
Exploring the Potential of Phytocompounds for Targeting Epigenetic Mechanisms in Rheumatoid Arthritis: An In Silico Study Using Similarity Indexing

 , , , ,
, , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Similarity Indexing
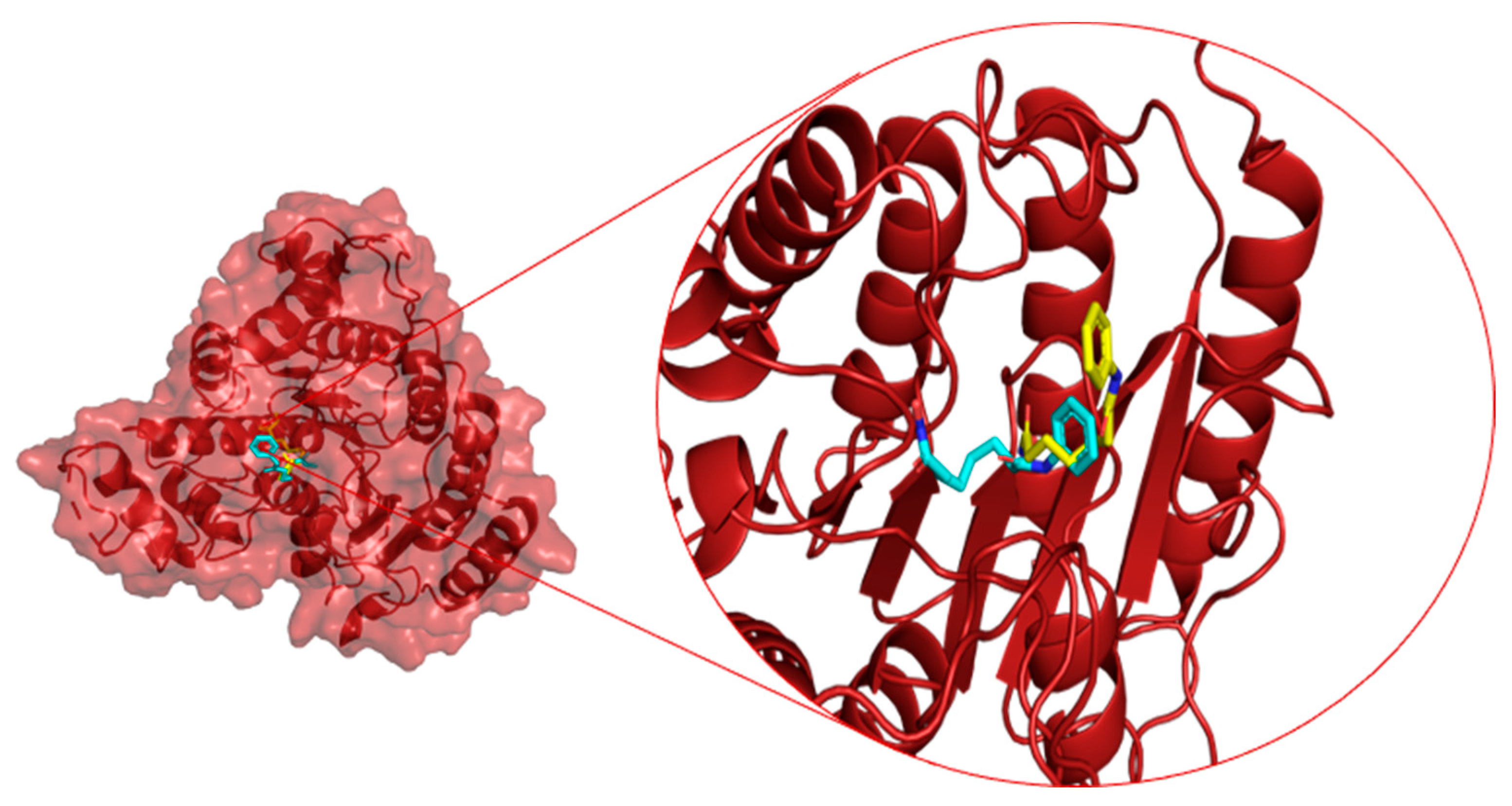
2.2. Binding Site Assignment
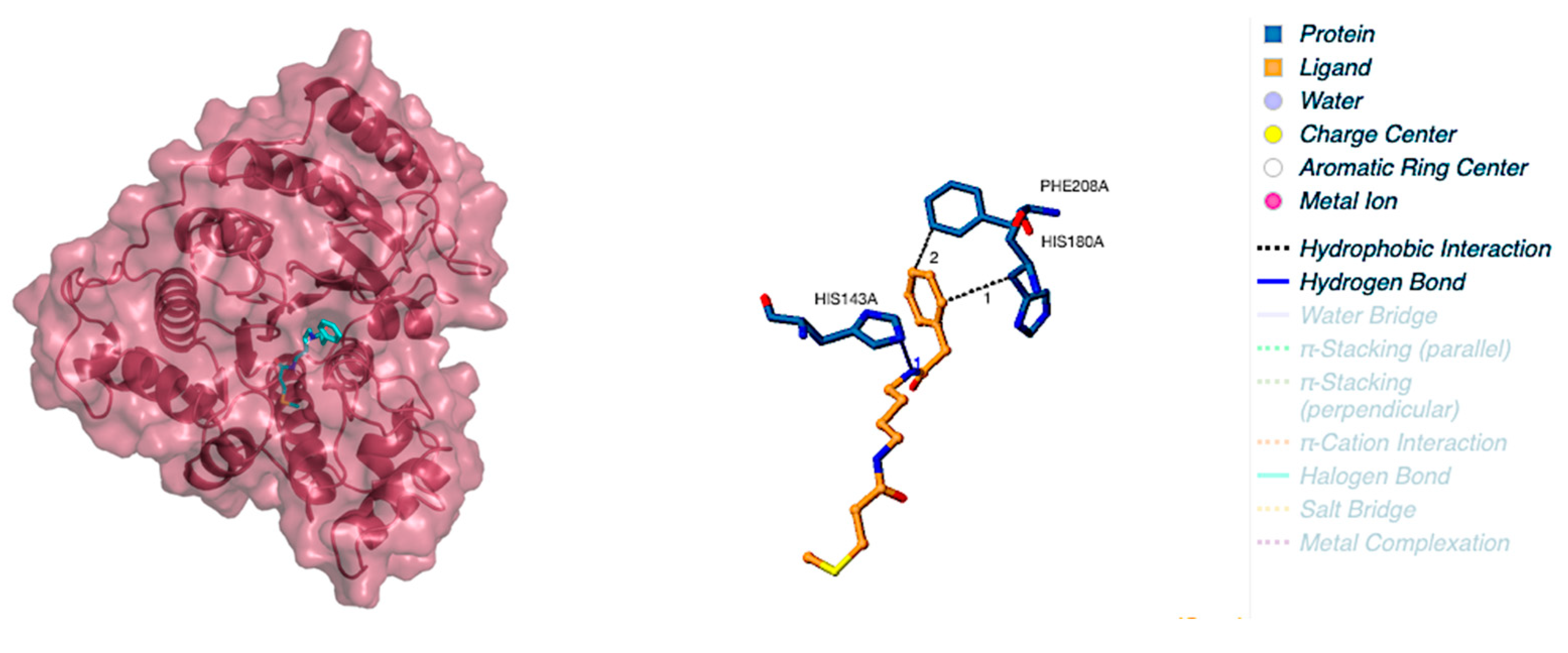
2.3. Protein–Ligand Interactions
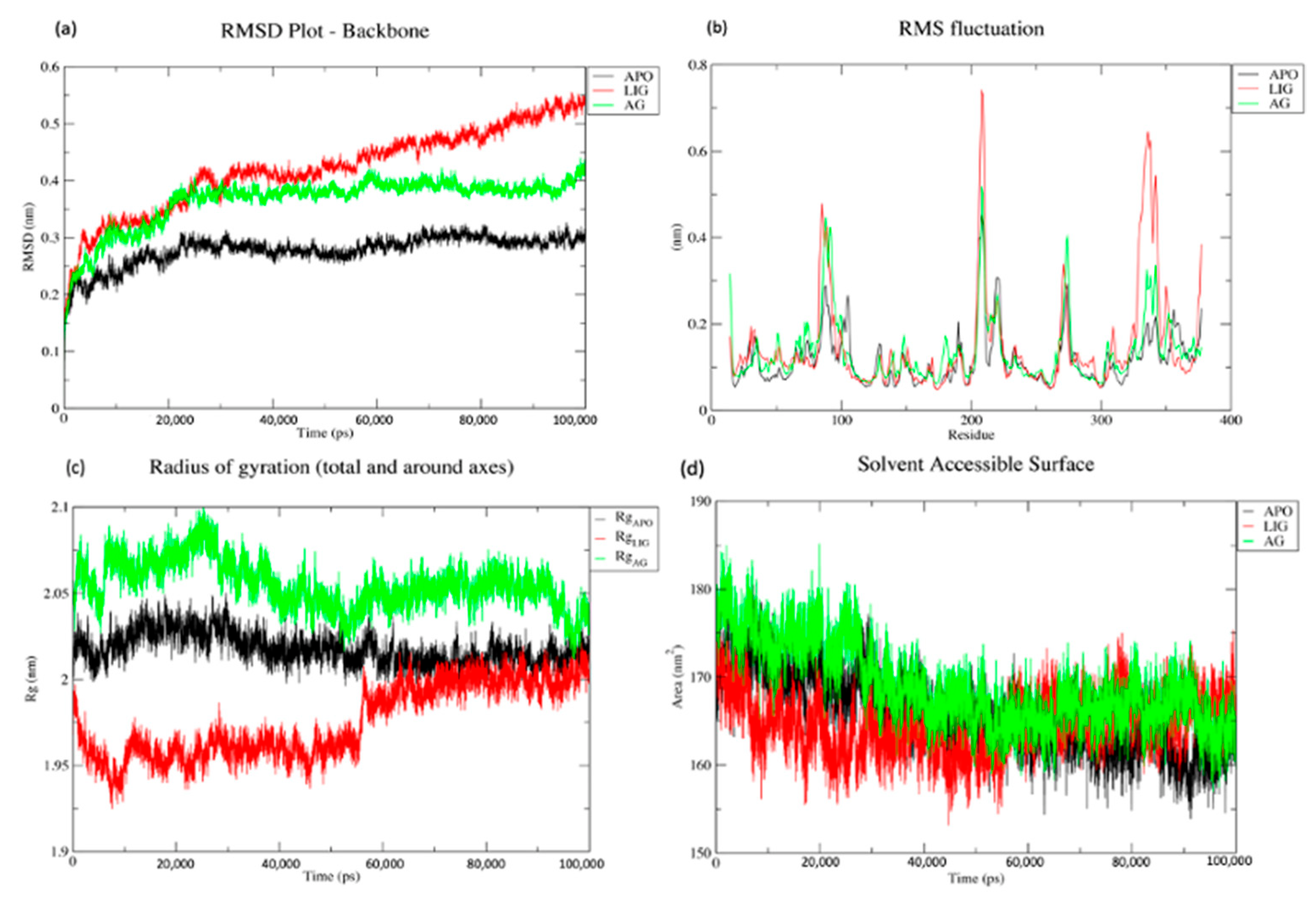
2.4. Molecular Dynamics and Simulation
2.5. Trajectory Analysis of APO, LIG and AG Systems
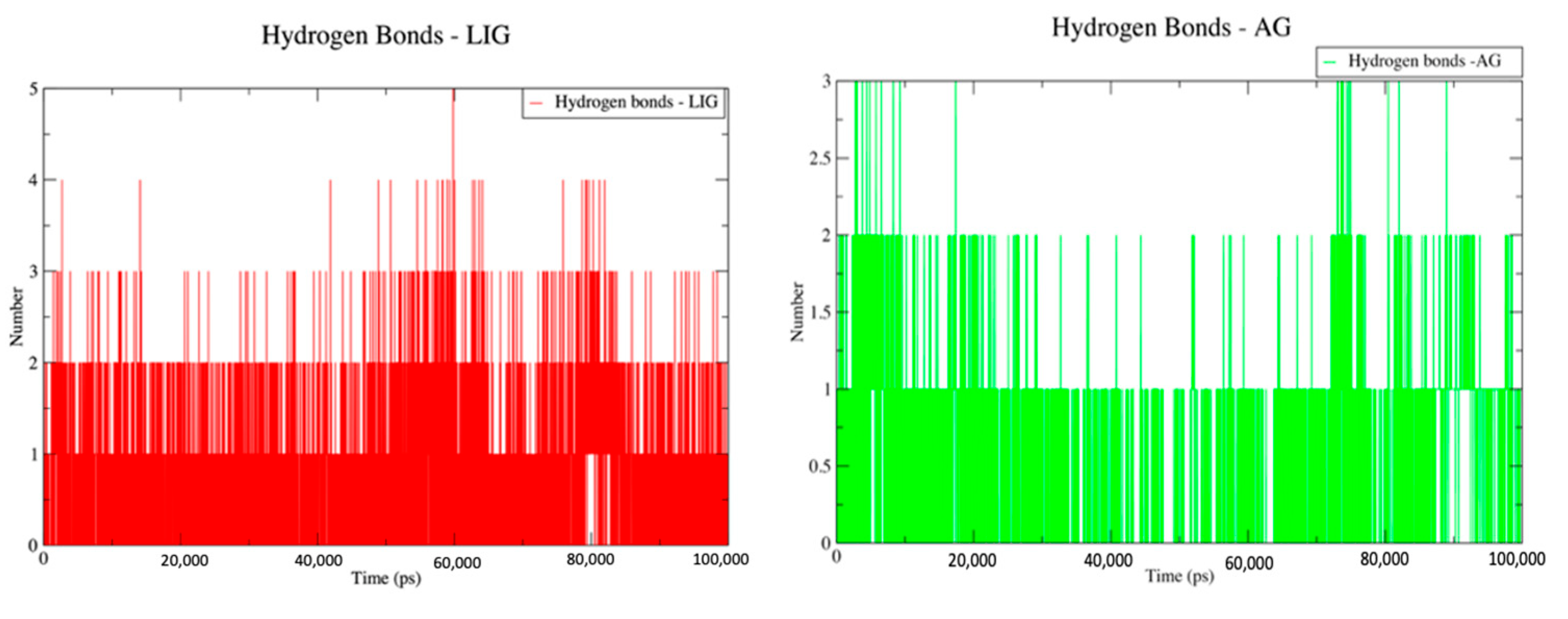
2.6. Hydrogen Bond Counts for LIG and AG Systems
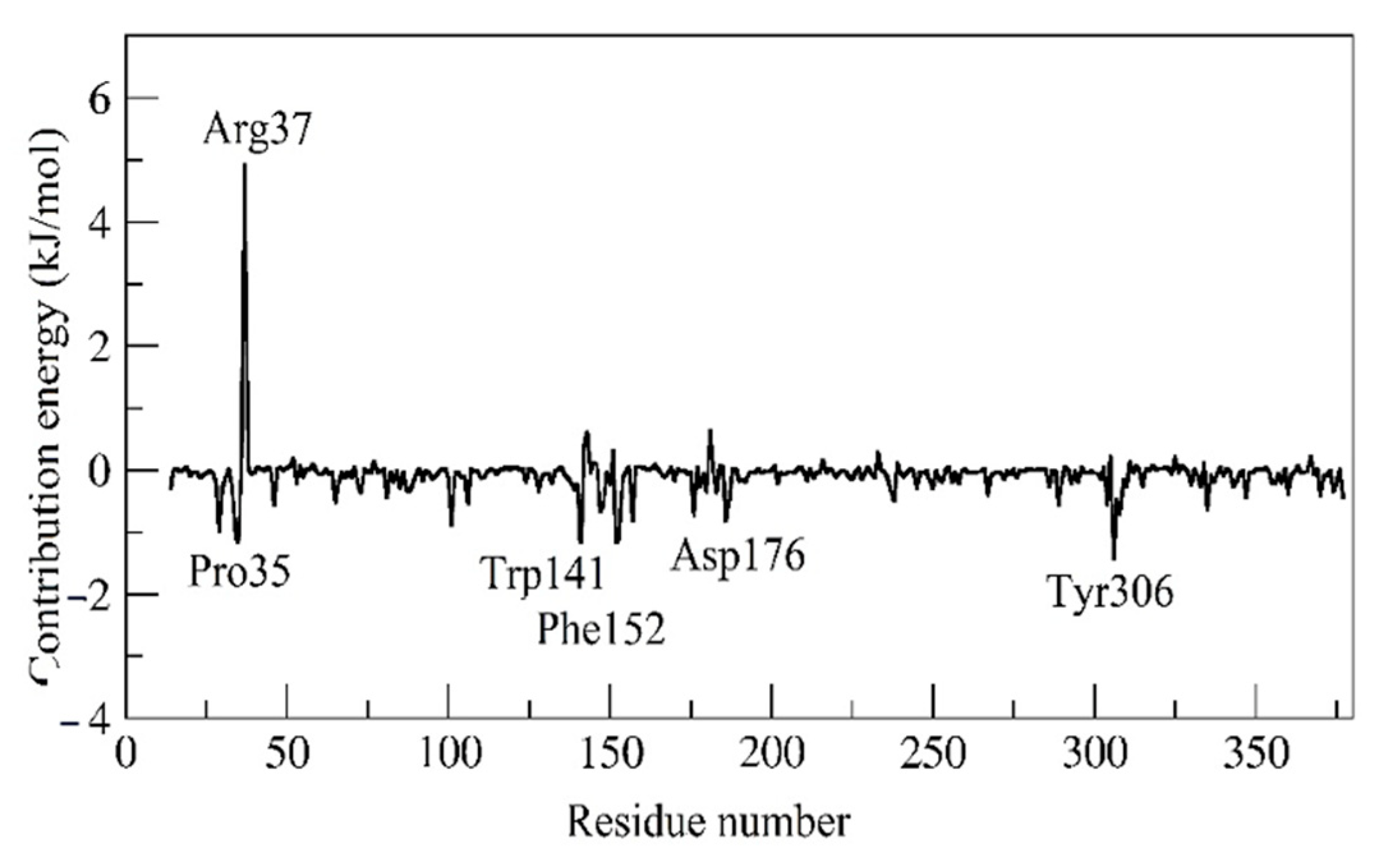
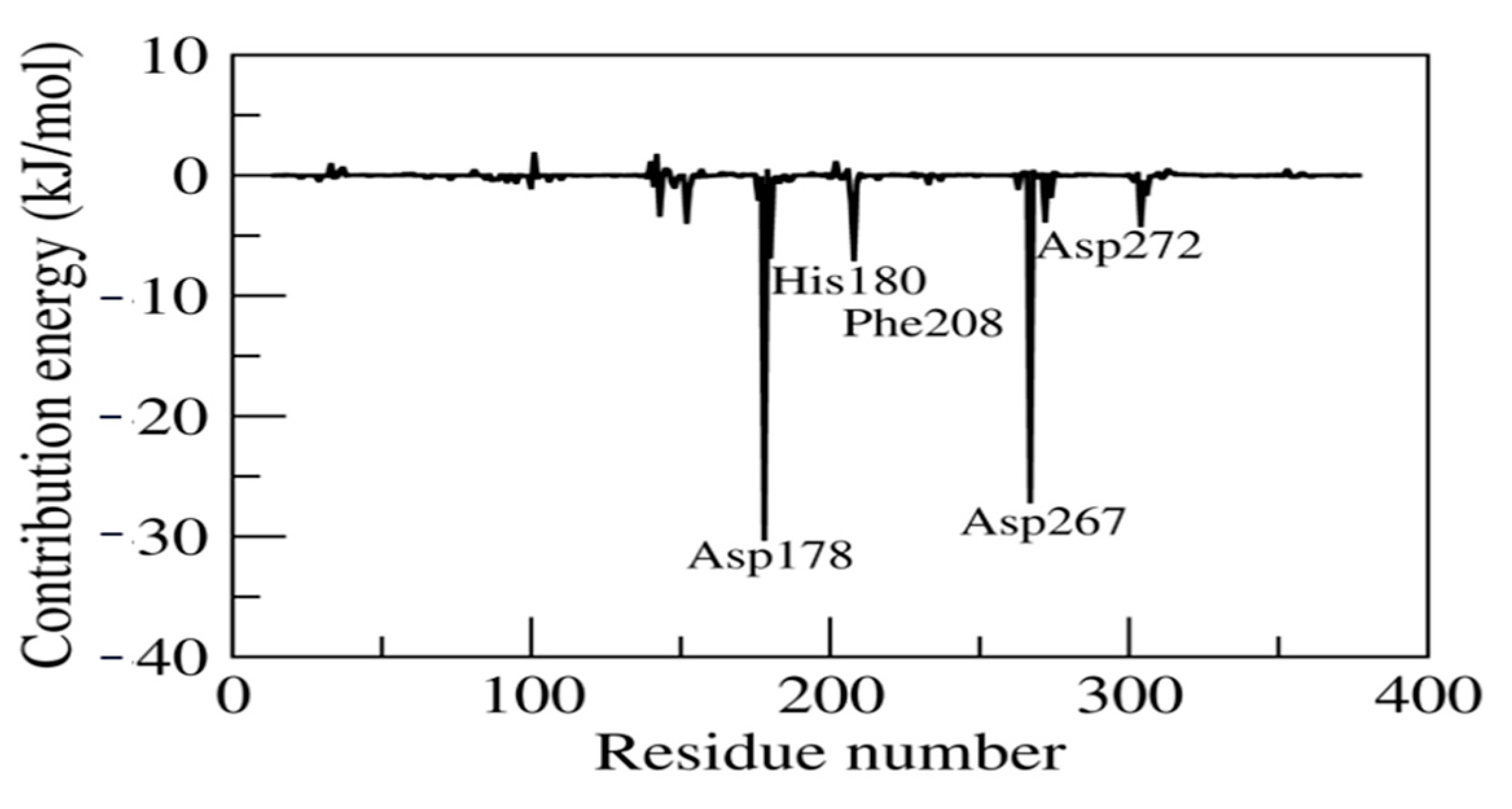
2.7. MM/PBSA and Residual Decomposition Energy
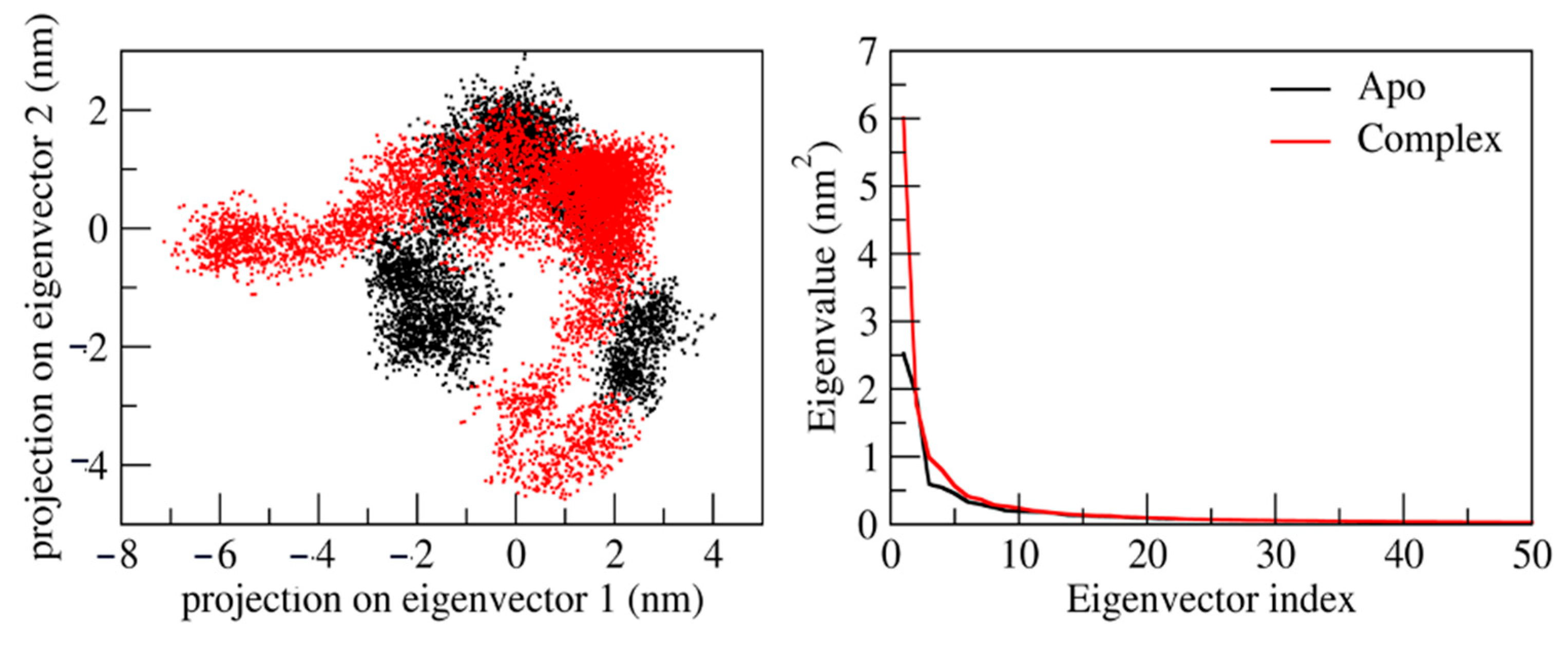
2.8. Principal Component Analysis
3. Discussion
4. Materials and Methods
4.1. Similarity Searching
4.2. Docking Studies
4.3. Molecular Dynamics Studies
4.4. MM/PBSA and Residual Decomposition Energy
4.5. Principal Component Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smolen, J.; Aletaha, D. The Burden of Rheumatoid Arthritis and Access to Treatment: A Medical Overview. Eur. J. Health Econ. 2008, 8, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Zhu, S.; Wang, J. A Protocol for Humanized Synovitis Mice Model. Am. J. Clin. Exp. Immunol. 2019, 8, 47–52. [Google Scholar] [PubMed]
- Ospelt, C.; Gay, S. The Role of Resident Synovial Cells in Destructive Arthritis. Best Pract. Res. Clin. Rheumatol. 2008, 22, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of Epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.; Winkler, J. Cloning and Characterization of a Novel Human Class I Histone Deacetylase That Functions as a Transcription Repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef] [Green Version]
- Van den Wyngaert, I.; de Vries, W.; Kremer, A.; Neefs, J.; Verhasselt, P.; Luyten, W.H.; Kass, S.U. Cloning and Characterization of Human Histone Deacetylase 8. FEBS Lett. 2000, 478, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Buggy, J.J.; Sideris, M.L.; Mak, P.; Lorimer, D.D.; McIntosh, B.; Clark, J.M. Cloning and Characterization of a Novel Human Histone Deacetylase, HDAC8. Biochem. J. 2000, 350 Pt 1, 199–205. [Google Scholar] [CrossRef]
- Lee, H.; Rezai-Zadeh, N.; Seto, E. Negative Regulation of Histone Deacetylase 8 Activity by Cyclic AMP-Dependent Protein Kinase A. Mol. Cell. Biol. 2004, 24, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Dowling, D.P.; Gantt, S.L.; Gattis, S.G.; Fierke, C.A.; Christianson, D.W. Structural Studies of Human Histone Deacetylase 8 and Its Site-Specific Variants Complexed with Substrate and Inhibitors. Biochemistry 2008, 47, 13554–13563. [Google Scholar] [CrossRef] [Green Version]
- Vannini, A.; Volpari, C.; Gallinari, P.; Jones, P.; Mattu, M.; Carfí, A.; De Francesco, R.; Steinkühler, C.; Di Marco, S. Substrate Binding to Histone Deacetylases as Shown by the Crystal Structure of the HDAC8-Substrate Complex. EMBO Rep. 2007, 8, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2018, 27, 501–507. [Google Scholar] [CrossRef]
- Yang, C.; Li, D.; Teng, D.; Zhou, Y.; Zhang, L.; Zhong, Z.; Yang, G.-J. Epigenetic Regulation in the Pathogenesis of Rheumatoid Arthritis. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Khare, C.P. Indian Medicinal Plants. Springer New York: New York, NY, USA, 2007; ISBN 978-0-387-70637-5. [Google Scholar]
- Willett, P. The Evaluation of Retrieval Effectiveness in Chemical Database Searching. In Charting a New Course: Natural Language Processing and Information Retrieval; Springer-Verlag: Berlin/Heidelberg, Germany; pp. 239–254.
- Bender, A.; Glen, R.C. Molecular Similarity: A Key Technique in Molecular Informatics. Org. Biomol. Chem. 2004, 2, 3204. [Google Scholar] [CrossRef]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, V.; Sethumadhavan, R. Drug Resistance Mechanism of PncA in Mycobacterium Tuberculosis. J. Biomol. Struct. Dyn. 2014, 32, 209–221. [Google Scholar] [CrossRef]
- Kumar, S.; Bhardwaj, V.K.; Singh, R.; Purohit, R. Structure Restoration and Aggregate Inhibition of V30M Mutant Transthyretin Protein by Potential Quinoline Molecules. Int. J. Biol. Macromol. 2023, 231, 123318. [Google Scholar] [CrossRef]
- Singh, R.; Purohit, R. Computational Analysis of Protein-Ligand Interaction by Targeting a Cell Cycle Restrainer. Comput. Methods Programs Biomed. 2023, 231, 107367. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Vilar, S.; Uriarte, E.; Santana, L.; Lorberbaum, T.; Hripcsak, G.; Friedman, C.; Tatonetti, N.P. Similarity-Based Modeling in Large-Scale Prediction of Drug-Drug Interactions. Nat. Protoc. 2014, 9, 2147–2163. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, V.; Gopalakrishnan, C.; Sethumadhavan, R. Pathological Role of a Point Mutation (T315I) in BCR-ABL1 Protein—A Computational Insight. J. Cell. Biochem. 2018, 119, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Harneti, D.; Supratman, U. Phytochemistry and Biological Activities of Aglaia Species. Phytochemistry 2021, 181, 112540. [Google Scholar] [CrossRef]
- Bordeleau, M.-E.; Robert, F.; Gerard, B.; Lindqvist, L.; Chen, S.M.H.; Wendel, H.-G.; Brem, B.; Greger, H.; Lowe, S.W.; Porco, J.A.; et al. Therapeutic Suppression of Translation Initiation Modulates Chemosensitivity in a Mouse Lymphoma Model. J. Clin. Investig. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgers, L.D.; Fürst, R. Natural Products as Drugs and Tools for Influencing Core Processes of Eukaryotic MRNA Translation. Pharmacol. Res. 2021, 170, 105535. [Google Scholar] [CrossRef] [PubMed]
- Paritala, V.; Chiruvella, K.K.; Thammineni, C.; Ghanta, R.G.; Mohammed, A. Phytochemicals and Antimicrobial Potentials of Mahogany Family. Rev. Bras. Farmacogn. 2015, 25, 61–83. [Google Scholar] [CrossRef] [Green Version]
- Bernhart, E.; Stuendl, N.; Kaltenegger, H.; Windpassinger, C.; Donohue, N.; Leithner, A.; Lohberger, B. Histone Deacetylase Inhibitors Vorinostat and Panobinostat Induce G1 Cell Cycle Arrest and Apoptosis in Multidrug Resistant Sarcoma Cell Lines. Oncotarget 2017, 8, 77254–77267. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.-Z.; Xu, X. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PLoS One 2013, 8, e62839. [Google Scholar] [CrossRef] [Green Version]
- Gillet, V.J.; Willett, P.; Bradshaw, J. Similarity Searching Using Reduced Graphs. J. Chem. Inf. Comput. Sci. 2003, 43, 338–345. [Google Scholar] [CrossRef]
- Vogt, M.; Bajorath, J. Similarity Searching for Potent Compounds Using Feature Selection. J. Chem. Inf. Model. 2013, 53, 1613–1619. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Similarity Searching. WIREs Comput. Mol. Sci. 2011, 1, 260–282. [Google Scholar] [CrossRef]
- Godden, J.W.; Xue, L.; Bajorath, J. Combinatorial Preferences Affect Molecular Similarity/Diversity Calculations Using Binary Fingerprints and Tanimoto Coefficients. J. Chem. Inf. Comput. Sci. 2000, 40, 163–166. [Google Scholar] [CrossRef]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural Snapshots of Human HDAC8 Provide Insights into the Class I Histone Deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Samdani, A.; Vetrivel, U. POAP: A GNU Parallel Based Multithreaded Pipeline of Open Babel and AutoDock Suite for Boosted High Throughput Virtual Screening. Comput. Biol. Chem. 2018, 74, 39–48. [Google Scholar] [CrossRef]
- Krivák, R.; Hoksza, D. P2Rank: Machine Learning Based Tool for Rapid and Accurate Prediction of Ligand Binding Sites from Protein Structure. J. Cheminform. 2018, 10, 39. [Google Scholar] [CrossRef]
- Koppal, V.V.; Melavanki, R.; Kusanur, R.; Bagewadi, Z.K.; Yaraguppi, D.A.; Deshpande, S.H.; Patil, N.R. Investigation of the Fluorescence Turn-off Mechanism, Genome, Molecular Docking In Silico and In Vitro Studies of 2-Acetyl-3 H -Benzo[ f ] Chromen-3-One. ACS Omega 2022, 7, 23759–23770. [Google Scholar] [CrossRef]
- Patil, V.S.; Harish, D.R.; Vetrivel, U.; Roy, S.; Deshpande, S.H.; Hegde, H.V. Hepatitis C Virus NS3/4A Inhibition and Host Immunomodulation by Tannins from Terminalia Chebula: A Structural Perspective. Molecules 2022, 27, 1076. [Google Scholar] [CrossRef]
- Singh, R.; Bhardwaj, V.K.; Purohit, R. Computational Targeting of Allosteric Site of MEK1 by Quinoline-based Molecules. Cell Biochem. Funct. 2022, 40, 481–490. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Wong-Ekkabut, J.; Karttunen, M. Assessment of Common Simulation Protocols for Simulations of Nanopores, Membrane Proteins, and Channels. J. Chem. Theory Comput. 2012, 8, 2905–2911. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose–Hoover Thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Dewdney, T.G.; Wang, Y.; Kovari, I.A.; Reiter, S.J.; Kovari, L.C. Reduced HIV-1 Integrase Flexibility as a Mechanism for Raltegravir Resistance. J. Struct. Biol. 2013, 184, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Yaraguppi, D.A.; Bagewadi, Z.K.; Deshpande, S.H.; Chandramohan, V. In Silico Study on the Inhibition of UDP-N-Acetylglucosamine 1-Carboxy Vinyl Transferase from Salmonella Typhimurium by the Lipopeptide Produced from Bacillus Aryabhattai. Int. J. Pept. Res. Ther. 2022, 28, 80. [Google Scholar] [CrossRef]
- Khanal, P.; Patil, V.S.; Bhandare, V.V.; Dwivedi, P.S.R.; Shastry, C.S.; Patil, B.M.; Gurav, S.S.; Harish, D.R.; Roy, S. Computational Investigation of Benzalacetophenone Derivatives against SARS-CoV-2 as Potential Multi-Target Bioactive Compounds. Comput. Biol. Med. 2022, 146, 105668. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa —A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Khanal, P.; Patil, V.S.; Bhandare, V.V.; Patil, P.P.; Patil, B.M.; Dwivedi, P.S.R.; Bhattacharya, K.; Harish, D.R.; Roy, S. Systems and in Vitro Pharmacology Profiling of Diosgenin against Breast Cancer. Front. Pharmacol. 2023, 13. [Google Scholar] [CrossRef]
- DasNandy, A.; Patil, V.S.; Hegde, H.V.; Harish, D.R.; Roy, S. Elucidating Type 2 Diabetes Mellitus Risk Factor by Promoting Lipid Metabolism with Gymnemagenin: An in Vitro and in Silico Approach. Front. Pharmacol. 2022, 13. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | SAHA | Aglaithioduline |
|---|---|---|
| Molecular Weight | 264.325 | 306.431 |
| LogP | 2.4711 | 2.1184 |
| Rotatable Bonds | 8 | 9 |
| Acceptors | 3 | 3 |
| Donors | 3 | 2 |
| 2D Structure |  |  |
| X | Y | Z | Residues |
|---|---|---|---|
| 31.5106 | −2.4886 | −9.5104 | ASP101 SER138 GLY139 GLY140 TRP141 HIS142 HIS143 GLY151 PHE152 CYS153 TYR154 HIS180 GLY206 PHE207 PHE208 MET274 GLY303 GLY304 TYR306 LEU31 LYS33 ILE34 ARG37 |
| Compound Name | Docking Score (Kcal/mol) | Interaction Sites | |
|---|---|---|---|
| Hydrophobic Interactions | Hydrogen Bonds | ||
| Aglaithioduline | −8.5 | HIS180, PHE208 | HIS143 |
| SAHA | −8.7 | TYR100, PHE152, PHE208 | HIS180 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshpande, S.H.; Bagewadi, Z.K.; Khan, T.M.Y.; Mahnashi, M.H.; Shaikh, I.A.; Alshehery, S.; Khan, A.A.; Patil, V.S.; Roy, S. Exploring the Potential of Phytocompounds for Targeting Epigenetic Mechanisms in Rheumatoid Arthritis: An In Silico Study Using Similarity Indexing. Molecules 2023, 28, 2430. https://doi.org/10.3390/molecules28062430
Deshpande SH, Bagewadi ZK, Khan TMY, Mahnashi MH, Shaikh IA, Alshehery S, Khan AA, Patil VS, Roy S. Exploring the Potential of Phytocompounds for Targeting Epigenetic Mechanisms in Rheumatoid Arthritis: An In Silico Study Using Similarity Indexing. Molecules. 2023; 28(6):2430. https://doi.org/10.3390/molecules28062430
Chicago/Turabian StyleDeshpande, Sanjay H., Zabin K. Bagewadi, T. M. Yunus Khan, Mater H. Mahnashi, Ibrahim Ahmed Shaikh, Sultan Alshehery, Aejaz A. Khan, Vishal S. Patil, and Subarna Roy. 2023. "Exploring the Potential of Phytocompounds for Targeting Epigenetic Mechanisms in Rheumatoid Arthritis: An In Silico Study Using Similarity Indexing" Molecules 28, no. 6: 2430. https://doi.org/10.3390/molecules28062430