues, A.E. Adsorption of basic dyes on granular activated carbon and natural zeolite. Water Res. 2001, 35, 3357–3366. [Google Scholar] [CrossRef]
- Kuo, W.S.; Ho, P.H. Solar photocatalytic decolorization of methylene blue in water. Chemosphere 2001, 45, 77–83. [Google Scholar] [CrossRef]
- Galindo, C.; Jacques, P.; Kalt, A. Photooxidation of the phenylazonaphthol AO20 on TiO2: Kinetic and mechanistic investigations. Chemosphere 2001, 45, 997–1005. [Google Scholar] [CrossRef]
- Kuo, W.G. Decolorizing dye wastewater with Fenton’s reagent. Water Res. 1992, 26, 881–886. [Google Scholar] [CrossRef]
- Balanosky, E.; Fernadez, J.; Kiwi, J.; Lopez, A. Degradation of membrane concentrates of the textile industry by Fenton like reactions in iron-free solutions at biocompatible pH values (pH approximate to 7–8). Water Sci. Technol. 1999, 40, 417–424. [Google Scholar] [CrossRef]
- Feng, W.; Nansheng, D.; Yuegang, Z. Discoloration of dye solutions induced by solar photolysis of ferrioxalate in aqueous solutions. Chemosphere 1999, 39, 2079–2085. [Google Scholar] [CrossRef]
- Bandara, J.; Morrison, C.; Kiwi, J.; Pulgarin, C.; Peringer, P. Degradation/decoloration of concentrated solutions of Orange II. Kinetics and quantum yield for sunlight induced reactions via Fenton type reagents. J. Photochem. Photobiol. A Chem. 1996, 99, 57–66. [Google Scholar] [CrossRef]
- Kang, S.F.; Liao, C.H.; Po, S.T. Decolorization of textile wastewater by photo-fenton oxidation technology. Chemosphere 2000, 41, 1287–1294. [Google Scholar] [CrossRef]
- Arslan, I.; Akmehmet, T.; Tuhkamen, T. Advanced Oxidation of Synthetic Dyehouse Effluent by O3, H2O2/O3 and H2O2/UV Processes. Environ. Technol. 1999, 20, 921–931. [Google Scholar] [CrossRef]
- Ince, N.H.; Gonenc, D.T. Treatability of a Textile Azo Dye by UV/H2O2. Environ. Technol. 1997, 18, 179–185. [Google Scholar] [CrossRef]
- Vinodgopal, K.; Kamat, P. Combine electrochemistry with photocatalysis. Chemtech 1996, 26, 18–22. [Google Scholar]
- Lizama, C.; Yeber, M.C.; Freer, J.; Baeza, J.; Mansilla, H.D. Reactive dyes decolouration by TiO2 photo-assisted catalysis. Water Sci. Technol. 2001, 44, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Khataee, A.R.; Kasiri, M.B. Photocatalytic degradation of organic dyes in the presence of nanostructured titanium dioxide: Influence of the chemical structure of dyes. J. Mol. Catal. A Chem. 2010, 328, 8–26. [Google Scholar] [CrossRef]
- Comparelli, R.; Fanizza, E.; Curri, M.L.; Cozzoli, P.D.; Mascolo, G.; Passino, R.; Agostiano, A. Photocatalytic degradation of azo dyes by organic-capped anatase TiO2 nanocrystals immobilized onto substrates. Appl. Catal. B Environ. 2005, 55, 81–91. [Google Scholar] [CrossRef]
- Konstantinou, I.K.; Albanis, T.A. TiO2-assisted photocatalytic degradation of azo dyes in aqueous solution: Kinetic and mechanistic investigations: A review. Appl. Catal. B Environ. 2004, 49, 1–14. [Google Scholar] [CrossRef]
- Pant, B.; Ojha, G.P.; Kuk, Y.-S.; Kwon, O.H.; Park, Y.W.; Park, M. Synthesis and Characterization of ZnO-TiO2/Carbon Fiber Composite with Enhanced Photocatalytic Properties. Nanomaterials 2020, 10, 1960. [Google Scholar] [CrossRef]
- Stylidi, M.; Kondarides, D.I.; Verykios, X.E. Pathways of solar light-induced photocatalytic degradation of azo dyes in aqueous TiO2 suspensions. Appl. Catal. B Environ. 2003, 40, 271–286. [Google Scholar] [CrossRef]
- Tanaka, K.; Padermpole, K.; Hisanaga, T. Photocatalytic degradation of commercial azo dyes. Water Res. 2000, 34, 327–333. [Google Scholar] [CrossRef]
- Lachheb, H.; Puzenat, E.; Houas, A.; Ksibi, M.; Elaoui, E.; Guillard, G.; Hermann, J.M. Photocatalytic degradation of various types of dyes (Alizarin S, Crocein Orange G, Methyl Red, Congo Red, Methylene Blue) in water by UV-irradiated titania. Appl. Catal. B: Environ. 2002, 39, 75–90. [Google Scholar] [CrossRef]
- Gouvea, C.A.K.; Wypych, F.; Moraes, S.G.; Duran, N.; Nagata, N.; Peralta-Zamora, P. Semiconductor-assisted photocatalytic degradation of reactive dyes in aqueous solution. Chemosphere 2000, 40, 433–440. [Google Scholar] [CrossRef]
- Palmisano, G.; Garcia-Lopez, E.; Marcì, G.; Loddo, V.; Yurdakal, S.; Augugliaro, V.; Palmisano, L. Advances in selective conversions by heterogeneous photocatalysis. Chem. Commun. 2010, 46, 7074–7089. [Google Scholar] [CrossRef]
- Kou, J.; Lu, C.; Wang, J.; Chen, Y.; Xu, Z.; Varma, R.S. Selectivity Enhancement in Heterogeneous Photocatalytic Transformations. Chem. Rev. 2017, 117, 1445–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, Y.; Hirakawa, H.; Togawa, Y.; Sugano, Y.; Ichikawa, S.; Hirai, T. Rutile Crystallites Isolated from Degussa (Evonik) P25 TiO2: Highly Efficient Photocatalyst for Chemoselective Hydrogenation of Nitroaromatics. ACS Catal. 2013, 3, 2318–2326. [Google Scholar] [CrossRef]
- Imamura, K.; Yoshikawa, T.; Hashimoto, K.; Kominami, H. Stoichiometric production of aminobenzenes and ketones by photocatalytic reduction of nitrobenzenes in secondary alcoholic suspension of titanium(IV) oxide under metal-free conditions. App. Catal. B Environ. 2013, 134–135, 193–197. [Google Scholar] [CrossRef]
- Molinari, A.; Maldotti, A.; Amadelli, R. Probing the Role of Surface Energetics of Electrons and their Accumulation in Photoreduction Processes on TiO2. Chem. Eur. J. 2014, 20, 7759–7765. [Google Scholar] [CrossRef]
- Molinari, A.; Mazzanti, M.; Fogagnolo, M. Photocatalytic Selective Reduction by TiO2 of 5-Nitrosalicylic Acid Ethyl Ester: A Mild Route to Mesalazine. Catal. Lett. 2020, 150, 1072–1080. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Pant, B.; Park, M.; Park, S.J. Recent Advances in TiO2 Films Prepared by Sol-Gel Methods for Photocatalytic Degradation of Organic Pollutants and Antibacterial Activities. Coatings 2019, 9, 613. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian Inc.: Wallingford, CT, USA, 2016.
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Ronconi, F.; Santoni, M.P.; Nastasi, F.; Bruno, G.; Argazzi, R.; Berardi, S.; Caramori, S.; Bignozzi, C.A.; Campagna, S. Charge injection into nanostructured TiO2 electrodes from the photogenerated reduced form of a new Ru(II) polypyridine compound: The “anti-biomimetic” mechanism at work. Dalton Trans. 2016, 45, 14109–14123. [Google Scholar] [CrossRef] [PubMed]
- Molinari, A.; Maldotti, A.; Amadelli, R. Effect of the electrolyte cations on photoinduced charge transfer at TiO2. Catal. Today 2017, 281, 71–77. [Google Scholar] [CrossRef]
- Bahnemann, D.W.; Henglein, A.; Lilie, J.; Spanhel, L. Flash photolysis observation of the absorption spectra of trapped positive holes and electrons in colloidal titanium dioxide. J. Phys. Chem. 1984, 88, 709–711. [Google Scholar] [CrossRef]
- Lawless, D.; Serpone, N.; Meisel, D. Role of hydroxyl radicals and trapped holes in photocatalysis. A pulse radiolysis study. J. Phys. Chem. 1991, 95, 5166–5170. [Google Scholar] [CrossRef]
- Bahnemann, D.W.; Henglein, A.; Spanhel, L. Detection of the intermediates of colloidal TiO2 catalyzed photoreactions. Faraday Discuss. 1984, 78, 151–163. [Google Scholar] [CrossRef]
- Bahnemann, D.W.; Hilgendorff, M.; Memming, R. Charge Carrier Dynamics at TiO2 Particles: Reactivity of Free and Trapped Holes. J. Phys. Chem. B 1997, 101, 4265–4275. [Google Scholar] [CrossRef]
- Molinari, A.; Argazzi, R.; Maldotti, A. Photocatalysis with Na4W10O32 in water system: Formation and reactivity of OH radicals. J. Mol. Catal. A Chem. 2013, 372, 23–28. [Google Scholar] [CrossRef]
- Maldotti, A.; Amadelli, R.; Carassiti, V.; Molinari, A. Catalytic oxygenation of cyclohexane by photoexcited (nBu4N)4W10O32: The role of radicals. Inorg. Chim. Acta 1997, 256, 309–312. [Google Scholar] [CrossRef]
- Buettner, G.R. Spin trapping: ESR parameters of spin adducts. Free Rad. Biol. Med. 1987, 3, 259–303. [Google Scholar] [PubMed]
- Molinari, A.; Samiolo, L.; Amadelli, R. EPR spin trapping evidence of radical intermediates in the photo-reduction of bicarbonate/CO2 in TiO2 aqueous suspensions. Photochem. Photobiol. Sci. 2015, 14, 1039–1046. [Google Scholar]
- Czili, H.; Horvath, A. Applicability of coumarin for detecting and measuring hydroxyl radicals generated by photoexcitation of TiO2 nanoparticles. Appl. Catal. B Environ. 2008, 81, 295–302. [Google Scholar]
- Zhang, J.; Nosaka, Y. Quantitative Detection of OH Radicals for Investigating the Reaction Mechanism of Various Visible-Light TiO2 Photocatalysts in Aqueous Suspension. J. Phys. Chem. C 2013, 117, 1383–1391. [Google Scholar]
- Xie, S.; Huang, P.; Kruzic, J.J.; Zeng, X.; Qian, H. A highly efficient degradation mechanism of methyl orange using Fe-based metallic glass powders. Sci. Rep. 2010, 6, 21947. [Google Scholar] [CrossRef] [Green Version]
- Molinari, A.; Sarti, E.; Marchetti, N.; Pasti, L. Degradation of emerging concern contaminants in water by heterogeneous photocatalysis with Na4W10O32. Appl. Catalys. B Environ. 2017, 203, 9–17. [Google Scholar]
- Pasti, L.; Sarti, E.; Martucci, A.; Marchetti, N.; Stevanin, C.; Molinari, A. An advanced oxidation process by photoexcited heterogeneous sodium decatungstate for the degradation of drugs present in aqueous environment. Appl. Catal. B Environ. 2018, 239, 345–351. [Google Scholar]
- Vinodgopal, K.; Bedja, I.; Hotchandani, S.; Kamat, P.V. A Photocatalytic Approach for the Reductive Decolorization of Textile Azo Dyes in Colloidal Semiconductor Suspensions. Langmuir 1994, 10, 1767–1771. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzanti, M.; Caramori, S.; Fogagnolo, M.; Cristino, V.; Molinari, A. Turning Waste into Useful Products by Photocatalysis with Nanocrystalline TiO2 Thin Films: Reductive Cleavage of Azo Bond in the Presence of Aqueous Formate. Nanomaterials 2020, 10, 2147. https://doi.org/10.3390/nano10112147
Mazzanti M, Caramori S, Fogagnolo M, Cristino V, Molinari A. Turning Waste into Useful Products by Photocatalysis with Nanocrystalline TiO2 Thin Films: Reductive Cleavage of Azo Bond in the Presence of Aqueous Formate. Nanomaterials. 2020; 10(11):2147. https://doi.org/10.3390/nano10112147
Chicago/Turabian StyleMazzanti, Michele, Stefano Caramori, Marco Fogagnolo, Vito Cristino, and Alessandra Molinari. 2020. "Turning Waste into Useful Products by Photocatalysis with Nanocrystalline TiO2 Thin Films: Reductive Cleavage of Azo Bond in the Presence of Aqueous Formate" Nanomaterials 10, no. 11: 2147. https://doi.org/10.3390/nano10112147