
Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads

1
Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD 4072, Australia
2
Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Queensland, Brisbane, QLD 4072, Australia
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(7), 3189; https://doi.org/10.3390/molecules28073189
Submission received: 16 March 2023
/
Revised: 27 March 2023
/
Accepted: 29 March 2023
/
Published: 3 April 2023
(This article belongs to the Section Medicinal Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Bioactive peptides are a highly abundant and diverse group of molecules that exhibit a wide range of structural and functional variation. Despite their immense therapeutic potential, bioactive peptides have been traditionally perceived as poor drug candidates, largely due to intrinsic shortcomings that reflect their endogenous heritage, i.e., short biological half-lives and poor cell permeability. In this review, we examine the utility of molecular engineering to insert bioactive sequences into constrained scaffolds with desired pharmaceutical properties. Applying lessons learnt from nature, we focus on molecular grafting of cyclic disulfide-rich scaffolds (naturally derived or engineered), shown to be intrinsically stable and amenable to sequence modifications, and their utility as privileged frameworks in drug design.
1. Introduction
Peptide therapeutics have been an area of considerable interest in recent years due to their unique structural and functional features that make them an excellent starting point in drug design [1,2,3]. Falling between the two major categories of therapeutics—small molecules (<500 Da) and protein-based biologics (>5000 Da)—peptides are able to simultaneously exhibit the high specificity and efficacy of biologics, allowing for selective targeting of conventionally “undruggable” protein-protein interactions [4], whilst maintaining the lower production cost and complexity of small molecules [5]. However, despite their potent activity and specificity, linear peptide sequences have been historically limited by their intrinsic in vivo instability [6]. One approach to circumvent this innate shortcoming is to combine bioactive linear peptides with stable molecular scaffolds, generating grafted products with the desired properties of both parent compounds. In terms of peptide drug design, cyclic disulfide-rich peptides are particularly attractive frameworks due to their exceptional stability and desirable drug-like properties.
Compared to their linear counterparts, cyclic peptides can, in favorable cases, exhibit improved pharmaceutical properties, including enhanced stability, specificity, bioavailability, and membrane permeability [7,8,9]. These improvements in drug-like qualities are largely due to the closed structural conformation adopted upon cyclization, resulting in sizeable free-energy barriers between alternative backbone conformations [10]. Whilst cyclization does not eliminate clearance due to glomerular filtration, the induced rigidity, in combination with the removal of terminal residues, allows cyclic peptides to circumvent the typically short in vivo half-life of linear peptides resulting from degradation by blood serum endo- and exopeptidases [11,12]. Furthermore, the limited conformational flexibility of the preorganized ring archetype reduces entropic binding costs, promoting greater specificity and binding affinity towards receptor and protein targets [13]. Recent reviews have provided detailed analyses of macrocyclic peptides as drug candidates [14,15,16,17,18].
In the case of cyclic disulfide-rich peptides, the combination of cyclic backbone and disulfide crosslinks affords exceptional stability, particularly in resistance to extreme pH and temperature [19,20]. However, these entropic effects are system dependent, varying with the number of disulfide bonds and their topological arrangement. In general, disulfide bonds are shown to enhance thermodynamic stability by limiting conformational freedom, with system entropy decreasing proportionally with an increasing number of disulfide bonds [21,22]. In this review, we focus specifically on head-to-tail cyclized disulfide-rich peptides, both native and engineered, and their utility as bioactive frameworks. Our coverage is limited to examples our laboratory has direct experience with and is not intended to be a comprehensive examination of all naturally occurring disulfide-rich or cyclic peptide families. Additional classes of natural peptides are covered in other recent articles [23,24,25,26]. Similarly, other classes of engineered peptides not covered here, such as stapled peptides, are described in more detail in other recent reports [27,28,29,30,31].
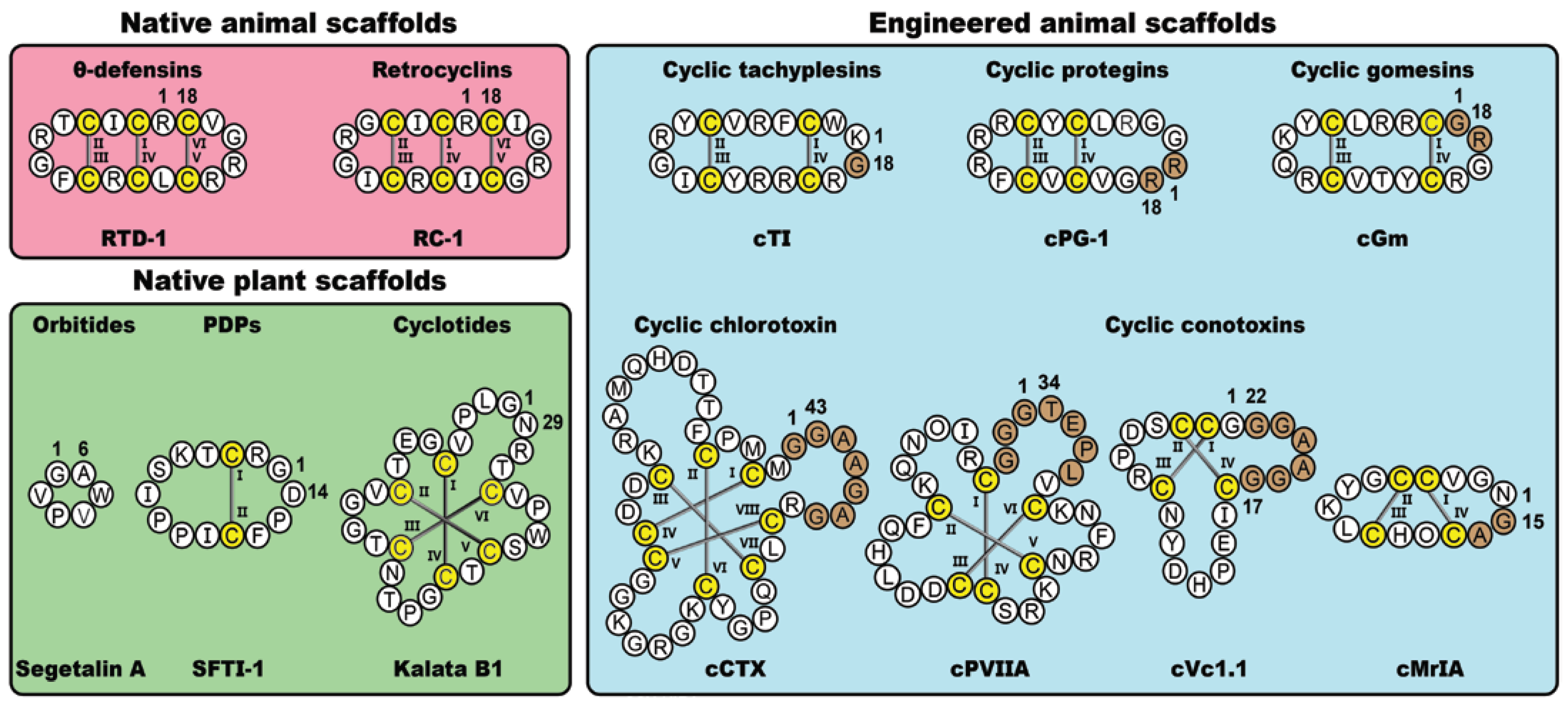
In nature, cyclic disulfide-rich peptides represent a diverse class of bioactive molecules found in plants and animals. Here, we focus on five native cyclic disulfide-rich scaffolds, namely the orbitides, PawS-derived peptides (PDPs), cyclotides, θ-defensins and retrocyclins. Alongside these native plant- and animal-derived macrocycles, we will also examine five classes of disulfide-rich peptides that have been engineered via non-native cyclization junctions and epitope inclusions, namely the conotoxins, chlorotoxins, tachyplesins, protegrins and gomesins. Figure 1 shows prototypic examples from the various classes of frameworks examined in this review, highlighting key structural variations and sites of non-natural modifications. As is apparent from the figure, these molecules offer a diverse range of scaffolds that are amenable to a range of molecular engineering applications, including molecular grafting.
2. Molecular Grafting
Molecular grafting is a drug design approach that involves the insertion of a bioactive sequence into a constrained scaffold with desired pharmaceutical properties. The overarching goal of this paradigm is to construct novel molecules that retain both the biological activity and structure of the grafted epitope, whilst preserving the stability of the molecular framework [32,33,34]. This process typically employs chemical techniques, such as residue mutagenesis, or library-based methods, such as recombinant display. Similar to horticultural grafting, this approach aims to produce an entity better than the sum of its parts. Due to their exceptional stability profile, cyclic disulfide-rich peptides have been widely used in a range of molecular grafting experiments, resulting in exciting developments in several therapeutic fields, including the treatment of cancer, chronic pain, neurodegeneration, and obesity [35]. For a more comprehensive review of molecular grafting utilizing disulfide-rich peptides we recommend the following recent perspective articles [32,35].
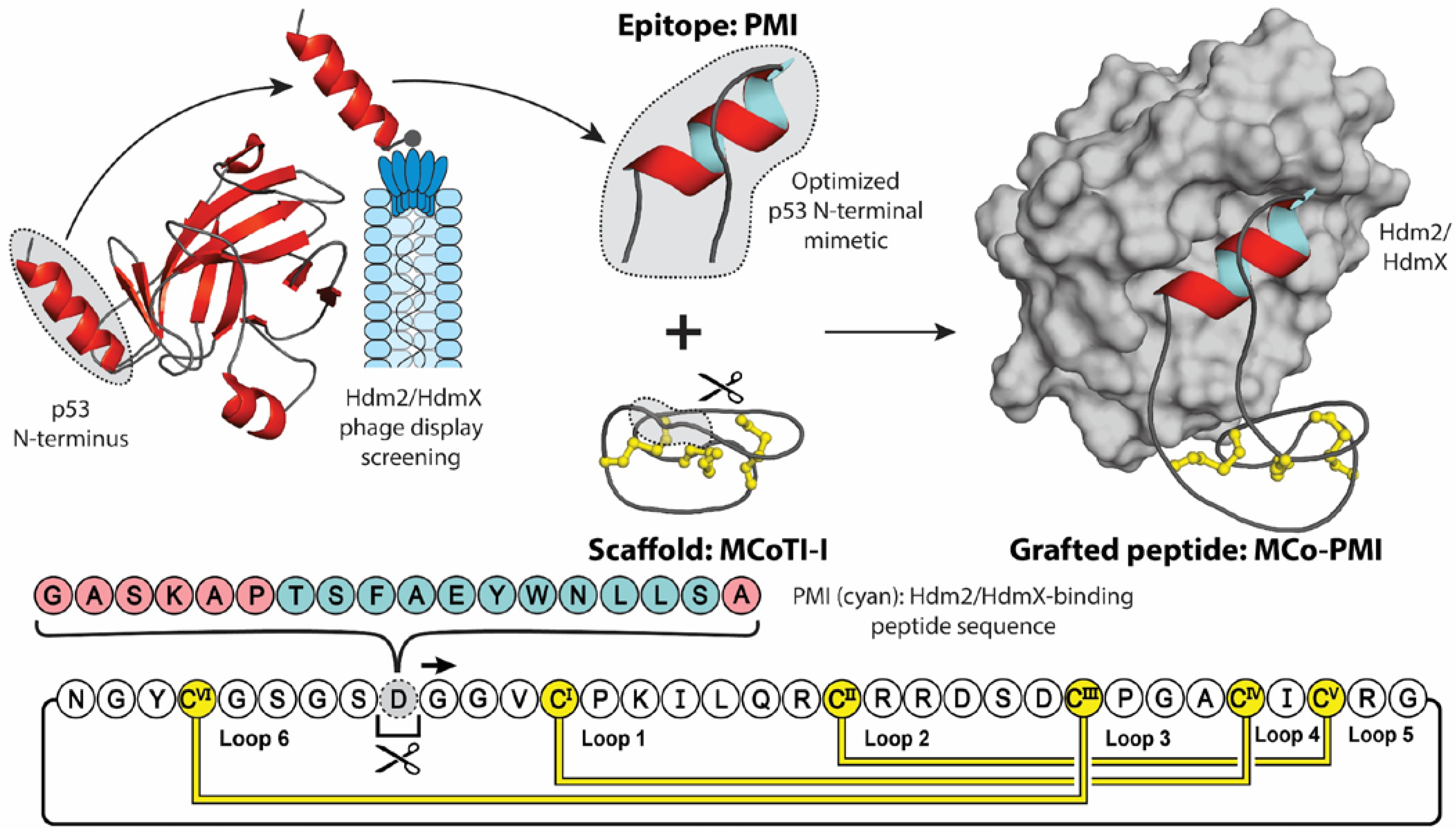
In conjugation with providing stability, molecular grafting can also transfer additional properties of the parent scaffold, including cell-penetrating, anti-microbial, anti-cancer, and analgesic activities [36]. Figure 2 highlights an elegant molecular grafting example, where Ji et al. engineered an α-helical Hdm2/HdmX-binding (human double minute 2 or X protein) peptide (PMI) into the cyclotide MCoTI-I [37,38]. Hdm2 and HdmX are two oncoprotein homologues, commonly overexpressed in tumor cells, that negatively regulate the activity and stability of the tumor-suppressing protein p53, through binding of p53′s N-terminal transactivation domain [39,40]. Short peptides derived from this helical domain, and variants optimized through phage display screening, have been shown to antagonize the intracellular interaction between p53 and Hdm2 and/or HdmX with low nanomolar affinities, stabilizing p53 proteins and reducing the viability of cancer cells expressing wild type p53 [38,41]. However, due to their peptidyl nature, these mimics displayed poor stability and bioavailability [42]. To counteract these intrinsic shortcomings, Ji et al. grafted the PMI sequence into the solvent exposed loop 6 of the MCoTI-I, a privileged scaffold with reported cell-penetrating ability and exceptional stability [43]. Furthermore, to aid the stabilizing of the bioactive α-helical conformation, the authors utilized a flanking helix-stabilizing sequence derived from apamin, which is a bee-venom peptide [44]. The resulting grafted peptide, MCo-PMI, was found to not only retain the structure of the helical linear epitope and cyclotide scaffold, but to also retain the desired bioactivities of both parent molecules. Demonstrating low nanomolar in vitro affinity for both Hdm2 and HdmX (IC50 = 30 ± 5 and 163 ± 17 nm, respectively), high ex vivo human serum stability (τ1/2 = 30 ± 4 h) and potent cytotoxicity to wild type p53 cancer cell lines in vitro and in vivo [37].
Following this brief introduction to molecular grafting, in the remainder of this review we will examine selected classes of disulfide-rich peptides and expand in more detail on several examples that have utilized these molecular frameworks as drug leads. These include examples from native cyclic peptides and artificially cyclised peptides.
3. Plant-Derived Cyclic Peptides
3.1. Orbitides
Orbitides, comprising 5–12 amino acids, are a class of cyclic peptides isolated from a variety of plants, including species of the Verbenaceae, Schizandraceae, Rutaceae, Phytolaccaceae, Linaceae, Lamiaceae, Euphorbiaceae and Annonaceae families [45,46,47,48]. Despite their small size, this family of macrocycles display considerable sequence diversity, with approximately 200 orbitides identified so far [49], and a wide range of functional diversity, including anti-bacterial [50], anti-cancer [51], anti-malarial [52], enzymatic inhibition [53], immunosuppressive [54,55,56] and vasodilatory activities [57]. Unlike many macrocyclic peptides of similar size, which are assembled non-ribosomally, orbitides are direct gene products and are biosynthesized by genetic translation and processing of precursor proteins to produce the mature cyclic peptides. Since our focus in this review is on disulfide-rich peptides, and orbitides are conspicuously lacking in such disulfide bonds, we will not cover them further here apart from noting that extra detail may be found in several recent reviews [47,48,58].
3.2. PawS-Derived Peptides (PDPs)
The PDPs are a family of head-to-tail cyclized peptides found in species of the daisy family, Asteraceae [59]. Ribosomally synthesized as part of a precursor protein for seed storage albumins, PDPs are post-translationally excised and cyclized during proteolytic processing [60]. To date 23 unique PDP sequences have been identified, with 15 confirmed in planta [61]. Despite exhibiting considerable structural diversity, PDPs adopt a rigid well-defined conformation (excluding PDP-8), stabilized by an anti-parallel β-sheet and bridged by a disulfide bond [62,63]. PDP-23 remains the only known exception to this structural classification, adopting a V-shaped structure approximately twice that of typical PDP members (28 amino acids), and comprising two anti-parallel β-sheets stabilized by two disulfide bonds (CysI–CysII and CysIII–CysIV) [61]. Recently identified from the seeds of the Zinnia elegans, PDP-23 demonstrates an intriguing chameleonic-like ability to structurally adapt to different surroundings, exposing different levels of hydrophobicity depending on the conditions, allowing PDP-23 to effectively penetrate cells in a non-toxic manner [61].
The prototypical PDP member, sunflower trypsin inhibitor-1 (SFTI-1), is a broad range serine protease inhibitor, consisting of 14 amino acids, isolated from seeds of the common sunflower (Helianthus annuus) [64]. Despite its small size, SFTI-1 is homologous in sequence to the family of potent serine protease inhibitors, Bowman-Birk inhibitors (BBIs), and is the most potent known trypsin inhibitor with reported sub-nanomolar Ki values [65]. Typically comprising 60–70 amino acids, BBIs perform dual inhibition of trypsin and/or chymotrypsin via a β-hairpin loop motif that binds the protease catalytic sites [66]. Although biosynthetically unrelated, SFTI-1 demonstrates striking sequence and structural homology to these BBI bioactive segments [67]. In accordance with the Laskowski mechanism [68], SFTI-1 protease inhibition is mediated through the insertion of a substrate-like loop into the active site via the formation of an extended β-sheet, effectively blocking access of incoming substrates. The binding loop of SFTI-1 is subsequently cleaved at the scissile bond (Lys5-Ser6) generating an acyl–enzyme adduct [69]. However, the structural conformation and tight binding of the bound inhibitor prevent the completion of the standard catalytic cycle, such that hydrolysis does not occur. Instead, the neo-N-terminus is activated to attack the acyl–enzyme bond, regenerating the scissile bond and the cyclic inhibitor [70].
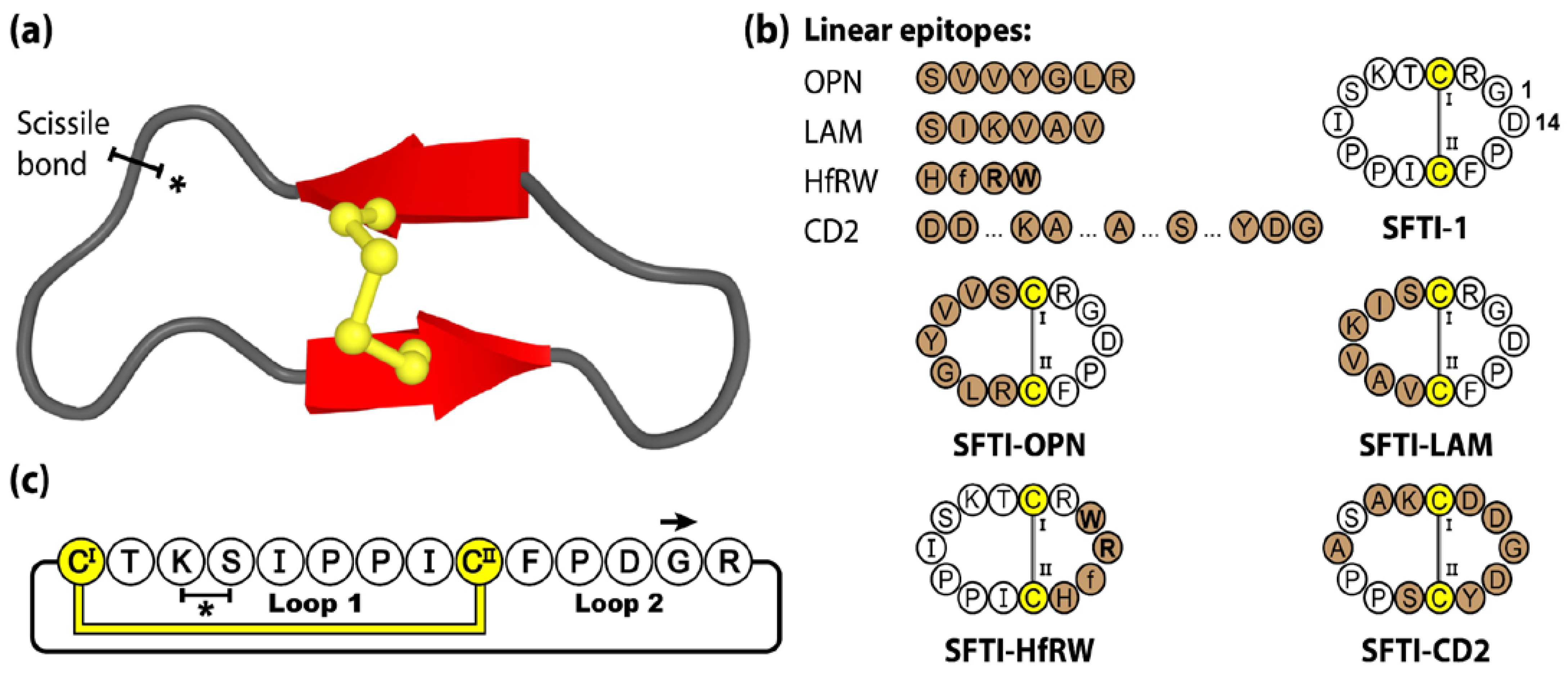
The cyclic backbone, disulfide bond and extensive hydrogen-bonding network of SFTI-1 confers a highly stable and rigid scaffold, readily accessible by chemical [71] and biological [72,73] means. Furthermore, with potent bioactivity towards a range of proteases, SFTI-1 was initially investigated for its anti-inflammatory and anti-cancer properties [65,74]. Over the years, the SFTI scaffold has been widely utilized as a molecular framework with numerous examples of sequence mutagenesis, epitope inclusions and library-based screening, and applications in the areas of autoimmune disease [75], cancer [76,77,78,79,80], cardiovascular and wound healing [81], neurological diseases [82,83] and inflammatory disorders [84,85]. For a more comprehensive review of SFTI-1 and its therapeutic applications we direct readers towards the following recent perspective article [62]. Figure 3 shows four linear epitopes and their subsequent SFTI-1 grafted products examined in our group. The bioactive epitopes include OPN and LAM, two proangiogenic peptide sequences [81], HfRW, a minimal MSH-derived core sequence shown to activate mammalian melanocortin receptors [86], and CD2, the adhesion domain sequence that forms the main CD2-CD58 binding interface, modulating cell adhesion between T-cells and epithelial cells [75].
3.3. Cyclotides
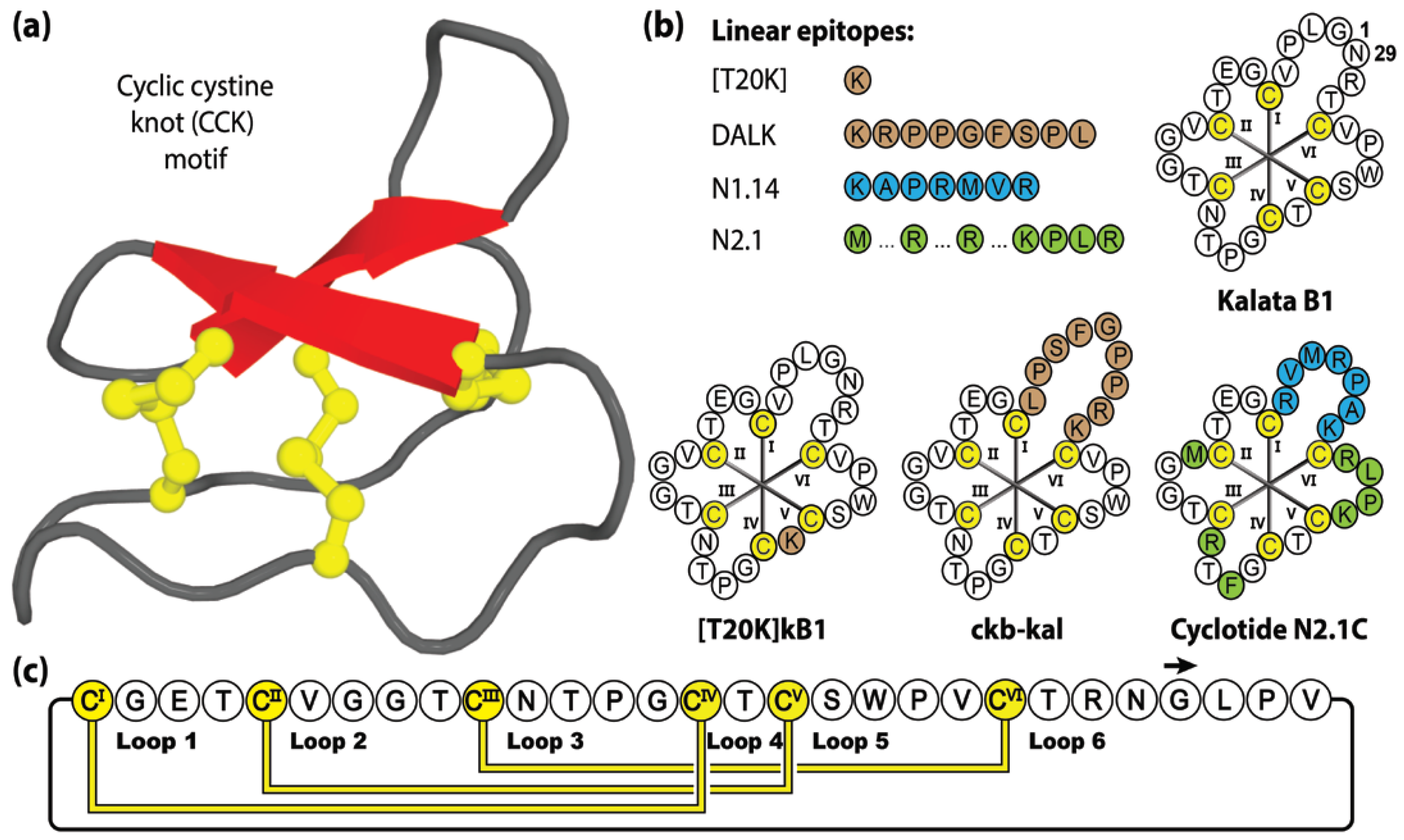
Cyclotides [87] are a large and well-studied family of macrocyclic peptides, characterized by a unique cyclic cystine knot (CCK) structural motif, comprising a head-to-tail cyclic backbone and interlocking arrangement of three disulfide bonds, that confers exceptional stability and resistance to chemical, thermal and enzymatic degradation [20]. This highly constrained structure provides an ultra-stable core that is decorated with six hypervariable backbone loops protruding between successive cysteine residues [88].
Comprising 28–37 amino acids, cyclotides have been classified into three subfamilies termed bracelet, Möbius or trypsin inhibitor cyclotides, with the prototypic or most widely studied members being cycloviolacin O1, kalata B1 and MCoTI-II, respectively [89]. To date, hundreds of cyclotides have been reported in species from five major plant families, namely Rubiaceae, Violaceae, Solanaceae, Cucurbitaceae and Fabaceae [90,91,92], or more commonly referred to as the coffee, violet, tomato, gourd, and legume families, respectively. Their sequences and structures are available on the online database CyBase www.cybase.org.au (accessed on 31 March 2023) [93]. Of these plant families, none have been exhaustively screened yet, but conservative estimates suggest that more than 50,000 native cyclotide variants await discovery [91,94].
This remarkable number of cyclotide variants is the result of the high sequence diversity present within the intra-cysteine backbone loops of the cyclotide framework, acting as a natural combinatory template (with >10 million sequence combinations estimated) [95]. Native cyclotides exhibit an extensive array of biological activities, including anti-HIV [96,97,98,99,100,101,102,103,104], anti-influenza [105], anti-microbial [106,107,108,109], anti-parasitic [52], uterotonic [110,111], anti-cancer [112,113,114,115,116] and immunosuppression activities [117,118,119,120]. With exceptional stability, tolerance to residue substitution and accessibility by chemical [121], biological [122] or plant-based [123] production methods, it is no surprise that cyclotides have been a popular drug design framework [88,124]. Figure 4 shows four linear epitopes and their subsequent grafted peptide sequences (based on the kalata B1 framework) that have been studied in our laboratory. The sequence inclusions include T20K, a single residue substitution that promotes immunomodulatory properties [125], DALK, a bradykinin B1 receptor antagonist [126] linked to chronic pain and inflammation, and N1.14 and N2.1, neurophilin-1 and -2 agonists sequences attained through two sequential generations of bacterial display libraries [127].
4. Animal-Derived Cyclic Peptides
4.1. θ-Defensins
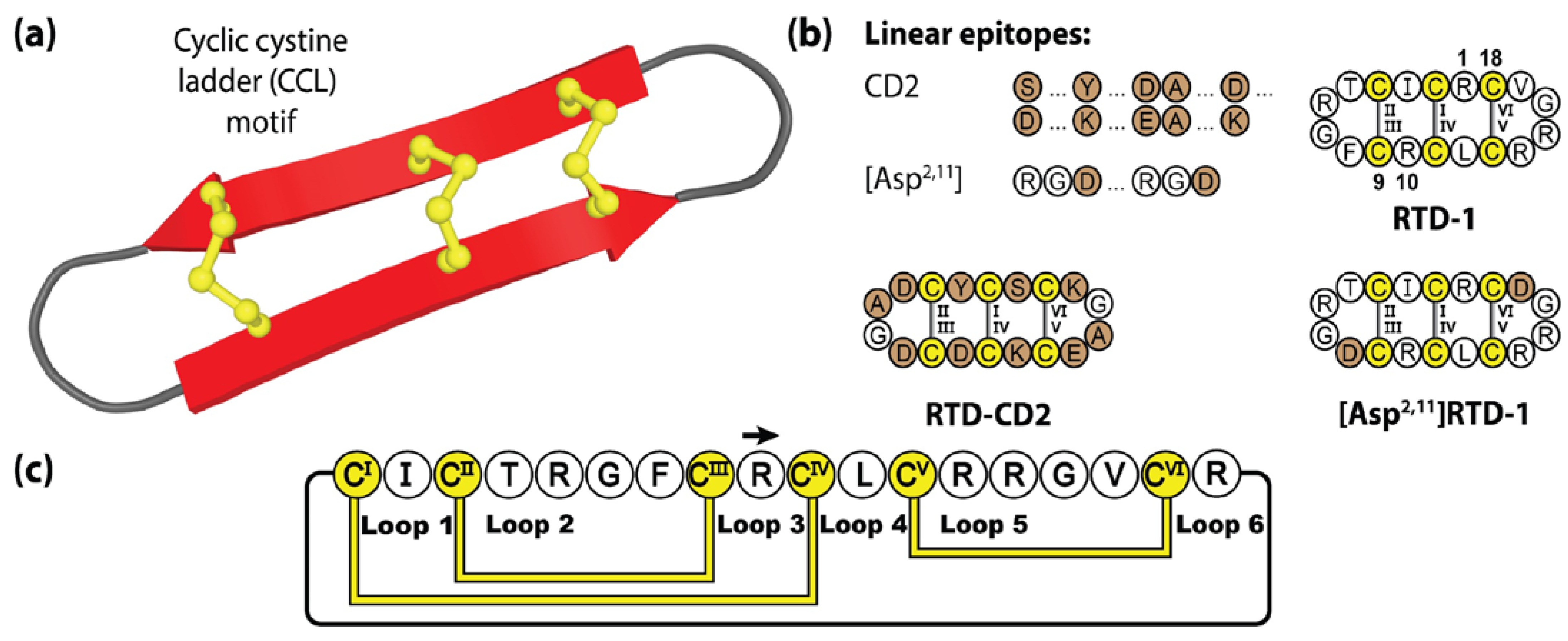
θ-defensins are a family of cysteine-rich peptides associated with the immune system in several species of primates [128,129]. Produced within leukocytes, θ-defensins are characterized by a cyclic cystine ladder (CCL) motif, comprising a head-to-tail cyclic peptide backbone and three disulfide bonds arranged in parallel [130]. This CCL motif creates a highly constrained system, with two adjoining anti-parallel β-strands, conferring excellent thermal and enzymatic stability [131]. In nature, θ-defensins are generated from the splicing of precursor gene products, truncated nonapeptide α-defensin genes termed demi-defensins, that are processed to form a backbone cyclic 18-amino acid product [132]. To date, θ-defensins are the only cyclic peptides native to animals, with six isolated from rhesus monkeys (RTD-1 to RTD-6) and five from baboon species (BTD-1 to BTD-5) [133]. Acting as immunomodulators within the innate immune system, θ-defensins suppress the production of proinflammatory cytokines [134]. The prototypic θ-defensin, RTD-1, contains 18 amino acids and demonstrates broad anti-microbial [128,135,136,137], anti-inflammatory [135], anti-fungal [128,138] and anti-viral activity [128,139]. In particular, RTD-1 has been shown to act as a prophylactic anti-viral in a mouse model of severe acute respiratory syndrome coronavirus (SARS-CoV) lung disease [139], with promising in silico investigations suggesting its utility as a COVID-19 treatment through furin inhibition [140].
Although not as widely utilized as the PDP or cyclotide frameworks, θ-defensins offer a number of therapeutically attractive features, including excellent resistance to protease digestion in biological fluids, minimal immunogenicity [135,141] and low haemolytic and cytotoxic activity [142]. Figure 5 illustrates two grafted θ-defensin scaffolds that have been used to host different bioactive epitopes, namely the CD2 binding domain [75] and the widely known RGD (Arg-Gly-Asp) integrin binding sequence, the latter readily incorporated into [Asp2,11]RTD-1 given the pre-existing native RG sequence present [143].
4.2. Retrocyclins
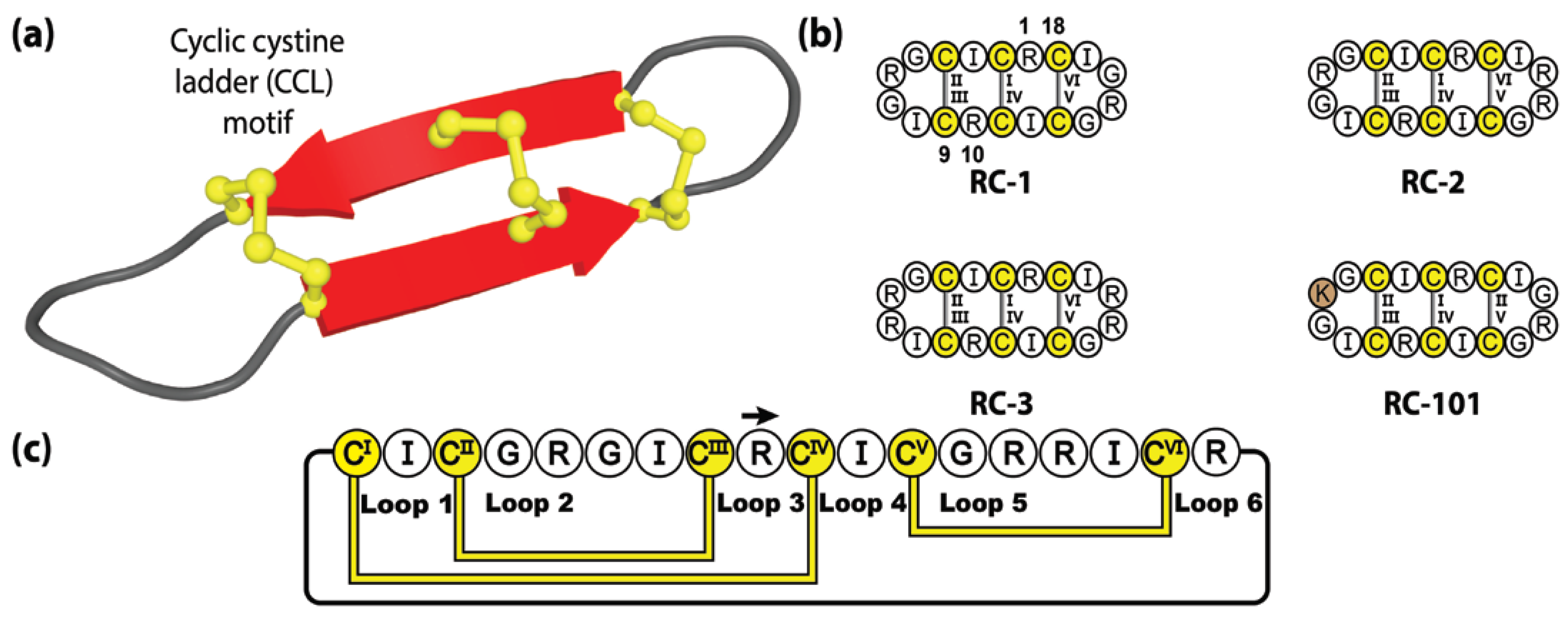
Despite their presence in several of our evolutionary cousins, including baboons, bonobos and macaques, humans do not produce θ-defensin peptides, even though our genome encodes for θ-defensin-like sequences [144]. Although these pseudogenes are transcribed to mRNA, they are not translated due to a premature stop codon upstream of the pro-peptide segment [129]. The putative coding regions of human θ-defensins have remained intact over the 7 million years of evolution from our primate cousins, with 89.4% sequence identity with the rhesus θ-defensin genes [145]. Accordingly, they are available as blueprints, and chemists have been able to synthetically resuscitate these genetically encoded sequences [145].
With the human genome encoding two distinct retrocyclin genes, three theoretical combinations of native retrocyclin peptides exist, with two homodimers, retrocyclin-1 (RC-1) and retrocyclin-3 (RC-3), and a heterodimer (RC-2) (see Figure 6). These native, although not naturally synthesized, cyclic peptides form an alternative sub-category of θ-defensin molecules called retrocyclins and exhibit exceptional activity against HIV-1 and bacterial agents [142]. Although retrocyclins have yet to be fully utilized as molecular frameworks, modest sequence mutagenesis experiments have been performed, generating several interesting retrocyclin variants, of which, RC-101 has garnered the most attention. Demonstrating significantly more potent activity against primary HIV type-1 isolates compared to RC-1, despite its sequence varying by a single amino acid substitution (Arg-to-Lys) [146]. Furthermore, RC-101 has been shown to destabilize SARS-CoV-2 Spike protein, inhibiting Spike-mediated membrane fusion and Spike/ACE2 interaction [147], thereby demonstrating the potential of retrocyclins as therapeutics tools in the development of new topical anti-viral drugs, for the treatment of HIV and COVID.
5. Engineered Cyclic Peptides
Naturally occurring peptides have benefited from millions of years of evolution during which their activity and stability have been stringently optimized, serendipitously endowing these native compounds with properties that are suitable for pharmaceutical exploitation. However, as mentioned earlier in this article, many native peptides are limited by one aspect that reflects their endogenous heritage, i.e., short biological half-lives when exogenously delivered. Applying the lessons learnt from studying naturally occurring cyclic scaffolds, an area of increasing interest is the re-engineering of acyclic bioactive peptides. By manufacturing engineered cyclic peptides, researchers have developed approaches to exploit the potent bioactivity of native acyclic compounds, further enhancing the pharmacological scope of these natural products, allowing them to hit targets which were not accessible in their natural environment. In this section, we outline the broad characteristics of such engineered cyclic scaffolds, all based upon animal-derived peptides, and describe the grafting strategy that was utilized in each case. The five molecular frameworks examined include, cyclic conotoxins, cyclic chlorotoxin, cyclic tachyplesins, cyclic protegrins, and cyclic gomesins.
5.1. Cyclic Conotoxins
Conotoxins are small peptide toxins, comprising 10–35 amino acids, found in the venom of marine cone snails of the Conus genus [148]. With individual species producing 50–1000 distinct variants and over 800 documented species, cone snails provide one of the highest known venom diversities, with current estimates of more than one million native conotoxins yet to be discovered [149]. Conotoxins adopt a wide range of compact structures, including motifs such as α-helices, β-sheets and β-turns, stabilized by multiple disulfide bridges and post-translational modifications [150]. These bioactive protein-like structures target a range of ion channels, receptors and transporters found throughout the nervous systems with high potency and exquisite selectivity [151]. Unsurprisingly, this extensive source of bioactive compounds has attracted considerable attention, with one such peptide (ω-conotoxin, MVIIA) being an FDA approved product and several native conotoxins having undergone clinical trials [152,153]. However, due to their natural biophysical properties, these acyclic peptides have been typically hampered by poor drug-like qualities. In the case of MVIIA the poor drug-like properties are overcome via an intrathecal delivery route.
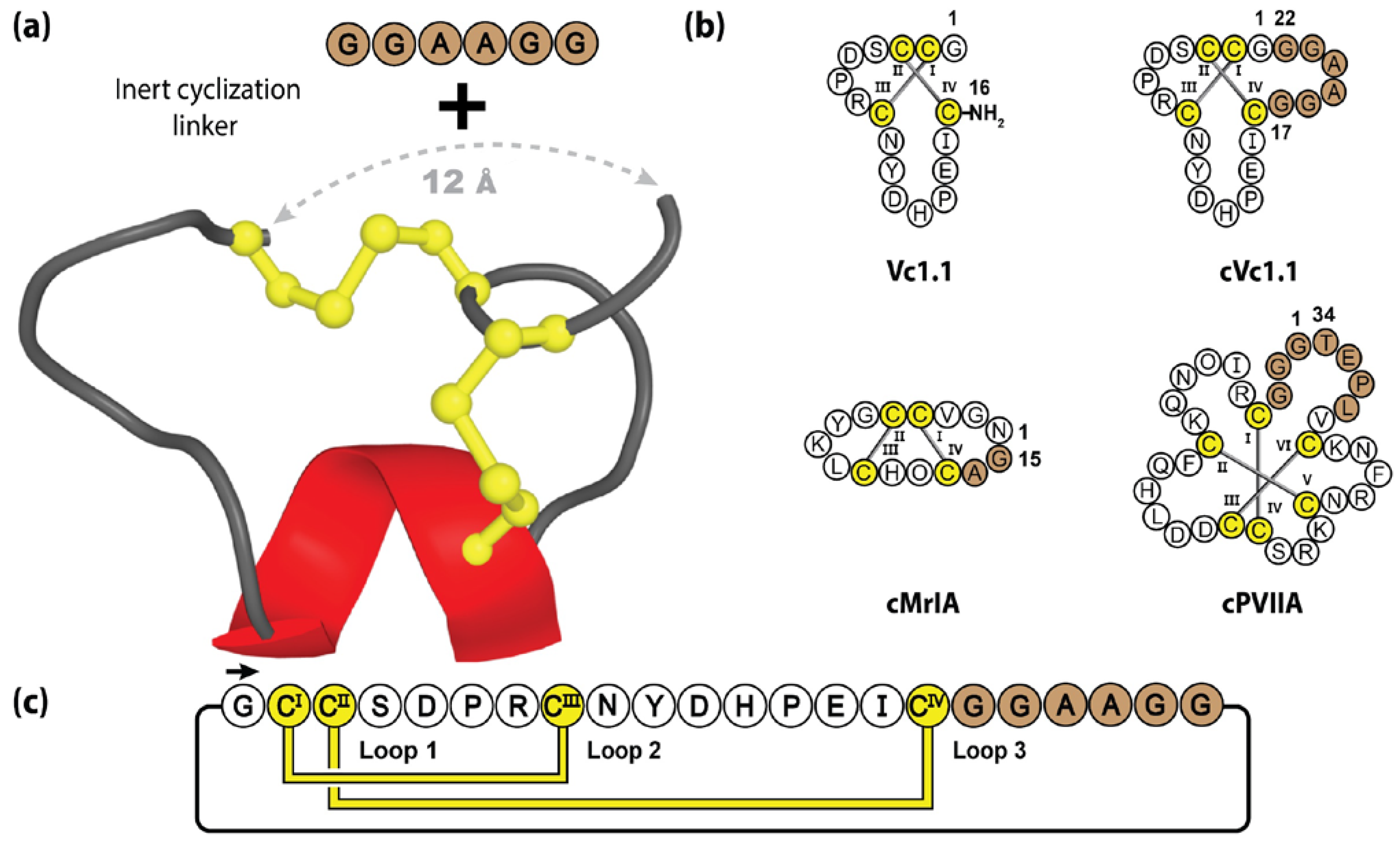
One example studied extensively in our laboratory is Vc1.1, an α-conotoxin, comprising 16 amino acids, that forms a small α-helix and two disulfide bridges [154]. Initially discovered during the PCR screening of cDNAs isolated from the venom ducts of Conus victoriae, Vc1.1 was originally characterized by its ability to inhibit nicotinic acetylcholine receptors and attracted considerable therapeutic interest for its potent analgesic activity [155,156]. However, as is the case with many nature-derived peptide molecules, synthetic Vc1.1’s utility was initially limited by its lack of oral activity [156]. To confer greater biological stability and drug-like qualities, molecular engineering was performed to manufacture a cyclic Vc1.1 (cVc1.1) variant, through the incorporation of a six-residue linker, spanning the 12 Å distance between the N- and C-termini (see Figure 7). Contrary to standard molecular grafting, the incorporated epitope linker was not bioactive but rather contained inert Gly and Ala residues [120]. The backbone cyclization resulted in significant pharmacological improvements, with increased intestinal fluid and serum stability, and oral activity in a rat pain model [157,158]—with MALDI imaging revealing cVc1.1 in the GI tract for >4 h post-oral dosing [159]. Furthermore, when tested in a rat CCI-model of neuropathic pain, cVc1.1 induced 120 times more potent analgesia than gabapentin, which is the gold standard for neuropathic pain [160]. It should be noted that successful cyclization was reliant upon the use of an appropriate linker length, as too short or long sequences can disrupt the native conformation and eliminate biological activity [160].
5.2. Cyclic Chlorotoxin
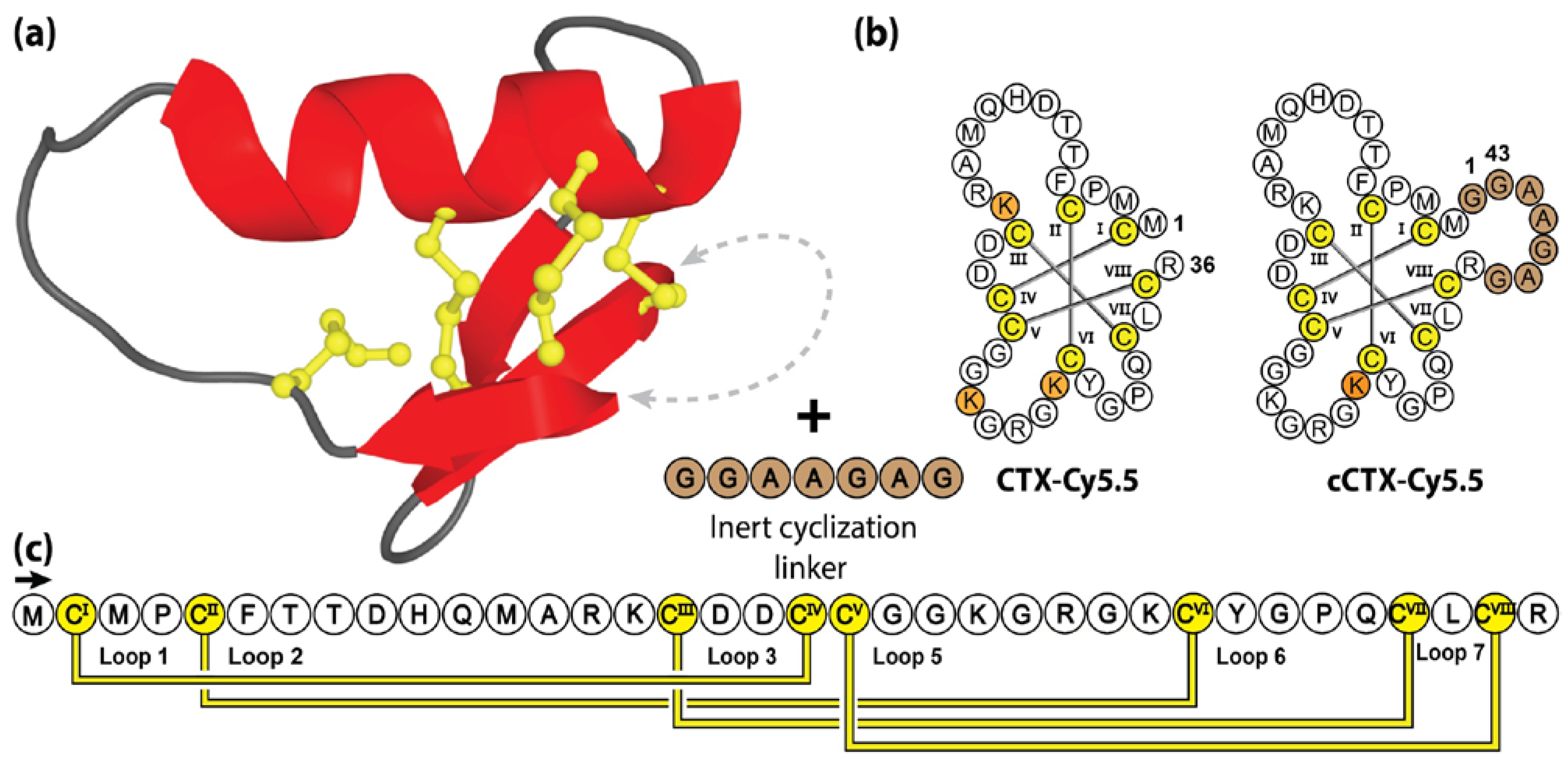
Chlorotoxin (CTX) is a 36 amino acid disulfide-rich peptide isolated from the venom of the deathstalker scorpion Leiurus quinquestriatus [161]. Adopting a compact knotted topology, chlorotoxin is characterized by four disulfide bonds, three small anti-parallel β-strands and a single α-helix [162]. Chlorotoxin has garnered significant therapeutic interest due to its ability to preferentially bind cancer cells, putatively mediated by selective interaction with matrix metallopreoteinase-2 isoforms, which are upregulated in gliomas and other solid tumors [163]. Nonetheless, the efficacy of chlorotoxin in binding cancer cells is clear and a number of articles have been published describing the potential of chlorotoxin as an imaging agent, as well as a platform for targeted cancer treatments [162,164,165,166,167].
One such example is “Tumor paint”, a bio-conjugate that combines the targeted tumor-binding properties of chlorotoxin, with a near infrared fluorescent dye to visualize tumors in real-time during surgery [168,169,170]. In early studies, this compound was manufactured by conjugating Cyanine5.5 (Cy5.5) dye with native lysine residues (Lys15, Lys23 or Lys27). Utilizing three molar equivalents of NHS-ester modified Cy5.5 dye, this approach was inherently non-specific, resulting in a mixture of mono-, di- and tri-labelled peptides [162]. Interestingly, the bio-conjugation resulted in 75–85% mono-labelled Lys27, along with small amounts of Lys15 and Lys23 labelled chlorotoxin [171]. With mono-labelled compounds preferred for FDA approval and commercialization, subsequent studies substituting Lys15 and Lys23 with alanine or arginine residues were conducted to obtain Lys27 mono-labelled chlorotoxin [171].
Surprisingly, that study also showed that mono-labelling was also possible through the incorporation of a 7-residue linker between the free N- and C-termini. As illustrated in Figure 8, without altering the native lysine residues, Cy5.5 conjugation using the engineered cyclic chlorotoxin variant (cCTX), resulted in a homogenous product with only Lys27 labelled. Unfortunately, despite showing limited improvements in ex vivo serum stability (70% to 90% intact peptide following 24 h incubation in human plasma), cCTX-Cy5.5 demonstrated reduced in vivo serum half-life compared to the linear CTX-Cy5.5 variant (τ1/2 = 11 h and 14 h, respectively) [171].
5.3. Cyclic Tachyplesins
Tachyplesins are a family of six host-defense peptides (TI to TVI) isolated from different species of horseshoe crab [172]. These cationic agents have broad anti-microbial properties, with reported activity against Gram-positive and Gram-negative bacteria, fungi, and cancer cells [173]. Similar to other host-defense peptides, tachyplesins exhibit an amphipathic secondary structure, with positively charged and hydrophobic residues segregated into distinct clusters [173]. These distinct regions allow for preferential binding to the anionic surfaces of microbes or cancer cells, followed by membrane insertion and subsequent cell death [174,175].
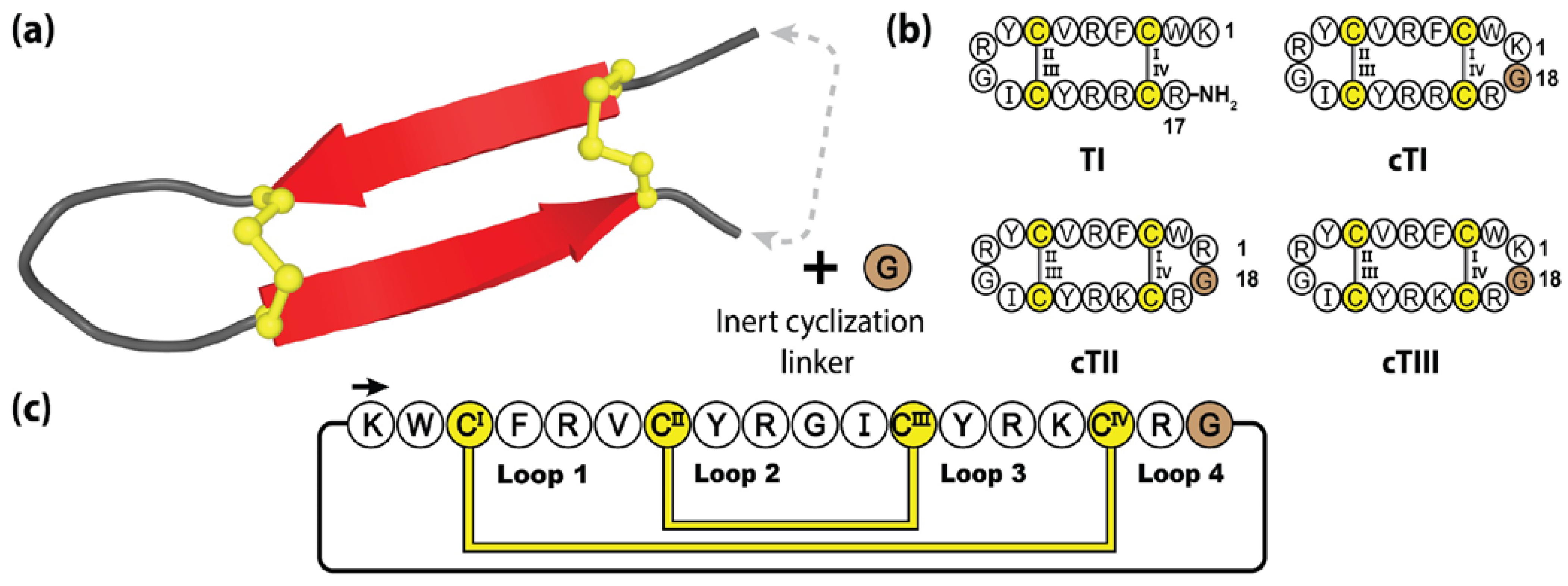
Comprising 17-residues and a α-amidated C-terminus, tachyplesins are organized in a β-hairpin structure, constrained by two disulfide bonds [176]. Interestingly, this disulfide connectivity positions the N- and C-termini in close proximity, making the tachyplesin family readily amendable to engineered backbone cyclization [177]. Figure 9 highlights the method followed by Vernen et al. to manufacture three cyclic tachyplesin variants (cTI, cTII and cTIII), through the insertion of a linker comprising a single glycine residue [173]. This small epitope inclusion simultaneously bridged the termini gap and produced a symmetrical structure with 18 amino acids, closely resembling that of the θ-defensins. Following cyclization, these engineered variants displayed promising pharmacological improvements with respect to serum stability and reduced red blood cell toxicity, whilst maintaining potent anti-cancer and anti-microbial activity [173].
5.4. Cyclic Protegrins
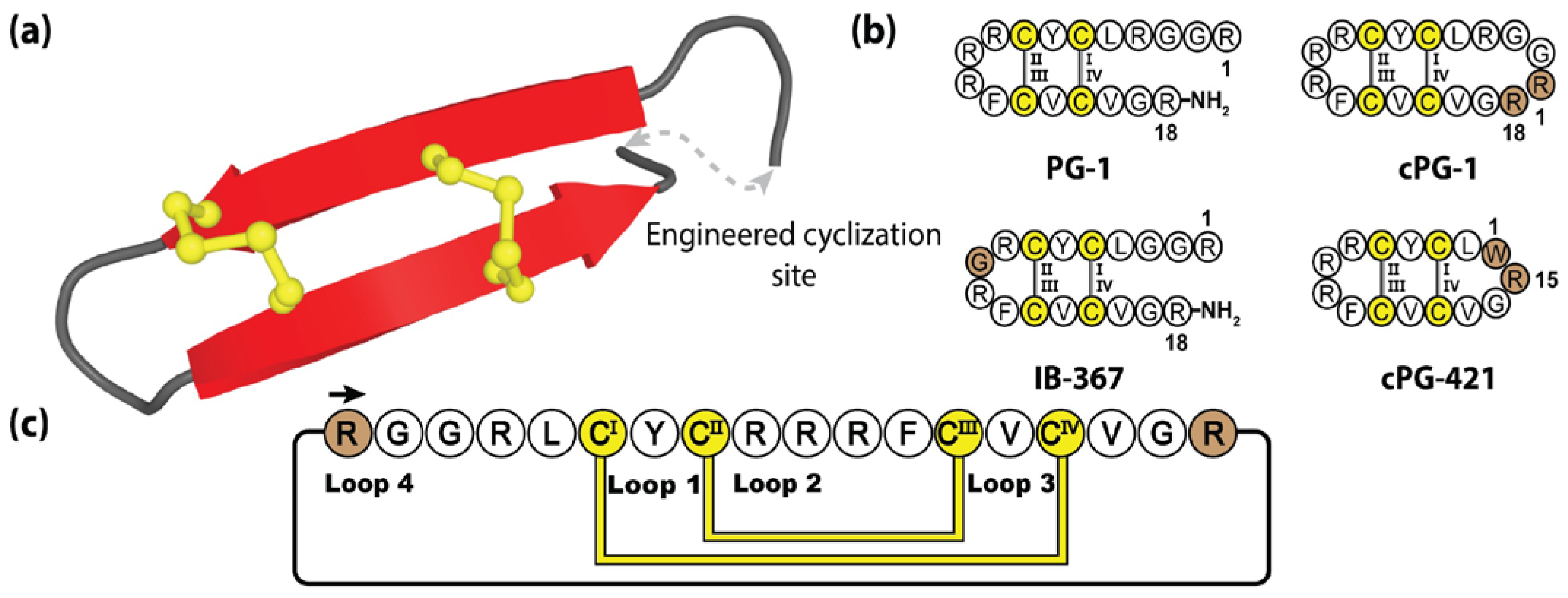
Protegrins are a family of secreted anti-microbial peptides found in porcine leukocytes, involved in defending various tissues from infection [178]. To date, five protegrins have been identified (PG-1 to PG-5), with reported activities against bacteria, fungi, and some envelope viruses [179]. Comprising 16–18 amino acids, protegrins bear a striking resemblance to tachyplesins, adopting two anti-parallel β-strands stabilized by two cystine bridges [180]. Furthermore, they demonstrate a similar mode of action, with the cationic and amphipathic protegrin composition performing targeted membrane disruption, via induced pore formation and subsequent cell death [181].
Protegrins and their synthetic congeners have demonstrated considerable pharmacological potential with IB-367, a truncated protegrin derivative, having undergone phase III clinical trials under the name Iseganan, as a topical antibiotic treatment. Although, ultimately these trials were terminated or had remained in limbo for the past 10 years [182,183,184]. Figure 10 illustrates the prototypical protegrin PG-1 and three non-native variants, two of which are re-engineered cyclic peptides generated through direct N-to-C cyclization. The cyclization loops of both cPG-1 and cPG-421 (also referred to as IB-200 and IB-421), containing 7 and 10 amino acids, respectively, displayed minimal structural perturbance as a result of this engineered junction [185,186]. The biological activity of these variants was mildly improved over their linear counterparts, along with the cyclic peptide analogues demonstrating improved resistance to enzymatic degradation [186].
5.5. Cyclic Gomesins
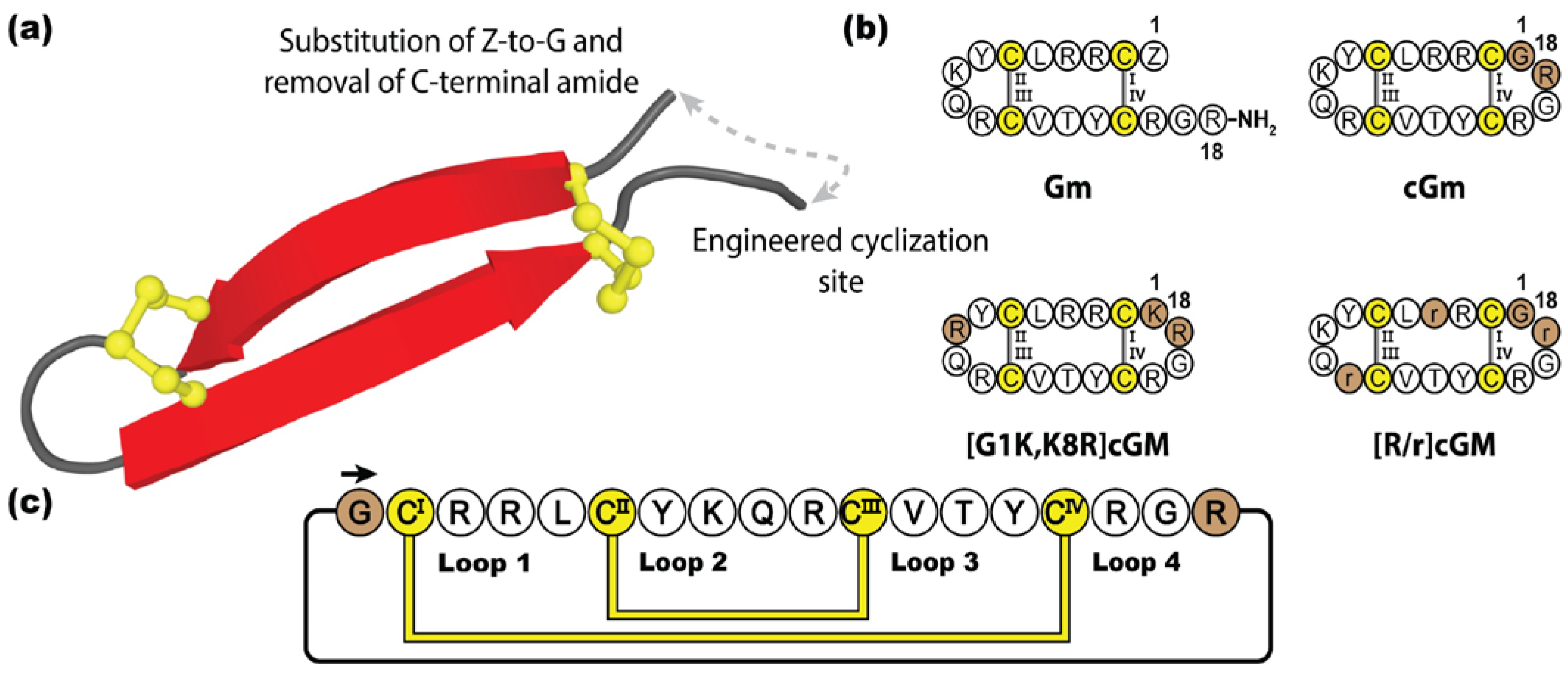
Gomesin (Gm) is an antimicrobial peptide isolated from the haemocytes of Brazilian tarantula Acanthoscurria gomesiana [187]. Initially identified for its role in the innate immune system fighting infection [188], gomesin has since been reported to have a myriad of therapeutic properties, such as cytotoxic activity against bacteria, fungi, parasites, and cancer cells [187,189,190,191]. Although not as potent, gomesin is related to the tachyplesin family, sharing considerable sequence and structural homology, adopting a β-hairpin conformation stabilized by a disulfide bond at each end [192]. Containing 18 amino acids, gomesin is highly cationic and amphipathic, although to a lesser degree than tachyplesin and protegrin, with post-translationally protected termini, including an N-terminal pyroglutamic acid and C-terminal amide [193,194]. In contrast to many other anti-microbial peptides, gomesin causes substantially lower levels of haemolysis, and hence is highly valued in the development of treatments for microbial infections and cancer [189]. Several studies have examined the re-engineering of gomesin for improved stability and bioactivity [195], with backbone cyclization shown to not only improve the in vitro stability of cyclic gomesin (cGm), but also to enhance cytotoxic activity against cancer cells lines, whilst maintaining its native fold [196].
Figure 11 illustrates how Chan et al. engineered cGm by substituting the original pyroglutamic acid for a glycine residue and introducing a cyclic backbone junction [196]. Following this study, several cyclic gomesin variants have subsequently been designed and synthesized with various pharmacological improvements. In particular, [G1K,K8R]cGm has demonstrated promising therapeutic potential, boasting ten times more potent anti-bacterial activity than gomesin or cGM, against a range of Gram-positive and Gram-negative bacteria (including Staphylococcus aureus and Escherichia coli, respectively), with no increase in haemolytic activity [197]. Additionally, [G1K,K8R]cGm has also demonstrated potent activity against S. aureus biofilms, being able to kill embedded bacterial cells in a concentration-dependent manner [198]. In an alternative approach, instead of focusing on the innate potent bioactivity of cGm, Benfield et al. developed a non-disruptive, non-toxic cGm analogue, [R/r]cGm, by substituting D-amino acids within the scaffold, creating a targeted delivery mechanism of therapeutic cargos into cancer cells, without compromising healthy cells [199].
6. Conclusions
This article highlights that naturally occurring cyclic peptides from plants and animals are stable scaffolds that have a wide range of potential applications in drug design. These naturally occurring cyclic peptides appear to have evolved to become highly resistant to proteases, and accordingly have further inspired chemists in the artificial cyclization of natural peptides to improve their properties. While we have focused on peptides that have been studied in our laboratory in recent years, there are many other examples in the literature of natural peptides as sources of inspiration in drug design.
Author Contributions
Conceptualization, T.J.T. and D.J.C.; methodology, T.J.T.; software, T.J.T.; validation, T.J.T.; formal analysis, T.J.T.; investigation, T.J.T.; resources, T.D. and D.J.C.; data curation, T.J.T.; writing—original draft preparation, T.J.T.; writing—review and editing, T.J.T., T.D. and D.J.C.; visualization, T.J.T.; supervision, T.D. and D.J.C.; project administration, T.D. and D.J.C.; funding acquisition, T.D. and D.J.C. All authors have read and agreed to the published version of the manuscript.
Funding
We thank the many colleagues cited in the references for their valuable contributions to the field of natural product peptide therapeutics. Work in our laboratory is supported by the Australian Research Council (ARC) Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012) and an ARC Discovery grant (DP200101299). DJC is supported by an NHMRC grant 2009564.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Robyn Craik for manuscript proofreading.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Craik, D.J.; Kan, M.W. How can we improve peptide drug discovery? Learning from the past. Expert Opin. Drug Discov. 2021, 16, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Philippe, G.J.B.; Craik, D.J.; Henriques, S.T. Converting peptides into drugs targeting intracellular protein-protein interactions. Drug Discov. Today 2021, 26, 1521–1531. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Tapeinou, A.; Matsoukas, M.T.; Simal, C.; Tselios, T. Review cyclic peptides on a merry-go-round; towards drug design. Biopolymers 2015, 104, 453–461. [Google Scholar] [CrossRef]
- Wang, W.; Khojasteh, S.C.; Su, D. Biosynthetic Strategies for Macrocyclic Peptides. Molecules 2021, 26, 3338. [Google Scholar] [CrossRef]
- McHugh, S.M.; Rogers, J.R.; Yu, H.; Lin, Y.-S. Insights into how cyclic peptides switch conformations. J. Chem. Theory Comput. 2016, 12, 2480–2488. [Google Scholar] [CrossRef]
- Abdalla, M.A.; McGaw, L.J. Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef] [Green Version]
- Räder, A.F.B.; Reichart, F.; Weinmüller, M.; Kessler, H. Improving oral bioavailability of cyclic peptides by N-methylation. Bioorgan. Med. Chem. 2018, 26, 2766–2773. [Google Scholar] [CrossRef]
- Chow, H.Y.; Zhang, Y.; Matheson, E.; Li, X. Ligation technologies for the synthesis of cyclic peptides. Chem. Rev. 2019, 119, 9971–10001. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef]
- Chia, L.Y.; Kumar, P.V.; Maki, M.A.A.; Ravichandran, G.; Thilagar, S. A Review: The Antiviral Activity of Cyclic Peptides. Int. J. Pept. Res. Ther. 2023, 29, 7. [Google Scholar] [CrossRef]
- Ramadhani, D.; Maharani, R.; Gazzali, A.M.; Muchtaridi, M. Cyclic Peptides for the Treatment of Cancers: A Review. Molecules 2022, 27, 4428. [Google Scholar] [CrossRef]
- Lai, S.; Zhang, Q.; Jin, L. Natural and Man-Made Cyclic Peptide-Based Antibiotics. Antibiotics 2022, 12, 42. [Google Scholar] [CrossRef]
- Barkan, D.T.; Cheng, X.L.; Celino, H.; Tran, T.T.; Bhandari, A.; Craik, C.S.; Sali, A.; Smythe, M.L. Clustering of disulfide-rich peptides provides scaffolds for hit discovery by phage display: Application to interleukin-23. BMC Bioinform. 2016, 17, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef]
- Fass, D. Disulfide bonding in protein biophysics. Annu. Rev. Biophys. 2012, 41, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Manteca, A.; Alonso-Caballero, A.; Fertin, M.; Poly, S.; De Sancho, D.; Perez-Jimenez, R. The influence of disulfide bonds on the mechanical stability of proteins is context dependent. J. Biol. Chem. 2017, 292, 13374–13380. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hu, S.; Jian, W.; Xie, C.; Yang, X. Plant antimicrobial peptides: Structures, functions, and applications. Bot. Stud. 2021, 62, 5. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ding, J.; Liao, C.; Xu, J.; Liu, X.; Lu, W. Defensins: The natural peptide antibiotic. Adv. Drug Deliv. Rev. 2021, 179, 114008. [Google Scholar] [CrossRef] [PubMed]
- Heilbronner, S.; Krismer, B.; Brotz-Oesterhelt, H.; Peschel, A. The microbiome-shaping roles of bacteriocins. Nat. Rev. Microbiol. 2021, 19, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Cristofori-Armstrong, B.; Israel, M.R.; Nixon, S.A.; Vetter, I.; King, G.F. Animal toxins—Nature’s evolutionary-refined toolkit for basic research and drug discovery. Biochem. Pharmacol. 2020, 181, 114096. [Google Scholar] [CrossRef]
- Morrison, C. Constrained peptides’ time to shine? Nat. Rev. Drug Discov. 2018, 17, 531–533. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.; Zhang, W.D.; Hu, H.G. Stapled Helical Peptides Bearing Different Anchoring Residues. Chem. Rev. 2020, 120, 10079–10144. [Google Scholar] [CrossRef]
- Bluntzer, M.T.J.; O’Connell, J.; Baker, T.S.; Michel, J.; Hulme, A.N. Designing stapled peptides to inhibitprotein-protein interactions: An analysis of successes in a rapidly changing field. Pept. Sci. 2021, 113, e24191. [Google Scholar] [CrossRef]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Domling, A. Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef]
- Moiola, M.; Memeo, M.G.; Quadrelli, P. Stapled Peptides—A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24, 3654. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.K.; Craik, D.J. Linking molecular evolution to molecular grafting. J. Biol. Chem. 2021, 296, 100425. [Google Scholar] [CrossRef]
- Kashmiri, S.V.; De Pascalis, R.; Gonzales, N.R.; Schlom, J. SDR grafting—A new approach to antibody humanization. Methods 2005, 36, 25–34. [Google Scholar] [CrossRef]
- Jones, N.C. Gene expression: Negative regulation of enhancers. Nature 1986, 321, 202–223. [Google Scholar] [CrossRef]
- Wang, C.K.; Craik, D.J. Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat. Chem. Biol. 2018, 14, 417–427. [Google Scholar] [CrossRef]
- Craik, D.J.; Lee, M.-H.; Rehm, F.B.H.; Tombling, B.; Doffek, B.; Peacock, H. Ribosomally-synthesised cyclic peptides from plants as drug leads and pharmaceutical scaffolds. Bioorganic Med. Chem. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Ji, Y.; Majumder, S.; Millard, M.; Borra, R.; Bi, T.; Elnagar, A.Y.; Neamati, N.; Shekhtman, A.; Camarero, J.A. In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide. J. Am. Chem. Soc. 2013, 135, 11623–11633. [Google Scholar] [CrossRef] [Green Version]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef] [Green Version]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef]
- Rosal, R.; Brandt-Rauf, P.; Pincus, M.R.; Wang, H.; Mao, Y.; Li, Y.; Fine, R.L. The role of alpha-helical structure in p53 peptides as a determinant for their mechanism of cell death: Necrosis versus apoptosis. Adv. Drug Deliv. Rev. 2005, 57, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.; Elnagar, A.Y.O.; Hamm-Alvarez, S.F.; Camarero, J.A. Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J. Control. Release 2011, 155, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Pazgier, M.; Liu, M.; Lu, W.Y.; Lu, W. Apamin as a template for structure-based rational design of potent peptide activators of p53. Angew. Chem. Int. Ed. Engl. 2009, 48, 8712–8715. [Google Scholar] [CrossRef] [PubMed]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef]
- Ramalho, S.D.; Wang, C.K.; King, G.J.; Byriel, K.A.; Huang, Y.; Bolzani, V.S.; Craik, D.J. Synthesis, racemic X-ray crystallographic, and permeability studies of bioactive orbitides from jatropha species. J. Nat. Prod. 2018, 81, 2436–2445. [Google Scholar] [CrossRef]
- Shim, Y.Y.; Young, L.W.; Arnison, P.G.; Gilding, E.; Reaney, M.J. Proposed systematic nomenclature for orbitides. J. Nat. Prod. 2015, 78, 645–652. [Google Scholar] [CrossRef]
- Ramalho, S.D.; Pinto, M.E.F.; Ferreira, D.; Bolzani, V.S. Biologically active orbitides from the euphorbiaceae family. Planta Med. 2018, 84, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Chekan, J.R.; Estrada, P.; Covello, P.S.; Nair, S.K. Characterization of the macrocyclase involved in the biosynthesis of RiPP cyclic peptides in plants. Proc. Natl. Acad. Sci. USA 2017, 114, 6551–6556. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, S.C.; Nobre, T.M.; Volpati, D.; Ciancaglini, P.; Cilli, E.M.; Lorenzon, E.N.; Oliveira, O.N., Jr. The importance of cyclic structure for labaditin on its antimicrobial activity against Staphylococcus aureus. Colloids Surf. B Biointerfaces 2016, 148, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Okinyo-Owiti, D.P.; Dong, Q.; Ling, B.; Jadhav, P.D.; Bauer, R.; Maley, J.M.; Reaney, M.J.T.; Yang, J.; Sammynaiken, R. Evaluating the cytotoxicity of flaxseed orbitides for potential cancer treatment. Toxicol. Rep. 2015, 2, 1014–1018. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.E.; Batista, J.M., Jr.; Koehbach, J.; Gaur, P.; Sharma, A.; Nakabashi, M.; Cilli, E.M.; Giesel, G.M.; Verli, H.; Gruber, C.W.; et al. Ribifolin, an orbitide from Jatropha ribifolia, and its potential antimalarial activity. J. Nat. Prod. 2015, 78, 374–380. [Google Scholar] [CrossRef]
- Altei, W.F.; Picchi, D.G.; Abissi, B.M.; Giesel, G.M.; Flausino, O., Jr.; Reboud-Ravaux, M.; Verli, H.; Crusca, E., Jr.; Silveira, E.R.; Cilli, E.M.; et al. Jatrophidin I, a cyclic peptide from Brazilian Jatropha curcas L.: Isolation, characterization, conformational studies and biological activity. Phytochemistry 2014, 107, 91–96. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Cebrat, M.; Siemion, I.Z.; Kay, J.E. Cyclolinopeptide A (CLA) mediates its immunosuppressive activity through cyclophilin-dependent calcineurin inactivation. FEBS Lett. 1997, 418, 224–227. [Google Scholar] [CrossRef] [Green Version]
- Thell, K.; Hellinger, R.; Schabbauer, G.; Gruber, C.W. Immunosuppressive peptides and their therapeutic applications. Drug Discov. Today 2014, 19, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Katarzyńska, J.; Mazur, A.; Rudzinska, E.; Artym, J.; Zimecki, M.; Jankowski, S.; Zabrocki, J. Cyclolinopeptide derivatives modify methotrexate-induced suppression of the humoral immune response in mice. Eur. J. Med. Chem. 2011, 46, 4608–4617. [Google Scholar] [CrossRef]
- Morita, H.; Eda, M.; Iizuka, T.; Hirasawa, Y.; Sekiguchi, M.; Yun, Y.S.; Itokawa, H.; Takeya, K. Structure of a new cyclic nonapeptide, segetalin F, and vasorelaxant activity of segetalins from Vaccaria segetalis. Bioorganic Med. Chem. Lett. 2006, 16, 4458–4461. [Google Scholar] [CrossRef]
- Shim, Y.Y.; Song, Z.L.; Jadhav, P.D.; Reaney, M.J.T. Orbitides from flaxseed (Linum usitatissimum L.): A comprehensive review. Trends Food Sci. Technol. 2019, 93, 197–211. [Google Scholar] [CrossRef]
- Franke, B.; Mylne, J.S.; Rosengren, K.J. Buried treasure: Biosynthesis, structures and applications of cyclic peptides hidden in seed storage albumins. Nat. Prod. Rep. 2018, 35, 137–146. [Google Scholar] [CrossRef]
- Mylne, J.S.; Colgrave, M.L.; Daly, N.L.; Chanson, A.H.; Elliott, A.G.; McCallum, E.J.; Jones, A.; Craik, D.J. Albumins and their processing machinery are hijacked for cyclic peptides in sunflower. Nat. Chem. Biol. 2011, 7, 257–259. [Google Scholar] [CrossRef]
- Payne, C.D.; Franke, B.; Fisher, M.F.; Hajiaghaalipour, F.; McAleese, C.E.; Song, A.; Eliasson, C.; Zhang, J.; Jayasena, A.S.; Vadlamani, G.; et al. A chameleonic macrocyclic peptide with drug delivery applications. Chem. Sci. 2021, 12, 6670–6683. [Google Scholar] [CrossRef] [PubMed]
- de Veer, S.J.; White, A.M.; Craik, D.J. Sunflower Trypsin Inhibitor-1 (SFTI-1): Sowing Seeds in the Fields of Chemistry and Biology. Angew. Chem. Int. Ed. Engl. 2021, 60, 8050–8071. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.G.; Franke, B.; Armstrong, D.A.; Craik, D.J.; Mylne, J.S.; Rosengren, K.J. Natural structural diversity within a conserved cyclic peptide scaffold. Amino Acids 2017, 49, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Luckett, S.; Garcia, R.S.; Barker, J.J.; Konarev, A.V.; Shewry, P.R.; Clarke, A.R.; Brady, R.L. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J. Mol. Biol. 1999, 290, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Qi, R.F.; Song, Z.W.; Chi, C.W. Structural features and molecular evolution of Bowman-Birk protease inhibitors and their potential application. Acta Biochim. Biophys. Sin. 2005, 37, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Birk, Y. The Bowman-Birk inhibitor trypsin- and chymotrypsin-inhibitor from soybeans. Int. J. Pept. Protein Res. 1985, 25, 113–131. [Google Scholar] [CrossRef]
- Jaulent, A.M.; Leatherbarrow, R.J. Design, synthesis and analysis of novel bicyclic and bifunctional protease inhibitors. Protein Eng. Des. Sel. 2004, 17, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, M.; Qasim, M.A. What can the structures of enzyme-inhibitor complexes tell us about the structures of enzyme substrate complexes? Biochim. Biophys. Acta 2000, 1477, 324–337. [Google Scholar] [CrossRef]
- de Veer, S.J.; Swedberg, J.E.; Akcan, M.; Rosengren, K.J.; Brattsand, M.; Craik, D.J.; Harris, J.M. Engineered protease inhibitors based on sunflower trypsin inhibitor-1 (SFTI-1) provide insights into the role of sequence and conformation in Laskowski mechanism inhibition. Biochem. J. 2015, 469, 243–253. [Google Scholar] [CrossRef]
- de Veer, S.J.; Ukolova, S.S.; Munro, C.A.; Swedberg, J.E.; Buckle, A.M.; Harris, J.M. Mechanism-based selection of a potent kallikrein-related peptidase 7 inhibitor from a versatile library based on the sunflower trypsin inhibitor SFTI-1. Pept. Sci. 2013, 100, 510–518. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Chen, C.C.; He, Y.; Yu, M.; Xu, L.; Tian, C.L.; Guo, Q.X.; Shi, J.; Zhang, M.; Li, Y.M. Efficient synthesis of trypsin inhibitor SFTI-1 via intramolecular ligation of peptide hydrazide. Tetrahedron Lett. 2014, 55, 2883–2886. [Google Scholar] [CrossRef]
- Li, Y.; Aboye, T.; Breindel, L.; Shekhtman, A.; Camarero, J.A. Efficient recombinant expression of SFTI-1 in bacterial cells using intein-mediated protein trans-splicing. Biopolymers 2016, 106, 818–824. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.A.; Yap, K.; Poth, A.G.; Gilding, E.K.; Swedberg, J.E.; Poon, S.; Qu, H.; Durek, T.; Harris, K.; Anderson, M.A.; et al. Rapid and Scalable Plant-Based Production of a Potent Plasmin Inhibitor Peptide. Front. Plant Sci. 2019, 10, 602. [Google Scholar] [CrossRef]
- Northfield, S.E.; Wang, C.K.; Schroeder, C.I.; Durek, T.; Kan, M.W.; Swedberg, J.E.; Craik, D.J. Disulfide-rich macrocyclic peptides as templates in drug design. Eur. J. Med. Chem. 2014, 77, 248–257. [Google Scholar] [CrossRef]
- Sable, R.; Durek, T.; Taneja, V.; Craik, D.J.; Pallerla, S.; Gauthier, T.; Jois, S. Constrained cyclic peptides as immunomodulatory inhibitors of the CD2:CD58 protein-protein interaction. ACS Chem. Biol. 2016, 11, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Swedberg, J.E.; Li, C.Y.; de Veer, S.J.; Wang, C.K.; Craik, D.J. Design of potent and selective cathepsin g inhibitors based on the sunflower trypsin inhibitor-1 scaffold. J. Med. Chem. 2017, 60, 658–667. [Google Scholar] [CrossRef]
- Quimbar, P.; Malik, U.; Sommerhoff, C.P.; Kaas, Q.; Chan, L.Y.; Huang, Y.H.; Grundhuber, M.; Dunse, K.; Craik, D.J.; Anderson, M.A.; et al. High-affinity cyclic peptide matriptase inhibitors. J. Biol. Chem. 2013, 288, 13885–13896. [Google Scholar] [CrossRef] [Green Version]
- Fittler, H.; Avrutina, O.; Empting, M.; Kolmar, H. Potent inhibitors of human matriptase-1 based on the scaffold of sunflower trypsin inhibitor. J. Pept. Sci. 2014, 20, 415–420. [Google Scholar] [CrossRef]
- Chan, L.Y.; Craik, D.J.; Daly, N.L. Cyclic thrombospondin-1 mimetics: Grafting of a thrombospondin sequence into circular disulfide-rich frameworks to inhibit endothelial cell migration. Biosci. Rep. 2015, 35, e00270. [Google Scholar] [CrossRef]
- Jendrny, C.; Beck-Sickinger, A.G. Inhibition of kallikrein-related peptidases 7 and 5 by grafting serpin reactive-center loop sequences onto sunflower trypsin inhibitor-1 (SFTI-1). ChemBioChem 2016, 17, 719–726. [Google Scholar] [CrossRef]
- Chan, L.Y.; Gunasekera, S.; Henriques, S.T.; Worth, N.F.; Le, S.J.; Clark, R.J.; Campbell, J.H.; Craik, D.J.; Daly, N.L. Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood 2011, 118, 6709–6717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yamaguchi, S.; Nagamune, T. Sortase A-mediated synthesis of ligand-grafted cyclized peptides for modulating a model protein-protein interaction. Biotechnol. J. 2015, 10, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Northfield, S.E.; Huang, Y.H.; Ramos, M.C.; Craik, D.J. Inhibition of tau aggregation using a naturally-occurring cyclic peptide scaffold. Eur. J. Med. Chem. 2016, 109, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobos Caceres, C.; Bansal, P.S.; Navarro, S.; Wilson, D.; Don, L.; Giacomin, P.; Loukas, A.; Daly, N.L. An engineered cyclic peptide alleviates symptoms of inflammation in a murine model of inflammatory bowel disease. J. Biol. Chem. 2017, 292, 10288–10294. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Taichi, M.; Wei, N.; Yang, H.; Luo, K.Q.; Tam, J.P. An orally active bradykinin B1 receptor antagonist engineered as a bifunctional chimera of sunflower trypsin inhibitor. J. Med. Chem. 2017, 60, 504–510. [Google Scholar] [CrossRef]
- Durek, T.; Cromm, P.M.; White, A.M.; Schroeder, C.I.; Kaas, Q.; Weidmann, J.; Ahmad Fuaad, A.; Cheneval, O.; Harvey, P.J.; Daly, N.L.; et al. Development of Novel Melanocortin Receptor Agonists Based on the Cyclic Peptide Framework of Sunflower Trypsin Inhibitor-1. J. Med. Chem. 2018, 61, 3674–3684. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef]
- de Veer, S.J.; Kan, M.W.; Craik, D.J. Cyclotides: From structure to function. Chem. Rev. 2019, 119, 12375–12421. [Google Scholar] [CrossRef]
- Huang, Y.H.; Du, Q.; Craik, D.J. Cyclotides: Disulfide-rich peptide toxins in plants. Toxicon 2019, 172, 33–44. [Google Scholar] [CrossRef]
- Poth, A.G.; Colgrave, M.L.; Philip, R.; Kerenga, B.; Daly, N.L.; Anderson, M.A.; Craik, D.J. Discovery of cyclotides in the Fabaceae plant family provides new insights into the cyclization, evolution, and distribution of circular proteins. ACS Chem. Biol. 2011, 6, 345–355. [Google Scholar] [CrossRef]
- Gruber, C.W.; Elliott, A.G.; Ireland, D.C.; Delprete, P.G.; Dessein, S.; Göransson, U.; Trabi, M.; Wang, C.K.; Kinghorn, A.B.; Robbrecht, E.; et al. Distribution and evolution of circular miniproteins in flowering plants. Plant Cell 2008, 20, 2471–2483. [Google Scholar] [CrossRef] [Green Version]
- Poth, A.G.; Mylne, J.S.; Grassl, J.; Lyons, R.E.; Millar, A.H.; Colgrave, M.L.; Craik, D.J. Cyclotides associate with leaf vasculature and are the products of a novel precursor in Petunia (Solanaceae). J. Biol. Chem. 2012, 287, 27033–27046. [Google Scholar] [CrossRef] [Green Version]
- Kaas, Q.; Craik, D.J. Analysis and classification of circular proteins in CyBase. Biopolym. Pept. Sci. 2010, 94, 584–591. [Google Scholar] [CrossRef]
- Burman, R.; Gruber, C.W.; Rizzardi, K.; Herrmann, A.; Craik, D.J.; Gupta, M.P.; Göransson, U. Cyclotide proteins and precursors from the genus Gloeospermum: Filling a blank spot in the cyclotide map of Violaceae. Phytochemistry 2010, 71, 13–20. [Google Scholar] [CrossRef]
- Craik, D.J.; Cemazar, M.; Wang, C.K.; Daly, N.L. The cyclotide family of circular miniproteins: Nature’s combinatorial peptide template. Biopolymers 2006, 84, 250–266. [Google Scholar] [CrossRef]
- Wang, C.K.; Colgrave, M.L.; Gustafson, K.R.; Ireland, D.C.; Göransson, U.; Craik, D.J. Anti-HIV cyclotides from the Chinese medicinal herb Viola yedoensis. J. Nat. Prod. 2008, 71, 47–52. [Google Scholar] [CrossRef]
- Daly, N.L.; Gustafson, K.R.; Craik, D.J. The role of the cyclic peptide backbone in the anti-HIV activity of the cyclotide kalata B1. FEBS Lett. 2004, 574, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Daly, N.L.; Clark, R.J.; Plan, M.R.; Craik, D.J. Kalata B8, a novel antiviral circular protein, exhibits conformational flexibility in the cystine knot motif. Biochem. J. 2006, 393, 619–626. [Google Scholar] [CrossRef]
- Gerlach, S.L.; Yeshak, M.; Göransson, U.; Roy, U.; Izadpanah, R.; Mondal, D. Cycloviolacin O2 (CyO2) suppresses productive infection and augments the antiviral efficacy of nelfinavir in HIV-1 infected monocytic cells. Biopolym. Pept. Sci. 2013, 100, 471–479. [Google Scholar] [CrossRef]
- Hallock, Y.F.; Sowder, R.C.; Pannell, L.K.; Hughes, C.B.; Johnson, D.G.; Gulakowski, R.; Cardellina, J.H.; Boyd, M.R. Cycloviolins A-D, anti-HIV macrocyclic peptides from Leonia cymosa. J. Org. Chem. 2000, 65, 124–128. [Google Scholar] [CrossRef]
- Bokesch, H.R.; Pannell, L.K.; Cochran, P.K.; Sowder, R.C.; McKee, T.C.; Boyd, M.R. A novel anti-HIV macrocyclic peptide from Palicourea condensata. J. Nat. Prod. 2001, 64, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Colgrave, M.L.; Daly, N.L.; Rosengren, K.J.; Gustafson, K.R.; Craik, D.J. Isolation and characterization of novel cyclotides from Viola hederaceae: Solution structure and anti-HIV activity of vhl-1, a leaf-specific expressed cyclotide. J. Biol. Chem. 2005, 280, 22395–22405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ireland, D.C.; Wang, C.K.; Wilson, J.A.; Gustafson, K.R.; Craik, D.J. Cyclotides as natural anti-HIV agents. Biopolym. Pept. Sci. 2008, 90, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, K.R.; Sowder, R.C.; Henderson, L.E.; Parsons, I.C.; Kashman, Y.; Cardellina, J.H.; McMahon, J.B.; Buckheit, R.W.; Pannell, L.K.; Boyd, M.R. Circulins A and B: Novel HIV-inhibitory macrocyclic peptides from the tropical tree Chassalia parvifolia. J. Am. Chem. Soc. 1994, 116, 9337–9338. [Google Scholar] [CrossRef]
- Liu, M.Z.; Yang, Y.; Zhang, S.X.; Tang, L.; Wang, H.M.; Chen, C.J.; Shen, Z.F.; Cheng, K.D.; Kong, J.Q.; Wang, W. A cyclotide against influenza A H1N1 virus from Viola yedoensis. Acta Pharm. Sin. 2014, 49, 905–912. [Google Scholar]
- Fensterseifer, I.C.; Silva, O.N.; Malik, U.; Ravipati, A.S.; Novaes, N.R.F.; Miranda, P.R.R.; Rodrigues, E.A.; Moreno, S.E.; Craik, D.J.; Franco, O.L. Effects of cyclotides against cutaneous infections caused by Staphylococcus aureus. Peptides 2015, 63, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.P.; Lu, Y.A.; Yang, J.L.; Chiu, K.W. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc. Natl. Acad. Sci. USA 1999, 96, 8913–8918. [Google Scholar] [CrossRef] [Green Version]
- Gran, L.; Sletten, K.; Skjeldal, L. Cyclic peptides from Oldenlandia affinis DC. Molecular and biological properties. Chem. Biodivers. 2008, 5, 2014–2022. [Google Scholar] [CrossRef]
- Pränting, M.; Lööv, C.; Burman, R.; Göransson, U.; Andersson, D.I. The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria. J. Antimicrob. Chemother. 2010, 65, 1964–1971. [Google Scholar] [CrossRef]
- Gran, L. On the effect of a polypeptide isolated from “Kalata-Kalata” (Oldenlandia affinis DC) on the oestrogen dominated uterus. Acta Pharmacol. Toxicol. 1973, 33, 400–408. [Google Scholar] [CrossRef]
- Koehbach, J.; O’Brien, M.; Muttenthaler, M.; Miazzo, M.; Akcan, M.; Elliott, A.G.; Daly, N.L.; Harvey, P.J.; Arrowsmith, S.; Gunasekera, S.; et al. Oxytocic plant cyclotides as templates for peptide G protein-coupled receptor ligand design. Proc. Natl. Acad. Sci. USA 2013, 110, 21183–21188. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, S.L.; Rathinakumar, R.; Chakravarty, G.; Göransson, U.; Wimley, W.C.; Darwin, S.P.; Mondal, D. Anticancer and chemosensitizing abilities of cycloviolacin O2 from Viola odorata and psyle cyclotides from Psychotria leptothyrsa. Biopolym. Pept. Sci. 2010, 94, 617–625. [Google Scholar] [CrossRef]
- Tang, J.; Wang, C.K.; Pan, X.; Yan, H.; Zeng, G.; Xu, W.; He, W.; Daly, N.L.; Craik, D.J.; Tan, N. Isolation and characterization of cytotoxic cyclotides from Viola tricolor. Peptides 2010, 31, 1434–1440. [Google Scholar] [CrossRef]
- Svangård, E.; Göransson, U.; Hocaoglu, Z.; Gullbo, J.; Larsson, R.; Claeson, P.; Bohlin, L. Cytotoxic cyclotides from Viola tricolor. J. Nat. Prod. 2004, 67, 144–147. [Google Scholar] [CrossRef]
- Herrmann, A.; Burman, R.; Mylne, J.S.; Karlsson, G.; Gullbo, J.; Craik, D.J.; Clark, R.J.; Göransson, U. The alpine violet, Viola biflora, is a rich source of cyclotides with potent cytotoxicity. Phytochemistry 2008, 69, 939–952. [Google Scholar] [CrossRef]
- Gerlach, S.L.; Burman, R.; Bohlin, L.; Mondal, D.; Göransson, U. Isolation, characterization, and bioactivity of cyclotides from the Micronesian plant Psychotria leptothyrsa. J. Nat. Prod. 2010, 73, 1207–1213. [Google Scholar] [CrossRef]
- Gründemann, C.; Koehbach, J.; Huber, R.; Gruber, C.W. Do plant cyclotides have potential as immunosuppressant peptides? J. Nat. Prod. 2012, 75, 167–174. [Google Scholar] [CrossRef]
- Grundemann, C.; Thell, K.; Lengen, K.; Garcia-Kaufer, M.; Huang, Y.H.; Huber, R.; Craik, D.J.; Schabbauer, G.; Gruber, C.W. Cyclotides suppress human T-lymphocyte proliferation by an interleukin 2-dependent mechanism. PLoS ONE 2013, 8, e68016. [Google Scholar] [CrossRef]
- Wieczorek, Z.; Siemion, I.Z.; Zimecki, M.; Bolewska-Pedyczak, E.; Wieland, T. Immunosuppressive activity in the series of cycloamanide peptides from mushrooms. Peptides 1993, 14, 1–5. [Google Scholar] [CrossRef]
- Hellinger, R.; Koehbach, J.; Fedchuk, H.; Sauer, B.; Huber, R.; Gruber, C.W.; Gründemann, C. Immunosuppressive activity of an aqueous Viola tricolor herbal extract. J. Ethnopharmacol. 2014, 151, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Bi, T.; Camarero, J.A. Chemical and biological production of cyclotides. Adv. Bot. Res. 2015, 76, 271–303. [Google Scholar] [PubMed] [Green Version]
- Yap, K.; Du, J.; Rehm, F.B.H.; Tang, S.R.; Zhou, Y.; Xie, J.; Wang, C.K.; de Veer, S.J.; Lua, L.H.L.; Durek, T.; et al. Yeast-based bioproduction of disulfide-rich peptides and their cyclization via asparaginyl endopeptidases. Nat. Protoc. 2021, 16, 1740–1760. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.A.; Xie, J.; Nguyen, L.T.T.; Wang, X.; Yap, K.; Harvey, P.J.; Gilding, E.K.; Craik, D.J. Plant-based production of an orally active cyclotide for the treatment of multiple sclerosis. Transgenic Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Camarero, J.A.; Campbell, M.J. The potential of the cyclotide scaffold for drug development. Biomedicines 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Colgrave, M.L.; Clark, R.J.; Kotze, A.C.; Craik, D.J. Lysine-scanning mutagenesis reveals a previously unidentified amendable face of the cyclotide kalata B1 for the optimisation of nematocidal activity. J. Biol. Chem. 2010, 285, 10797–10805. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.T.T.; Rowlands, D.K.; Wong, C.H.; Lo, T.W.C.; Nguyen, G.K.T.; Li, H.Y.; Tam, J.P. Orally active peptidic bradykinin B-1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem. Int. Ed. 2012, 51, 5620–5624. [Google Scholar] [CrossRef]
- Getz, J.A.; Cheneval, O.; Craik, D.J.; Daugherty, P.S. Design of a cyclotide antagonist of neuropilin-1 and -2 that potently inhibits endothelial cell migration. ACS Chem. Biol. 2013, 8, 1147–1154. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Yuan, J.; Osapay, G.; Osapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Conibear, A.C.; Craik, D.J. The chemistry and biology of theta defensins. Angew. Chem. Int. Ed. 2014, 53, 10612–10623. [Google Scholar] [CrossRef]
- Conibear, A.C.; Rosengren, K.J.; Harvey, P.J.; Craik, D.J. Structural characterization of the cyclic cystine ladder motif of theta-defensins. Biochemistry 2012, 51, 9718–9726. [Google Scholar] [CrossRef]
- Conibear, A.C.; Rosengren, K.J.; Daly, N.L.; Henriques, S.T.; Craik, D.J. The cyclic cystine ladder in theta-defensins is important for structure and stability, but not antibacterial activity. J. Biol. Chem. 2013, 288, 10830–10840. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, R.I.; Cole, A.M.; Selsted, M.E. θ-Defensins: Cyclic peptides with endless potential. J. Biol. Chem. 2012, 287, 27014–27019. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.E.; Osapay, G.; Tran, P.A.; Yuan, J.; Selsted, M.E. Isolation, synthesis, and antimicrobial activities of naturally occurring theta-defensin isoforms from baboon leukocytes. Infect. Immun. 2008, 76, 5883–5891. [Google Scholar] [CrossRef] [Green Version]
- Bensman, T.J.; Jayne, J.G.; Sun, M.; Kimura, E.; Meinert, J.; Wang, J.C.; Schaal, J.B.; Tran, D.; Rao, A.P.; Akbari, O.; et al. Efficacy of rhesus theta-defensin-1 in experimental models of Pseudomonas aeruginosa lung infection and inflammation. Antimicrob. Agents Chemother. 2017, 61, e00154-17. [Google Scholar] [CrossRef] [Green Version]
- Schaal, J.B.; Tran, D.; Tran, P.; Osapay, G.; Trinh, K.; Roberts, K.D.; Brasky, K.M.; Tongaonkar, P.; Ouellette, A.J.; Selsted, M.E. Rhesus macaque theta defensins suppress inflammatory cytokines and enhance survival in mouse models of bacteremic sepsis. PLoS ONE 2012, 7, e51337. [Google Scholar] [CrossRef]
- Wilmes, M.; Stockem, M.; Bierbaum, G.; Schlag, M.; Gotz, F.; Tran, D.Q.; Schaal, J.B.; Ouellette, A.J.; Selsted, M.E.; Sahl, H.G. Killing of staphylococci by theta-defensins involves membrane impairment and activation of autolytic enzymes. Antibiotics 2014, 3, 617–631. [Google Scholar] [CrossRef] [Green Version]
- Tongaonkar, P.; Trinh, K.K.; Schaal, J.B.; Tran, D.; Gulko, P.S.; Ouellette, A.J.; Selsted, M.E. Rhesus macaque theta-defensin RTD-1 inhibits proinflammatory cytokine secretion and gene expression by inhibiting the activation of NF-kappaB and MAPK pathways. J. Leukoc. Biol. 2015, 98, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.; Tran, P.; Roberts, K.; Osapay, G.; Schaal, J.; Ouellette, A.; Selsted, M.E. Microbicidal properties and cytocidal selectivity of rhesus macaque theta defensins. Antimicrob. Agents Chemother. 2008, 52, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Wohlford-Lenane, C.L.; Meyerholz, D.K.; Perlman, S.; Zhou, H.; Tran, D.; Selsted, M.E.; McCray, P.B., Jr. Rhesus theta-defensin prevents death in a mouse model of severe acute respiratory syndrome coronavirus pulmonary disease. J. Virol. 2009, 83, 11385–11390. [Google Scholar] [CrossRef] [Green Version]
- Negahdaripour, M.; Rahbar, M.R.; Mosalanejad, Z.; Gholami, A. Theta-Defensins to Counter COVID-19 as Furin Inhibitors: In Silico Efficiency Prediction and Novel Compound Design. Comput. Math. Methods Med. 2022, 2022, 9735626. [Google Scholar] [CrossRef]
- Oh, Y.T.; Tran, D.; Buchanan, T.A.; Selsted, M.E.; Youn, J.H. theta-Defensin RTD-1 improves insulin action and normalizes plasma glucose and FFA levels in diet-induced obese rats. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E154–E160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penberthy, W.T.; Chari, S.; Cole, A.L.; Cole, A.M. Retrocyclins and their activity against HIV-1. Cell. Mol. Life Sci. 2011, 68, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C.; Bochen, A.; Rosengren, K.J.; Stupar, P.; Wang, C.; Kessler, H.; Craik, D.J. The cyclic cystine ladder of theta-defensins as a stable, bifunctional scaffold: A proof-of-concept study using the integrin-binding RGD motif. ChemBioChem 2014, 15, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, N.; Cole, A.L.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Stuchlik, O.; Pohl, J.; Cole, A.M. Reawakening retrocyclins: Ancestral human defensins active against HIV-1. PLoS Biol. 2009, 7, e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.M.; Hong, T.; Boo, L.M.; Nguyen, T.; Zhao, C.; Bristol, G.; Zack, J.A.; Waring, A.J.; Yang, O.O.; Lehrer, R.I. Retrocyclin: A primate peptide that protects cells from infection by T- and M-tropic strains of HIV-1. Proc. Natl. Acad. Sci. USA 2002, 99, 1813–1818. [Google Scholar] [CrossRef] [Green Version]
- Owen, S.M.; Rudolph, D.L.; Wang, W.; Cole, A.M.; Waring, A.J.; Lal, R.B.; Lehrer, R.I. RC-101, a retrocyclin-1 analogue with enhanced activity against primary HIV type 1 isolates. AIDS Res. Hum. Retrovir. 2004, 20, 1157–1165. [Google Scholar] [CrossRef]
- Kudryashova, E.; Zani, A.; Vilmen, G.; Sharma, A.; Lu, W.; Yount, J.S.; Kudryashov, D.S. Inhibition of SARS-CoV-2 Infection by Human Defensin HNP1 and Retrocyclin RC-101. J. Mol. Biol. 2022, 434, 167225. [Google Scholar] [CrossRef]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; de Santos, V.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar] [CrossRef]
- Jin, A.H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Bjorn-Yoshimoto, W.E.; Ramiro, I.B.L.; Yandell, M.; McIntosh, J.M.; Olivera, B.M.; Ellgaard, L.; Safavi-Hemami, H. Curses or Cures: A Review of the Numerous Benefits Versus the Biosecurity Concerns of Conotoxin Research. Biomedicines 2020, 8, 235. [Google Scholar] [CrossRef]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [Green Version]
- Pope, J.E.; Deer, T.R. Ziconotide: A clinical update and pharmacologic review. Expert Opin. Pharmacother. 2013, 14, 957–966. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone Snails: A big store of conotoxins for novel drug discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.J.; Fischer, H.; Nevin, S.T.; Adams, D.J.; Craik, D.J. The synthesis, structural characterization, and receptor specificity of the alpha-conotoxin Vc1.1. J. Biol. Chem. 2006, 281, 23254–23263. [Google Scholar] [CrossRef] [Green Version]
- Sandall, D.W.; Satkunanathan, N.; Keays, D.A.; Polidano, M.A.; Liping, X.; Pham, V.; Down, J.G.; Khalil, Z.; Livett, B.G.; Gayler, K.R. A novel alpha-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 2003, 42, 6904–6911. [Google Scholar] [CrossRef]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005, 1059, 149–158. [Google Scholar] [CrossRef]
- Clark, R.J.; Akcan, M.; Kaas, Q.; Daly, N.L.; Craik, D.J. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon 2012, 59, 446–455. [Google Scholar] [CrossRef]
- Castro, J.; Grundy, L.; Deiteren, A.; Harrington, A.M.; O’Donnell, T.; Maddern, J.; Moore, J.; Garcia-Caraballo, S.; Rychkov, G.Y.; Yu, R.; et al. Cyclic analogues of alpha-conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain. Br. J. Pharmacol. 2018, 175, 2384–2398. [Google Scholar] [CrossRef] [Green Version]
- Poth, A.G.; Chiu, F.C.K.; Stalmans, S.; Hamilton, B.R.; Huang, Y.-H.; Shackleford, D.M.; Patil, R.; Le, T.T.; Kan, M.-W.; Durek, T.; et al. Effects of backbone cyclization on the pharmacokinetics and drug efficiency of the orally active analgesic conotoxin cVc1.1. Med. Drug Discov. 2021, 10, 100087. [Google Scholar] [CrossRef]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 2010, 49, 6545–6548. [Google Scholar] [CrossRef]
- DeBin, J.A.; Maggio, J.E.; Strichartz, G.R. Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. Am. J. Physiol. 1993, 264, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Dardevet, L.; Rani, D.; Aziz, T.A.; Bazin, I.; Sabatier, J.M.; Fadl, M.; Brambilla, E.; De Waard, M. Chlorotoxin: A helpful natural scorpion peptide to diagnose glioma and fight tumor invasion. Toxins 2015, 7, 1079–1101. [Google Scholar] [CrossRef] [PubMed]
- Čemažar, M.; Kwon, S.; Mahatmanto, T.; Ravipati, A.S.; Craik, D.J. Discovery and applications of disulfide-rich cyclic peptides. Curr. Top. Med. Chem. 2012, 12, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Mamelak, A.N.; Jacoby, D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef]
- Fu, Y.; An, N.; Li, K.; Zheng, Y.; Liang, A. Chlorotoxin-conjugated nanoparticles as potential glioma-targeted drugs. J. Neurooncol. 2012, 107, 457–462. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhao, J.; Qiao, W.; Chen, K. Recent advances in diagnosis and treatment of gliomas using chlorotoxin-based bioconjugates. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 385–405. [Google Scholar]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12, eaaw2672. [Google Scholar] [CrossRef]
- Veiseh, M.; Gabikian, P.; Bahrami, S.B.; Veiseh, O.; Zhang, M.; Hackman, R.C.; Ravanpay, A.C.; Stroud, M.R.; Kusuma, Y.; Hansen, S.J.; et al. Tumor paint: A chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007, 67, 6882–6888. [Google Scholar] [CrossRef] [Green Version]
- Butte, P.V.; Mamelak, A.; Parrish-Novak, J.; Drazin, D.; Shweikeh, F.; Gangalum, P.R.; Chesnokova, A.; Ljubimova, J.Y.; Black, K. Near-infrared imaging of brain tumors using the Tumor Paint BLZ-100 to achieve near-complete resection of brain tumors. Neurosurg. Focus 2014, 36, E1. [Google Scholar] [CrossRef] [Green Version]
- Fidel, J.; Kennedy, K.C.; Dernell, W.S.; Hansen, S.; Wiss, V.; Stroud, M.R.; Molho, J.I.; Knoblaugh, S.E.; Meganck, J.; Olson, J.M.; et al. Preclinical Validation of the Utility of BLZ-100 in Providing Fluorescence Contrast for Imaging Spontaneous Solid Tumors. Cancer Res. 2015, 75, 4283–4291. [Google Scholar] [CrossRef] [Green Version]
- Akcan, M.; Stroud, M.R.; Hansen, S.J.; Clark, R.J.; Daly, N.L.; Craik, D.J.; Olson, J.M. Chemical re-engineering of chlorotoxin improves bioconjugation properties for tumor imaging and targeted therapy. J. Med. Chem. 2011, 54, 782–787. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus), isolation and chemical structure. J. Biol. Chem. 1988, 263, 16709–16713. [Google Scholar] [CrossRef]
- Vernen, F.; Harvey, P.J.; Dias, S.A.; Veiga, A.S.; Huang, Y.H.; Craik, D.J.; Lawrence, N.; Troeira Henriques, S. Characterization of tachyplesin peptides and their cyclized analogues to improve antimicrobial and anticancer properties. Int. J. Mol. Sci. 2019, 20, 4184. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Liu, C.; Qi, J.; Shan, B.; Ma, Y. Tachyplesin causes membrane instability that kills multidrug-resistant bacteria by inhibiting the 3-ketoacyl carrier protein reductase FabG. Front. Microbiol. 2018, 9, 825–838. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.P.; Lu, Y.A.; Yang, J.L. Marked increase in membranolytic selectivity of novel cyclic tachyplesins constrained with an antiparallel two-beta strand cystine knot framework. Biochem. Biophys. Res. Commun. 2000, 267, 783–790. [Google Scholar] [CrossRef]
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, D.A.; Hurst, M.A.; Fujii, C.A.; Kung, A.H.; Ho, J.F.; Cheng, F.C.; Loury, D.J.; Fiddes, J.C. Protegrin-1: A broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob. Agents Chemother. 1997, 41, 1738–1742. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, R.L.; Dieckmann, T.; Harwig, S.S.; Lehrer, R.I.; Eisenberg, D.; Feigon, J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996, 3, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Sokolov, Y.; Mirzabekov, T.; Martin, D.W.; Lehrer, R.I.; Kagan, B.L. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta 1999, 1420, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellm, L.; Lehrer, R.I.; Ganz, T. Protegrins: New antibiotics of mammalian origin. Expert Opin. Investig. Drugs 2000, 9, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Miller, C.B.; Hurd, D.D.; Wingard, J.R.; Fleming, T.R.; Sonis, S.T.; Bradford, W.Z.; Pulliam, J.G.; Anaissie, E.J.; Beveridge, R.A.; et al. A phase III, randomized, double-blind, placebo-controlled, multinational trial of iseganan for the prevention of oral mucositis in patients receiving stomatotoxic chemotherapy (PROMPT-CT trial). Leuk. Lymphoma 2003, 44, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Elad, S.; Epstein, J.B.; Raber-Durlacher, J.; Donnelly, P.; Strahilevitz, J. The antimicrobial effect of Iseganan HCl oral solution in patients receiving stomatotoxic chemotherapy: Analysis from a multicenter, double-blind, placebo-controlled, randomized, phase III clinical trial. J. Oral Pathol. Med. 2012, 41, 229–234. [Google Scholar] [CrossRef]
- Tam, J.P.; Wu, C.; Yang, J.L. Membranolytic selectivity of cystine-stabilized cyclic protegrins. Eur. J. Biochem. 2000, 267, 3289–3300. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Falla, T.J.; Liu, H.; Hurst, M.A.; Fujii, C.A.; Mosca, D.A.; Embree, J.R.; Loury, D.J.; Radel, P.A.; Chang, C.C.; et al. Development of protegrins for the treatment and prevention of oral mucositis: Structure-activity relationships of synthetic protegrin analogues. Biopolymers 2004, 55, 88–98. [Google Scholar] [CrossRef]
- Silva, P.I., Jr.; Daffre, S.; Bulet, P. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptides of the tachyplesin family. J. Biol. Chem. 2000, 275, 33464–33470. [Google Scholar] [CrossRef] [Green Version]
- Fukuzawa, A.H.; Vellutini, B.C.; Lorenzini, D.M.; Silva, P.I., Jr.; Mortara, R.A.; da Silva, J.M.; Daffre, S. The role of hemocytes in the immunity of the spider Acanthoscurria gomesiana. Dev. Comp. Immunol. 2008, 32, 716–725. [Google Scholar] [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.; Cooper, M.A. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of beta-hairpin peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef]
- Moreira, C.K.; Rodrigues, F.G.; Ghosh, A.; Varotti Fde, P.; Miranda, A.; Daffre, S.; Jacobs-Lorena, M.; Moreira, L.A. Effect of the antimicrobial peptide gomesin against different life stages of Plasmodium spp. Exp. Parasitol. 2007, 116, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, F.M.; Daffre, S.; Maldonado, R.A.; Miranda, A.; Nimrichter, L.; Rodrigues, M.L. Gomesin, a peptide produced by the spider Acanthoscurria gomesiana, is a potent anticryptococcal agent that acts in synergism with fluconazole. FEMS Microbiol. Lett. 2007, 274, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Fazio, M.A.; Jouvensal, L.; Vovelle, F.; Bulet, P.; Miranda, M.T.; Daffre, S.; Miranda, A. Biological and structural characterization of new linear gomesin analogues with improved therapeutic indices. Biopolymers 2007, 88, 386–400. [Google Scholar] [CrossRef]
- Ikonomopoulou, M.P.; Fernandez-Rojo, M.A.; Pineda, S.S.; Cabezas-Sainz, P.; Winnen, B.; Morales, R.A.V.; Brust, A.; Sanchez, L.; Alewood, P.F.; Ramm, G.A.; et al. Gomesin inhibits melanoma growth by manipulating key signaling cascades that control cell death and proliferation. Sci. Rep. 2018, 8, 11519–11633. [Google Scholar] [CrossRef] [Green Version]
- Mandard, N.; Bulet, P.; Caille, A.; Daffre, S.; Vovelle, F. The solution structure of gomesin, an antimicrobial cysteine-rich peptide from the spider. Eur. J. Biochem. 2002, 269, 1190–1198. [Google Scholar] [CrossRef]
- Tanner, J.D.; Deplazes, E.; Mancera, R.L. The biological and biophysical properties of the spider peptide gomesin. Molecules 2018, 23, 1733. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.Y.; Zhang, V.M.; Huang, Y.H.; Waters, N.C.; Bansal, P.S.; Craik, D.J.; Daly, N.L. Cyclization of the antimicrobial peptide gomesin with native chemical ligation: Influences on stability and bioactivity. ChemBioChem 2013, 14, 617–624. [Google Scholar] [CrossRef]
- Henriques, S.T.; Lawrence, N.; Chaousis, S.; Ravipati, A.S.; Cheneval, O.; Benfield, A.H.; Elliott, A.G.; Kavanagh, A.M.; Cooper, M.A.; Chan, L.Y.; et al. Redesigned spider peptide with improved antimicrobial and anticancer properties. ACS Chem. Biol. 2017, 12, 2324–2334. [Google Scholar] [CrossRef]
- Dias, S.A.; Pinto, S.N.; Silva-Herdade, A.S.; Cheneval, O.; Craik, D.J.; Coutinho, A.; Castanho, M.; Henriques, S.T.; Veiga, A.S. A designed cyclic analogue of gomesin has potent activity against Staphylococcus aureus biofilms. J. Antimicrob. Chemother. 2022, 77, 3256–3264. [Google Scholar] [CrossRef]
- Benfield, A.H.; Defaus, S.; Lawrence, N.; Chaousis, S.; Condon, N.; Cheneval, O.; Huang, Y.H.; Chan, L.Y.; Andreu, D.; Craik, D.J.; et al. Cyclic gomesin, a stable redesigned spider peptide able to enter cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183480. [Google Scholar] [CrossRef]
Figure 1.
Selected classes of native and engineered cyclic disulfide-rich peptides derived from plants and animals, with representative examples from each class. Cysteine residues are numbered and highlighted in yellow, with engineered cyclization linkers represented by brown shading.
Figure 1.
Selected classes of native and engineered cyclic disulfide-rich peptides derived from plants and animals, with representative examples from each class. Cysteine residues are numbered and highlighted in yellow, with engineered cyclization linkers represented by brown shading.

Figure 2.
Molecular grafting onto a cyclic peptide scaffold to stabilize a biologically active peptide sequence. In a recent study, the MCoTI-I scaffold was used to stabilize the helical Hdm2/HdmX-binding peptide PMI (cyan), with an apamin-derived linker (pink), for the treatment of cancer. The PMI epitope was derived from the N-terminal domain of p53 and optimized through phage display screening. When grafted onto the MCoTI-I framework, the resulting grafted peptide MCo-PMI displayed high serum stability and potent intracellular activity.
Figure 2.
Molecular grafting onto a cyclic peptide scaffold to stabilize a biologically active peptide sequence. In a recent study, the MCoTI-I scaffold was used to stabilize the helical Hdm2/HdmX-binding peptide PMI (cyan), with an apamin-derived linker (pink), for the treatment of cancer. The PMI epitope was derived from the N-terminal domain of p53 and optimized through phage display screening. When grafted onto the MCoTI-I framework, the resulting grafted peptide MCo-PMI displayed high serum stability and potent intracellular activity.

Figure 3.
Structure and sequence of SFTI-1, the prototypic PDP member, and four grafted examples. (a) Three-dimensional structure of SFTI-1, with scissile bond signified by a * symbol. (b) Peptide sequences of SFTI-1 and four engineered SFTI variants. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of SFTI-1. Cysteine residues are numbered and highlighted in yellow, with engineered epitope sequences represented by brown shading. D-amino acids are designated with lowercase lettering and N-methylated amino acids by bolded letters.
Figure 3.
Structure and sequence of SFTI-1, the prototypic PDP member, and four grafted examples. (a) Three-dimensional structure of SFTI-1, with scissile bond signified by a * symbol. (b) Peptide sequences of SFTI-1 and four engineered SFTI variants. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of SFTI-1. Cysteine residues are numbered and highlighted in yellow, with engineered epitope sequences represented by brown shading. D-amino acids are designated with lowercase lettering and N-methylated amino acids by bolded letters.

Figure 4.
Structure and sequence of kalata B1, prototypic Möbius cyclotide member, and three grafted examples. (a) Three-dimensional structure of Möbius cyclotide kalata B1. (b) Peptide sequences of kalata B1 and four bioactive epitope examples, with their corresponding three grafted cyclotide products. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of kalata B1. Cysteine residues are numbered and highlighted in yellow, with non-native epitope inclusions represented by brown, blue or green shading. Blue and green shading distinguishing the first (N1.14) and second generations (N2.1) of bioactive epitope sequences generated via sequential bacterial display libraries, respectively.
Figure 4.
Structure and sequence of kalata B1, prototypic Möbius cyclotide member, and three grafted examples. (a) Three-dimensional structure of Möbius cyclotide kalata B1. (b) Peptide sequences of kalata B1 and four bioactive epitope examples, with their corresponding three grafted cyclotide products. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of kalata B1. Cysteine residues are numbered and highlighted in yellow, with non-native epitope inclusions represented by brown, blue or green shading. Blue and green shading distinguishing the first (N1.14) and second generations (N2.1) of bioactive epitope sequences generated via sequential bacterial display libraries, respectively.

Figure 5.
Structure and sequence of the θ-defensin RTD-1 and two grafted examples. (a) Three-dimensional structure of RTD-1. (b) Peptide sequences of RTD-1 and two engineered variants. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of RTD-1. Cysteine residues are numbered and highlighted in yellow, with engineered epitope sequences represented by brown shading.
Figure 5.
Structure and sequence of the θ-defensin RTD-1 and two grafted examples. (a) Three-dimensional structure of RTD-1. (b) Peptide sequences of RTD-1 and two engineered variants. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of RTD-1. Cysteine residues are numbered and highlighted in yellow, with engineered epitope sequences represented by brown shading.

Figure 6.
Structure and sequence of the genetically encoded, but untranslated, retrocyclin family. (a) Three-dimensional structure of retrocyclin RC-2. (b) Peptide sequences of three natively encoded retrocyclins and the engineered retrocyclin variant RC-101. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of retrocyclin RC-2. Cysteine residues are numbered and highlighted in yellow, with the non-native residue depicted by brown shading.
Figure 6.
Structure and sequence of the genetically encoded, but untranslated, retrocyclin family. (a) Three-dimensional structure of retrocyclin RC-2. (b) Peptide sequences of three natively encoded retrocyclins and the engineered retrocyclin variant RC-101. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of retrocyclin RC-2. Cysteine residues are numbered and highlighted in yellow, with the non-native residue depicted by brown shading.

Figure 7.
Structure and sequence of α-conotoxin Vc1.1 and three engineered cyclic conotoxin variants. (a) Three-dimensional structure of synthetic Vc1.1, with engineered inert cyclization linker. (b) Peptide sequence of Vc1.1 and three different engineered cyclic conotoxin variants. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of cVc1.1. Cysteine residues are numbered and highlighted in yellow, with the engineered sequences represented by brown shading.
Figure 7.
Structure and sequence of α-conotoxin Vc1.1 and three engineered cyclic conotoxin variants. (a) Three-dimensional structure of synthetic Vc1.1, with engineered inert cyclization linker. (b) Peptide sequence of Vc1.1 and three different engineered cyclic conotoxin variants. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of cVc1.1. Cysteine residues are numbered and highlighted in yellow, with the engineered sequences represented by brown shading.

Figure 8.
Structure and sequence of CTX and its cyclic analogue. (a) Three-dimensional structure of CTX, with engineered cyclization linker. (b) Peptide sequences of CTX and its engineered cyclic variant cCTX. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of CTX. Cysteine residues are numbered and highlighted in yellow, with non-native residues and sites of lysine Cy5.5 conjugation indicated by brown and orange shading, respectively.
Figure 8.
Structure and sequence of CTX and its cyclic analogue. (a) Three-dimensional structure of CTX, with engineered cyclization linker. (b) Peptide sequences of CTX and its engineered cyclic variant cCTX. (c) Amino acid sequence, disulfide bond connectivity and loop nomenclature of CTX. Cysteine residues are numbered and highlighted in yellow, with non-native residues and sites of lysine Cy5.5 conjugation indicated by brown and orange shading, respectively.

Figure 9.
(a) Three-dimensional structure of the prototypical tachyplesin TI, with engineered cyclization linker. (b) The native peptide sequence of TI and three engineered cyclic tachyplesin variants. (c) Representation of the amino acid sequence, disulfide bond connectivity and loop nomenclature of cTI. Cysteine residues are numbered and highlighted in yellow, with the engineered cyclizing epitope inclusions represented by brown shading.
Figure 9.
(a) Three-dimensional structure of the prototypical tachyplesin TI, with engineered cyclization linker. (b) The native peptide sequence of TI and three engineered cyclic tachyplesin variants. (c) Representation of the amino acid sequence, disulfide bond connectivity and loop nomenclature of cTI. Cysteine residues are numbered and highlighted in yellow, with the engineered cyclizing epitope inclusions represented by brown shading.

Figure 10.
(a) Three-dimensional structure of the prototypical protegrin PG-1, with engineering cyclization site. (b) The native peptide sequence of PG-1 and three engineered protegrins variants [163,167]. (c) Depiction of the amino acid sequence and disulfide bond connectivity of the cyclic protegrin cPG-1. Cysteine residues are numbered and highlighted in yellow, with the engineered sequences and junction points represented by brown shading.
Figure 10.
(a) Three-dimensional structure of the prototypical protegrin PG-1, with engineering cyclization site. (b) The native peptide sequence of PG-1 and three engineered protegrins variants [163,167]. (c) Depiction of the amino acid sequence and disulfide bond connectivity of the cyclic protegrin cPG-1. Cysteine residues are numbered and highlighted in yellow, with the engineered sequences and junction points represented by brown shading.

Figure 11.
(a) Three-dimensional structure of Gm and its engineered cyclization site. (b) Peptide sequences of Gm and three engineered Gm variants. (c) Amino acid sequence of cGm, depicting disulfide bond connectivity and loop nomenclature. Cysteine residues are numbered and highlighted in yellow, with engineered cyclization sites and modifications indicated with brown shading.
Figure 11.
(a) Three-dimensional structure of Gm and its engineered cyclization site. (b) Peptide sequences of Gm and three engineered Gm variants. (c) Amino acid sequence of cGm, depicting disulfide bond connectivity and loop nomenclature. Cysteine residues are numbered and highlighted in yellow, with engineered cyclization sites and modifications indicated with brown shading.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tyler, T.J.; Durek, T.; Craik, D.J. Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads. Molecules 2023, 28, 3189. https://doi.org/10.3390/molecules28073189
AMA Style
Tyler TJ, Durek T, Craik DJ. Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads. Molecules. 2023; 28(7):3189. https://doi.org/10.3390/molecules28073189
Chicago/Turabian StyleTyler, Tristan J., Thomas Durek, and David J. Craik. 2023. "Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads" Molecules 28, no. 7: 3189. https://doi.org/10.3390/molecules28073189