
Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines
by
 ,
,
Yan Chen
1,2,3,
Jian-Qiang Zhao
2,
Yan-Ping Zhang
2,
Ming-Qiang Zhou
1,2,3,
Xiao-Mei Zhang
1,4,* and
Wei-Cheng Yuan
1,2,* 1
National Engineering Research Center of Chiral Drugs, Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, China
2
Innovation Research Center of Chiral Drugs, Institute for Advanced Study, Chengdu University, Chengdu 610106, China
3
University of Chinese Academy of Sciences, Beijing 100049, China
4
Department of Chemistry, Xihua University, Chengdu 610039, China
*
Authors to whom correspondence should be addressed.
Molecules 2023, 28(6), 2765; https://doi.org/10.3390/molecules28062765
Submission received: 13 February 2023
/
Revised: 17 March 2023
/
Accepted: 17 March 2023
/
Published: 19 March 2023
(This article belongs to the Special Issue Recent Advances of Catalytic Asymmetric Synthesis)
Abstract
:Catalytic asymmetric dearomative [3+2] cycloaddition of α-imino γ-lactones with either 3-nitroindoles or 2-nitrobenzofurans by using a chiral copper complex as the catalyst was developed. A wide range of structurally diverse polyheterocyclic compounds containing spirocyclic-fused butyrolactone-pyrrolidine-indoline and butyrolactone–pyrrolidine–dihydrobenzofuran skeletons could be smoothly obtained with excellent results (>99:1 dr and 98% ee). The potential synthetic applications of this methodology were also demonstrated by the scale-up experiment and by the diverse transformations of one product. This method is characterized by high asymmetric induction, wide functional group tolerance and scalability, and attractive product diversification.
1. Introduction
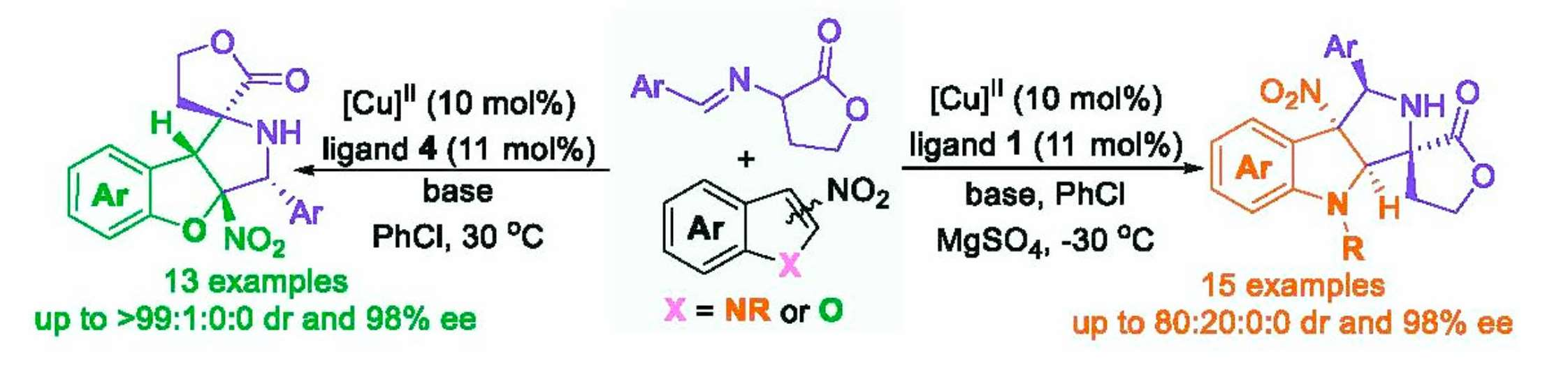
The privileged skeletons with multiple contiguous stereogenic centers are widely encountered in natural alkaloids and synthetic compounds with important biological profiles [1,2,3,4,5,6,7]. As shown in Figure 1, the dispiro–butyrolactone–pyrrolidine-fused oxindole derivative A exhibits significant anticancer activity [8]. Compound B has been used as one of the key building blocks in the total synthesis of the complex marine alkaloid (-)-Sarain A, which was reported by Overman and coworkers [9]. The spiro-heterocyclic compound C was used to prepare the alkaloid Cephalotaxine [10]. These bioactive molecules or key intermediates of natural products contain a chiral spirocyclic butyrolactone–pyrrolidine moiety as their structural core. In this context, the exploration of facile and efficient enantioselective approaches for the construction of optically pure spirocyclic butyrolactone–pyrrolidine compounds with innovative structures, which may be potentially applied as pharmaceutical agents, would bring some unique benefits to drug discovery and is thus particularly appealing.
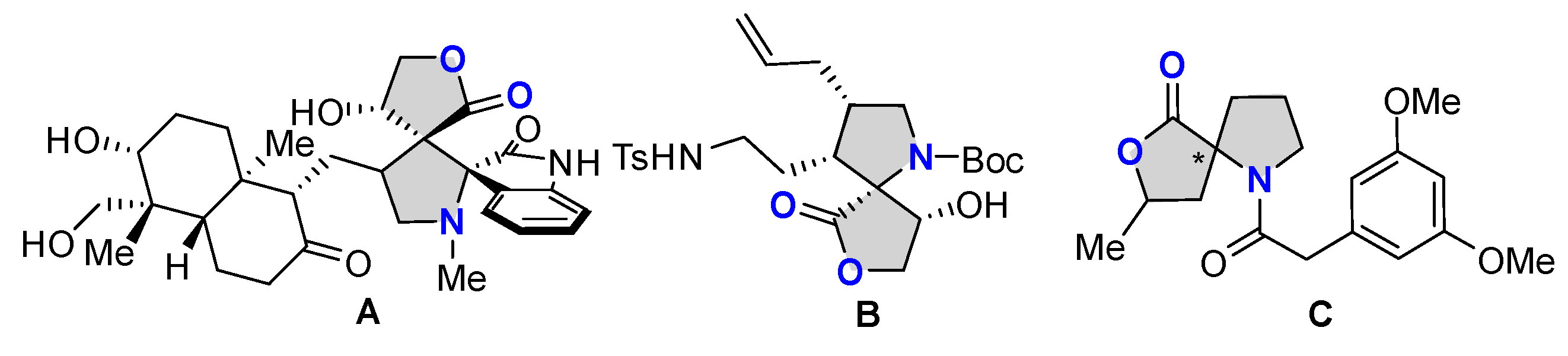
Catalytic asymmetric dearomatization reaction has emerged as one of the powerful strategies to access enantioenriched three-dimensional complex frameworks from readily available planar aromatic molecules [11,12,13,14,15,16,17]. In this aspect, considerable efforts have been devoted to exploring various catalytic asymmetric dearomatization reactions, particularly with the electron-rich heteroaromatic compounds as reactants relying on their intrinsic nucleophilic character [18,19,20]. In sharp contrast, the research with respect to electron-deficient heteroaromatic compounds remains relatively underdeveloped. Because the first palladium-catalyzed asymmetric dearomative [3+2] cycloaddition reaction of simple nitroarenes and trimethylenemethane was reported in 2014 by Trost and coworkers [21], the research on the catalytic asymmetric dearomatization reaction of electron-deficient nitroheteroarenes, including nitroindoles, nitrobenzofurans, and nitrobenzothiophenes, had attracted great interest from the synthetic organic chemistry community [22,23,24,25,26]. In this research area, the use of electron-deficient nitroheteroarenes as dipolarophiles for the asymmetric dearomative [3+2] cycloaddition reaction with various azomethine ylides has been sporadically reported. On the basis of a comprehensive and systematic literature research, it was found that only three types of azomethine ylide precursors have been explored for the asymmetric dearomative cycloaddition reactions with different electron-deficient nitroheteroarenes [27,28,29,30,31,32,33,34,35,36]. With glycine- or alanine-derived imino esters as 1,3-dipoles, the asymmetric dearomative [3+2] cycloaddition reactions of 3-nitroindoles and 2-nitrobenzofurans were realized by using different chiral copper complexes as catalysts from the groups of Arai, Stanley, and Guo, respectively (Scheme 1a) [27,28,29]. In addition, using cyclic azomethine ylides as 1,3-dipoles and 2-nitrobenzofurans as dipolarophiles, the corresponding asymmetric dearomative [3+2] cycloaddition reactions were reported by Guo and Wang, respectively (Scheme 1b) [30,31]. Beyond the two above-mentioned types of azomethine ylides, employing N-2,2,2-trifluoroethylisatin ketimines as a new type of highly active azomehthine ylide precursors for the catalytic enantioselective dearomative 1,3-dipolar cycloaddition reaction with diverse nitroheteroarenes was also implemented by several research groups (Scheme 1c) [32,33,34,35,36]. Despite these successful precedents, considering the great application potential and the promising prospect of polyheterocyclic compounds with structural diversity in drug discovery programs, exploiting new types of azomethine ylides which enable the enantioselective dearomative cycloaddition reaction of diverse electron-deficient nitroheteroarenes for accessing elaborated heterocycles with creative frameworks is still in high demand.
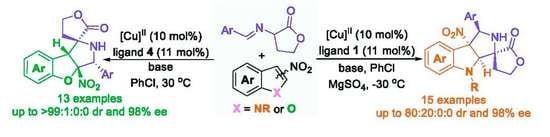
α-Imino γ-lactones as azomethine ylide precursors have proven to be a type of efficient 1,3-dipole that could be used in catalytic enantioselective [3+2] cycloadditon reactions with various dipolarophiles for the construction of spirocyclic butyrolactone–pyrrolidine skeletons [37,38,39,40]. Enlightened by the previous works on the asymmetric cycloaddition reaction with α-imino γ-lactones as 1,3-dipoles, we speculated that the asymmetric dearomative cycloaddition reaction of α-imino γ-lactones and electron-deficient nitroheteroarenes would take place in the presence of a suitable chiral copper complex as the catalyst. As a continuation of our research interest in developing catalytic asymmetric dearomatization reactions of electron-deficient nitroheteroarenes [41,42,43,44,45,46,47], we recently developed an efficient enantioselective dearomative [3+2] cycloaddition of α-imino γ-lactones and 3-nitroindoles with a chiral copper complex as the catalyst, providing a facile access to spirocyclic butyrolactone–pyrrolidine–indoline derivatives in up to 67% yield for the major diastereomer with 80:20:0:0 dr and 98% ee (Scheme 1d). Moreover, the similar catalytic system was also extended to the dearomative [3+2] cycloaddition reaction of 2-nitrobenzofurans for the construction of spirocyclic butyrolactone–pyrrolidine–dihydrobenzofuran compounds in up to 86% yield for the major diastereomer with >99:1 dr and 98% ee (Scheme 1d). Herein, we wish to report the details of our study on this subject.
2. Results and Discussion
2.1. Optimization Studies
Initially, we investigated the catalytic asymmetric dearomative [3+2] cycloaddition reaction of 3-nitroindole 1a and α-imino γ-lactone 2a with Cs2CO3 as the base and methyl tert-butyl ether (MTBE) as the solvent for screening the optimal reaction conditions. As shown in Table 1, with the complex of Cu(CH3CN)4PF6 and (S)-BINAP-L1 as the catalyst, the reaction of 1a and 2a smoothly proceeded, giving the desired product 3a in 59% yield for the major diastereomer with moderate diastereoselectivity and 60% ee value (Table 1, entry 1). When the Pybox-L2 or (S)-iPr-PHOX-L3 as the chiral ligand for the reaction was examined, product 3a was obtained with only 42% ee and 35% ee, respectively (Table 1, entries 2 and 3). We also screened other BINAP derivatives as ligands, but no improved results were observed [48]. In the presence of (S)-BINAP-L1, replacing the metal salt Cu(CH3CN)4PF6 with CuBr, Cu(OAc)2, or Cu(OTf)2, we found that the reaction with Cu(OTf)2 was able to give product 3a with the best results (52% yield for the major diastereomer, 57:12:31:0 dr, and 81% ee) (Table 1, entry 6) under the identical conditions (Table 1, entries 4–6). Afterward, a survey of solvents including THF, CH2Cl2, toluene, PhCl, and xylene was implemented, and it indicated PhCl was the best candidate to generate product 3a in 45% yield for the major diastereomer, with 55:16:29:0 dr and 85% ee (Table 1, entry 10 vs. entries 7–9 and 11). Then, with anhydrous MgSO4 as an additive, a slightly improved enantioselectivity to 89% ee could be observed (Table 1, entry 12). Running the reaction at 0 °C, 3a could be obtained in 49% yield for the major diastereomer with 91% ee value but a prolonged reaction time to 120 h was needed (Table 1, entry 13). Ultimately, we were delighted to find that performing the reaction in PhCl with anhydrous MgSO4 as an additive at −30 °C for 120 h, the reaction proceeded well to provide 3a in 61% yield for the major diastereomer with good diastereoselectivity and up to 98% ee (Table 1, entry 14).
2.2. Substrate Scope Studies
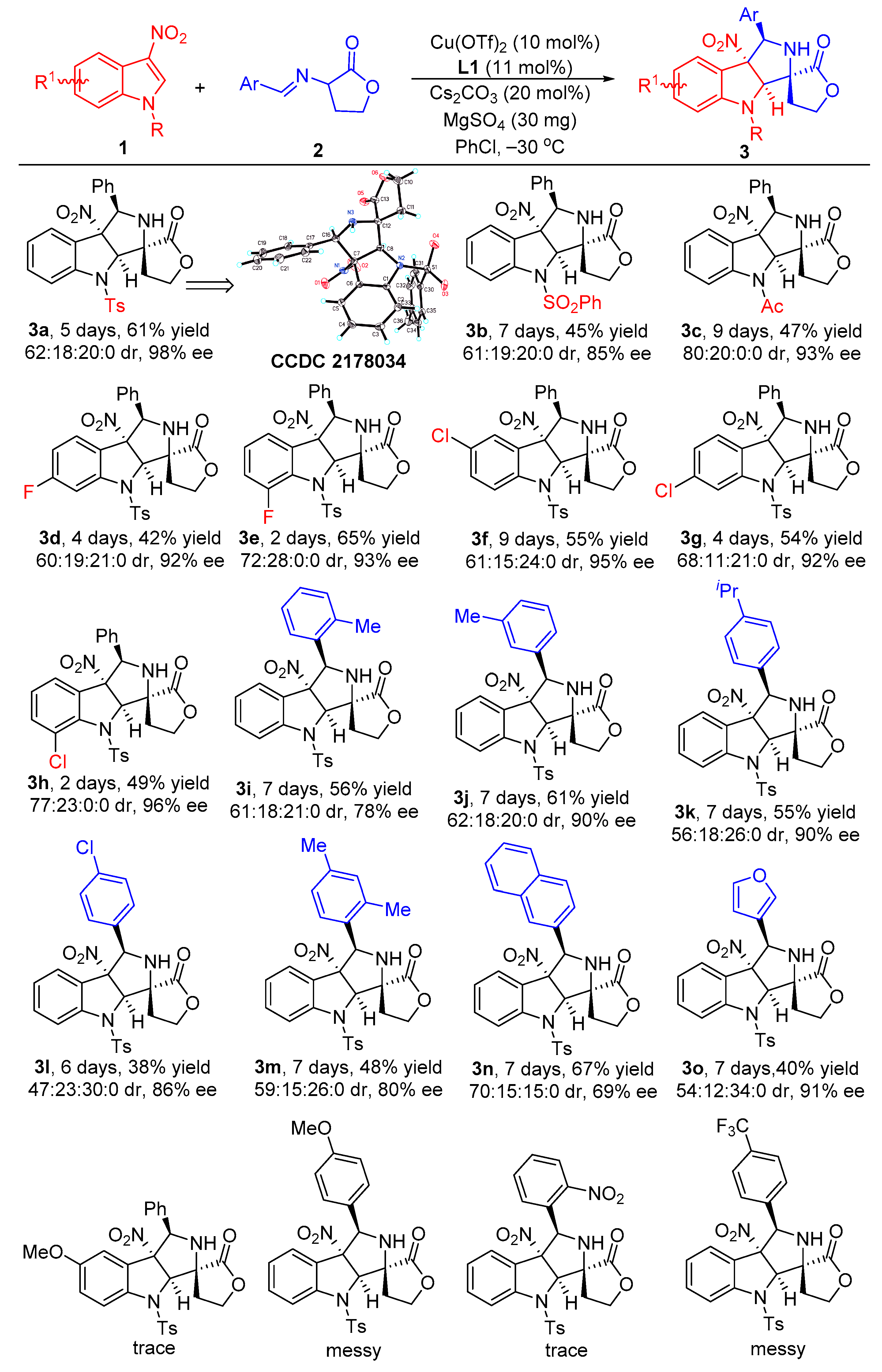
With the optimized reaction conditions in hand, we set out to investigate the substrate scope and generality of the copper-catalyzed asymmetric dearomative [3+2] cycloaddition reaction of different 3-nitroindoles 1 and diverse α-imino γ-lactones 2. As shown in Scheme 2, the 3-nitroindoles bearing different N-protecting groups, such as Ts-, Bs- and Ac-, were suitable substrates in reaction with α-imino γ-lactone 2a for generating the corresponding products 3a–c in 45–61% yields with 85–98% ee for their major diastereomers. In addition, installing different electron-withdrawing groups into the aromatic ring of N-Ts-3-nitroindoles, regardless of their positions on the aromatic ring, was not detrimental to reactivities and stereoselectivities of the dearomative [3+2] cycloaddition, affording major diastereomers products 3d–h in 42–65% yields with acceptable diastereoselectivities and excellent ee values (92–96% ee). However, for the reaction between 5-OMe substituted 3-nitroindole with α-imino γ-lactone 2a, only a trace amount of product could be observed. On the other hand, the investigation of the α-imino γ-lactones was also conducted by using the standard reaction conditions. Both electron-donating and electron-withdrawing substituents at different positions on the phenyl ring of α-imino γ-lactones were well tolerated in reaction with 3-nitroindole 1a, providing the desired cycloaddition products 3i–l in 38–61% yields with good diastereoselectivities and 78–90% ee for their major diastereomers. Moreover, the disubstituted substrate also could smoothly react with 3-nitroindole 1a to give the corresponding product 3m in 48% yield for the major diastereomer with 59:15:26:0 dr and 80% ee. As for bulker naphthyl-substituted α-imino γ-lactone, the developed catalytic system also showed good compatibility, furnishing the product 3n in 67% yield with good stereoselectivity. Additionally, the heteroaromatic substrate, such as 3-fury-substituted α-imino γ-lactone, also efficiently worked in this asymmetric dearomative [3+2] cycloaddition reaction, providing the desired product 3o in 40% yield for the major diastereomer with 54:12:34:0 dr and 91% ee. To our disappointment, the α-imino γ-lactones 2 with strong EDG (-OMe) or EWG groups (–NO2 and –CF3) in the phenyl ring by reacting with 3-nitroindole 1 became messy or showed very poor reactivity, and no expected products could be obtained. The absolute and relative configuration of product 3a was unambiguously determined to be C7R, C8R, C12S, and C16R by single crystal X-ray analysis, the configurations of all other products in Scheme 2 were assigned by analogy due to their formation via a common reaction pathway [49].
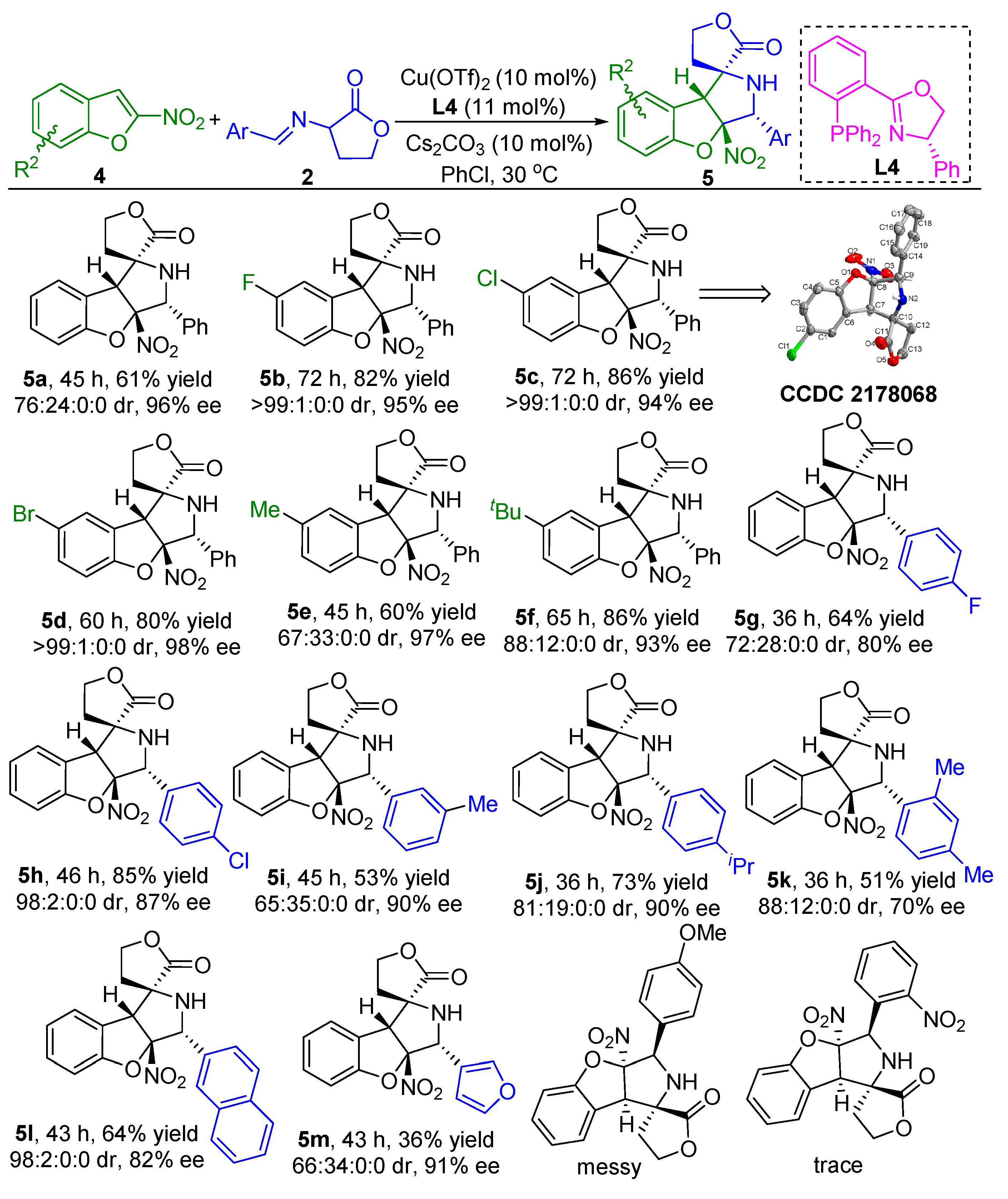
Having established a general scope to the catalytic asymmetric dearomative [3+2] cycloaddition reaction between 3-nitroindoles 1 and α-imino γ-lactones 2, we next attempted to apply the similar catalytic system to the asymmetric dearomative [3+2] cycloaddition reaction between α-imino γ-lactones 2 and 2-nitrobenzofuran substrates 4 (for details of the optimization of conditions, see the Supporting Information). As illustrated in Scheme 3, with the complex of Cu(OTf)2 and (S)-Ph-PHOX ligand L4 as the catalyst, Cs2CO3 as the base, the asymmetric dearomative [3+2] cycloaddition reaction of 2-nitrobenzofuran 4a and α-imino γ-lactone 2a smoothly proceeded in PhCl at 30 °C, furnishing the desired product 5a in 61% yield for the major diastereomer with 76:24:0:0 dr and 96% ee. As for the 2-nitrobenzofurans bearing various electron-withdrawing groups such as F-, Cl-, and Br- on the aryl ring, these substrates could work well in reaction with α-imino γ-lactone 2a under the standard conditions to give products 5b–d in 80–86% yields with excellent stereoselectivities (>99:1:0:0 dr and 94–98% ee). Likewise, 2-nitrobenzofurans bearing an electron-donating group were also viable for the developed protocol, as demonstrated in the synthesis of products 5e and 5f in good yields with 97% ee and 93% ee, respectively. On the other hand, the asymmetric dearomative [3+2] cycloaddition reactions of diverse α-imino γ-lactones containing either electron-withdrawing or electron-donating groups on the aryl ring with 2-nitrobenzofuran 4a could smoothly occur under the standard conditions, delivering the corresponding dearomative cycloaddition products 5g–j in 53–85% yields with good stereoselectivities for their major diastereomers (up to 98:2:0:0 dr and 90% ee). In addition, the α-imino γ-lactone with a double substitution pattern could also smoothly react with 2-nitrobenzofuran 4a under the standard conditions to generate product 5k in 51% yield for the major diastereomer with 88:12:0:0 and 70% ee. The bulky naphthyl group was also tolerated well with the catalytic system, resulting in the formation of product 5l in moderate yield with excellent diastereoselectivity and good enantioselectivity. Moreover, the 3-fury substituted substrate could smoothly react with 2-nitrobenzofuran 4a, delivering the corresponding product 5m in moderate yield with 66:34:0:0 dr and 91% ee. However, when para-OMe- or ortho-NO2-substituted α-imino γ-lactone was tested, no expected products could be obtained. The absolute and relative configuration of product 5c was determined to be C5R, C6R, C7R, and C8R by single crystal X-ray analysis, the configurations of all other products in Scheme 3 were assigned by analogy due to their formation via a common reaction pathway [49].
2.3. Scale-Up Experiment and the Versatile Transformations of Product 3a
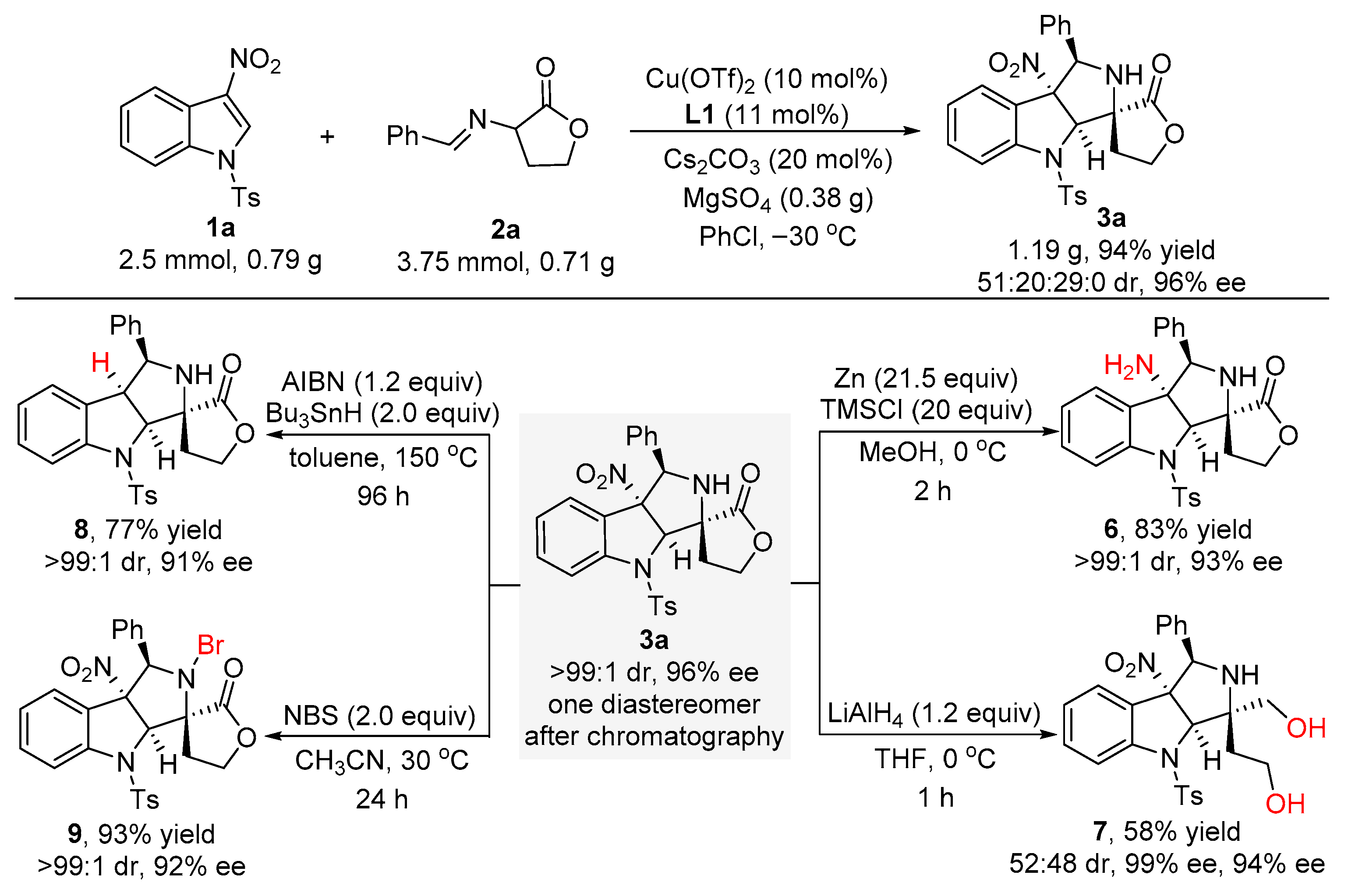
In order to demonstrate the potential synthetic application of the developed catalytic asymmetric dearomative [3+2] cycloaddition reaction, a scale-up experiment between 3-nitroindole 1a and α-imino γ-lactone 2a was conducted under the standard conditions. As shown in Scheme 4, the gram scale reaction was able to proceed to completion at −30 °C after 120 h, smoothly affording the cycloaddition product 3a in 94% yield for the sum of the diastereomers with 51:20:29:0 dr and 96% ee. To our delight, the major diastereomer of product 3a could be easily isolated by flash column chromatography on silica gel (0.6 g, 48% yield) without any loss of the enantioselectivity (>99:1 dr and 96% ee). Moreover, different synthetic transformations of 3a to other heterocyclic compounds were also performed (Scheme 4). Treating 3a with newly activated zinc powder and TMSCl in MeOH solvent for two hours, the nitro group of 3a could be converted into an amine group to give compound 6 in 83% yield with >99:1 dr and 93% ee. Reduction of 3a with LiAlH4 in THF for one hour gave rise to the formation of compound 7 in 58% yield with 52:48 dr and excellent enantioselectivities (99% ee and 94% ee for the diastereomers, respectively). In addition, 3a was subjected to tributyltin hydrogen and AIBN in toluene at 150 °C for 96 h in a sealed tube, thus affording compound 8 in 77% yield with >99:1 dr and 91% ee via a radical denitration process. Ultimately, the bromination reaction of 3a with N-bromosuccinimide (NBS) in CH3CN solvent could smoothly occur at 30 °C to produce compound 9 in 93% yield with >99:1 dr and 92% ee.
2.4. Proposed Mechanism for the Catalytic Asymmetric Dearomative [3+2] Cycloaddition
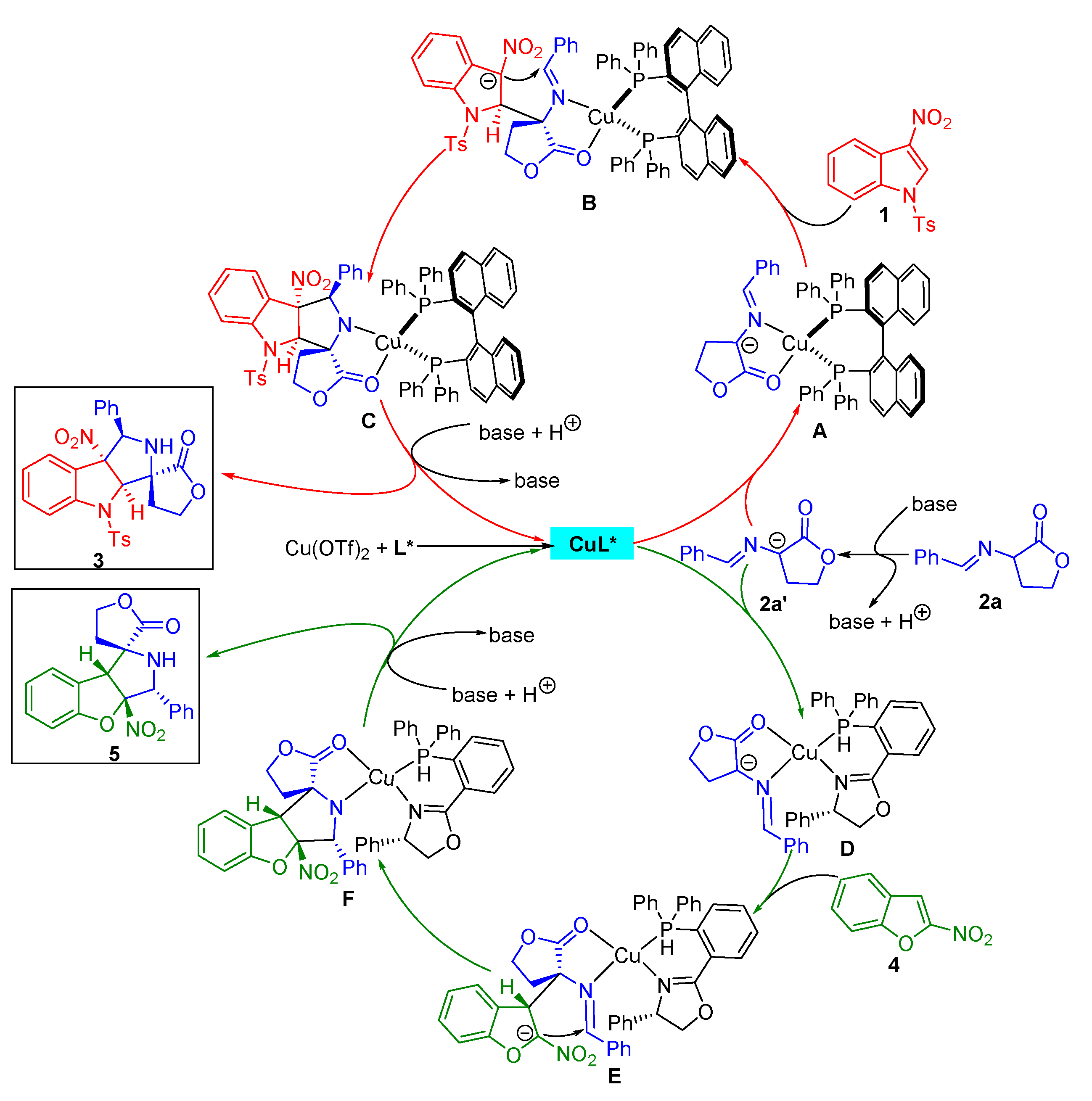
Based on our experimental results and previous related reports [27,28,29,30,31,37,38,39,40], a plausible catalytic mechanism involving a stepwise reaction process was assumed to explain the stereoselectivity of the asymmetric dearomative [3+2] cycloaddition of α-imino γ-lactones with electron-deficient nitroheteroarenes (Scheme 5). For the reaction with 3-nitroindoles as partners, the in situ-formed azomethine ylide 2a’ is coordinated to the chiral CuL* complex, leading to the active species A. Under the chiral environmental control, the α-carbon anion of 2a’ approaches the Re face of the C2-position of 3-nitroindoles 1, resulting in the intermediate B. Subsequently, the C3-positon carbon anion of 1 attacks the Si face of the C=N of azomethine ylide to deliver the species C, which is protonated, giving rise to the formation of products 3 with the specific stereochemical outcome and regenerating the chiral catalyst for the next cycle. Likewise, for the reaction with 2-nitrobenzofurans as partners, the active species D is formed from the azomethine ylide 2a’ and the chiral CuL* complex. The α-carbon anion of 2a’ attacks the Si face of the C3-position of 2-nitrobenzofurans to form intermediate E, which undergoes an intramolecular cyclization from the C2-positon carbon anion of 4 to the Si face of the C=N of azomethine ylide to generate species F. Then, a protonation of F leads to the formation of products 5 and the release of a chiral catalyst.
3. Materials and Methods
3.1. General Information
Reagents were purchased from commercial sources and were used as received unless mentioned otherwise. Reactions were monitored by thin-layer chromatography (TLC). 1H NMR and 13C NMR spectra were recorded in CDCl3 and DMSO-d6. 1H NMR chemical shifts are reported in ppm relative to tetramethylsilane (TMS) with the solvent resonance employed as the internal standard (CDCl3 at 7.26 ppm and DMSO-d6 at 2.50 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants (Hz), and integration. 13C NMR chemical shifts are reported in ppm from tetramethylsilane (TMS) with the solvent resonance as the internal standard (CDCl3 at 77.20 ppm and DMSO-d6 at 39.52 ppm). The enantiomeric excesses were determined by chiral high-performance liquid chromatography (HPLC) analysis. High-performance liquid chromatography analysis (HPLC) was performed on Shimadzu SCL-10AVP HPLC systems consisting of a LC-10AD pump and an SPD-10A detector, measured at 254 nm. High-resolution mass spectrometer (HRMS) was recorded on Bruker Q TOF. Optical rotations were measured with a Perkin-Elmer-341 polarimeter. Melting points were recorded on a Büchi Melting Point B-545.
3.2. General Experimental Procedure for the Catalytic Asymmetric Dearomative [3+2] Cycloaddtion Reaction of α-imino γ-lactones with 3-nitroindoles for the Synthesis of Products 3 (Scheme 2)
MgSO4 (30 mg), ligand L1 (11 mol%, 0.11 equiv), and Cu(OTf)2 (10 mol%, 0.1 equiv) were added to a Schlenk tube containing a magnetic stir bar under an argon atmosphere. chlorobenzene (1.0 mL) was added to the tube and the mixture was stirred for 1.0 h at room temperature. Then, in the resulting solution, a mixture including α-imino γ-lactones (0.3 mmol, 1.5 equiv), Cs2CO3 (20 mol%, 0.20 equiv), 3-nitroindoles (0.2 mmol, 1.0 equiv), and chlorobenzene (1.0 mL) was subsequently added under an argon atmosphere at −30 °C. After being stirred for the appropriate reaction time, the reaction mixture was quenched with water. The organic layer was extracted with dichloromethane three times. The collected organic layer was dried over anhydrous Na2SO4. After the removal of the solvent under reduced pressure, the diastereomeric ratio was determined by 1H NMR analysis. The resulting crude mixture was purified by silica gel column chromatography to afford cycloadducts 3. The enantiomeric excesses of product 3 were determined by chiral stationary phase HPLC using a Chiralpak AD-H, AS-H, IA, IB, and Chiralpak IC column.
3.3. General Experimental Procedure for the Catalytic Asymmetric Dearomative [3+2] Cycloaddtion Reaction of α-imino γ-lactones with 2-nitrobenzofurans for the Synthesis of Products 5 (Scheme 3)
Ligand L4 (11 mol%, 0.11 equiv) and Cu(OTf)2 (10 mol%, 0.10 equiv) were added to a Schlenk tube containing a magnetic stir bar under an argon atmosphere. Chlorobenzene (1.0 mL) was added to the tube and the mixture was stirred for 1.0 h at room temperature. Then, in the resulting solution, a mixture including α-imino γ-lactones (0.3 mmol, 1.5 equiv), Cs2CO3 (10 mol%, 0.10 equiv), 2-nitrobenzofurans (0.2 mmol, 1.0 equiv), and chlorobenzene (1.0 mL) were subsequently added under an argon atmosphere at 30 °C. After being stirred for the appropriate reaction time, the reaction mixture was quenched with water. The organic layer was extracted three times with dichloromethane. The collected organic layer was dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the resulting crude mixture was purified by silica gel column chromatography to afford cycloadducts 5. The diastereomeric ratio and the enantiomeric excess values of product 5 were determined by chiral stationary phase HPLC using a Chiralpak AD-H, AS-H, IA, IB, and Chiralpak IC column.
3.4. Procedure for the Catalytic Asymmetric Synthesis of Product 3a on a 2.5 mmol Scale
MgSO4 (375 mg), ligand L1 (11 mol%, 171 mg), and Cu(OTf)2 (10 mol%. 90.5 mg) were added to a Schlenk tube containing a magnetic stir bar under an argon atmosphere. chlorobenzene (12.5 mL) was added to the tube and the mixture was stirred for 1.0 h at room temperature. In the resulting solution, the mixture including α-imino γ-lactone 2a (3.75 mmol, 0.71 g), Cs2CO3 (20 mol%, 163 mg), 3-nitroindole 1a (2.5 mmol, 0.79 g), and chlorobenzene (12.5 mL) were subsequently added under an argon atmosphere at −30 °C. After being stirred for 120 h, the reaction mixture was quenched with water. The organic layer was extracted three times with dichloromethane. The collected organic layer was dried over anhydrous Na2SO4. After the removal of the solvent under reduced pressure, the diastereomeric ratio was determined as 51:20:29:0 dr by 1H NMR analysis. The resulting crude mixture was purified by silica gel column chromatography using petroleum ether/ethyl acetate (8:1) as the eluent to afford cycloadduct 3a for the sum of diastereomers (1.19 g, 94% yield, 51:20:29:0 dr, and 96% ee).
3.5. Procedure for the Synthesis of Compound 6
Newly activated zinc powder (140.6 mg, 2.15 mmol, 21.5 equiv) was slowly added into a solution of 3a (>99:1 dr, 96% ee) (50.6 mg, 0.10 mmol, 1.0 equiv) and trimethylsilyl chloride (0.25 mL, 2.00 mmol, 20.0 equiv) in 1.0 mL of methanol at 0 °C. After the mixture was stirred at 0 °C for 2 h, the suspension was filtered and washed with methanol two times (2 × 5.0 mL) and dichloromethane two times (2 × 5.0 mL), respectively. After the filtrate was washed with saturated NaHCO3 aqueous two times (2 × 15 mL), the organic phase was separated while the aqueous phase was extracted with dichloromethane three times (3 × 15 mL). The combined organic phase was washed with brine three times (3 × 15 mL) and then dried with anhydrous Na2SO4. Subsequently, the organic phase was concentrated under reduced pressure. The residue was subjected to column chromatography over silica gel using petroleum ether/ethyl acetate (4:1) as the eluent to give compound 6 as a white solid (83% yield, >99:1 dr, and 93% ee).
3.6. Procedure for the Synthesis of Compound 7
LiAlH4 (4.5 mg, 0.12 mmol, 1.2 equiv) was cautiously added into the solution of compound 3a (>99:1 dr, 96% ee) (50.6 mg, 0.1 mmol, 1.0 equiv) in the 1.0 mL of THF at 0 °C. After the mixture was stirred at 0 °C for 1.0 h, the resulting mixture was quenched with 10 mL of 0.1 mol/L NaOH solution and extracted with dichloromethane three times (3 × 15 mL). The organic phase was isolated and washed with brine three times (3 × 15 mL), and then dried with anhydrous Na2SO4. Subsequently, the organic phase was concentrated under reduced pressure. The residue was subjected to column chromatography over silica gel using petroleum ether/ethyl acetate (2:1) (about 200 mL) as eluent, and then using MeOH as an eluent to give compound 7 as a white solid (58% yield, 52:48 dr, and 99% ee/94% ee).
3.7. Procedure for the Synthesis of Compound 8
Tributyltin hydride (54 µL, 58.2 mg, 0.2 mmol, 2.0 equiv) was added to a solution of compound 3a (>99:1 dr, 96% ee) (50.6 mg, 0.1mol, 1.0 equiv) and AIBN (azobisisobutyronitrile) (19.7 mg, 0.12 µmol, 1.2 equiv) in toluene (1.0 mL). Then, the reaction mixture in a sealed tube was stirred at 150 °C for 96 h. Subsequently, the reaction mixture was cooled to room temperature, the solution was concentrated under reduced pressure. The residue was subjected to column chromatography over silica gel using petroleum ether/ethyl acetate (7:1) as an eluent to give compound 8 as a white solid (77% yield, >99:1 dr, and 91% ee).
3.8. Procedure for the Synthesis of Compound 9
NBS (N-Bromosuccinimide) (35.6 mg, 0.2 mmol, 2.0 equiv) was added into the solution of compound 3a (>99:1 dr, 96% ee) (50.6 mg, 0.1 mmol, 1.0 equiv) in the 1.0 mL of MeCN, and the reaction mixture was stirred at 30 °C for 24 h. Subsequently, the resulting mixture was quenched with 10 mL of saturated NH4Cl solution and extracted with dichloromethane three times (3 × 15 mL). The combined organic phase was washed with brine three times (3 × 15 mL) and then dried with anhydrous Na2SO4. Subsequently, the organic phase was concentrated under reduced pressure. The residue was subjected to column chromatography over silica gel using petroleum ether/ethyl acetate (6:1) as an eluent to give compound 9 as a white solid (93% yield, >99:1 dr, and 92% ee).
4. Conclusions
In summary, we have successfully developed a copper-catalyzed asymmetric dearomative [3+2] cycloaddition reaction of α-imino γ-lactones with nitroheteroarenes including 3-nitroindoles and 2-nitrobenzofurans. A wide range of structurally diverse polyheterocyclic compounds containing spirocyclic-fused butyrolactone–pyrrolidine–indoline and butyrolactone–pyrrolidine–dihydrobenzofuran skeletons bearing four contiguous chiral stereocenters could be smoothly obtained with excellent results (>99:1 dr and 98% ee) under mild reaction conditions. Importantly, these innovative polyheterocyclic compounds would be potentially promising for high-throughout drug screening due to the intriguing fusion of three privileged butyrolactone, pyrrolidine, and indoline/dihydrobenzofuran subunits into one molecular architecture. The potential synthetic applications of the dearomative [3+2] cycloaddition reaction were also demonstrated by the scale-up experiment and the diverse transformations of one product to diverse heterocyclic compounds. More importantly, this developed protocol also shows distinct features including high asymmetric induction, broad functional group tolerance and scalability, and attractive product diversification. The application of the asymmetric dearomative cycloaddition reaction of electron-deficient nitroheteroarenes toward library synthesis and subsequent biological evaluation of its members is underway.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28062765/s1, characterization data for obtained products; X-ray data for products 3a and 5c; copies of 1H, 13C NMR, and HPLC spectra.
Author Contributions
Conceptualization, X.-M.Z. and W.-C.Y.; methodology, Y.C.; investigation, J.-Q.Z., Y.-P.Z. and M.-Q.Z.; writing—original draft preparation, J.-Q.Z. and W.-C.Y.; writing—review and editing, X.-M.Z. and W.-C.Y.; supervision, X.-M.Z. and W.-C.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of China, grant numbers 22271027, 22171029, 21901024, and 21871252; the Sichuan Science and Technology Program, grant number 2021YFS0315; and the Talent Program of Chengdu University, grant numbers 2081919035 and 2081921038.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the required data are reported in the manuscript and Supplementary Materials.
Acknowledgments
This work was performed using the equipment of Chengdu University and Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References and Notes
- Lee, K.K.; Zhou, B.N.; Kingston, D.G.I.; Vaisberg, A.J.; Hammond, G.B. Bioactive Indole Alkaloids from the Bark of Uncaria guianensis. Planta Med. 1999, 65, 759–760. [Google Scholar] [CrossRef]
- Kang, T.-H.; Matsumoto, K.; Murakami, Y.; Takayama, H.; Kitajima, M.; Aimi, N.; Watanabe, H. Pteropodine and isopteropodine positively modulate the function of rat muscarinic M1 and 5-HT2 receptors expressed in Xenopus oocyte. Eur. J. Pharmacol. 2002, 444, 39–45. [Google Scholar] [CrossRef]
- Raj, A.A.; Raghunathan, R.; SrideviKumari, M.R.; Raman, N. Synthesis, Antimicrobial and Antifungal Activity of a New Class of Spiro pyrrolidines. Bioorg. Med. Chem. 2003, 11, 407–419. [Google Scholar] [CrossRef]
- Dzedulionytė, K.; Veikšaitė, M.; Morávek, V.; Malinauskienė, V.; Račkauskienė, G.; Šačkus, A.; Žukauskaitė, A.; Arbačiauskienė, E. Convenient Synthesis of N-Heterocycle-Fused Tetrahydro-1,4-diazepinones. Molecules 2022, 27, 8666. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Huang, J.; Yao, M.; Xu, X. Recent Advances in Nitrene/Alkyne Metathesis Cascade Reaction. Chin. J. Org. Chem. 2022, 42, 344–352. [Google Scholar] [CrossRef]
- Philippov, I.; Gatilov, Y.; Sonina, A.; Vorob’ev, A. Oxidative [3+2] Cycloaddition of Alkynylphosphonates with Heterocyclic N-Imines: Synthesis of Pyrazolo [1,5-a]Pyridine-3-phosphonates. Molecules 2022, 27, 7913. [Google Scholar] [CrossRef]
- Dong, S.; Fu, X.; Xu, X. [3+2]-Cycloaddition of Catalytically Generated Pyridinium Ylide: A General Access to Indolizine Derivatives. Asian J. Org. Chem. 2020, 9, 1133–1143. [Google Scholar] [CrossRef]
- Hazra, A.; Bharitkar, Y.P.; Chakraborty, D.; Mondal, S.K.; Singal, N.; Mondal, S.; Maity, A.; Paira, R.; Banerjee, S.; Mondal, N.B. Regio- and Stereoselective Synthesis of a Library of Bioactive Dispiro-Oxindolo/Acenaphthoquino Andrographolides via 1,3-Dipolar Cycloaddition Reaction Under Microwave Irradiation. ACS Comb. Sci. 2013, 15, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.H.; Chua, P.; Downham, R.; Douglas, C.J.; Garg, N.K.; Hiebert, S.; Jaroch, S.; Matsuoka, R.T.; Middleton, J.A.; Ng, F.W.; et al. Total Synthesis of (-)-Sarain A. J. Am. Chem. Soc. 2007, 129, 11987–12002. [Google Scholar] [CrossRef]
- Sun, M.-R.; Lu, H.-T.; Wang, Y.-Z.; Yang, H.; Liu, H.-M. Highly Efficient Formal Synthesis of Cephalotaxine, Using the Stevens Rearrangement-Acid Lactonization Sequence as A Key Transformation. J. Org. Chem. 2009, 74, 2213–2216. [Google Scholar] [CrossRef]
- Zhuo, C.-X.; Zhang, W.; You, S.-L. Catalytic Asymmetric Dearomatization Reactions. Angew. Chem. Int. Ed. 2012, 51, 12662–12686. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.-X.; Zheng, C.; You, S.-L. Transition-Metal-Catalyzed Asymmetric Allylic Dearomatization Reactions. Acc. Chem. Res. 2014, 47, 2558–2573. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, S.-L. Catalytic Asymmetric Dearomatization by Transition-Metal Catalysis: A Method for Transformations of Aromatic Compounds. Chem 2016, 1, 830–857. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.-T.; Zhang, L.; You, S.-L. Recent Progress on Gold-catalyzed Dearomatization Reactions. Acta Chim. Sinica 2017, 75, 419–438. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; You, S.-L. Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products. Nat. Prod. Rep. 2019, 36, 1589–1605. [Google Scholar] [CrossRef]
- Xia, Z.-L.; Xu-Xu, Q.-F.; Zheng, C.; You, S.-L. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem. Soc. Rev. 2020, 49, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, S.-L. Advances in Catalytic Asymmetric Dearomatization. ACS Cent. Sci. 2021, 7, 432–444. [Google Scholar] [CrossRef]
- Wu, W.-T.; Zhang, L.; You, S.-L. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev. 2016, 45, 1570–1580. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Yin, B. Recent Developments in Transition Metal-Catalyzed Dearomative Cyclizations of Indoles as Dipolarophiles for the Construction of Indolines. Adv. Synth. Catal. 2019, 361, 405–425. [Google Scholar] [CrossRef]
- Sheng, F.-T.; Wang, J.-Y.; Tan, W.; Zhang, Y.-C.; Shi, F. Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives. Org. Chem. Front. 2020, 7, 3967–3998. [Google Scholar] [CrossRef]
- Trost, B.M.; Ehmke, V.; O’Keefe, B.M.; Bringley, D.A. Palladium-Catalyzed Dearomative Trimethylenemethane Cycloaddition Reactions. J. Am. Chem. Soc. 2014, 136, 8213–8216. [Google Scholar] [CrossRef]
- Cerveri, A.; Bandini, M. Recent Advances in the Catalytic Functionalization of “Electrophilic” Indoles. Chin. J. Chem. 2020, 38, 287–294. [Google Scholar] [CrossRef]
- Rkein, B.; Bigot, A.; Birbaum, L.; Manneveau, M.; De Paolis, M.; Legros, J.; Chataigner, I. Reactivity of 3-nitroindoles with electron-rich species. Chem. Commun. 2021, 57, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.R.; Baire, B. Recent Dearomatization Strategies of Benzofurans and Benzothiophenes. Asian J. Org. Chem. 2021, 10, 932–948. [Google Scholar] [CrossRef]
- Wang, N.; Ren, J.; Li, K. Dearomatization of Nitro(hetero)arenes through Annulation. Eur. J. Org. Chem. 2022, 2022, e202200039. [Google Scholar] [CrossRef]
- Li, Y.-L.; Wang, K.-K.; He, X.-L. Recent Progress of Electron-Withdrawing-Group-Tethered Arenes Involved Asymmetric Nucleophilic Aromatic Functionalizations. Adv. Synth. Catal. 2022, 364, 3630–3650. [Google Scholar] [CrossRef]
- Awata, A.; Arai, T. PyBidine/Copper Catalyst: Asymmetric exo′-Selective [3+2] Cycloaddition using Imino Ester and Electrophilic Indole. Angew. Chem. Int. Ed. 2014, 53, 10462–10465. [Google Scholar] [CrossRef]
- Gerten, A.L.; Stanley, L.M. Enantioselective dearomative [3+2] cycloadditions of indoles with azomethine ylides derived from alanine imino esters. Org. Chem. Front. 2016, 3, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Niu, H.-Y.; Wang, D.-C.; Yang, X.-H.; Qu, G.-R.; Guo, H.-M. Facile synthesis of chiral [2,3]-fused hydrobenzofuran via asymmetric Cu(I)-catalyzed dearomative 1,3-dipolar cycloaddition. Chem. Commun. 2019, 55, 553–556. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.-C.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M. Enantioselective Synthesis of Fused Polycyclic Tropanes via Dearomative [3+2] Cycloaddition Reactions of 2-Nitrobenzofurans. Org. Lett. 2020, 22, 164–167. [Google Scholar] [CrossRef]
- Tan, J.-P.; Li, X.; Chen, Y.; Rong, X.; Zhu, L.; Jiang, C.; Xiao, K.; Wang, T. Highly stereoselective construction of polycyclic benzofused tropane scaffolds and their latent bioactivities: Bifunctional phosphonium salt-enabled cyclodearomatization process. Sci. China Chem. 2020, 63, 1091–1099. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, X.; Zhang, H.; Tan, J.-P.; Ren, X.; Gao, G.; Wang, T. Bifunctinoal Phosphonium Salt-Catalyzed Asymmetric Cyclodearomatization of 2-Nitroindoles and 2-Nitrobenzofurans for Constructing CF3-Containing Spiro-Polycycles. Adv. Synth. Catal. 2021, 363, 3115–3120. [Google Scholar] [CrossRef]
- Saktura, M.; Skrzyńska, A.; Frankowski, S.; Wódka, S.; Albrecht, Ł. Asymmetric Dearomative (3+2)-Cycloaddition Involving Nitro-Substituted Benzoheteroarenes under H-Bonding Catalysis. Molecules 2021, 26, 4992. [Google Scholar] [CrossRef]
- Zhao, J.-Q.; Zhou, S.; Yang, L.; Du, H.-Y.; You, Y.; Wang, Z.-H.; Zhou, M.-Q.; Yuan, W.-C. Catalytic Asymmetric Dearomative 1,3-Dipolar Cycloaddition of 2-Nitrobenzothiophenes and Isatin-Derived Azomethine Ylides. Org. Lett. 2021, 23, 8600–8605. [Google Scholar] [CrossRef]
- Zhou, P.; Yi, Y.; Hua, Y.-Z.; Jia, S.-K.; Wang, M.-C. Dinuclear Zinc Catalyzed Enantioselective Dearomatization [3+2] Annulation of 2-Nitrobenzofurans and 2-Nitrobenzothiophenes. Chem. Eur. J. 2021, 28, e202103688. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-J.; Zhao, J.-Q.; Lai, Y.-Q.; You, Y.; Wang, Z.-H.; Yuan, W.-C. Organocatalyzed asymmetric dearomative 1,3-dipolar cycloaddition of 2-nitrobenzofurans and N-2,2,2-trifluoroethylisatin ketimines. Chirality 2022, 34, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.-C.; Zhou, X.-J.; Zhao, J.-Q.; Chen, Y.-Z.; You, Y.; Wang, Z.-H. Catalytic Enantioselective Dearomatization/Rearomatization of 2-Nitroindoles to Access 3-Indolyl-3′-Aryl-/Alkyloxindoles: Application in the Formal Synthesis of Cyclotryptamine Alkaloids. Org. Lett. 2020, 22, 7088–7093. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-Q.; Zhou, S.; Wang, Z.-H.; You, Y.; Chen, S.; Liu, X.-L.; Zhou, M.-Q.; Yuan, W.-C. Catalytic asymmetric dearomative [4 + 2] annulation of 2-nitrobenzofurans and 5H-thiazol-4-ones: Stereoselective construction of dihydrobenzofuran-bridged polycyclic skeletons. Org. Chem. Front. 2021, 8, 6330–6336. [Google Scholar] [CrossRef]
- Zhao, J.-Q.; Zhou, S.; Qian, H.-L.; Wang, Z.-H.; Zhang, Y.-P.; You, Y.; Yuan, W.-C. Higher-order [10 + 2] cycloaddition of 2-alkylidene-1-indanones enables the dearomatization of 3-nitroindoles: Access to polycyclic cyclopenta[b]indoline derivatives. Org. Chem. Front. 2022, 9, 3322–3327. [Google Scholar] [CrossRef]
- Yuan, W.-C.; Chen, X.-M.; Zhao, J.-Q.; Zhang, Y.-P.; Wang, Z.-H.; You, Y. Ag-Catalyzed Asymmetric Interrupted Barton-Zard Reaction Enabling the Enantioselective Dearomatization of 2- and 3-Nitroindoles. Org. Lett. 2022, 24, 826–831. [Google Scholar] [CrossRef]
- Liu, T.-L.; He, Z.-L.; Tao, H.-Y.; Wang, C.-J. Stereoselective Construction of Spiro(butyrolactonepyrrolidines) by Highly Efficient Copper(I)/TF-BiphamPhos-Catalyzed Asymmetric 1,3-Dipolar Cycloaddition. Chem. Eur. J. 2012, 18, 8042–8046. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.-L.; Huang, H.; Wang, C.-J. Catalytic Asymmetric Construction of Spiro(γ-butyrolactam-γ-butyrolactone) Moieties through Sequential Reactions of Cyclic Imino Esters with Morita–Baylis–Hillman Bromides. Chem. Eur. J. 2012, 18, 12614–14628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, X.-M.; Dong, W.-P.; Zhua, L.-P.; Wang, R. Efficient construction of highly functionalized spiro[γ-butyrolactone-pyrrolidin-3,3′-oxindole] tricyclic skeletons via an organocatalytic 1,3-dipolar cycloaddition. Chem. Commun. 2013, 49, 3458–3460. [Google Scholar] [CrossRef]
- Cayuelas, A.; Ortiz, R.; Nájera, C.; Sansano, J.M.; Larrañaga, O.; de Cózar, A.; Cossío, F.P. Enantioselective Synthesis of Polysubstituted Spiro-nitroprolinates Mediated by a (R,R)-Me-DuPhos·AgF-Catalyzed 1,3-Dipolar Cycloaddition. Org. Lett. 2016, 18, 2926–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.-W.; Li, C.; Zhao, B.-J.; Lan, L.; Zhang, M.; Zhou, Z.-M. Diastereoselective construction of spirocyclic pyrrolidines bearing two quaternary centers via CuII-P, N-Ligand catalyzed 1,3-dipolar cycloaddition. Tetrahedron 2017, 73, 923–930. [Google Scholar] [CrossRef]
- Chen, N.; Zhu, L.; Gan, L.; Liu, Z.; Wang, R.; Cai, X.; Jiang, X. Asymmetric Synthesis of Bispiro[γ-butyrolactone-pyrrolidin-4,4′-pyrazolone] Scaffolds Containing Two Quaternary Spirocenters via an Organocatalytic 1,3-Dipolar Cycloaddition. Eur. J. Org. Chem. 2018, 2018, 2939–2943. [Google Scholar] [CrossRef]
- Guo, D.-G.; Li, Z.; Han, X.-X.; Zhang, L.; Zhang, M.; Liu, X.-L. Decarboxylative, Diastereoselective and exo-Selective 1,3-Dipolar Cycloaddition for Diversity-Oriented Construction of Structural Spiro[Butyrolactone–Pyrrolidine–Chromanone] Hybrids. Synlett 2021, 32, 1447–1452. [Google Scholar]
- According to reviewer’s require, we screened other BINAP derivatives ligands with Cu(CH3CN)4PF6 as the metal source in PhCl at rt for 72 h ((R)-Tol-BINAP: 33% yield, 45:39:16:0 dr, 69% ee; (S)-Di(3,5-xylyl)-BINAP: 21% yield, 70:10:20:0 dr, 45% ee; (R)-Segphos: 15% yield, 83:8:9:0 dr, 73% ee).
- CCDC 2178034 (3a) and CCDC 2178068 (5c) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif, or by emailing [email protected].
Figure 1.
Selected bioactive compound and the key intermediate of nature products containing a chiral spirocyclic butyrolactone-pyrrolidine moiety as the structural core.
Figure 1.
Selected bioactive compound and the key intermediate of nature products containing a chiral spirocyclic butyrolactone-pyrrolidine moiety as the structural core.

Scheme 1.
Profile of catalytic asymmetric dearomative [3+2] cycloaddition reaction of electron-deficient nitroheteroarenes and diverse azomethine ylides.
Scheme 1.
Profile of catalytic asymmetric dearomative [3+2] cycloaddition reaction of electron-deficient nitroheteroarenes and diverse azomethine ylides.

Scheme 2.
Substrate scope investigation of the catalytic asymmetric dearomtive [3+2] cycloaddition reaction of 3-nitroindoles and α-imino γ-lactones. Reaction conditions: the reactions were carried out with 3-nitroindoles 1 (0.2 mmol, 1.0 equiv), α-imino γ-lactones 2 (0.3 mmol, 1.5 equiv), Cu(OTf)2 (10 mol%), L1 (11 mol%), Cs2CO3 (20 mol%), and MgSO4 (30 mg) in 2.0 mL of PhCl as the solvent at −30 °C under argon atmosphere. The yields refer to the isolated yield of the major diastereomers. The dr values were determined by 1H NMR spectroscopy of the crude mixture. The ee values were determined by chiral high-performance liquid chromatography (HPLC) analysis after purification of the diastereomers.
Scheme 2.
Substrate scope investigation of the catalytic asymmetric dearomtive [3+2] cycloaddition reaction of 3-nitroindoles and α-imino γ-lactones. Reaction conditions: the reactions were carried out with 3-nitroindoles 1 (0.2 mmol, 1.0 equiv), α-imino γ-lactones 2 (0.3 mmol, 1.5 equiv), Cu(OTf)2 (10 mol%), L1 (11 mol%), Cs2CO3 (20 mol%), and MgSO4 (30 mg) in 2.0 mL of PhCl as the solvent at −30 °C under argon atmosphere. The yields refer to the isolated yield of the major diastereomers. The dr values were determined by 1H NMR spectroscopy of the crude mixture. The ee values were determined by chiral high-performance liquid chromatography (HPLC) analysis after purification of the diastereomers.

Scheme 3.
Substrate scope investigation of the catalytic asymmetric dearomtive [3+2] cycloaddition reaction of 2-nitrobenzofurans and α-imino γ-lactones. Reaction conditions: the reactions were carried out with 2-nitrobenzofurans 4 (0.2 mmol, 1.0 equiv), α-imino γ-lactones 2 (0.3 mmol, 1.5 equiv), Cu(OTf)2 (10 mol%), L4 (11 mol%), and Cs2CO3 (10 mol%) in 2.0 mL of PhCl as the solvent at 30 °C under argon atmosphere. The yields refer to the isolated yield of the major diastereomers. The dr values and ee values were determined by chiral high-performance liquid chromatography (HPLC) analysis.
Scheme 3.
Substrate scope investigation of the catalytic asymmetric dearomtive [3+2] cycloaddition reaction of 2-nitrobenzofurans and α-imino γ-lactones. Reaction conditions: the reactions were carried out with 2-nitrobenzofurans 4 (0.2 mmol, 1.0 equiv), α-imino γ-lactones 2 (0.3 mmol, 1.5 equiv), Cu(OTf)2 (10 mol%), L4 (11 mol%), and Cs2CO3 (10 mol%) in 2.0 mL of PhCl as the solvent at 30 °C under argon atmosphere. The yields refer to the isolated yield of the major diastereomers. The dr values and ee values were determined by chiral high-performance liquid chromatography (HPLC) analysis.

Scheme 4.
Scale-up experiment and further transformations of compound 3a.

Scheme 5.
Proposed mechanism for the catalytic asymmetric dearomative [3+2] cycloaddition of α-imino γ-lactones with 3-nitroindoles and 2-nitrobenzofurans.
Scheme 5.
Proposed mechanism for the catalytic asymmetric dearomative [3+2] cycloaddition of α-imino γ-lactones with 3-nitroindoles and 2-nitrobenzofurans.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of reaction conditions [a].

| Entry | [Cu] | Ligand | Solvent | Time (h) | Yield (%) [b] | dr [c] | ee (%) [d] |
|---|---|---|---|---|---|---|---|
| 1 | Cu(CH3CN)4PF6 | L1 | MTBE | 72 | 94 (59) | 63:15:22:0 | 60 |
| 2 | Cu(CH3CN)4PF6 | L2 | MTBE | 72 | 96 (64) | 67:20:13:0 | 42 |
| 3 | Cu(CH3CN)4PF6 | L3 | MTBE | 66 | 93 (47) | 50:15:35:0 | 35 |
| 4 | CuBr | L1 | MTBE | 70 | 85 (54) | 63:11:26:0 | 21 |
| 5 | Cu(OAc)2 | L1 | MTBE | 70 | 72 (47) | 65:10:24:0 | 31 |
| 6 | Cu(OTf)2 | L1 | MTBE | 70 | 91 (52) | 57:12:31:0 | 81 |
| 7 | Cu(OTf)2 | L1 | THF | 68 | 87 (60) | 69:9:22:0 | 17 |
| 8 | Cu(OTf)2 | L1 | CH2Cl2 | 68 | 86 (52) | 61:11:28:0 | 46 |
| 9 | Cu(OTf)2 | L1 | toluene | 68 | 92 (46) | 50:15:35:0 | 78 |
| 10 | Cu(OTf)2 | L1 | PhCl | 68 | 82 (45) | 55:16:29:0 | 85 |
| 11 [e] | Cu(OTf)2 | L1 | PhCl | 90 | 99 (51) | 52:16:32:0 | 89 |
| 12 [e,f] | Cu(OTf)2 | L1 | PhCl | 120 | 99 (49) | 49:18:33:0 | 91 |
| 13 [e,g] | Cu(OTf)2 | L1 | PhCl | 120 | 99 (61) | 62:18:20:0 | 98 |
[a] Unless otherwise noted, the reaction was carried out with 1a (0.2 mmol, 1.0 equiv), 2a (0.3 mmol, 1.5 equiv), Cu salt (10 mol%), ligand (11 mol%), and Cs2CO3 (20 mol%) in 2.0 mL of solvent at room temperature under argon atmosphere. [b] Isolated yield of the sum of the diastereomers. The yield in the bracket refers to the isolated yield of the major diastereomer. [c] The dr values were determined by 1H NMR. [d] The ee values were determined by chiral high-performance liquid chromatography (HPLC) analysis after purification of the diastereomers. [e] 30 mg MgSO4 was added to the reaction mixture as an additive. [f] The reaction was run at 0 °C. [g] The reaction was run at −30 °C.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, Y.; Zhao, J.-Q.; Zhang, Y.-P.; Zhou, M.-Q.; Zhang, X.-M.; Yuan, W.-C. Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines. Molecules 2023, 28, 2765. https://doi.org/10.3390/molecules28062765
AMA Style
Chen Y, Zhao J-Q, Zhang Y-P, Zhou M-Q, Zhang X-M, Yuan W-C. Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines. Molecules. 2023; 28(6):2765. https://doi.org/10.3390/molecules28062765
Chicago/Turabian StyleChen, Yan, Jian-Qiang Zhao, Yan-Ping Zhang, Ming-Qiang Zhou, Xiao-Mei Zhang, and Wei-Cheng Yuan. 2023. "Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines" Molecules 28, no. 6: 2765. https://doi.org/10.3390/molecules28062765