
Formation, Characterization, and Bonding of cis- and trans-[PtCl2{Te(CH2)6}2], cis-trans-[Pt3Cl6{Te(CH2)6}4], and cis-trans-[Pt4Cl8{Te(CH2)6}4]: Experimental and DFT Study †
 , , and
, , and
Abstract
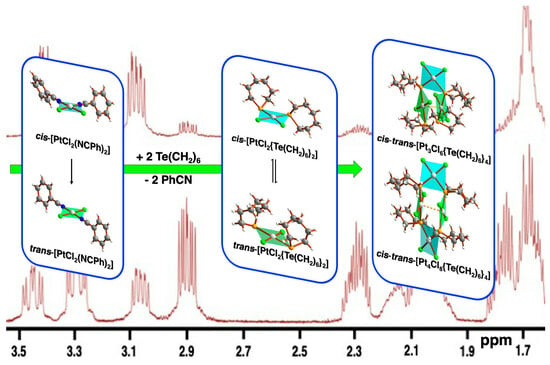
:
1. Introduction
2. Results and Discussion
2.1. General
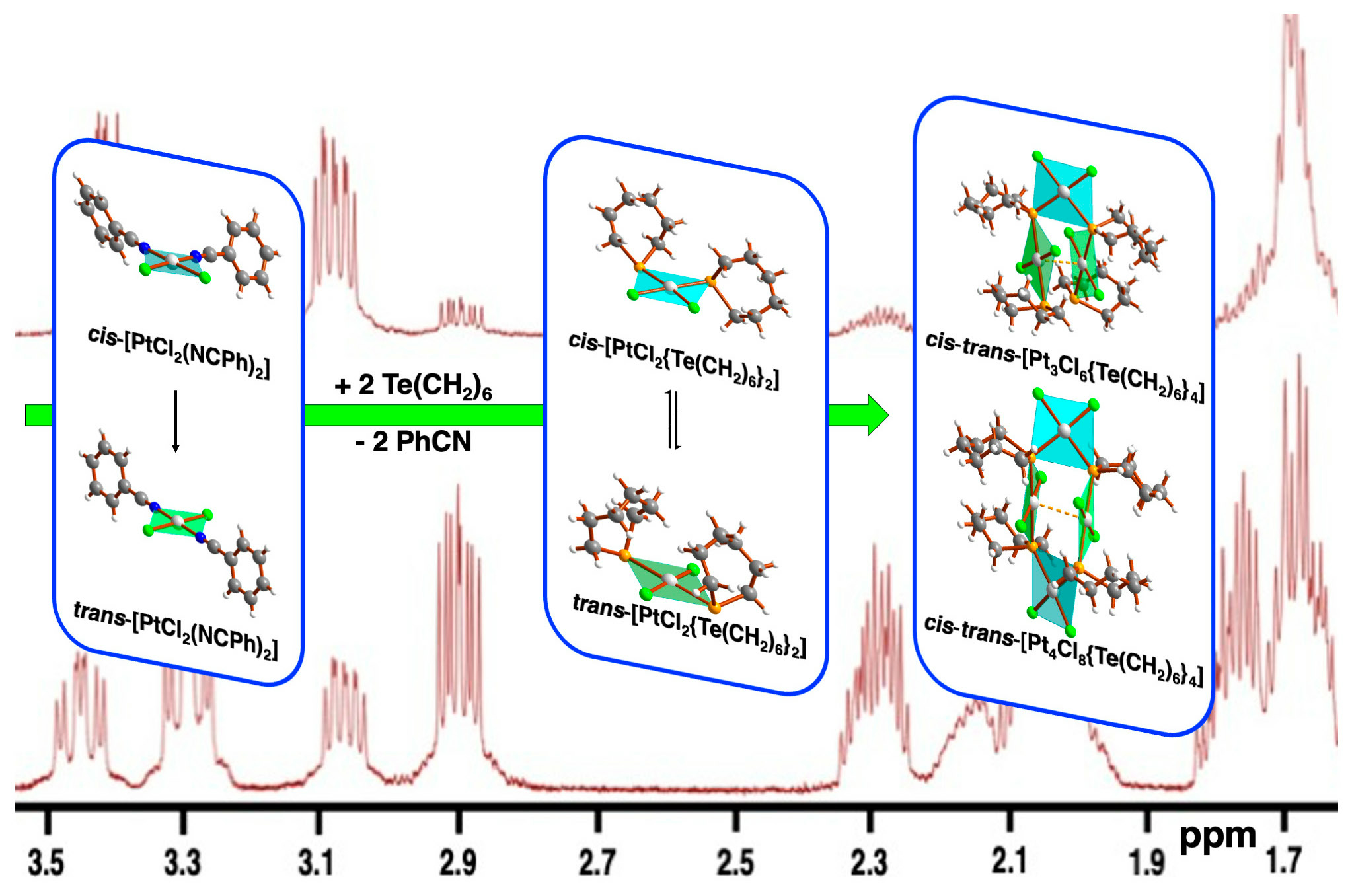
2.2. NMR Spectroscopy
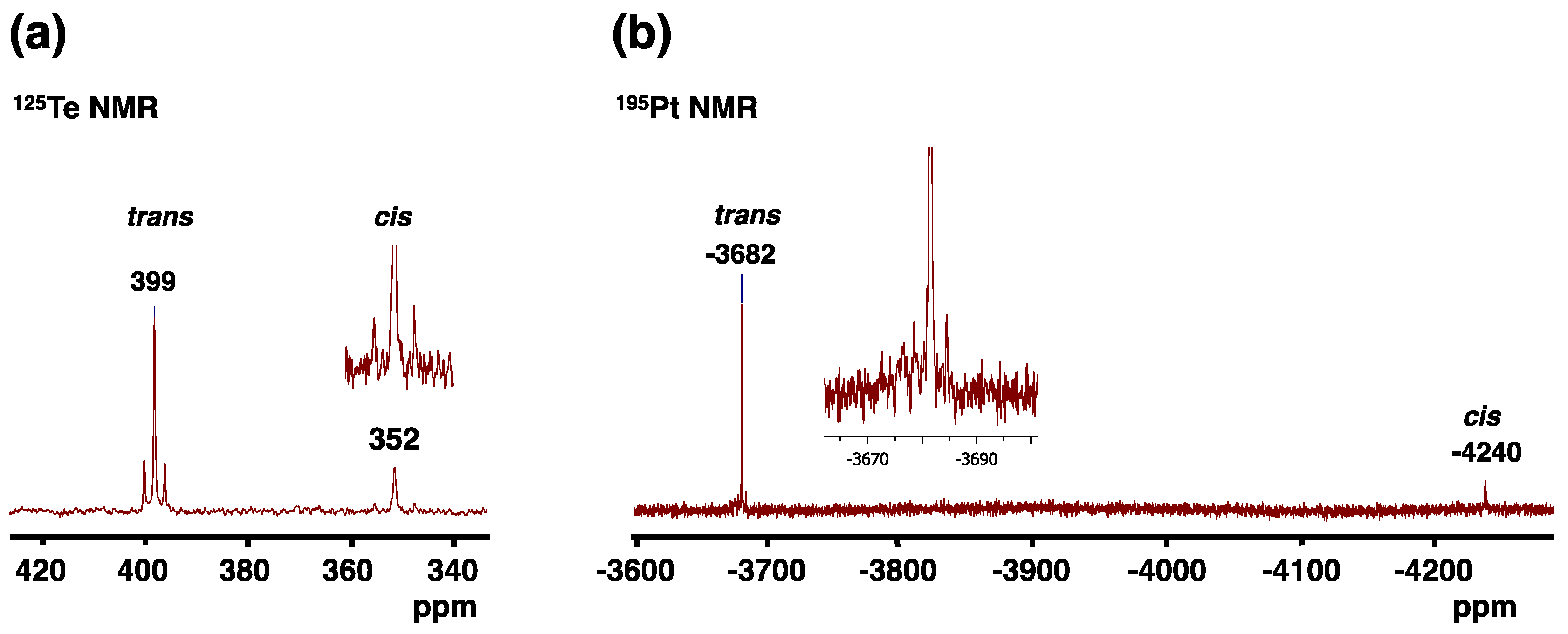
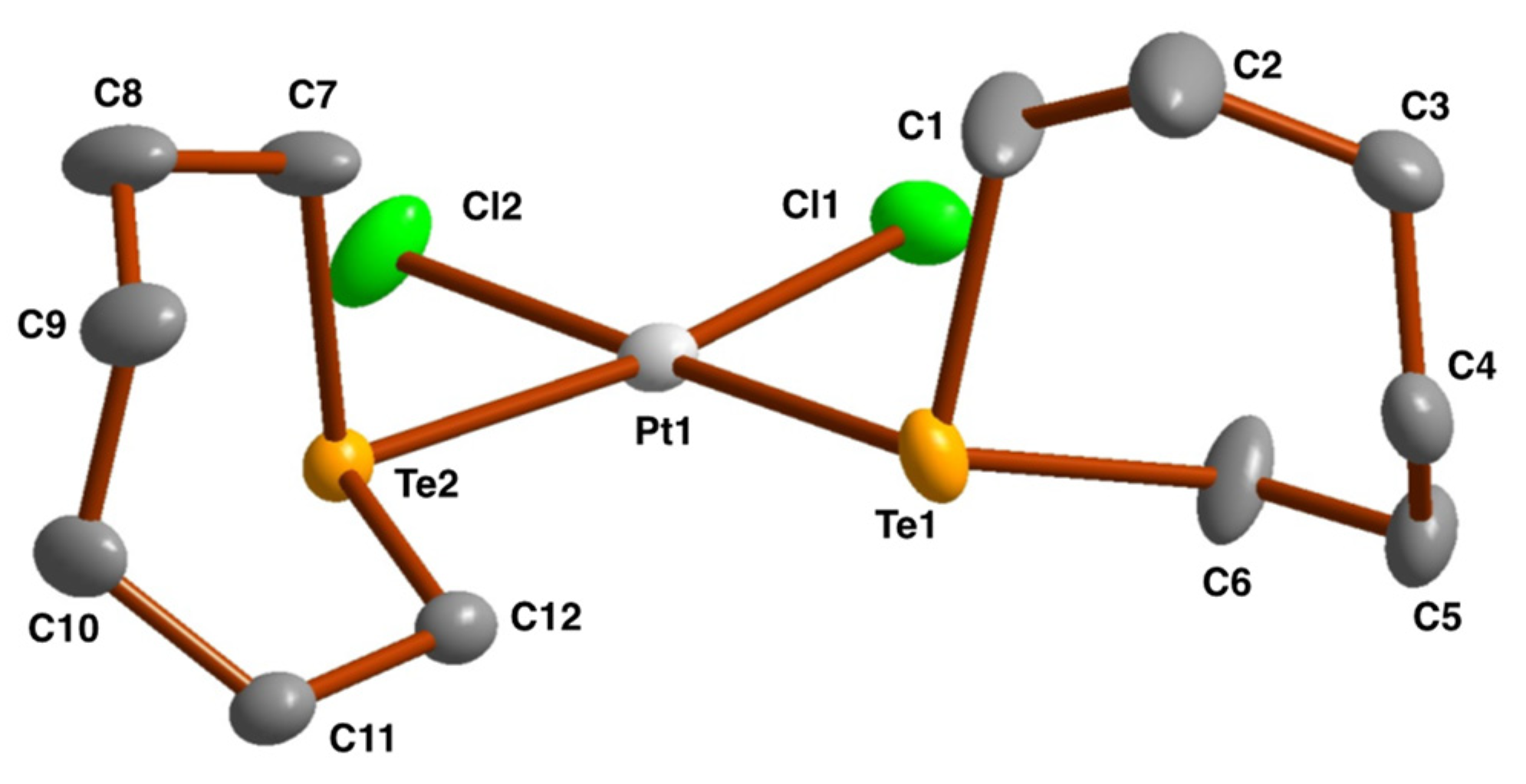
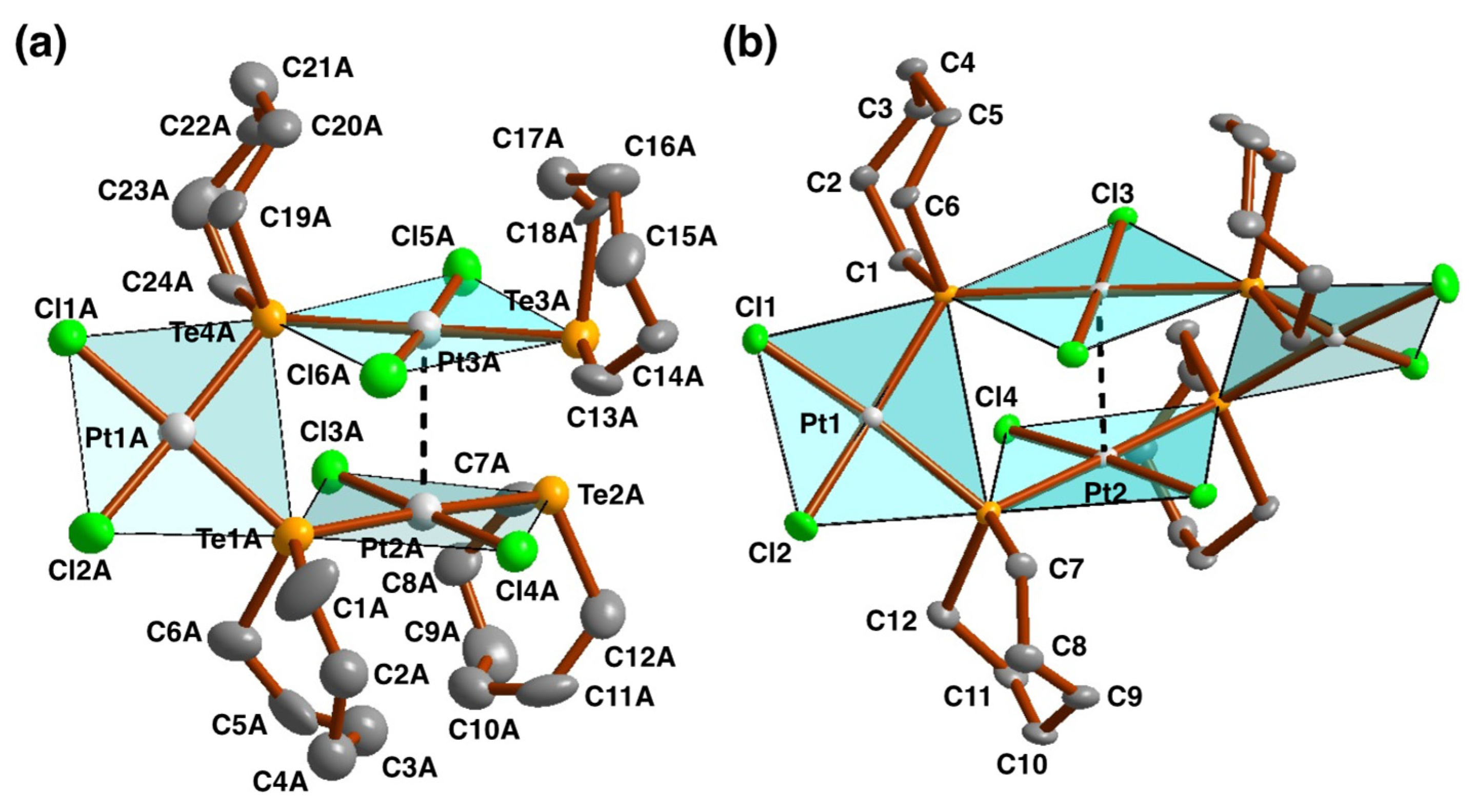
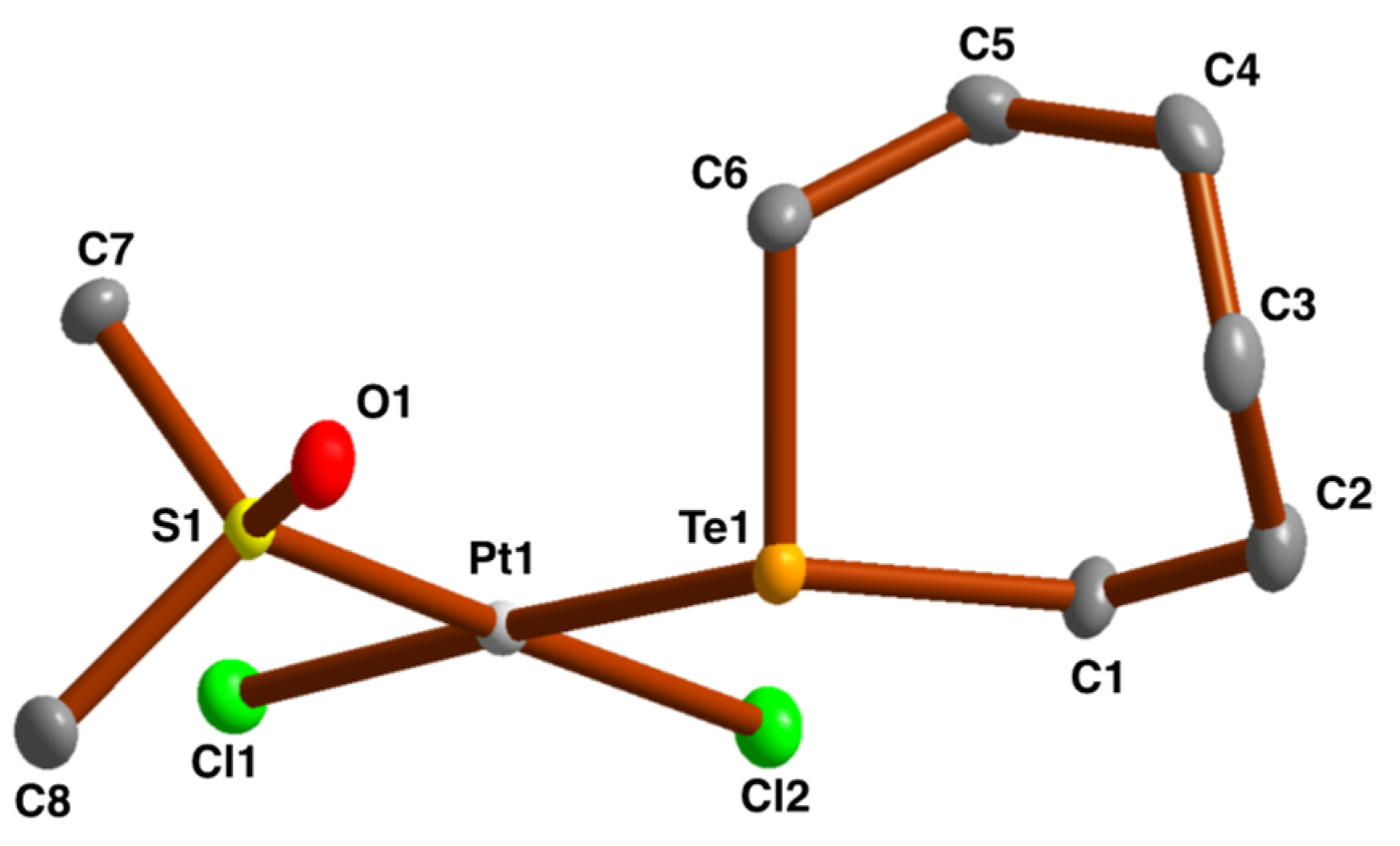
2.3. Crystal and Molecular Structures
2.4. Density Functional Theory (DFT) Computations
2.4.1. General
2.4.2. Optimized Geometries
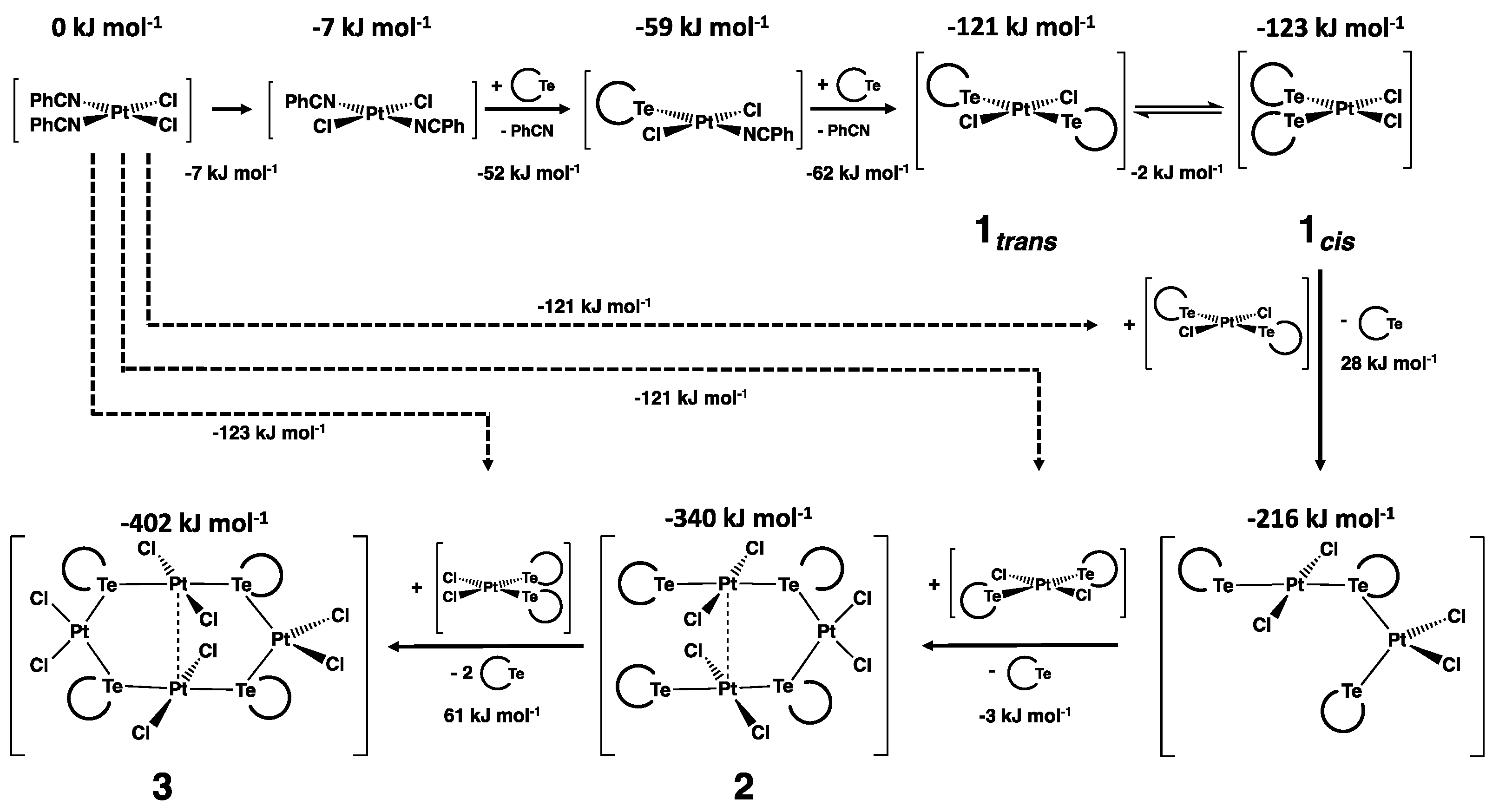
2.4.3. Cis-trans Isomerization
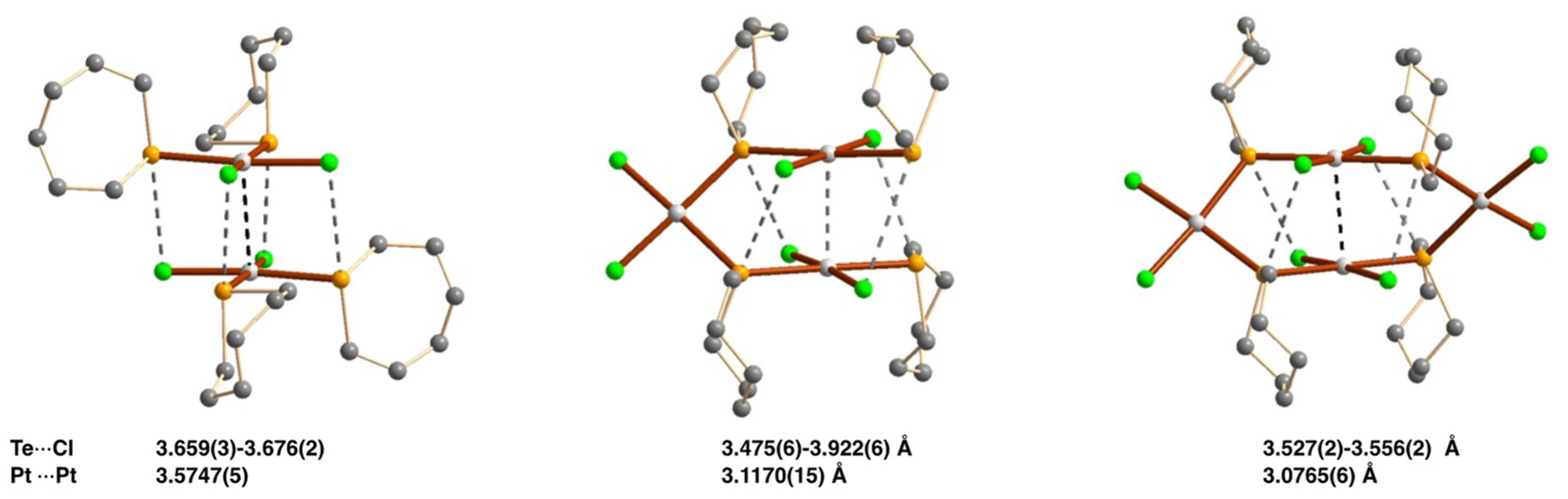
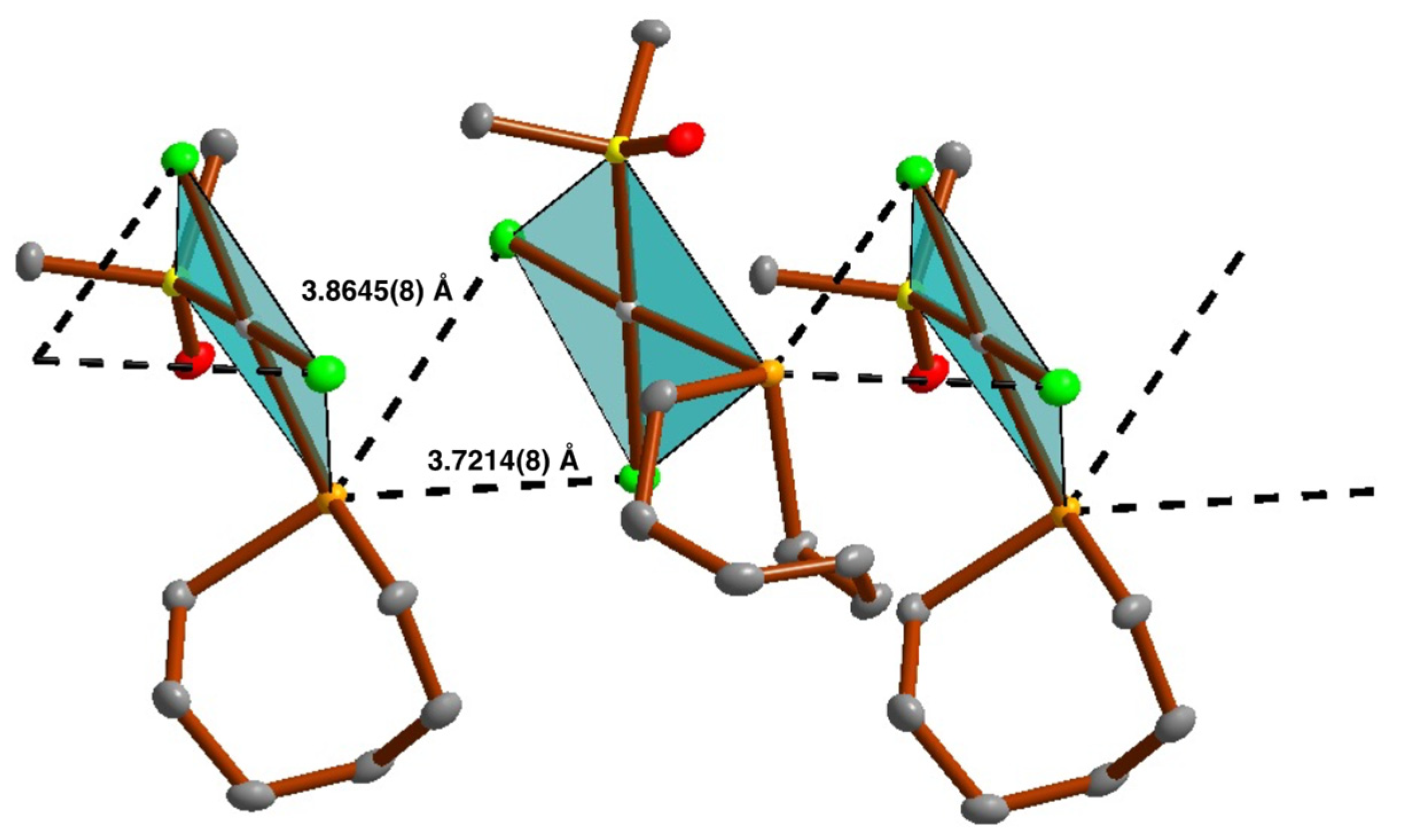
2.4.4. Chalcogen Bonding and Metallophilic Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Expl. | PBE0-D3/def2-TZVP | QTAIM | |||||
|---|---|---|---|---|---|---|---|---|
| ρ (e Å–3) | DI | ∇2ρ (e Å–5) | Gb (kJ mol−1 bohr–3) a | Vb (kJ mol−1 bohr–3) a | Eint (kJ mol−1 bohr–3) a, b | |||
| 1cis | ||||||||
| Pt-Te(cis) | 2.5145(7)–2.5266(6) | 2.537–2.548 | 0.619/0.628 | 0.98/1.03 | 1.15/1.18 | 130/133 | −228/−234 | −114/−117 |
| Pt⋯Pt | 3.5747(5) | 3.403 | 0.122 | 0.18 | 1.0 | 29 | −31 | −16 |
| Te⋯Cl | 3.659(3)–3.676(2) | 3.516–3.531 | 0.074–0.068 | 0.10 | 0.74–0.67 | 14 | −13–(−14) | −7 |
| 2 | ||||||||
| Pt-Te(cis) | 2.5140(15)–2.5219(16) | 2.515–2.515 | 0.630–0.634 | 0.94 | 1.83–1.85 | 152–153 | −254–(−256) | −127–(−128) |
| Pt-Te(trans) | 2.5560(15)–2.5774(15) | 2.572–2.581 | 0.578–0.588 | 0.81–0.92 | 1.13–1.77 | 118–132 | −206–(−217) | −103–(−109) |
| Pt⋯Pt | 3.1170(11)–3.1499(13) | 3.167 | 0.172 | 0.26 | 1.42 | 44 | −50 | −25 |
| Te⋯Cl | 3.481(7)–3.964(6) | 3.508–3.628 | 0.074–0.061 | 0.10–0.07 | 0.71–0.59 | 13–17 | −11–(−14) | −6–(−7) |
| 3 | ||||||||
| Pt-Te(cis) | 2.5043(6)–2.5226(6) | 2.510 | 0.638 | 0.95 | 1.88 | 155 | −259 | −130 |
| Pt-Te(trans) | 2.5547(6)–2.5577(6) | 2.561 | 0.595 | 0.84 | 1.55 | 132 | −222 | −111 |
| Pt⋯Pt | 3.0764(6) | 3.075 | 0.202 | 0.30 | 1.71 | 54 | −62 | −31 |
| Te⋯Cl | 3.527(2)–3.556(2) | 3.613 | 0.061 | 0.06 | 0.61 | 14 | −11 | −6 |
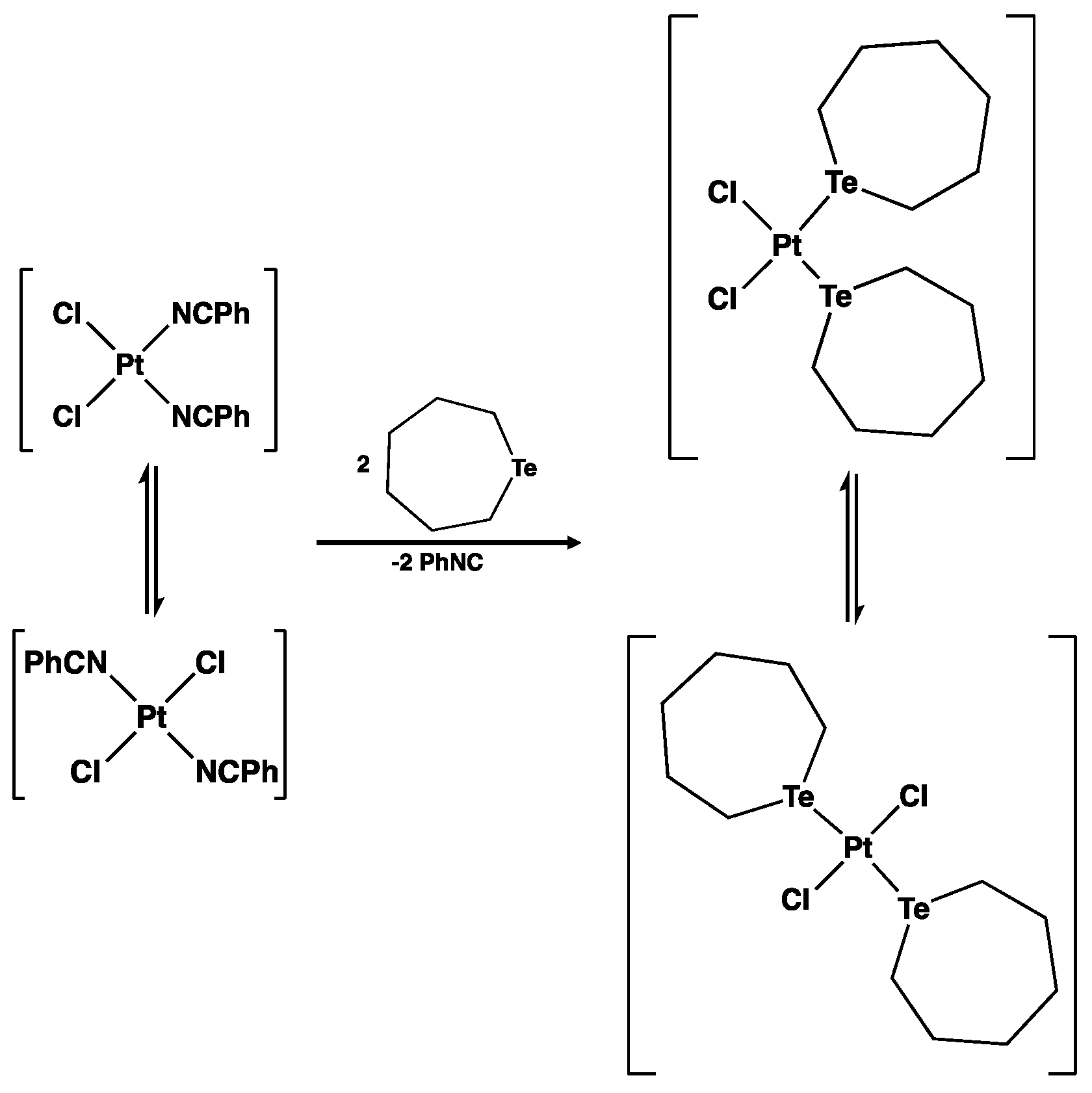
2.4.5. Formation of cis-trans-[Pt3Cl6{Te(CH2)6}4] and cis-trans-[Pt4Cl8{Te(CH2)6}4]
3. Materials and Methods
3.1. General Procedures
3.2. Spectroscopy
3.2.1. NMR Spectroscopy
3.2.2. Mass Spectrometry
3.3. X-ray Diffraction
3.4. Computational Details
3.5. Reaction of Te(CH2)6 with cis-[PtCl2(NCPh)2]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pedersen, C.J. Cyclic polyethers and their complexes with metal salts. J. Am. Chem. Soc. 1967, 89, 7017–7036. [Google Scholar] [CrossRef]
- Gokel, G.W.; Negin, S.; Catwell, R. Crown Ethers. In Comprehensive Supramolecular Chemistry II; Atwood, J.L., Gokel, G.W., Barbour, L.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 3, pp. 3–48. [Google Scholar]
- Levason, W.; Orchard, S.D.; Reid, G. Recent developments in the chemistry of selenoethers and telluroethers. Coord. Chem. Rev. 2002, 225, 159–199. [Google Scholar] [CrossRef]
- Segi, M. Acyclic dialkyl tellurides. Sci. Synth. 2007, 39, 1069–1082. [Google Scholar]
- Barton, A.J.; Genge, A.R.J.; Hill, N.J.; Levason, W.; Orchard, S.D.; Patel, B.; Reid, G.; Ward, A.J. Recent developments in thio-, seleno-, and telluroether ligand chemistry. Heteroat. Chem. 2002, 13, 550–560. [Google Scholar] [CrossRef]
- Panda, A. Chemistry of selena macrocycles. Coord. Chem. Rev. 2009, 253, 1056–1098. [Google Scholar] [CrossRef]
- Levason, W.; Reid, G. Recent developments in the chemistry of thio-, seleno-, and telluroethers. In Handbook of Chalcogen Chemistry; Devillanova, F.A., Ed.; RSC Publishing: Cambridge, UK, 2007; pp. 81–106. [Google Scholar]
- Levason, W.; Reid, G. Macrocyclic thio-, seleno- and telluroether ligands. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 399–410. [Google Scholar]
- Sommen, G.L. Rings containing selenium and tellurium. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsdenm, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 14, pp. 863–900. [Google Scholar]
- Levason, W.; Reid, G.; Zhang, W.-J. The chemistry of the p-block elements with thioether, selenoether and telluroether ligands. Dalton Trans. 2011, 40, 8491–8506. [Google Scholar] [CrossRef]
- Moorefield, C.N.; Newcombe, G.R. Eight-membered and larger rings. Progr. Heterocycl. Chem. 2020, 31, 649–669. [Google Scholar]
- Singh, A.K.; Sharma, S. Recent developments in the ligand chemistry of tellurium. Coord. Chem. Rev. 2000, 209, 49–98. [Google Scholar] [CrossRef]
- Levason, W.; Reid, G. Macrocyclic and polydentate thio- and seleno-ether ligand complexes of the p-block elements. J. Chem. Soc. Dalton Trans. 2001, 2953–2960. [Google Scholar] [CrossRef]
- Levason, W.; Reid, G. Early transition metal complexes of polydentate and macrocyclic thio- and seleno-ethers. J. Chem. Res. Synop. 2002, 467–472, 1001–1022. [Google Scholar] [CrossRef]
- Jain, V.K.; Chauhan, R.S. New vistas in the chemistry of platinum group metals with tellurium ligands. Coord. Chem. Rev. 2016, 306, 270–301. [Google Scholar] [CrossRef]
- Gahan, L.R. Cyclononanes: The extensive chemistry of fundamentally simple ligands. Coord. Chem. Rev. 2016, 311, 168–223. [Google Scholar] [CrossRef]
- Oilunkaniemi, R.; Laitinen, R.S. Synthesis and molecular structures of cyclic selenoethers and their derivatives. In Comprehensive Inorganic Chemistry III; Reedijk, J., Poeppelmeier, K.R., Laitinen, R.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; Volume 1, pp. 527–555. [Google Scholar]
- Laitinen, R.S.; Oilunkaniemi, R.; Weigand, W. Structure, bonding, and ligand chemistry of macrocyclic seleno- and telluroethers. In Chalcogen Chemistry: Fundamentals and Applications; Lippolis, V., Santi, C., Lenardão, E.J., Braga, A.L., Eds.; Royal Society of Chemistery: Cambridge, UK, 2023; pp. 530–566. [Google Scholar]
- Boyle, P.D.; Godfrey, S.M. The reactions of sulfur and selenium donor molecules with dihalogens and interhalogens. Coord. Chem. Rev. 2001, 223, 265–299. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Elder, P.J.W.; Vargas-Baca, I. A survey of tellurium-centered secondary-bonding supramolecular synthons. Coord. Chem. Rev. 2011, 255, 1426–1438. [Google Scholar] [CrossRef]
- du Mont, W.-W.; Hrib, C.G. Halogen-chalcogen X-E (X = F, Cl, Br, I.; E = S, Se, Te) chemistry. In Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, 2nd ed.; Devillanova, F.A., du Month, W.-W., Eds.; RSC Publishing: Cambridge, UK, 2013; Volume 2, pp. 273–316. [Google Scholar]
- Chivers, T.; Laitinen, R.S. Tellurium: A maverick among the chalcogens. Chem. Soc. Rev. 2015, 44, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Rominger, F.; Bleiholder, C. From noncovalent chalcogen-chalcogen interactions to supramolecular aggregates: Experiments and calculations. Chem. Rev. 2018, 118, 2010–2041. [Google Scholar] [CrossRef]
- Kolb, S.; Oliver, G.A.; Werz, D.B. Chemistry evolves, terms evolve, but phenomena do not evolve: From chalcogen-chalcogen interactions to chalcogen bonding. Angew. Chem. Int. Ed. 2020, 59, 22306–22310. [Google Scholar] [CrossRef]
- Kolb, S.; Oliver, G.A.; Werz, D.B. Chalcogen bonding in supramolecular structures, anion recognition, and catalysis. In Comprehensive Inorganic Chemistry III; Reedijk, J., Poeppelmeier, K.R., Laitinen, R.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; Volume 1, pp. 602–650. [Google Scholar]
- Morgan, G.T.; Burgess, H. XLIV. -cycloTelluropantane. J. Chem. Soc. 1928, 321–329. [Google Scholar] [CrossRef]
- Morgan, G.T.; Burstall, F.H. XXIV.-cycloTellurobutane (Tetrahydrotellurophen). J. Chem. Soc. 1931, 180–184. [Google Scholar] [CrossRef]
- Takaguchi, Y.; Horn, E.; Furukawa, N. Preparation and X-ray Structure Analysis of 1,1,5,5,9,9-Hexachloro-1,5,9-tritelluracyclododecane (Cl6([12]aneTe3)) and Its Redox Behavior. Organometallics 1996, 15, 5112–5115. [Google Scholar] [CrossRef]
- Rodewald, M.; Rautiainen, J.M.; Niksch, T.; Görls, H.; Oilunkaniemi, R.; Weigand, W.; Laitinen, R.S. Chalcogen-bonding interactions in telluroether heterocycles [Te(CH2)m]n (n = 1-4; m = 3-7). Chem. Eur. J. 2020, 26, 13806–13818. [Google Scholar] [CrossRef]
- Kemmitt, T.; Levason, W.; Oldroyd, R.D.; Webster, M. Palladium and platinum complexes of telluracyclopentane. Structure of trans-[Pd{Te(CH2)4}2Cl2]. Polyhedron 1992, 11, 2165–2169. [Google Scholar]
- Singh, A.K.; Kadarkaraisamy, M.; Husebye, S. The first metal complexes of 1,4-oxatellurane: Synthesis and crystal structure of its platinum(II) complex. J. Chem. Res. 2000, 64–65. [Google Scholar] [CrossRef]
- Hesford, M.J.; Levason, W.; Matthews, M.L.; Reid, G. Synthesis and complexation ofm the mixed tellurium-oxygen macrocycles 1-tellura-4,7,13,16-tetraoxacyclooctadecane, [18]aneO4Te2 and their selenium analogues. Dalton Trans. 2003, 2852–2858. [Google Scholar] [CrossRef]
- Devery, M.P.; Dickson, R.S.; Skelton, B.W.; White, A.H. Coordination and rearrangement of organic chalcogenides on a rhodium-rhodium bond: Reactions with strained-ring cyclic thioethers and with selenium and tellurium ligands. Organometallics 1999, 18, 5292–5298. [Google Scholar] [CrossRef]
- Levason, W.; Orchard, S.D.; Reid, G. Synthesis and properties of the first series of mixed thioether/telluroether macrocycles. Chem. Commun. 2001, 427–428. [Google Scholar] [CrossRef]
- Vigo, L.; Poropudas, M.J.; Salin, P.; Oilunkaniemi, R.; Laitinen, R.S. Versatile coordination chemistry of rhodium complexes containing the bis(trimethylsilylmethyl)tellane ligand. J. Organomet. Chem. 2009, 694, 2053–2060. [Google Scholar] [CrossRef]
- Vigo, L.; Salin, P.; Oilunkaniemi, R.; Laitinen, R.S. Formation and structural characterization of mercury complexes from Te{(R)(CH2SiMe3)} (R = Ph, CH2SiMe3) and HgCl2. J. Organomet. Chem. 2009, 694, 3134–3141. [Google Scholar] [CrossRef]
- Poropudas, M.J.; Vigo, L.; Oilunkaniemi, R.; Laitinen, R.S. Versatile Solid-State Coordination Chemistry of Telluroether Complexes of Silver(I) and Copper(I). Dalton Trans. 2013, 42, 16868–16877. [Google Scholar] [CrossRef]
- Taimisto, M.; Bajorek, T.; Rautiainen, J.M.; Pakkanen, T.A.; Oilunkaniemi, R.; Laitinen, R.S. Experimental and computational investigation on the formation pathway of [RuCl2(CO)2(ERR’)2] (E = S, Se, Te; R, R’ = Me, Ph) from [RuCl2(CO)3]2 and ERR’. Dalton Trans. 2022, 51, 11747–11757. [Google Scholar] [CrossRef]
- Taimisto, M.; Poropudas, M.J.; Rautiainen, J.M.; Oilunkaniemi, R.; Laitinen, R.S. Ruthenium-assisted tellurium abstraction in bis(thiophen-2-yl) ditelluride. Eur. J. Inorg. Chem. 2023, 26, e202200772. [Google Scholar] [CrossRef]
- Kemmitt, T.; Levason, W. Synthesis and multinuclear NMR studies of [M{o-C6H4(TeMe)2}X2] (M = Pd, Pt; X = Cl, Br, I). The presence of a characteristic ring contribution to 125Te chemical shifts. Inorg. Chem. 1990, 29, 731–735. [Google Scholar] [CrossRef]
- Vigo, L.; Oilunkaniemi, R.; Laitinen, R.S. Formation and characterization of platinum and palladium complexes of bis(trimethylsilylmethyl) tellane. Eur. J. Inorg. Chem. 2008, 284–290. [Google Scholar] [CrossRef]
- Knorr, M.; Guyon, F.; Jourdain, I.; Kneifel, S.; Frenzel, J.; Strohmann, C. (Phenylthiomethyl)silanes and (butyltelluromethyl)-silanes as novel bifunctional ligands for the construction of dithioether-, ditelluroether- and transition metal-silicon complexes. Inorg. Chim. Acta 2003, 350, 455–466. [Google Scholar] [CrossRef]
- Prasad, R.R.; Singh, H.B.; Butcher, R.J. Synthesis, structure and reactivity of b-chalcocyclohexanals: Dichalcogenides and chalcogenides. J. Organomet. Chem. 2016, 814, 42–56. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, J.S.; Butcher, R.J. N-{2-(4-methoxyphenyltelluro)ethyl}morpholine (L1) and its platinum(II) and ruthenium(II) complexes. Synthesis and crystal structure of L1 and trans-[PtCl2(L1)2]. Inorg. Chim. Acta 2001, 312, 163–169. [Google Scholar]
- Kolay, S.; Kumar, M.; Wadawale, A.; Das, D.; Jain, V.K. Role of anagostic interactions in cycloplatination of telluroethers: Synthesis and structural characterization. J. Organomet. Chem. 2015, 794, 40–47. [Google Scholar] [CrossRef]
- Kapoor, P.; Lövqvist, K.; Oskarsson, A. Cis/trans influences in platinum(II) complexes. X-ray crystal structures of cis-dichloro(dimethyl sulfide)(dimethyl sulfoxide)platinum(II) and cis-dichloro(dimethyl sulfide)(dimethyl phenyl phosphine)platinum(II). J. Mol. Struct. 1998, 470, 39–47. [Google Scholar] [CrossRef]
- ConQuest; Version 2022.3.0; Cambridge Crystallographic Data Center: Cambridge, UK, 2022.
- Oilunkaniemi, R.; Komulainen, J.; Laitinen, R.S.; Ahlgrén, M.; Pursiainen, J. Trends in the structure and bonding of [MCl2{(C4H3S)ECH3}2] (M = Pd, Pt; E = Te, Se). J. Organomet. Chem. 1998, 571, 129–138. [Google Scholar] [CrossRef]
- Levason, W.; Webster, M.; Mitchell, C.J. Structure of trans-diiodobis(methyl phenyl telluride)platinum(II). Acta Crystallogr. Sect. C 1992, 48, 1931–1933. [Google Scholar] [CrossRef]
- Drake, J.E.; Yang, J.; Khalid, A.; Srivastava, V.; Singh, A.K. Palladium(II) and platinum(II) complexes of bis (4-methoxyphenyltelluro) methane. Crystal structure of [{meso-(4-MeOC6H4Te)2CH2}(Ph2PCH2CH2PPh2)Pd(II)](ClO4)·H2O and [meso-(4-MeOC6H4Te) 2CH2]Pd(II)Cl2. Inorg. Chim. Acta 1997, 254, 57–62. [Google Scholar] [CrossRef]
- Kemmitt, T.; Levason, W.; Webster, M. Chelating ditelluroether complexes of palladium and platinum: Synthesis and multinuclear NMR Studies. Structure of dibromo(meso-l,3-bis(phenyltelluro)propane)palladium(II), [Pd{meso -PhTe(CH2)3TePh}Br2]. Inorg. Chem. 1989, 28, 692–696. [Google Scholar] [CrossRef]
- Rocha, R.W.; de Almeida, W.B. On the cis->trans isomerization of the square-planar [PtCl(SnCl3)(PH3)2] compound: Ab initio gas phase reaction mechanism and solvent effects using continuum models. J. Braz. Chem. Soc. 2000, 11, 112–120. [Google Scholar] [CrossRef]
- Redfield, D.A.; Nelson, J.H. Equilibrium energetics of cis-trans isomerization for two square-planar palladium(II)-Phosphine Complexes. Inorg. Chem. 1973, 12, 15–19. [Google Scholar] [CrossRef]
- Anderson, G.K.; Cross, R.J. Isomerisation mechanisms of square-planar complexes. J. Chem. Soc. Rev. 1980, 9, 185–215. [Google Scholar] [CrossRef]
- Lepetit, C.; Fau, P.; Fajerwerg, K.; Kahn, M.L.; Silvi, B. Topological analysis of the metal-metal bond: A tutorial review. Coord. Chem. Rev. 2017, 345, 150–181. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical bonds without bonding electron density—Does the difference electron-density analysis suffice for a description of the chemical bond? Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A description of the chemical bond in terms of local properties of electron density and energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H···F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Macchi, P.; Sironi, A. Chemical bonding in transition metal carbonyl clusters: Complementary analysis of theoretical and experimental electron densities. Coord. Chem. Rev. 2003, 238–239, 383–412. [Google Scholar] [CrossRef]
- Macchi, P.; Proserpio, D.M.; Sironi, A. Experimental Electron Density in a Transition Metal Dimer: Metal-Metal and Metal-Ligand Bonds. J. Am. Chem. Soc. 1998, 120, 13429–13435. [Google Scholar] [CrossRef]
- Firme, C.L.; Antunes, O.A.C.; Esteves, P.M. Relation between bond order and delocalization index of QTAIM. Chem. Phys. Lett. 2009, 468, 129–133. [Google Scholar] [CrossRef]
- Sivchik, V.; Kochetov, A.; Eskelinen, T.; Kisel, K.S.; Solomatina, A.I.; Grachova, E.V.; Tunik, S.P.; Hirva, P.; Koshevoy, I.O. Modulation of Metallophilic and π–π Interactions in Platinum Cyclometalated Luminophores with Halogen Bonding. Chem. Eur. J. 2021, 27, 1787–1794. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Spackman, M.A. How reliable are intermolecular interaction energies estimated from topological analysis of experimental electron densities? Cryst. Growth. Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature: Nuclear spin properties and conventions for chemical shifts. IUPAC recommendations 2001. Magn. Reson. Chem. 2002, 40, 489–505. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Nonius, B.V. COLLECT; Data Collection Software; Nonius BV: Delft, The Netherlands, 1998. [Google Scholar]
- Otwinowski, Z.; Minor, W. Macromolecular Crystallography, Part A, Methods in Enzymology; Carter, C.W., Sweet, R.M., Eds.; Academic Press: Cambridge, UK, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Kratzert, D.; Krossing, I. Recent improvements in DSR. J. Appl. Cryst. 2018, 51, 928–934. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett.1997, 78, 1396. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Maaninen, T.; Tuononen, H.M.; Kosunen, K.; Oilunkaniemi, R.; Hiitola, J.; Laitinen, R.; Chivers, T. Formation, structural characterization and calculated NMR chemical shifts of selenium-nitrogen compounds from SeCl4 and ArNHLi (Ar supermesityl, mesityl). Z. Anorg. Allg-. Chem. 2004, 630, 1947–1954. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104/1–154104/19. [Google Scholar] [CrossRef]
- Burns, L.A.; Vazquez-Mayagoitia, A.; Sumpter, B.G.; Sherrill, C.D. Density-functional approaches to noncovalent interactions: A comparison of dispersion corrections (DFT-D), exchange-hole dipole moment (XDM) theory, and specialized functions. J. Chem. Phys. 2011, 134, 084107/1–084107/25. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. TK Gristmill Software; Version 19.10.12; AIMAll: Overland Park, KS, USA, 2019; Available online: aim.tkgristmill.com (accessed on 9 February 2022).








| Compound | δ(125Te), ppm | 1JPtTe, Hz | δ(195Pt), ppm | Ref. | |||
|---|---|---|---|---|---|---|---|
| cis | trans | cis | trans | cis | trans | ||
| [PtCl2{Te(CH2)6}2] | 352 | 399 | 979 | 506 | −4240 | −3682 | This work |
| [PtCl2{Te(CH2)4}2] | 457 | 459 | 665 | 321 | −4251 | −3715 | [30] |
| [PtCl2(TeMe2)2] | 224 | 234 | 824 | 489 | −4351 | −3765 | [40] |
| [PtCl2{Te(CH2SiMe3)2}2] | 266 | 292 | 804 | 469 | −4236 | −3707 | [41] |
| [PtCl2{S(O)(CD3)2}{Te(CH2)6}] | 490 | 1052 | −3830 | This work | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodewald, M.; Rautiainen, J.M.; Görls, H.; Oilunkaniemi, R.; Weigand, W.; Laitinen, R.S. Formation, Characterization, and Bonding of cis- and trans-[PtCl2{Te(CH2)6}2], cis-trans-[Pt3Cl6{Te(CH2)6}4], and cis-trans-[Pt4Cl8{Te(CH2)6}4]: Experimental and DFT Study. Molecules 2023, 28, 7551. https://doi.org/10.3390/molecules28227551
Rodewald M, Rautiainen JM, Görls H, Oilunkaniemi R, Weigand W, Laitinen RS. Formation, Characterization, and Bonding of cis- and trans-[PtCl2{Te(CH2)6}2], cis-trans-[Pt3Cl6{Te(CH2)6}4], and cis-trans-[Pt4Cl8{Te(CH2)6}4]: Experimental and DFT Study. Molecules. 2023; 28(22):7551. https://doi.org/10.3390/molecules28227551
Chicago/Turabian StyleRodewald, Marko, J. Mikko Rautiainen, Helmar Görls, Raija Oilunkaniemi, Wolfgang Weigand, and Risto S. Laitinen. 2023. "Formation, Characterization, and Bonding of cis- and trans-[PtCl2{Te(CH2)6}2], cis-trans-[Pt3Cl6{Te(CH2)6}4], and cis-trans-[Pt4Cl8{Te(CH2)6}4]: Experimental and DFT Study" Molecules 28, no. 22: 7551. https://doi.org/10.3390/molecules28227551