
Identification and Pharmacological Characterization of a Low-Liability Antinociceptive Bifunctional MOR/DOR Cyclic Peptide

 , ,
, ,
Abstract
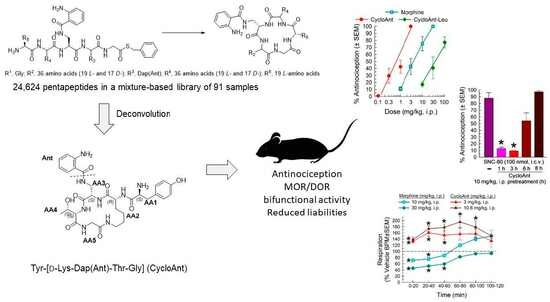
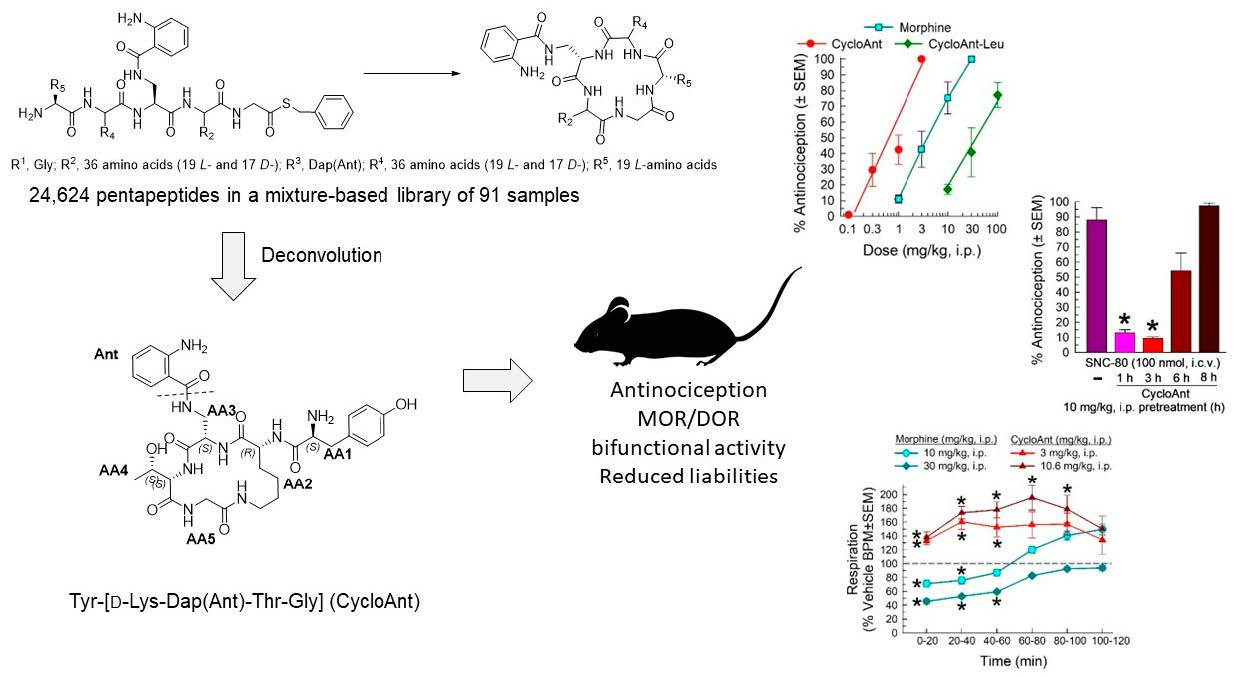
:
1. Introduction
2. Results
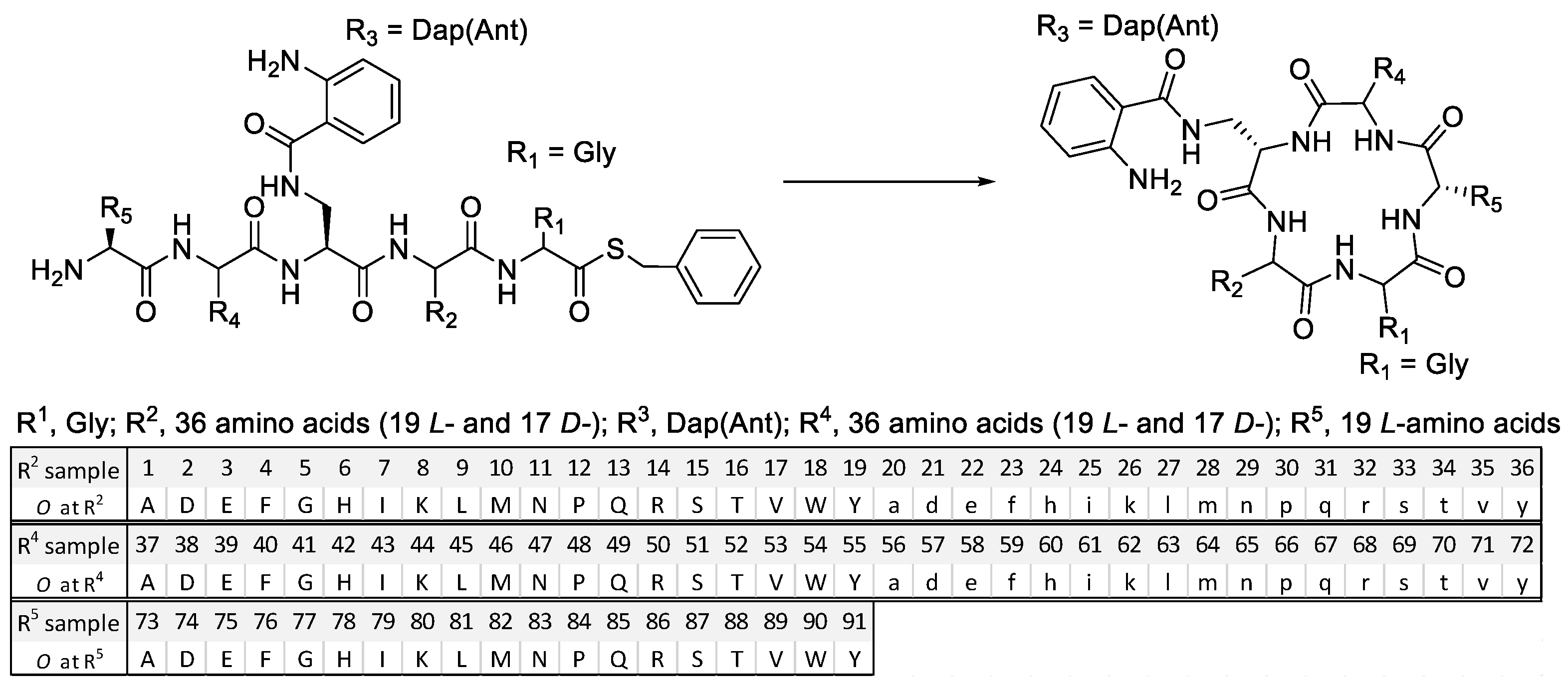
2.1. Mixture-Based Cyclic Peptide Library
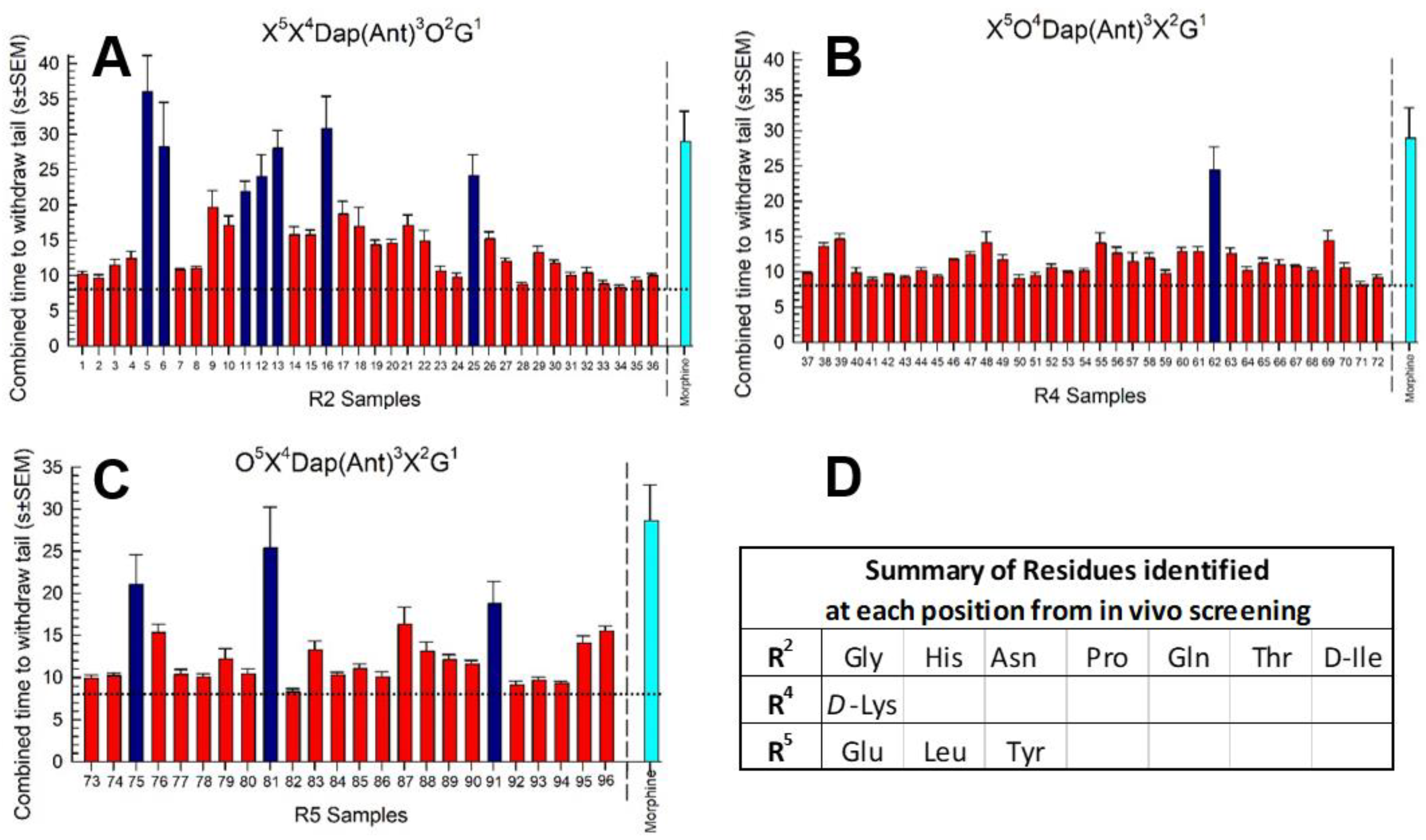
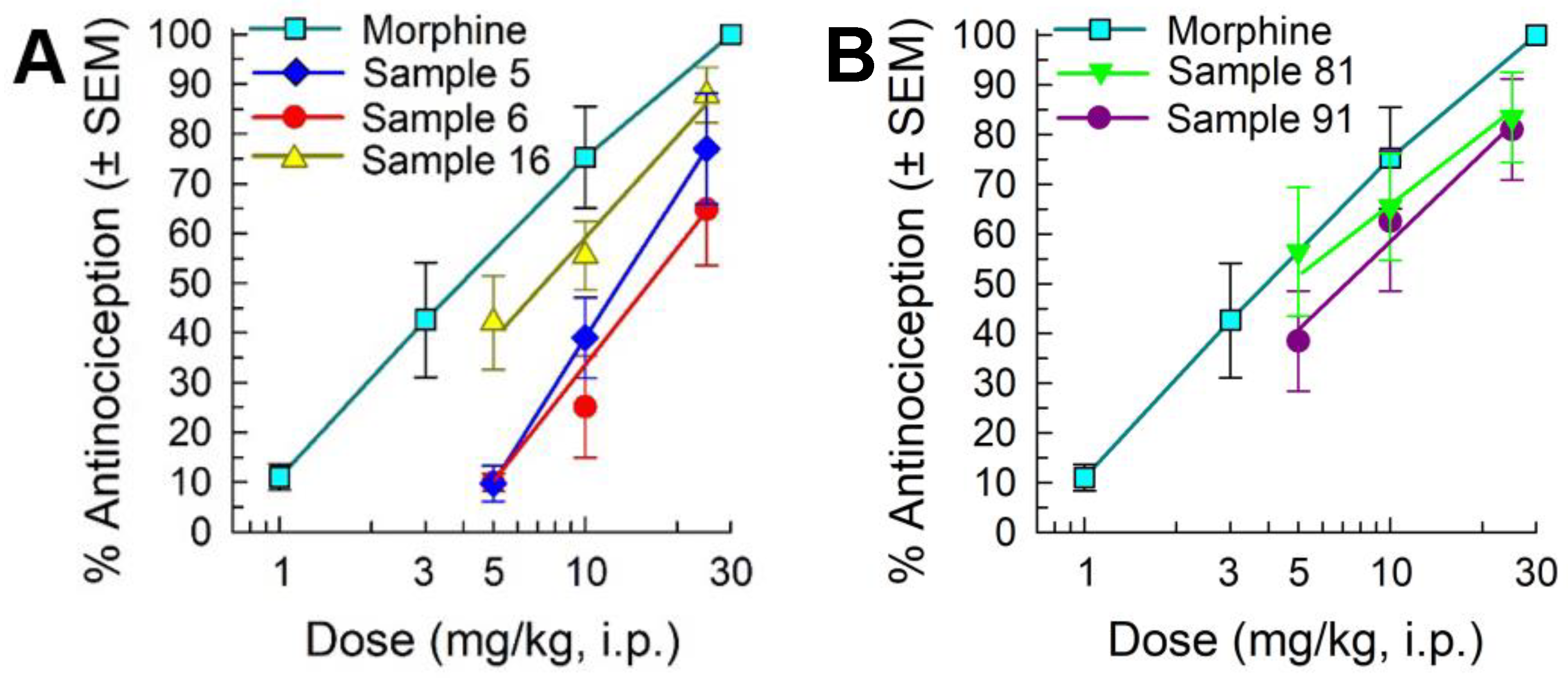
2.2. Library Deconvolution Using Direct In Vivo Screening
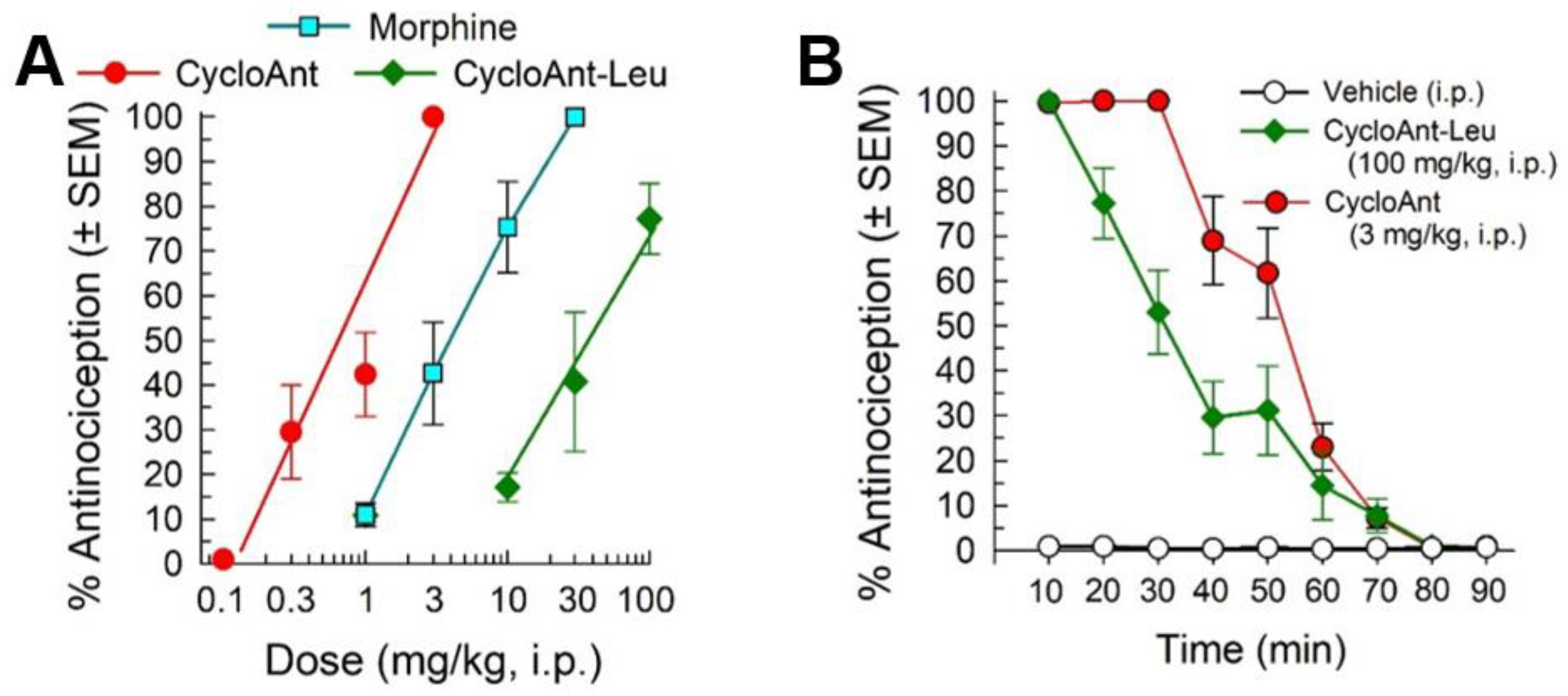
2.3. Synthesis of Cyclic Peptides Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) and Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu) for the Deconvolution of the Library
2.4. Screening of the Cyclic Peptide CycloAnt for Protein Interaction
2.5. Evaluation of the Cyclic Peptides for Their Antinociceptive Effects in Wild-Type Mice
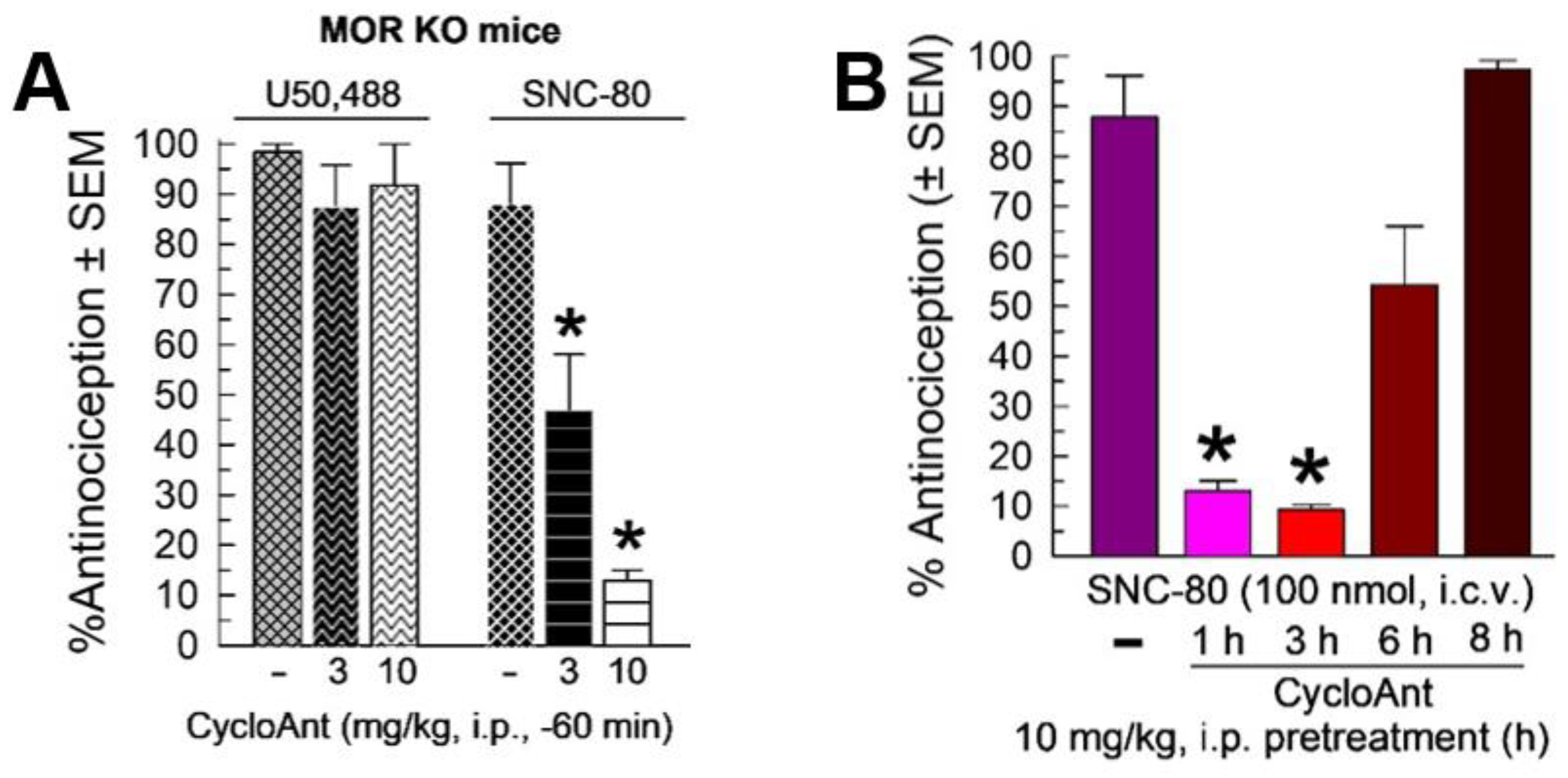
2.6. Evaluation of Opioid Receptor Involvement in CycloAnt Antinociception
2.7. Evaluation of Opioid-Receptor-Selective Antagonism Mediated by CycloAnt
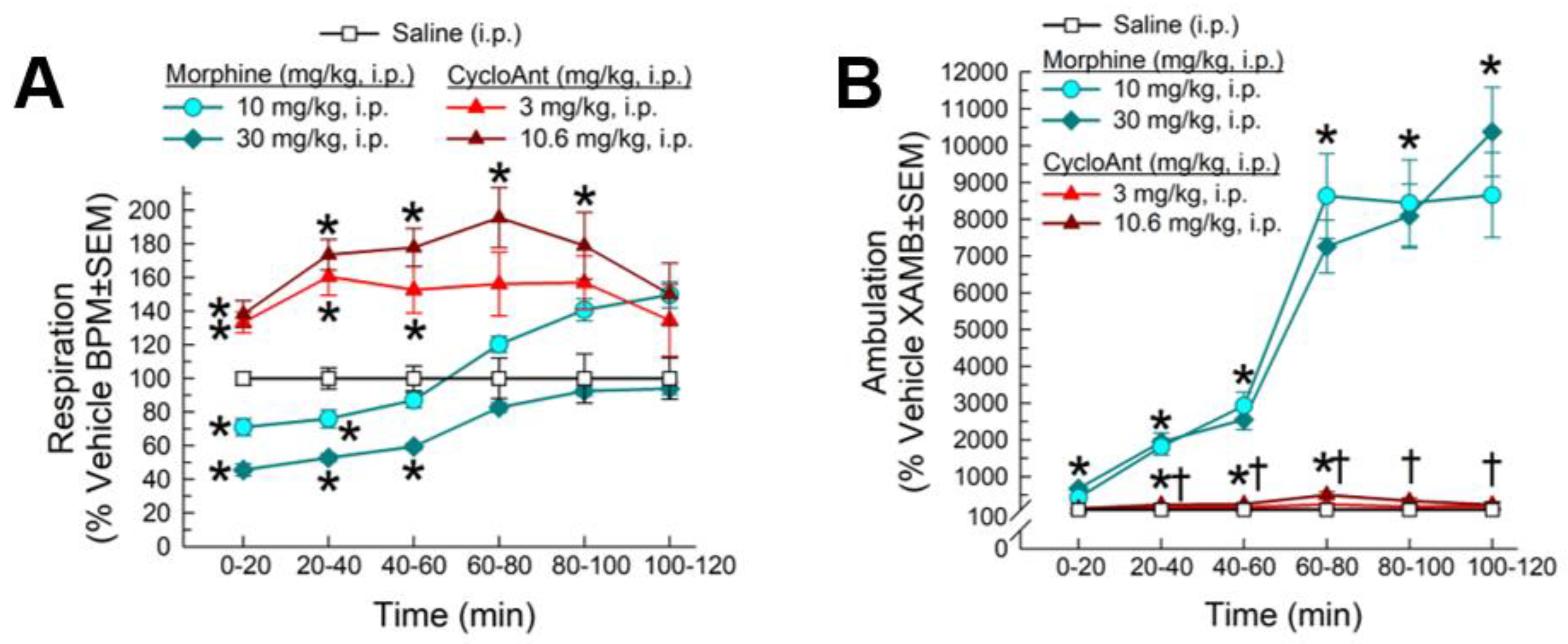
2.8. Evaluation of CycloAnt for Respiratory and Hyperlocomotor Effects
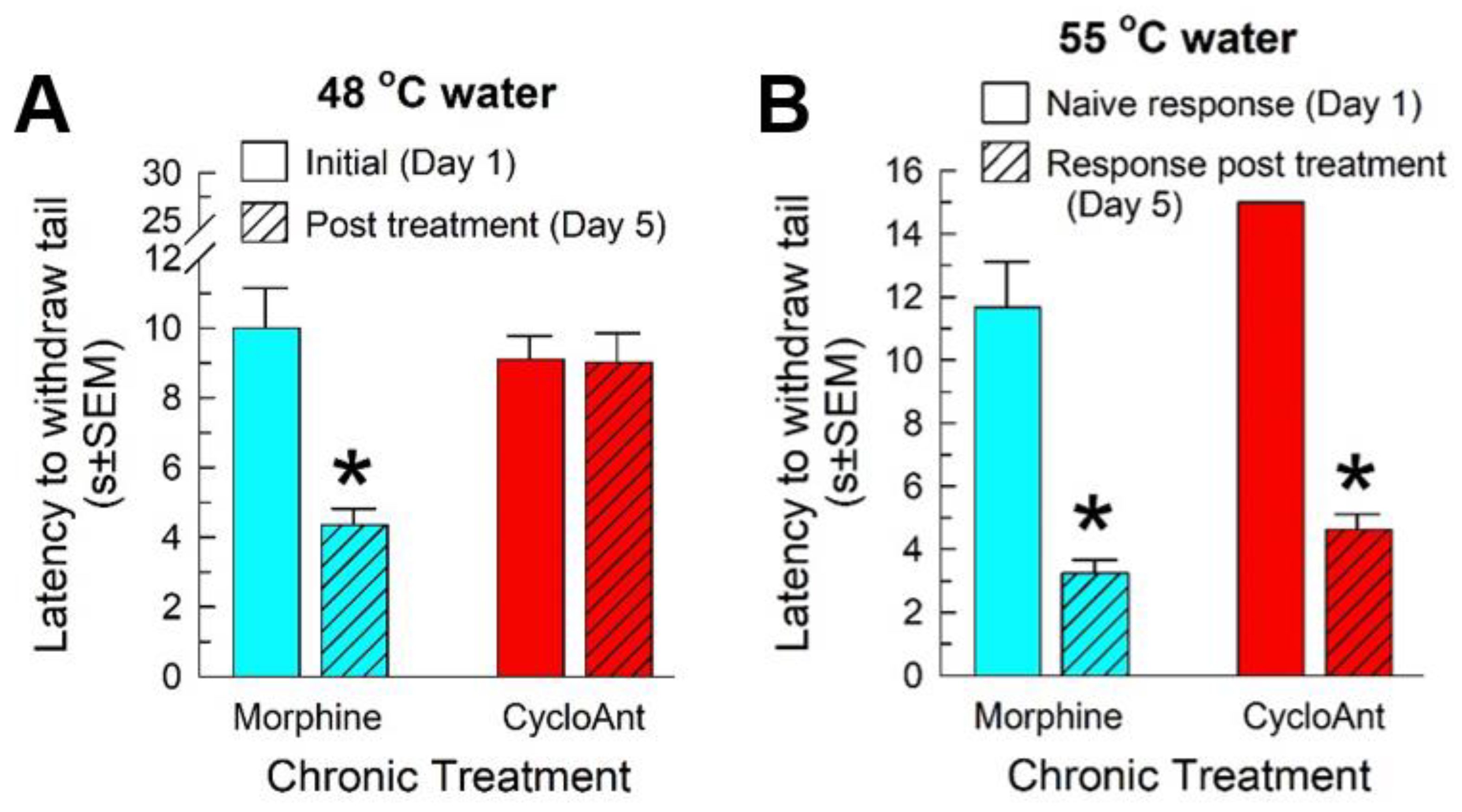
2.9. Evaluation of CycloAnt for Potential of Opioid-Induced Hyperalgesia (OIH), Tolerance and Dependence Development
3. Discussion
4. Materials and Methods
4.1. General Materials
4.2. Synthesis of the Mixture-Based Cyclic Peptide Library
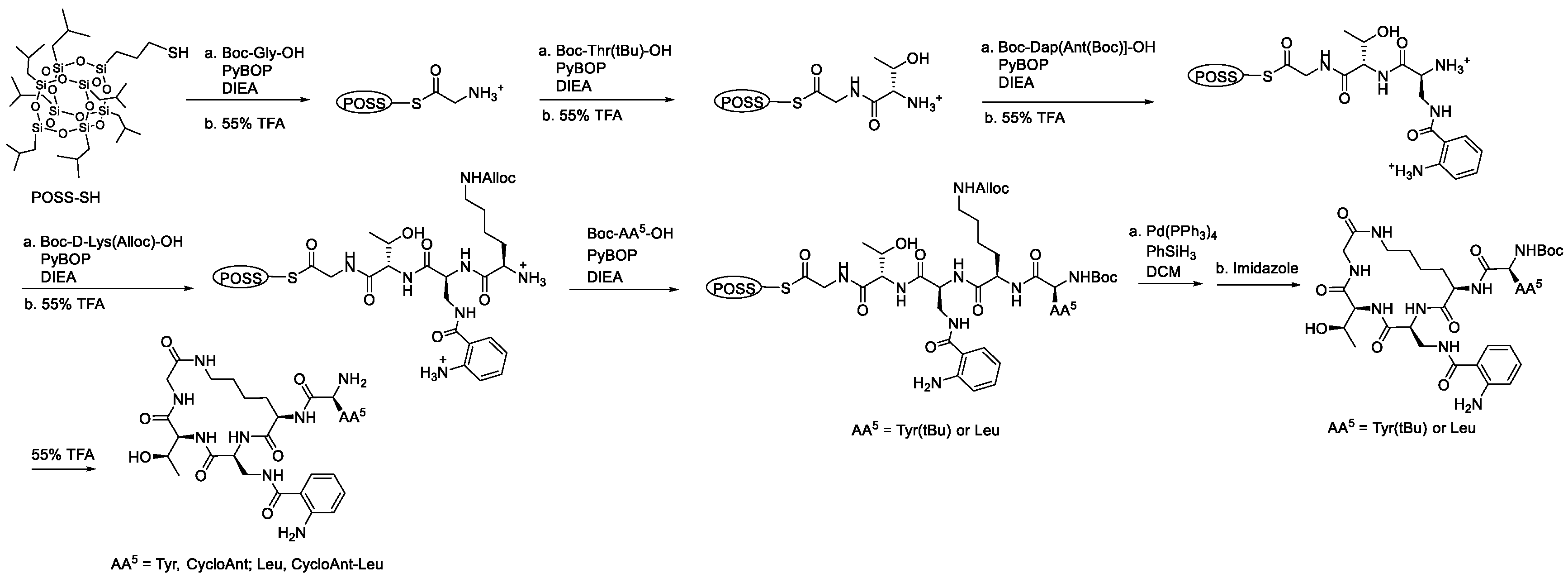
4.3. Solution-Phase Synthesis of Cyclic Peptides Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) and Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu)
4.3.1. Synthesis of Boc-Dap[Ant(Boc)]-OH
4.3.2. Synthesis of Linear Peptide POSS-Thioesters Boc-Tyr(tBu)-D-Lys(Alloc)-Dap(Ant)-Thr-Gly-S-POSS and Boc-Leu-D-Lys(Alloc)-Dap(Ant)-Thr-Gly-S-POSS
4.3.3. Synthesis of Cyclic Peptides Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) and Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu)
- Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt). 1H NMR (400 MHz, DMSO-d6) δ 0.99–1.22 (m, 2H), 1.03 (d, 3H, J = 6 Hz), 1.08–1.18 (m, 1H), 1.29–1.39 (m, 3H), 1.49–1.56 (m, 1H), 2.85–2.87 (m, 1H), 2.90 (m, 1H), 2.96–2.98 (m, 1H), 3.08–3.12 (m, 1H), 3.50 (td, 1H, J = 14, 6 Hz), 3.88 (d, 1H, J = 6.8 Hz), 3.90–3.94 (m, 1H), 3.99–4.02 (m, 2H), 4.08 (t, 1H, J = 6 Hz), 4.2–4.24 (m, 1H), 5.07 (d, 1H, J = 4.8 Hz), 6.34 (br.s, 2H), 6.49–6.52 (dd, 1H, J = 7.2, 1 Hz), 6.54 (d, 1H, J = 2 Hz), 6,72 (d, 3H, J = 8 Hz), 7.03 (d, 2 H, J = 8 Hz), 7.16 (td, 1H, J = 7.6, 1.6 Hz), 7.33 (t, 1H, J = 5.2 Hz), 7.46 (dd, 1H, J = 8, 1.2 Hz), 7.64 (d, 1H, J = 5.2 Hz), 7.72 (br.s, 1H), 8.13 (br.s, 2H), 8.29 (t, 1H, J = 5.6 Hz), 8.38 (t, 1H, J = 5.6 Hz), 8.43 (d, 1H, J = 7.2 Hz), 8.47 (d, 1H, J = 6.4 Hz), 9.39 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 20.1, 21.2, 28.1, 31.5, 36.8, 38.2, 40.7, 43.8, 53.6, 54.2, 56.4, 60.2, 66.3, 114.9, 115.3, 115.7, 115.8, 116.9, 118.7, 125.2, 128.8, 130.9, 132.5, 149.7, 157.1, 158.4, 158.7, 168.2, 169.2, 170.5(2), 170.6, 172.1. 19F NMR (376 MHz) δ-37.7 (s). [M + H]+ calculated 655.3; found 655.3
- Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu). 1H NMR (400 MHz, DMSO-d6) δ 0.92 (d, 6H, J = 8 Hz), 1.03 (d, 3H, J = 4 Hz), 1.29–1.32 (m, 2H), 1.38–1.39 (m, 2H), 1.57–1.65 (m, 5H), 2.96–2.99 (m, 1H), 3.12–3.16 (m, 1H), 3.36 (dd, 1H, J = 16, 4 Hz), 3.53 (m, 1H), 3.88 (dd, 1H, J = 16, 8 Hz), 3.93–4.00 (m, 1H), 4.02–4.04 (m, 2H), 4.12 (t, 1H, J = 8 Hz), 4.19 (t, 1H, J= 8 Hz), 6.51 (td, 1H, J = 8, 1 Hz), 6.73 (d, 1H, J = 8 Hz), 7.16 (td, 1 H, J = 8, 1 Hz), 7.41 (t, 1H, J = 8 Hz), 7.48 (dd, 1H, J = 7.6, 1.6 Hz), 7.66 (d, 1H, J = 8 Hz), 8.15 (br.s, 3H), 8.26 (t, 1H, J = 6 Hz), 8.38 (t, 1H, J = 5.6 Hz), 8.57 (d, 1H, J = 8 Hz), 8.83 (d, 1H, J = 6.8 Hz). 13C NMR (100 MHz, DMSO-d6) δ 20.1, 21.7, 22.1, 23.2, 24.1, 28.2, 31.6, 38.2, 40.4, 40.5, 43.8, 51.4, 54.4, 56.4, 60.0, 66.3, 114.7, 115.2, 115.8, 116.9, 118.8, 128.8, 132.5, 149.8, 158.4, 158.8, 169.2, 169.5, 170.5(2), 170.7, 172.4. [M + H]+ calculated 605.3; found 605.3.
4.4. Animals
4.5. Assessment of Opioid-Induced Hyperalgesia Using the 48 °C Tail-Withdrawal Assay in Mice
4.6. Antinociceptive Test Using the 55 °C Tail-Withdrawal Assay in Mice
4.7. Respiratory Depression and Hyperlocomotion
4.8. Antinociceptive Tolerance and Naloxone-Precipitated Opioid Withdrawal Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Alorfi, N.M. Pharmacological methods of pain management: Narrative review of medication used. Int. J. Gen. Med. 2023, 16, 3247–3256. [Google Scholar]
- Savage, S.R.; Kirsh, K.L.; Passik, S.D. Challenges in using opioids to treat pain in persons with substance use disorders. Addict. Sci. Clin. Pract. 2008, 4, 4–25. [Google Scholar] [CrossRef]
- Schiller, P.W. Opioid peptide-derived analgesics. AAPS J. 2005, 7, E560–E565. [Google Scholar] [PubMed]
- Webster, L.; Schmidt, W.K. Dilemma of addiction and respiratory depression in the treatment of pain: A prototypical endomorphin as a new approach. Pain Med. 2020, 21, 992–1004. [Google Scholar]
- DiMaio, J.; Nguyen, T.M.-D.; Lemieux, C.; Schiller, P.W. Synthesis and Pharmacological Characterization in Vitro of Cyclic Enkephalin Analogues: Effect of Conformational Constraints on Opiate Receptor Selectivity. J. Med. Chem. 1982, 25, 1432–1438. [Google Scholar] [CrossRef]
- Stoeber, M.; Jullié, D.; Lobingier, B.T.; Laeremans, T.; Steyaert, J.; Schiller, P.W.; Manglik, A.; Von Zastrow, M. A genetically encoded biosensor reveals location bias of opioid drug action. Neuron 2018, 98, 963–976. [Google Scholar] [PubMed]
- Tsvetanova, N.G.; Von Zastrow, M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol. 2014, 10, 1061–1065. [Google Scholar] [PubMed]
- Jong, Y.-J.I.; Sergin, I.; Purgert, C.A.; O’Malley, K.L. Location-dependent signaling of the group 1 metabotropic glutamate receptor mGlu5. Mol. Pharmacol. 2014, 86, 774–785. [Google Scholar]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Sierra, S.; Lueptow, L.; Gupta, A.; Gouty, S.; Margolis, E.B.; Cox, B.M.; Devi, L.A. Biased signaling by endogenous opioid peptides. Proc. Natl. Acad. Sci. USA 2020, 117, 11820–11828. [Google Scholar] [PubMed]
- Gallantine, E.L.; Meert, T.F. A Comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Clin. Pharmacol. Toxicol. 2005, 97, 39–51. [Google Scholar] [CrossRef]
- Zhang, X.; Arvidsson, L.B.; Elde, U.R.; Hökfelt, T. Localization and regulation of the delta-opioid receptor in dorsal root ganglia and spinal cord of the rat and monkey: Evidence for association with the membrane of large dense-core vesicles. Neuroscience 1998, 82, 1225–1242. [Google Scholar] [CrossRef]
- Mas-Orea, X.; Basso, L.; Blanpied, C.; Gaveriaux-Ruff, C.; Cenac, N.; Dietrich, G. Delta opioid receptors on nociceptive sensory neurons mediate peripheral endogenous analgesia in colitis. J. Neuroinflamm. 2022, 19, 7. [Google Scholar] [CrossRef]
- Conibear, A.E.; Asghar, J.; Hill, R.; Henderson, G.; Borbely, E.; Tekus, V.; Helyes, Z.; Palandri, J.; Bailey, C.; Starke, I.; et al. A novel G protein–biased agonist at the δ opioid receptor with analgesic efficacy in models of chronic pain. J. Pharmacol. Exp. Ther. 2020, 372, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Vekariya, R.H.; Lei, W.; Ray, A.; Saini, S.K.; Zhang, S.; Molnar, G.; Barlow, D.; Karlage, K.L.; Bilsky, E.J.; Houseknecht, K.L.; et al. Synthesis and structure–activity relationships of 5′-aryl-14-alkoxypyridomorphinans: Identification of a μ opioid receptor agonist/δ opioid receptor antagonist ligand with systemic antinociceptive activity and diminished opioid side effects. J. Med. Chem. 2020, 63, 7663–7694. [Google Scholar] [CrossRef]
- Faouzi, A.; Uprety, R.; Gomes, I.; Massaly, N.; Keresztes, A.I.; Le Rouzic, V.; Gupta, A.; Zhang, T.; Yoon, H.J.; Ansonoff, M.; et al. Synthesis and pharmacology of a novel μ–δ opioid receptor heteromer-selective agonist based on the carfentanyl template. J. Med. Chem. 2020, 63, 13618–13637. [Google Scholar] [CrossRef]
- Matsumoto, K.; Narita, M.; Muramatsu, N.; Nakayama, T.; Misawa, K.; Kitajima, M.; Tashima, K.; Devi, L.A.; Suzuki, T.; Takayama, H.; et al. Orally active opioid μ/δ dual agonist MGM-16, a derivative of the indole alkaloid mitragynine, exhibits potent antiallodynic effect on neuropathic pain in mice. J. Pharmacol. Exp. Ther. 2014, 348, 383–392. [Google Scholar] [CrossRef]
- Podolsky, A.T.; Sandweiss, A.; Hu, J.; Bilsky, E.J.; Cain, J.P.; Kumirov, V.K.; Lee, Y.S.; Hruby, V.J.; Vardanyan, R.S.; Vanderah, T.W. Novel fentanyl-based dual μ/δ-opioid agonists for the treatment of acute and chronic pain. Life Sci. 2013, 93, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Lenard, N.R.; Daniels, D.J.; Portoghese, P.S.; Roerig, S.C. Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur. J. Pharmacol. 2007, 566, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Dietis, N.; Niwa, H.; Tose, R.; McDonald, J.; Ruggieri, V.; Filaferro, M.; Vitale, G.; Micheli, L.; Ghelardini, C.; Salvadori, S.; et al. In vitro and in vivo characterization of the bifunctional mu and delta opioid receptor ligand UFP-505. Br. J. Pharmacol. 2018, 175, 2881–2896. [Google Scholar] [CrossRef]
- Stevenson, G.W.; Folk, J.E.; Rice, K.C.; Negus, S.S. Interactions between delta and mu opioid agonists in assays of schedule-controlled responding, thermal nociception, drug self-administration, and drug versus food choice in rhesus monkeys: Studies with SNC80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide] and heroin. J. Pharmacol. Exp. Ther. 2005, 314, 221–231. [Google Scholar]
- Lowery, J.J.; Raymond, T.J.; Giuvelis, D.; Bidlack, J.M.; Polt, R.; Bilsky, E.J. In vivo characterization of MMP-2200, a mixed δ/μ opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011, 336, 767–778. [Google Scholar] [CrossRef]
- Stevenson, G.W.; Luginbuhl, A.; Dunbar, C.; Lavigne, J.; Dutra, J.; Atherton, P.; Bell, B.; Cone, K.; Giuvelis, D.; Polt, R.; et al. The mixed-action delta/mu opioid agonist MMP-2200 does not produce conditioned place preference but does maintain drug self-administration in rats, and induces in vitro markers of tolerance and dependence. Pharmacol. Biochem. Behav. 2015, 132, 49–55. [Google Scholar] [CrossRef]
- Lei, W.; Vekariya, R.H.; Ananthan, S.; Streicher, J.M. A novel mu-delta opioid agonist demonstrates enhanced efficacy with reduced tolerance and dependence in mouse neuropathic pain models. J. Pain 2020, 21, 146–160. [Google Scholar] [CrossRef]
- Vicario, N.; Pasquinucci, L.; Spitale, F.M.; Chiechio, S.; Turnaturi, R.; Caraci, F.; Tibullo, D.; Avola, R.; Gulino, R.; Parenti, R.; et al. Simultaneous activation of mu and delta opioid receptors reduces allodynia and astrocytic connexin 43 in an animal model of neuropathic pain. Mol. Neurobiol. 2019, 56, 7338–7354. [Google Scholar] [CrossRef]
- Anand, J.P.; Kochan, K.E.; Nastase, A.F.; Montgomery, D.; Griggs, N.W.; Traynor, J.R.; Mosberg, H.I.; Jutkiewicz, E.M. In vivo effects of μ-opioid receptor agonist/δ-opioid receptor antagonist peptidomimetics following acute and repeated administration. Br. J. Pharmacol. 2018, 175, 2013–2027. [Google Scholar] [CrossRef]
- Nastase, A.F.; Anand, J.P.; Bender, A.M.; Montgomery, D.; Griggs, N.W.; Fernandez, T.J.; Jutkiewicz, E.M.; Traynor, J.R.; Mosberg, H.I. Dual pharmacophores explored via structure-activity relationship (SAR) matrix: Insights into potent, bifunctional opioid ligand design. J. Med. Chem. 2019, 62, 4193–4203. [Google Scholar] [CrossRef]
- Schiller, P.W.; Fundytus, M.E.; Merovitz, L.; Weltrowska, G.; Nguyen, T.M.-D.; Lemieux, C.; Chung, N.N.; Coderre, T.J. The opioid µ agonist/δ antagonist DIPP-NH2[Ψ] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999, 42, 7. [Google Scholar] [CrossRef]
- Ananthan, S. Opioid ligands with mixed μ/δ opioid receptor interactions: An emerging approach to novel analgesics. AAPS J. 2006, 8, E118–E125. [Google Scholar] [CrossRef]
- Ananthan, S.; Saini, S.K.; Dersch, C.M.; Xu, H.; McGlinchey, N.; Giuvelis, D.; Bilsky, E.J.; Rothman, R.B. 14-Alkoxy- and 14-acyloxypyridomorphinans: Mu agonist/delta antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 2012, 55, 8350–8363. [Google Scholar] [CrossRef]
- Li, Y.; Dooley, C.T.; Misler, J.A.; Debevec, G.; Giulianotti, M.A.; Cazares, M.E.; Maida, L.; Houghten, R.A. Fluorescent mu selective opioid ligands from a mixture based cyclic peptide library. ACS Comb. Sci. 2012, 14, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Reilley, K.J.; Giulianotti, M.; Dooley, C.T.; Nefzi, A.; McLaughlin, J.P.; Houghten, R.A. Identification of two novel, potent, low-liability antinociceptive compounds from the direct in vivo screening of a large mixture-based combinatorial library. AAPS J. 2010, 12, 318–329. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Banerjee, J.; Ganno, M.L.; Reilley, K.J.; Eans, S.O.; Mizrachi, E.; Gyanda, R.; Hoot, M.R.; Houghten, R.A.; McLaughlin, J.P. Discovery of novel antinociceptive α-conotoxin analogues from the direct in vivo screening of a synthetic mixture-based combinatorial library. ACS Comb. Sci. 2013, 15, 153–161. [Google Scholar] [CrossRef]
- Houghten, R.A.; Ganno, M.L.; McLaughlin, J.P.; Dooley, C.T.; Eans, S.O.; Santos, R.G.; Lavoi, T.; Nefzi, A.; Welmaker, G.; Giulianotti, M.A.; et al. Direct phenotypic screening in mice: Identification of individual, novel antinociceptive compounds from a library of 734 821 pyrrolidine bis-piperazines. ACS Comb. Sci. 2016, 18, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, Y.; Giulianotti, M.; Houghten, R.A. High throughput synthesis of peptide α-thioesters through the use of “volatilizable” support. J. Comb. Chem. 2008, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yongye, A.; Giulianotti, M.; Martinez-Mayorga, K.; Yu, Y.; Houghten, R.A. Synthesis of cyclic peptides through direct aminolysis of peptide thioesters catalyzed by imidazole in aqueous organic solutions. J. Comb. Chem. 2009, 11, 1066–1072. [Google Scholar] [CrossRef]
- Perry, K.; Sui, B.; Li, Y. Mercapto-functionalized polyhedral oligomeric silsesquioxane as a soluble support for the synthesis of peptide thioesters. Tetrahedron Lett. 2021, 85, 153483. [Google Scholar] [CrossRef]
- Li, Y.; Giulionatti, M.; Houghten, R.A. Macrolactonization of peptide thioesters catalyzed by imidazole and its application in the synthesis of Kahalalide B and analogues. Org. Lett. 2010, 12, 2250–2253. [Google Scholar] [CrossRef]
- Wilson, L.L.; Eans, S.O.; Cirino, T.J.; Stacy, H.M.; Simons, C.A.; Uprety, R.; Majumdar, S.; McLaughlin, J.P. Kratom alkaloids, natural and semi-synthetic, show less physical dependence and ameliorate opioid withdrawal. Cell Mol. Neurobiol. 2021, 41, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef]
- Singleton, S.; Bastista-Hon, D.T.; Edelsten, E.; McCaughey, K.S.; Camplisson, E.; Hales, T.G. TRV130 partial agonism and capacity to induce anti-nociceptive tolerance revealed through reducing available μ-opioid receptor number. Br. J. Pharmacol. 2021, 178, 1855–1868. [Google Scholar] [CrossRef]
- Kelly, E. Efficacy and ligand bias at the mu-opioid receptor. Br. J. Pharmacol. 2013, 169, 1430–1446. [Google Scholar] [CrossRef]
- Schiller, P.W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [Google Scholar] [CrossRef]
- Abdelhamid, E.E.; Sultana, M.; Portoghese, P.S.; Takemori, A.E. Selective blockage of delta-opioid receptors prevents the development of morphine-tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991, 258, 299–303. [Google Scholar]
- Fundytus, M.E.; Schiller, P.W.; Shapiro, M.; Weltrowska, G.; Coderre, T.J. Attenuation of Morphine Tolerance and Dependence with the Highly Selective Delta-Opioid Receptor Antagonist TIPPY. Eur. J. Pharmacol. 1995, 286, 105–108. [Google Scholar] [CrossRef]
- Scherrer, K.H.; Eans, S.O.; Medina, J.M.; Senadheera, S.N.; Khaliq, T.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Tryptophan substitution in CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]) introduces δ-opioid receptor antagonism, preventing antinociceptive tolerance and stress-induced reinstatement of extinguished cocaine-conditioned place preference. Pharmaceuticals 2023, 16, 1218–1238. [Google Scholar] [CrossRef]
- Sanchez-Blazquez, P.; Garcia-Espana, A.; Garzon, J. Antisense oligodeoxynucleotides to opioid mu and delta receptors reduced morphine dependence in mice: Role of delta-2 opioid receptors. J. Pharmacol. Exp. Ther. 1997, 280, 1423–1431. [Google Scholar]
- Kest, B.; Lee, C.E.; McLemore, G.L.; Inturrisi, C.E. An Antisense Oligodeoxynucleotide to the Delta Opioid Receptor (DOR-1) Inhibits Morphine Tolerance and Acute Dependence in Mice. Brain Res. Bull. 1996, 39, 185–188. [Google Scholar] [CrossRef]
- Zhu, Y.; King, M.A.; Schuller, A.G.P.; Nitsche, J.F.; Reidl, M.; Elde, R.P.; Unterwald, E.; Pasternak, G.W.; Pintar, J.E. Retention of Supraspinal Delta-like Analgesia and Loss of Morphine Tolerance in δ Opioid Receptor Knockout Mice. Neuron 1999, 24, 243–252. [Google Scholar] [CrossRef]
- Crain, S.M.; Shen, K.F. Acute thermal hyperalgesia elicited by low-dose morphine in normal mice is blocked by ultra-low-dose naltrexone, unmasking potent opioid analgesia. Brain Res. 2001, 888, 75–82. [Google Scholar] [CrossRef]
- Journigan, V.B.; Mésangeau, C.; Vyas, N.; Eans, S.O.; Cutler, S.J.; McLaughlin, J.P.; Mollereau, C.; McCurdy, C.R. Nonpeptide small molecule agonist and antagonist original leads for neuropeptide FF1 and FF2 receptors. J. Med. Chem. 2014, 57, 8903–9827. [Google Scholar] [CrossRef]
- McLaughlin, J.P.; Hill, K.P.; Jiang, Q.; Sebastian, A.; Archer, S.; Bidlack, J.M. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: In vivo and in vitro characterization of μ-selective agonist and antagonist activity. J. Pharmacol. Exp. Ther. 1999, 289, 304–311. [Google Scholar] [PubMed]
- Brice-Tutt, A.C.; Wilson, L.L.; Eans, S.O.; Stacy, H.M.; Simons, C.A.; Simpson, G.G.; Coleman, J.S.; Ferracane, M.J.; Aldrich, J.V.; McLaughlin, J.P. Multifunctional opioid receptor agonism and antagonism by a novel macrocyclic tetrapeptide prevents reinstatement of morphine-seeking behaviour. Br. J. Pharmacol. 2020, 177, 4209–4222. [Google Scholar] [CrossRef] [PubMed]
- Nomenclature Committee of IUB (NC-IUB) and IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Eur. J. Biochem. 1984, 138, 9–37.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measure | Treatment (Drug on Day 5, -120 min Prior to Naloxone) | Outcome | |||||
|---|---|---|---|---|---|---|---|
| CycloAnt (n = 9) | Saline (n = 10) ‡ | Morphine (n = 10) | (1-Way ANOVA) | ||||
| Ave | SEM | Ave | SEM | Ave | SEM | F(2,26) = | |
| Forepaw Tremor | 23.7 † | 6.85 | 20.2 | 80.4 | 66 * | 17.0 | 4.77, p = 0.02 |
| Wet Dog Shakes | 2.22 | 0.43 | 0.8 | 0.51 | 1.3 | 0.56 | 1.95, p = 0.16 |
| Straightening | 1.22 | 0.47 | 5.7 | 2.33 | 1.1 | 0.46 | 3.35, p = 0.05 |
| Number of soft stools | 6.67 * | 0.91 | 1.1 | 0.41 | 5.4 * | 0.78 | 16.6, p < 0.001 |
| Presence of diarrhea | yes | no | yes | ||||
| Jumping Frequency | 1.89 † | 1.31 | 0 | 0 | 41.6 * | 2.64 | 188.1, p < 0.001 |
| Rearing Frequency | 17.8 * | 5.94 | 55.4 | 6.59 | 22.2 * | 2.46 | 15.4, p < 0.0001 |
| Forepaw Licking Frequency | 11.2 † | 1.80 | 15.1 | 3.27 | 1.7 * | 1.00 | 9.59, p = 0.0008 |
| Teeth Chattering Frequency | 0 † | 0 | 0.4 | 0.27 | 9.5 * | 1.79 | 25.0, p < 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Eans, S.O.; Ganno-Sherwood, M.; Eliasof, A.; Houghten, R.A.; McLaughlin, J.P. Identification and Pharmacological Characterization of a Low-Liability Antinociceptive Bifunctional MOR/DOR Cyclic Peptide. Molecules 2023, 28, 7548. https://doi.org/10.3390/molecules28227548
Li Y, Eans SO, Ganno-Sherwood M, Eliasof A, Houghten RA, McLaughlin JP. Identification and Pharmacological Characterization of a Low-Liability Antinociceptive Bifunctional MOR/DOR Cyclic Peptide. Molecules. 2023; 28(22):7548. https://doi.org/10.3390/molecules28227548
Chicago/Turabian StyleLi, Yangmei, Shainnel O. Eans, Michelle Ganno-Sherwood, Abbe Eliasof, Richard A. Houghten, and Jay P. McLaughlin. 2023. "Identification and Pharmacological Characterization of a Low-Liability Antinociceptive Bifunctional MOR/DOR Cyclic Peptide" Molecules 28, no. 22: 7548. https://doi.org/10.3390/molecules28227548