
3-Nitroindoles Serving as N-Centered Nucleophiles for Aza-1,6-Michael Addition to para-Quinone Methides

 , and
, and
Abstract
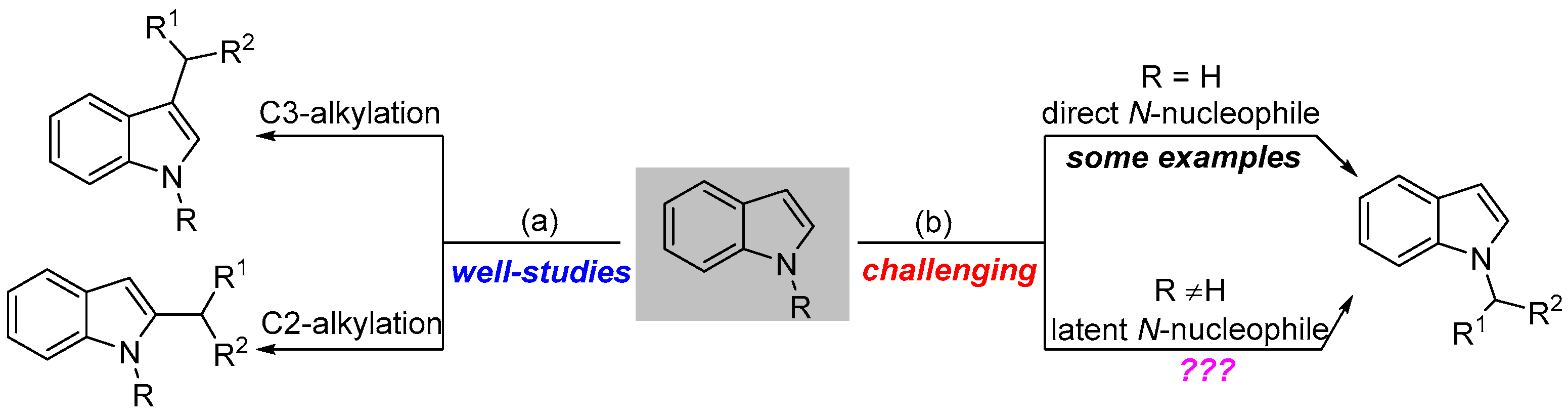
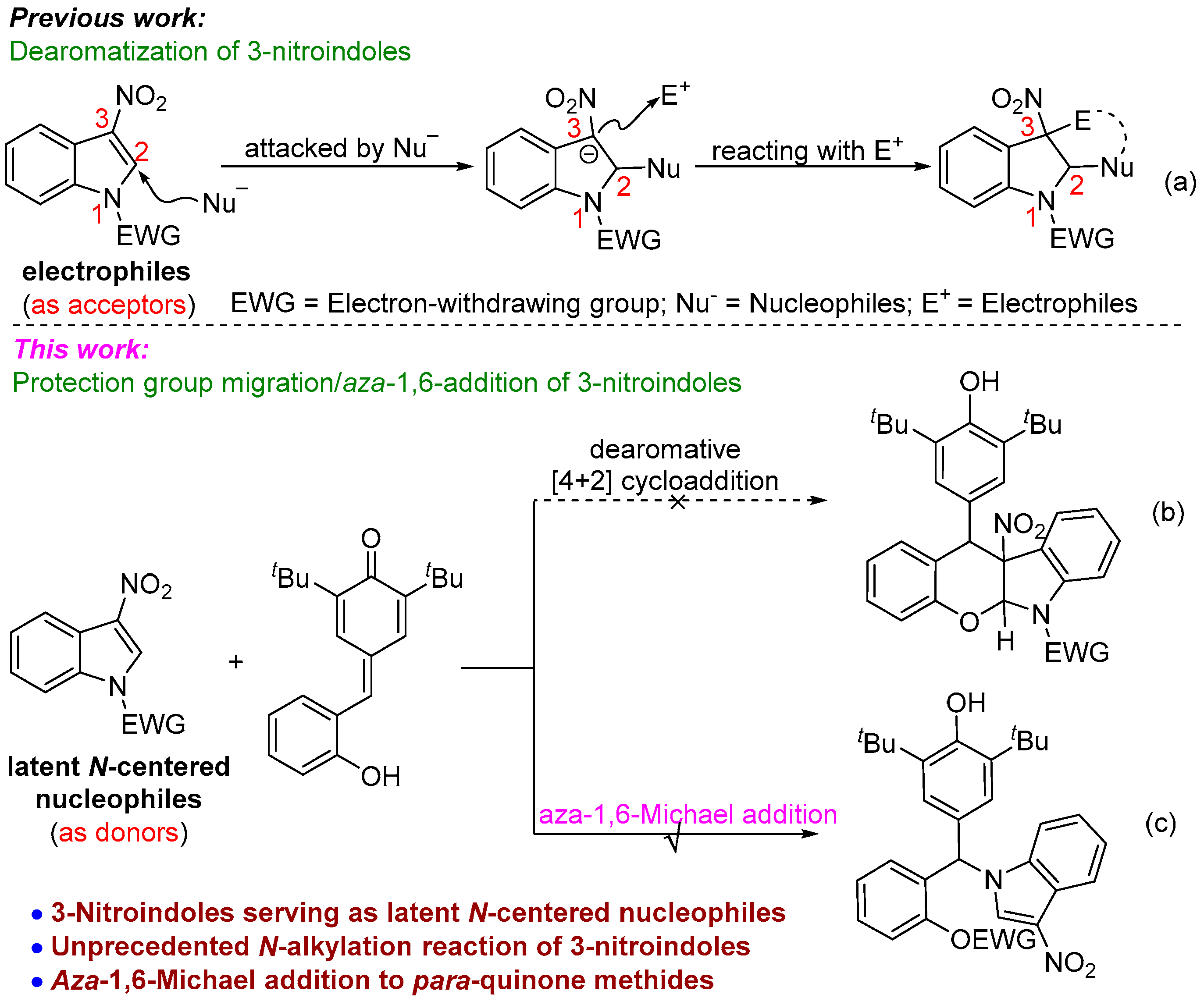
:1. Introduction
2. Results and Discussion
2.1. Optimization Studies
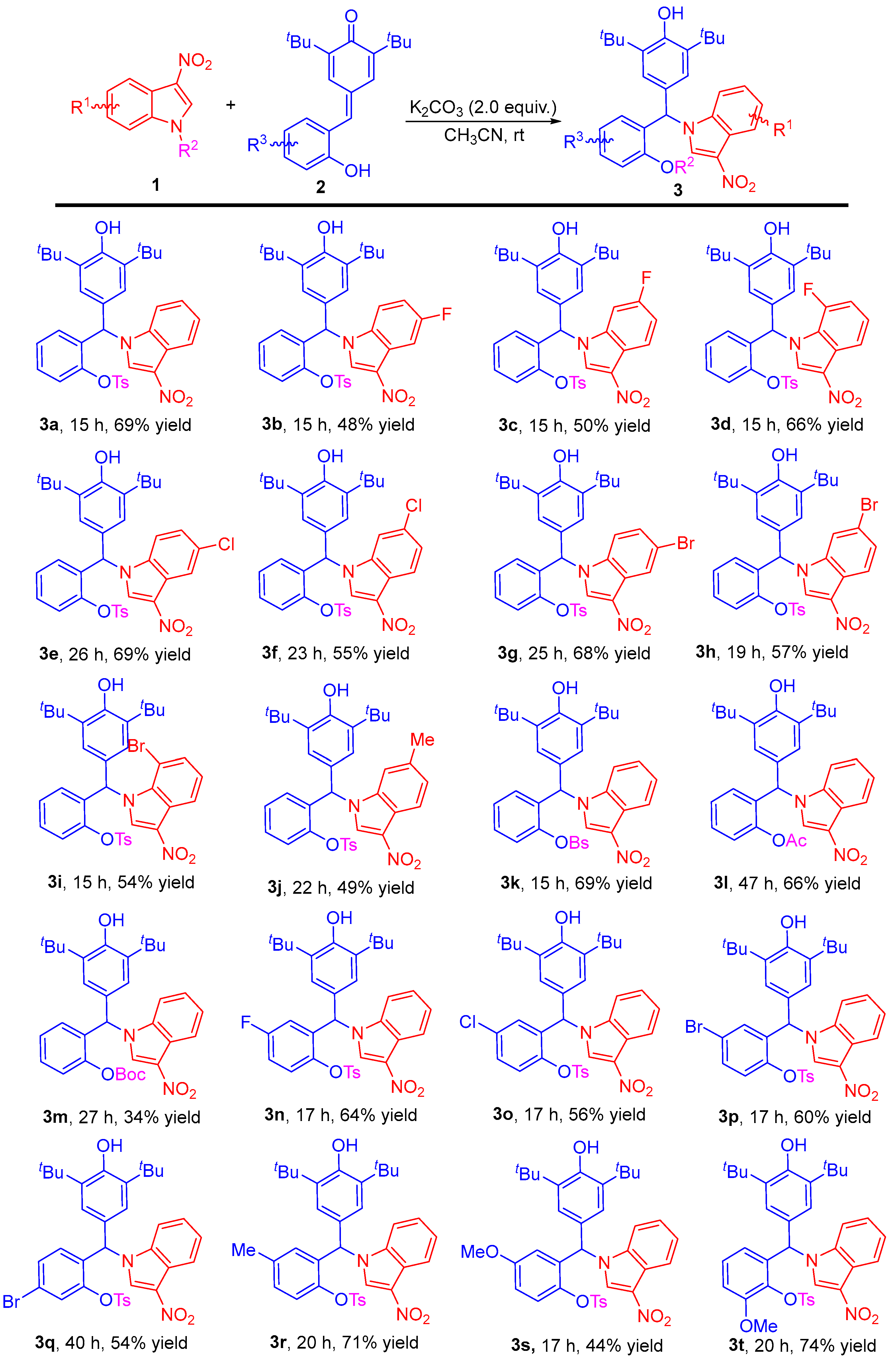
2.2. Substrate Scope Studies
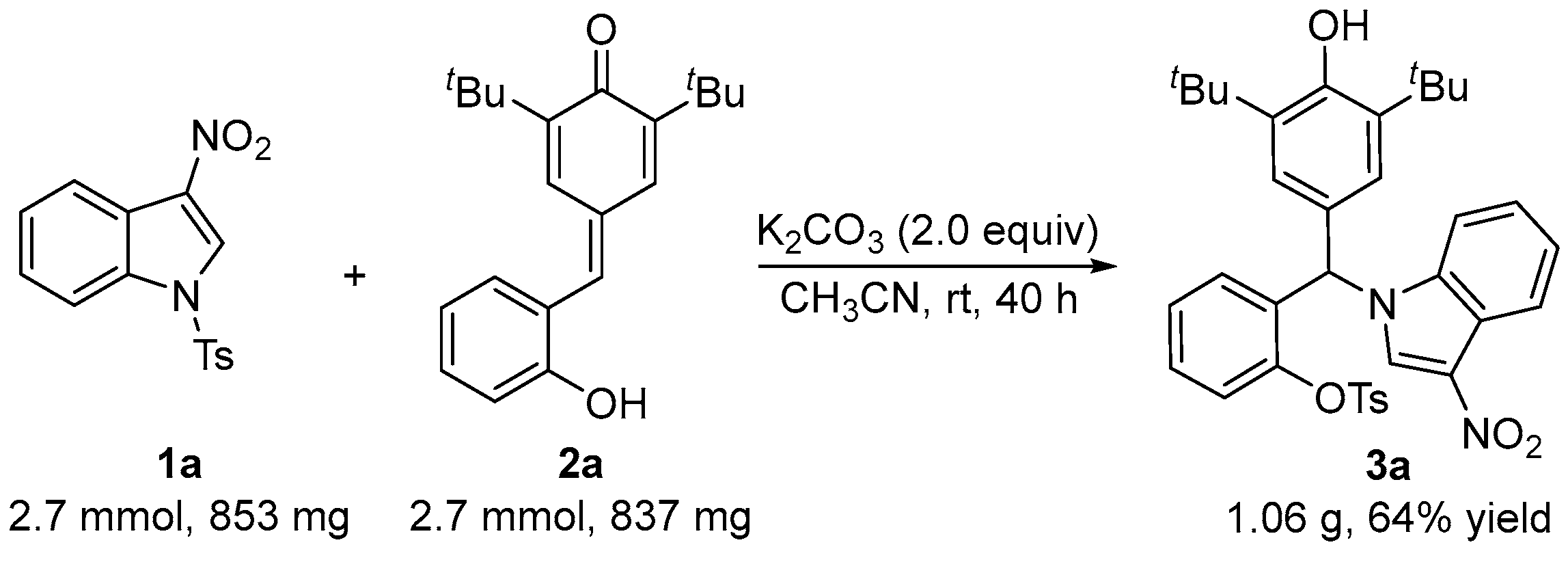
2.3. Scale-Up Experiment
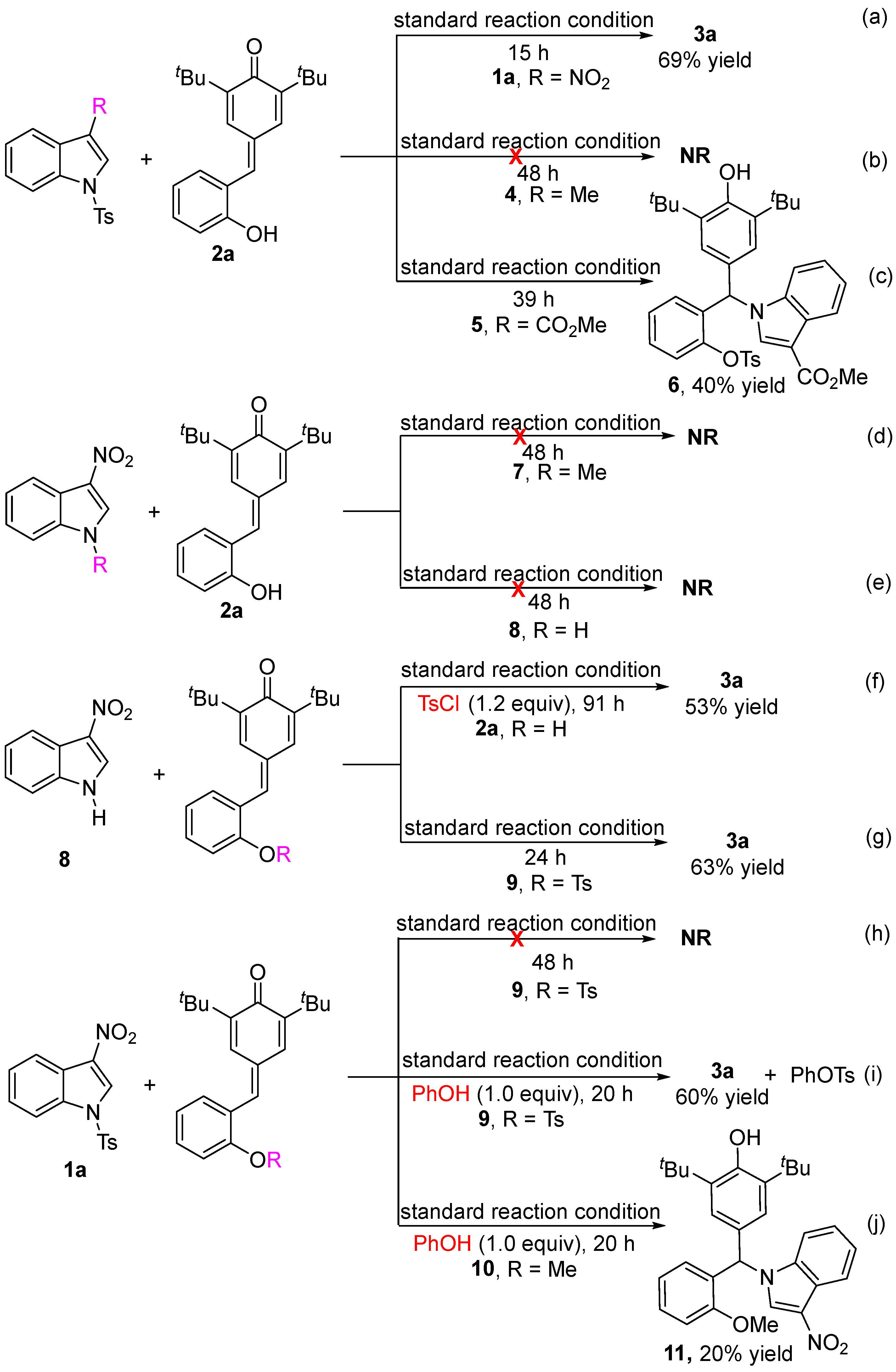
2.4. Control Experiments
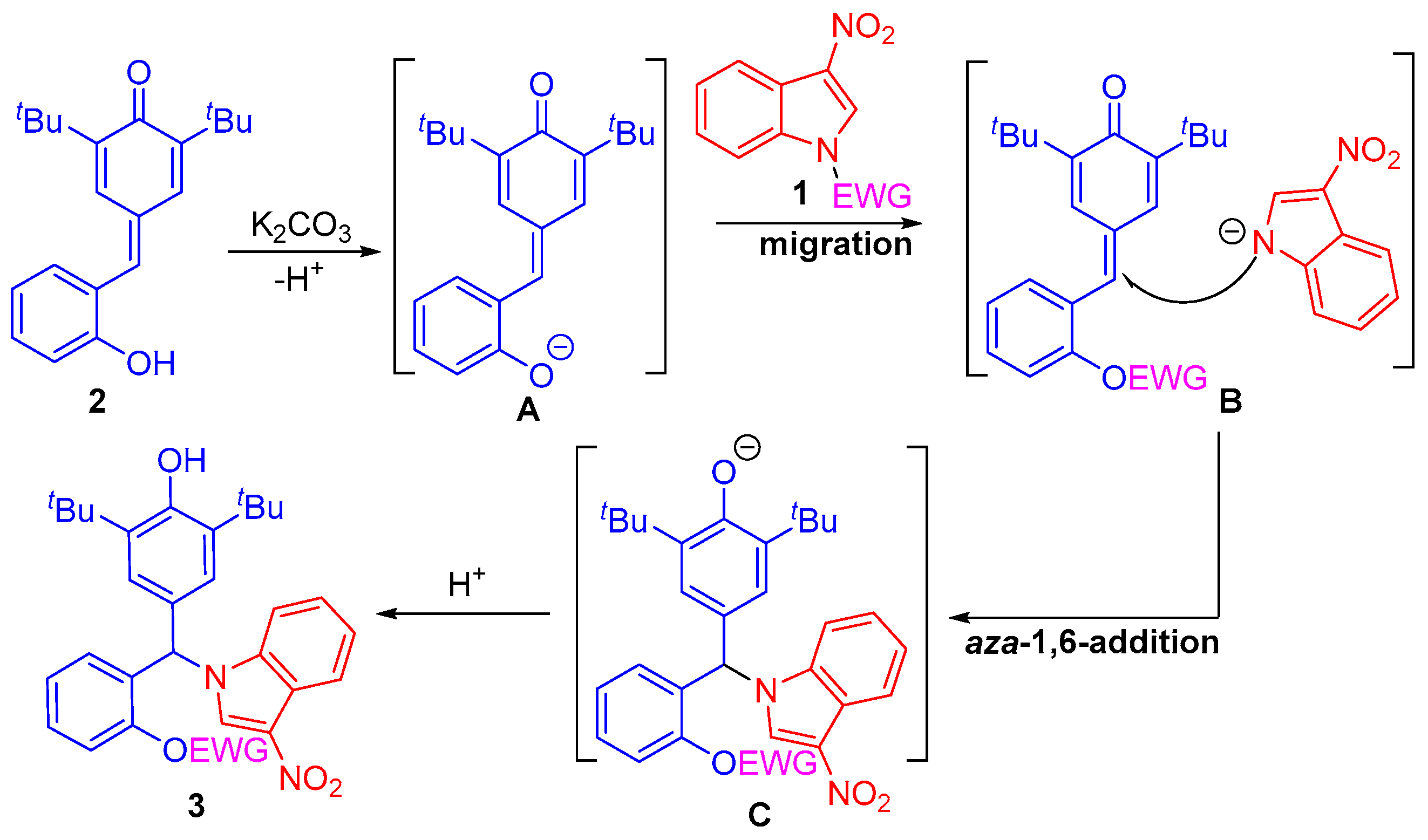
2.5. Plausible Reaction Mechanism
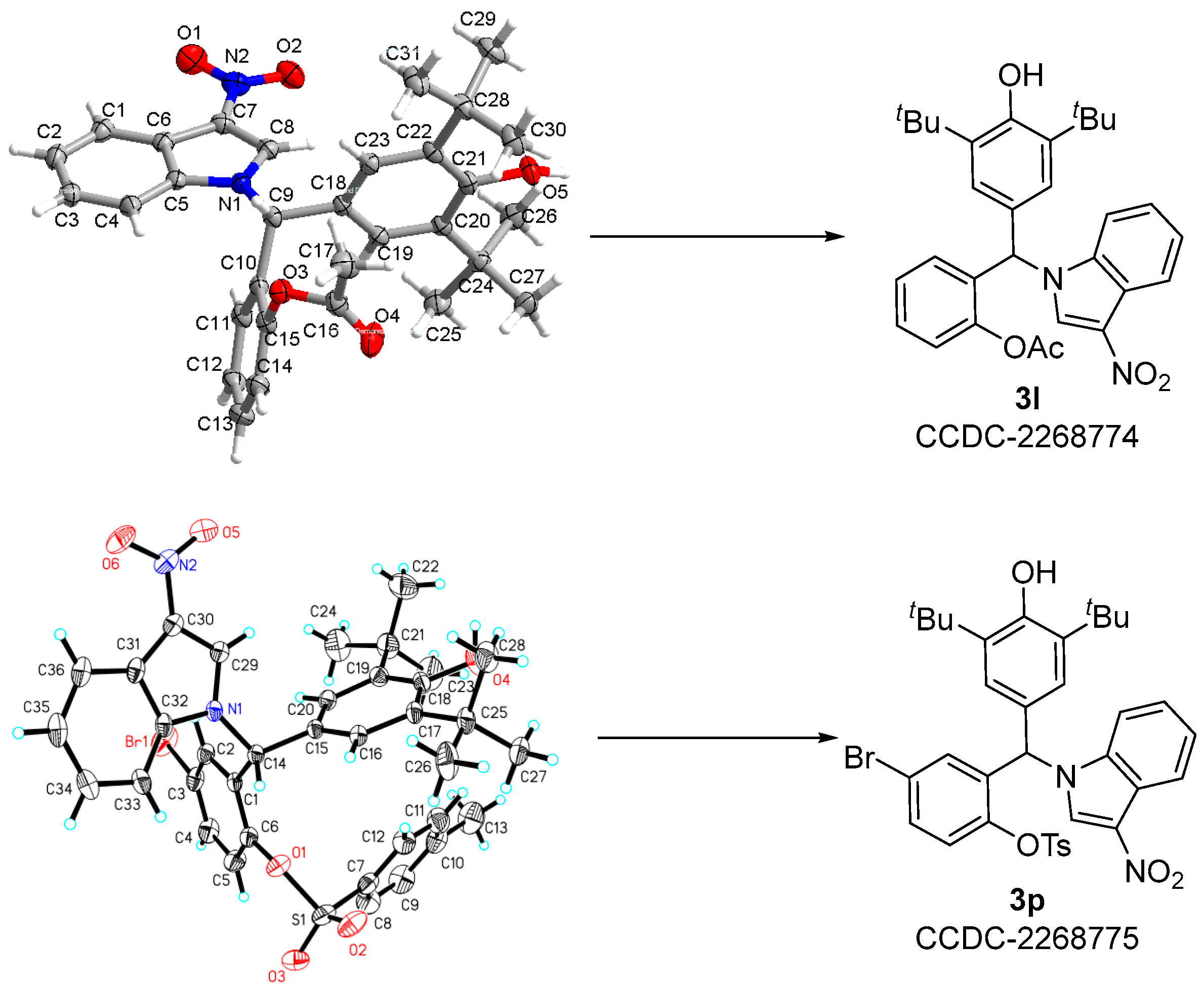
2.6. X-ray Crystallographic Structures
3. Materials and Methods
3.1. General Information
3.2. General Experimental Procedure for the N-Alkylation of 3-Nitroindoles with para-Quinone Methides for the Synthesis of N-Diarylmethylindole Derivatives 3 (Scheme 3)
3.3. The Experimental Procedure for Synthesis of Compound 6 (Scheme 5)
3.4. The Experimental Procedure for Synthesis of Compound 11 (Scheme 5)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Somei, M.; Yamada, F. Simple indole alkaloids and those with a non-rearranged monoterpenoid unit. Nat. Prod. Rep. 2005, 22, 73–103. [Google Scholar] [CrossRef] [PubMed]
- de Sa Alves, F.R.; Barreiro, E.J.; Fraga, C.A.M. From Nature to Drug Discovery: The Indole Scaffold as a ‘Privileged Structure’. Mini-Rev. Med. Chem. 2009, 9, 782–793. [Google Scholar] [CrossRef]
- Kochanowska-Karamyan, A.J.; Hamann, M.T. Marine indole alkaloids: Potential new drug leads for the control of depression and anxiety. Chem. Rev. 2010, 110, 4489–4497. [Google Scholar] [CrossRef] [Green Version]
- Sravanthi, T.V.; Manju, S.L. Indoles–A Promising Scaffold for Drug Development. Eur. J. Pharm. Sci. 2016, 91, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chadha, N.; Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 111691. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, G.R.; Kuethe, J.T. Practical Methodologies for the Synthesis of Indoles. Chem. Rev. 2006, 106, 2875–2911. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A. Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem. Int. Ed. 2009, 48, 9608–9644. [Google Scholar] [CrossRef]
- Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 2010, 39, 4449–4465. [Google Scholar] [CrossRef]
- Dalpozzo, R. Strategies for the asymmetric functionalization of indoles: An update. Chem. Soc. Rev. 2015, 44, 742–778. [Google Scholar] [CrossRef]
- Sheng, F.-T.; Wang, J.-Y.; Tan, W.; Zhang, Y.-C.; Shi, F. Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives. Org. Chem. Front. 2020, 7, 3967–3998. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Shi, F. Organocatalytic Atroposelective Synthesis of Indole Derivatives Bearing Axial Chirality: Strategies and Applications. Acc. Chem. Res. 2022, 55, 2562–2580. [Google Scholar] [CrossRef] [PubMed]
- You, S.-L.; Cai, Q.; Zeng, M. Chiral Brønsted acid catalyzed Friedel–Crafts alkylation reactions. Chem. Soc. Rev. 2009, 38, 2190–2201. [Google Scholar] [CrossRef]
- Sandtorv, A.H. Transition Metal-Catalyzed C–H Activation of Indoles. Adv. Synth. Catal. 2015, 357, 2403–2435. [Google Scholar] [CrossRef]
- Trubitsõn, D.; Kanger, T. Enantioselective Catalytic Synthesis of N-alkylated Indoles. Symmetry 2020, 12, 1184. [Google Scholar] [CrossRef]
- Ma, J.; Feng, R.; Dong, Z.-B. Recent Advances in Indole Synthesis and the Related Alkylation. Asian J. Org. Chem. 2023, 12, e202300092. [Google Scholar] [CrossRef]
- Chen, M.; Sun, J. Catalytic Asymmetric N-Alkylation of Indoles and Carbazoles through 1,6-Conjugate Addition of Aza-para-quinone Methides. Angew. Chem. Int. Ed. 2017, 56, 4583–4587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, B.; Chen, Z.; Hu, J.; Zeng, X.; Zhong, G. Chiral phosphoric acid catalyzed enantioselective N-alkylation of indoles with in situ generated cyclic N-acyl ketimines. Chem. Commun. 2018, 54, 9230–9233. [Google Scholar] [CrossRef]
- Allen, J.R.; Bahamonde, A.; Furukawa, Y.; Sigman, M.S. Enantioselective N-Alkylation of Indoles via an Intermolecular Aza-Wacker-Type Reaction. J. Am. Chem. Soc. 2019, 141, 8670–8674. [Google Scholar] [CrossRef]
- Gnanamani, E.; Yan, X.; Zare, R.N. Chemoselective N-Alkylation of Indoles in Aqueous Microdroplets. Angew. Chem. Int. Ed. 2020, 59, 3069–3072. [Google Scholar] [CrossRef]
- Clanton, N.A.; Spiller, T.E.; Ortiz, E.; Gao, Z.; Rodriguez-Poirier, J.M.; DelMonte, A.J.; Frantz, D.E. A Metal-Free Reductive N-Alkylation of Indoles with Aldehydes. Org. Lett. 2021, 23, 3233–3236. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, L.; Zhao, J. Chiral Phosphoric Acid-Catalyzed Chemo- and Enantioselective N-Alkylation of Indoles with Imines. Adv. Syn. Catal. 2022, 364, 4166–4172. [Google Scholar] [CrossRef]
- Zha, T.; Rui, J.; Zhang, Z.; Zhang, D.; Yang, Z.; Yu, P.; Wang, Y.; Peng, F.; Shao, Z. Direct Catalytic Asymmetric and Regiodivergent N1-and C3-Allenylic Alkylation of Indoles. Angew. Chem. Int. Ed. 2023, 62, e202300844. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Shurtleff, V.W.; Terrett, J.A.; Cuthbertson, J.D.; MacMillan, D.W.C. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science 2016, 352, 1304–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geri, J.B.; Wade Wolfe, M.M.; Szymczak, N.K. The Difluoromethyl Group as a Masked Nucleophile: A Lewis Acid/Base Approach. J. Am. Chem. Soc. 2018, 140, 9404–9408. [Google Scholar] [CrossRef]
- Lange, M.; Zi, Y.; Vilotijevic, I. Enantioselective Synthesis of Pyrrolizin-1-ones via Lewis Base Catalyzed N-Allylation of N-Silyl Pyrrole Latent Nucleophiles. J. Org. Chem. 2020, 85, 1259–1269. [Google Scholar] [CrossRef]
- Kumar, S.; Lange, M.; Zi, Y.; Görls, H.; Vilotijevic, I. Latent Pronucleophiles in Lewis Base Catalysis: Enantioselective Allylation of Silyl Enol Ethers with Allylic Fluorides. Chem. Eur. J. 2023, 29, e202300641. [Google Scholar] [CrossRef]
- Zi, Y.; Lange, M.; Schultz, C.; Vilotijevic, I. Latent Nucleophiles in Lewis Base Catalyzed Enantioselective N-Allylation of N-Heterocycles. Angew. Chem. Int. Ed. 2019, 58, 10727–10731. [Google Scholar] [CrossRef]
- Cerveri, A.; Bandini, M. Recent Advances in the Catalytic Functionalization of “Electrophilic” Indoles. Chin. J. Chem. 2020, 38, 287–294. [Google Scholar] [CrossRef]
- Rkein, B.; Bigot, A.; Birbaum, L.; Manneveau, M.; De Paolis, M.; Legros, J.; Chataigner, I. Reactivity of 3-nitroindoles with electron-rich species. Chem. Commun. 2021, 57, 27–44. [Google Scholar] [CrossRef]
- Nair, S.R.; Baire, B. Recent Dearomatization Strategies of Benzofurans and Benzothiophenes. Asian J. Org. Chem. 2021, 10, 932–948. [Google Scholar] [CrossRef]
- Wang, N.; Ren, J.; Li, K. Dearomatization of Nitro(hetero)arenes through Annulation. Eur. J. Org. Chem. 2022, 2022, e202200039. [Google Scholar] [CrossRef]
- Li, Y.-L.; Wang, K.-K.; He, X.-L. Recent Progress of Electron-Withdrawing-Group-Tethered Arenes Involved Asymmetric Nucleophilic Aromatic Functionalizations. Adv. Synth. Catal. 2022, 364, 3630–3650. [Google Scholar] [CrossRef]
- Jaworski, A.A.; Scheidt, K.A. Emerging Roles of in situ Generated Quinone Methides in Metal-Free Catalysis. J. Org. Chem. 2016, 81, 10145–10153. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, X.; Zhang, P.; Li, P. Recent Advances in the Catalytic Enantioselective Reactions of para-Quinone Methides. Chem.-Asian J. 2018, 13, 2350–2359. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Recent Developments in 1,6-Addition Reactions of para-Quinone Methides (p-QMs). Org. Chem. Front. 2020, 7, 1743–1778. [Google Scholar] [CrossRef]
- Wang, D.; Sun, J.; Yan, C.-G. Diastereoselective Synthesis of Spiro[chromane-3,3′-indolines] and Spiro[chromane-3,2′-indenes] via DBU Promoted Formal [4 + 2] Cycloaddition Reaction. Green Synth. Catal. 2022, 3, 53–58. [Google Scholar] [CrossRef]
- Zhao, K.; Zhi, Y.; Shu, T.; Valkonen, A.; Rissanen, K.; Enders, D. Organocatalytic Domino Oxa-Michael/1,6-Addition Reactions: Asymmetric Synthesis of Chromans Bearing Oxindole Scaffolds. Angew. Chem. Int. Ed. 2016, 55, 12104–12108. [Google Scholar] [CrossRef]
- Jiang, X.-L.; Wu, S.-F.; Wang, J.-R.; Mei, G.-J.; Shi, F. Catalytic Asymmetric [4 + 2] Cyclization of para-Quinone Methide Derivatives with 3-Alkyl-2-vinylindoles. Adv. Syn. Catal. 2018, 360, 4225–4235. [Google Scholar] [CrossRef]
- Xiang, M.; Li, C.-Y.; Song, X.-J.; Zou, Y.; Huang, Z.-C.; Li, X.; Tian, F.; Wang, L.-X. Organocatalytic and enantioselective [4 + 2] cyclization between hydroxymaleimides and ortho-hydroxyphenyl para-quinone methide-selective preparation of chiral hemiketals. Chem. Commun. 2020, 56, 14825–14828. [Google Scholar] [CrossRef]
- You, Y.; Li, T.-T.; Yuan, S.-P.; Xie, K.-X.; Wang, Z.-H.; Zhao, J.-Q.; Zhou, M.-Q.; Yuan, W.-C. Catalytic asymmetric [4 + 2] cycloaddition of 1-((2-aryl)vinyl)naphthalen-2-ols with in situ generated ortho-quinone methides for the synthesis of polysubstituted chromanes. Chem. Commun. 2020, 56, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Li, H.; Liang, G.; Pu, Q.; Bai, L.; Zhang, D.; Ye, Y.; Li, Y.; Zhou, J.; Zhou, H. Facile construction of dibenzodioxo[3.3.1]nonanes bearing spirocyclohexadienones via domino [4 + 2] cycloaddition/C(sp3)–H oxidative dehydrogenation coupling reactions. Org. Biomol. Chem. 2022, 20, 9392–9396. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xi, W.; Liu, S.; Yang, Y.; Yang, J.; Ding, H.; Wang, Z. HFIP-catalyzed highly diastereoselective formal [4 + 2] cyclization to synthesize difluorinated multisubstituted chromans using difluoroenoxysilanes as C2 synthons. Chin. Chem. Lett. 2022, 33, 3007–3011. [Google Scholar] [CrossRef]
- Li, H.-H.; Meng, Y.-N.; Chen, C.-M.; Wang, Y.-Q.; Zhang, Z.-X.; Xu, Z.; Zhou, B.; Ye, L.-W. Chiral Brønsted acid-catalyzed asymmetric intermolecular [4 + 2] annulation of ynamides with para-quinone methides. Sci. China Chem. 2023, 66, 1467–1473. [Google Scholar] [CrossRef]
- Zhao, J.-Q.; Zhou, S.; Wang, Z.-H.; You, Y.; Chen, S.; Liu, X.-L.; Zhou, M.-Q.; Yuan, W.-C. Catalytic asymmetric dearomative [4 + 2] annulation of 2-nitrobenzofurans and 5H-thiazol-4-ones: Stereoselective construction of dihydrobenzofuran-bridged polycyclic skeletons. Org. Chem. Front. 2021, 8, 6330–6336. [Google Scholar] [CrossRef]
- Zhao, J.-Q.; Zhou, S.; Qian, H.-L.; Wang, Z.-H.; Zhang, Y.-P.; You, Y.; Yuan, W.-C. Higher-order [10 + 2] cycloaddition of 2-alkylidene-1-indanones enables the dearomatization of 3-nitroindoles: Access to polycyclic cyclopenta[b]indoline derivatives. Org. Chem. Front. 2022, 9, 3322–3327. [Google Scholar] [CrossRef]
- Zhou, X.-J.; Zhao, J.-Q.; Lai, Y.-Q.; You, Y.; Wang, Z.-H.; Yuan, W.-C. Organocatalyzed asymmetric dearomative 1,3-dipolar cycloaddition of 2-nitrobenzofurans and N-2,2,2-trifluoroethylisatin ketimines. Chirality 2022, 34, 1019–1034. [Google Scholar] [CrossRef]
- Yuan, W.-C.; Chen, X.-M.; Zhao, J.-Q.; Zhang, Y.-P.; Wang, Z.-H.; You, Y. Ag-Catalyzed Asymmetric Interrupted Barton-Zard Reaction Enabling the Enantioselective Dearomatization of 2- and 3-Nitroindoles. Org. Lett. 2022, 24, 826–831. [Google Scholar] [CrossRef]
- Zhou, S.; Qian, H.-L.; Zhao, J.-Q.; You, Y.; Wang, Z.-H.; Yin, J.-Q.; Zhang, Y.-P.; Chen, M.-F.; Yuan, W.-C. Diastereoselective synthesis of polycyclic indolines via dearomative [4 + 2] cycloaddition of 3-nitroindoles with ortho-aminophenyl p-quinone methides. Org. Biomol. Chem. 2023, 21, 1373–1378. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | T (°C) | Time (h) | Yield [b] |
| 1 | DABCO | toluene | 50 | 168 | 44 |
| 2 | DBU | toluene | 50 | 68 | 24 |
| 3 | Na2CO3 | toluene | 50 | 145 | trace |
| 4 | K2CO3 | toluene | 50 | 26 | 60 |
| 5 | K2CO3 | CH2Cl2 | 50 | 88 | 57 |
| 6 | K2CO3 | THF | 50 | 63 | 58 |
| 7 | K2CO3 | EtOAc | 50 | 136 | 63 |
| 8 | K2CO3 | CH3CN | 50 | 23 | 67 |
| 9 | K2CO3 | MeOH | 50 | 20 | 19 |
| 10 | K2CO3 | CH3CN | rt | 23 | 71 |
| 11 [c] | K2CO3 | CH3CN | rt | 48 | 64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.-Q.; Wang, W.-J.; Zhou, S.; Xiao, Q.-L.; Xue, X.-S.; Zhang, Y.-P.; You, Y.; Wang, Z.-H.; Yuan, W.-C. 3-Nitroindoles Serving as N-Centered Nucleophiles for Aza-1,6-Michael Addition to para-Quinone Methides. Molecules 2023, 28, 5529. https://doi.org/10.3390/molecules28145529
Zhao J-Q, Wang W-J, Zhou S, Xiao Q-L, Xue X-S, Zhang Y-P, You Y, Wang Z-H, Yuan W-C. 3-Nitroindoles Serving as N-Centered Nucleophiles for Aza-1,6-Michael Addition to para-Quinone Methides. Molecules. 2023; 28(14):5529. https://doi.org/10.3390/molecules28145529
Chicago/Turabian StyleZhao, Jian-Qiang, Wen-Jie Wang, Shun Zhou, Qi-Lin Xiao, Xi-Sha Xue, Yan-Ping Zhang, Yong You, Zhen-Hua Wang, and Wei-Cheng Yuan. 2023. "3-Nitroindoles Serving as N-Centered Nucleophiles for Aza-1,6-Michael Addition to para-Quinone Methides" Molecules 28, no. 14: 5529. https://doi.org/10.3390/molecules28145529