
Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives

1
Department of Chemistry, College of Science, King Khalid University, P.O. Box 9004, Abha 61413, Saudi Arabia
2
Research Center for Advanced Materials Science, College of Science, King Khalid University, P.O. Box 9004, Abha 61413, Saudi Arabia
3
Soft Materials Research Laboratory, Discipline of Metallurgy Engineering and Materials Science, Indian Institute of Technology Indore, Indore 453552, India
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(24), 8672; https://doi.org/10.3390/molecules27248672
Submission received: 7 November 2022
/
Revised: 27 November 2022
/
Accepted: 1 December 2022
/
Published: 8 December 2022
(This article belongs to the Special Issue Computational Studies of Novel Function Materials)
Abstract
:A series of new benzothiazole-derived donor–acceptor-based compounds (Comp1–4) were synthesized and characterized with the objective of tuning their multifunctional properties, i.e., charge transport, electronic, and optical. All the proposed structural formulations (Comp1–4) were commensurate using FTIR, 1H NMR, 13C NMR, ESI-mass, UV–vis, and elemental analysis techniques. The effects of the electron-donating group (-CH3) and electron-withdrawing group (-NO2) on the optoelectronic and charge transfer properties were studied. The substituent effect on absorption was calculated at the TD-B3LYP/6-31+G** level in the gas and solvent phases. The effect of solvent polarity on the absorption spectra using various polar and nonpolar solvents, i.e., ethanol, acetone, DMF, and DMSO was investigated. Light was shed on the charge transport in benzothiazole compounds by calculating electron affinity, ionization potential, and reorganization energies. Furthermore, the synthesized compounds were used to prepare thin films on the FTO substrate to evaluate the charge carrier mobility and other related device parameters with the help of I-V characteristic measurements.
1. Introduction
Organic semiconductors have become indispensable because of their cheap, malleable, lightweight, and environmentally friendly properties [1,2,3,4,5,6,7,8]. Advances in organic semiconductor materials (OSMs) in organic field effect transistors (OFETs), photovoltaics, and organic thin film transistors (OTFTs) have motivated scientists to explore materials [9,10,11,12,13,14,15,16,17,18,19,20,21]. The versatility of OSMs allows many interesting properties to be tuned because organic materials such as benzothiazole have promising applications in fluorescence sensors, OLEDs, solar cells, OFETs, OTFTs, etc., and have attracted great interest [22,23,24,25,26].
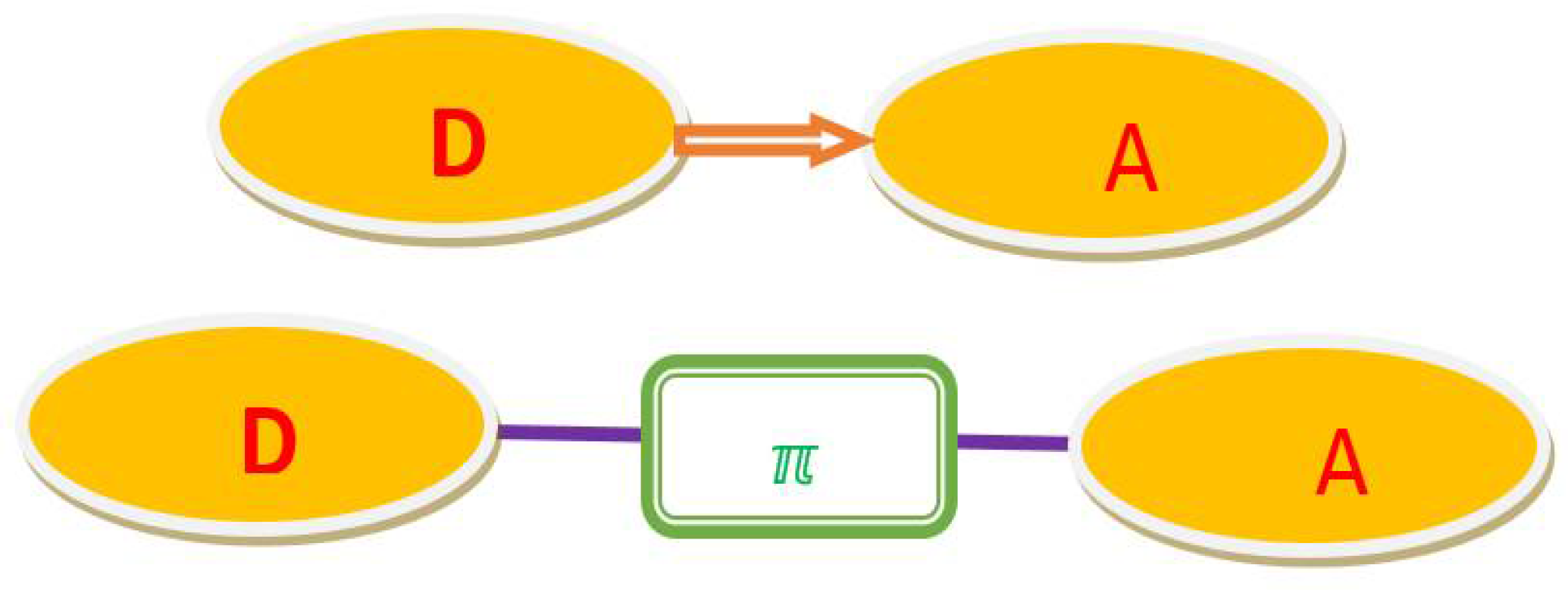
Low band gap materials have attracted the attention of the scientific community because of their high tendency to harvest photons of visible wavelength, redox-tunable quality, and use in ambipolar transistors [27,28,29,30,31,32,33]. Essentially, a promising strategy is to develop molecules that carry electron-donating moiety (D) and electron-acceptor moiety (A) that can match the charge carrier injection and receive both holes and electrons. The most important materials to date have been modifications of the HOMO/LUMO (highest/lowest occupied/unoccupied molecular orbital) of the molecule, which can be obtained by the simple introduction of the suitable D and A units in the molecule [34,35,36]. In such compounds, the interaction of the HOMO, which donates the electron (D), and the LUMO, which accepts the electron (A), results in a narrowing of the gap [37]. At present, HOMO and LUMO molecule levels can be controlled, making those systems more attractive for use in OSMs [38]. The benzothiazole group has been described as a valuable receptor. It provides a D-π-A compound that exhibits two-photon absorption (TPA) compared with asymmetrical chromophores [39]. The donor–acceptor (DA) model system diagram is illustrated in Figure 1. By adopting the benzothiazole unit as a well-made acceptor, the changes made by various donors can further increase the TPA.
The computational probing of the electronic structure of compounds has made a significant contribution to estimating known and unpredicted properties. In this context and to guide the integration of smaller energy gap materials, computational methods have been widely used to predict energy gaps [40,41,42]. Computational chemistry is accompanied by a variety of applications in fields such as materials science, energy, engineering, nanoscience, etc. [43]. The density functional theory (DFT) seems to be a good choice and has been established as an effective means of investigating the electronic, charge transfer, and physical properties associated with various materials. The DFT method provides the most accurate results for electronic, optical, and charge transport properties that are in the closest agreement with experimental evidence [44,45,46]. In addition, computational calculations are extensively used to determine properties and provide spectral data. There are many studies on the calculation using DFT of the various properties of certain thiophene and benzothiazoles, such as ultraviolet–visible spectroscopy (UV–vis), HOMO-LUMO energy, the molecular electrostatic potential, and charge distribution [47,48].
We synthesized various dyes aiming to investigate their OFET properties. First-principle methods were used to discover the impact of electron-donating -CH3 and -NO2 groups on the charge transport and optoelectronic properties. We anticipated that the substitution of positions 3 and 4 for electron-donating groups (EDGs) and position 5 for the electron-withdrawing group (-NO2) would boost properties of interest (Figure 2).
2. Methods
2.1. Computational Details
The optimization of ground state (S0) geometries of benzothiazole compounds was achieved by density functional theory (DFT) [49,50,51,52] at the B3LYP/6-31+G** level. The frequency calculations were performed at the B3LYP/6-31+G** level. There was no negative frequency obtained, which indicated that the optimized structures were suitable for further studies. Earlier studies demonstrated that B3LYP was able to calculate charge transport [53]. Here, electron affinity (EA), ionization potential (IP), and reorganization energy for hole/electron (λhole/λelec) values were computed at the B3LYP/6-31G** level. The optimization of geometries in an excited state (S1) was achieved by time-dependent DFT [54] at the TD-B3LYP/6-31+G** level. The absorption (λmax) was assessed at the TD-B3LYP/6-31+G** level in gas-phase and solvents (ethanol, acetone, DMF, and DMSO) with the Gaussian16 package [55] using self-consistent reaction field (SCRF) theory with a polarized continuum model (PCM) [56].
2.2. Experimental Details
All the characterization spectra of FTIR, 1HNMR, 13C NMR, and ESI-mass were incorporated in supporting files as Figures S1–S17.
2.2.1. Synthesis of Comp1
Methanolic solution of 4-methyl-2-thiophenecarboxaldehyde (0.332 g, 2 mmol) was added dropwise to the solution of 2-hydrazino benzothiazole (0.252 g, 2 mmol) dissolved in methanol. The resulting mixture was continuously stirred for 12 h under reflux conditions. The resulting pale yellow-colored precipitate of Comp1 was further filtered and washed with methanol and ether thrice (yield: 0.428 g, 78%).
1H NMR (500 MHz, DMSO, ppm): 2.21 (s, 3H, -CH3), 7.09 (s, 1H, thiophene-H), 7.19 (s, 1H, thiophene-H), 7.22 (s, 2H, Ar-H), 7.28 (s, 1H, Ar-H), 7.73 (s, 1H, Ar-H), 8.26 (s, 1H, =CH), 12.15 (s, 1H, -NH). 13C NMR (500 MHz, DMSO, ppm): 15.74, 121.98, 122.14, 123.79, 126.47, 132.23, 138.19, 144.10, 167.04. FTIR (ATR, cm−1): 1615 (C=N), 1575 (C=N; benzothiazole ring). UV–vis spectrum [(0.5 × 10−5 M; lmax, solvent), (e; L M−1 cm−1)]: 353 (acetone; 59,000), 360 (DMF; 59,000), 362 (DMSO; 58,500). Anal. calcd. for C13H11N3S2: C, 57.12; H, 4.06; N, 15.37; S, 23.46. Found C, 57.16; H, 4.10; N, 15.41; S, 23.42. HR-MS [M+H]+: 274 (calcd. 274).
2.2.2. Synthesis of Comp2
Ethanolic solution of 3-methyl-2-thiophenecarboxaldehyde (0.229 g, 1.815 mmol) was added dropwise to the solution of 2-hydrazino benzothiazole (0.300 g, 1.815 mmol) dissolved in ethanol in the presence of 5 drops of acetic acid. The resulting mixture was continuously stirred for 12 h under reflux conditions. The resulting off-white-colored precipitate of Comp2 was further filtered and washed with ethanol and ether (yield: 0.372 g, 75%).
1H NMR (500 MHz, DMSO, ppm): 2.38 (s, 3H, -CH3), 7.01 (s, 1H, Fural-H), 7.14 (s, 1H, Fural-H), 7.34 (s, 1H, Ar-H), 7.43 (s, 1H, Ar-H), 7.57 (s, 1H, Ar-H), 7.79 (s, 1H, Ar-H), 8.40 (s, 1H, =CH), 12.12 (s, 1H, -NH). FTIR (ATR, cm−1): 1616 (C=N), 1576 (C=N; benzothiazole ring). 13C NMR (500 MHz, DMSO, ppm): 14.52, 121.01, 121.24, 122.70, 124.52, 128.81, 131.31, 143.01, 167.01. UV–vis spectrum [(0.5 × 10−5 M; lmax, solvent), (ε; L M−1 cm−1)]: 352 (ethanol; 55,150), 353 (acetone; 55,150), 358 (DMF; 55,000), 361 (DMSO; 55,050). Anal. calcd. for C13H11N3S2: C, 57.12; H, 4.06; N, 15.37; S, 23.46. Found C, 57.11; H, 4.09; N, 15.41; S, 23.40. HR-MS [M+H]+: 274 (calcd. 274).
2.2.3. Crystal Structure of Comp2
Empirical formula: C13H11N3S2; formula weight: 273.37; crystal system: triclinic; space group P-1, unit cell dimensions a = 5.8300 (12) Å, α = 87.37 (3)°, b = 13.365 (3) Å, β = 82.25 (3)°, c = 16.684 (3) Å and γ = 83.70 (3)°; volume 1279.7 (5) Å3; Z = 4; density (calculated) 1.419 Mg/m3 (see Figure 3 and Figure 4).
2.2.4. Synthesis of Comp3
Ethanolic solution of 5-nitro-2-furaldehyde (0.256 g, 1.815 mmol) was added dropwise to the solution of 2-hydrazino benzothiazole (0.300 g, 1.815 mmol) dissolved in ethanol in the presence of 5-7 drops of acetic acid. The resulting mixture was continuously stirred for 10 h under reflux conditions. The resulting orange-colored precipitate of Comp3 was further filtered and washed with ethanol and ether (yield: 0.377 g, 72%).
1H NMR (500 MHz, DMSO, ppm): 7.16 (d, 2H, Ar-H), 7.33 (d, 1H, Ar-H), 7.45 (s, 1H, Ar-H), 7.79 (d, 2H, Fural-H), 8.10 (s, 1H, =CH), 12.71 (s, 1H, -NH). 13C NMR (500 MHz, DMSO, ppm): 108.99, 115.01, 121.91, 122.04, 126.42, 133.41, 148.35, 154.68, 156.52, 167.08. FTIR (ATR, cm−1): 1622 (C=N), 1581 (C=N, benzothiazole). UV–vis spectrum [(0.5 × 10−5 M; λmax, solvent), (ε; LM−1 cm−1)]: 430 (DMF; 57,500), 445 (DMSO; 57,400). Anal. calcd. for C13H11N3S2: C, 50.00; H, 2.80; N, 19.43; S, 11.12. Found C, 50.11; H, 2.83; N, 19.40; S, 11.15. HR-MS [M+Na]+: 311 (calcd. 311). [M+H]+; 289 (289).
2.2.5. Synthesis of Comp4
A solution mixture of 2-hydrazino benzothiazole (0.300 g, 1.815 mmol) with 5 drops of acetic acid was separately prepared and allowed to react with 5-methylfurfrualdehyde (0.200 g, 1.815 mmol). The resulting solution mixture was refluxed for 12 h to further obtain the light brown precipitate of compound Comp4, which was washed with ethanol and ether (yield: 0.355 g, 76%).
1H NMR (500 MHz, DMSO, ppm): 2.36 (s, 3H, -CH3), 6.26 (s, 1H, Fural-H), 6.74 (s, 1H, Fural-H), 7.08 (s, 1H, Ar-H), 7.30 (s, 1H, Ar-H), 7.38 (s, 1H, Ar-H), 7.74 (s, 1H, Ar-H), 7.93 (s, 1H, =CH), 12.11 (s, 1H, -NH). 13C NMR (500 MHz, DMSO, ppm): 14.17, 117.06, 122.00, 122.13, 131.52, 132.94, 139.85, 149.14, 152.20, 166.99. FTIR (ATR, cm−1): 1622 (C=N), 1607 (C=N, benzothiazole). UV–vis spectrum [(0.5 × 10−5 M; λmax, solvent), (ε; L M−1 cm−1)]: 343 (acetone; 59,000), 344 (ethanol; 59,000), 350 (DMF; 59,000), 353 (DMSO; 58,950). Anal. calcd. for C13H11N3OS: C, 60.68; H, 4.31; N, 16.33; S, 12.46. Found C, 60.26; H, 4.40; N, 16.30; S, 12.39. HR-MS [M+H]+: 258 (calcd. 258), [M+Na]+; 280 (calcd. 280).
3. Results and Discussion
3.1. Electronic Properties
The EHOMO, ELUMO, and Egap of S0 and S1 of the benzothiazole compounds are provided in Table 1. The trend in the S0 EHOMO was: Comp4 (−5.52) > Comp2 (−5.58) > Comp1 (−5.59) > Comp3 (−6.18). The trend in ELUMO at S0 was: Comp2 (−1.88) > Comp4 (−1.92) > Comp1 (−1.95) > Comp3 (−3.35). The effect of -NO2 in place of -CH3 in the furan-based derivative was more useful for tuning the energy values of frontier molecular orbitals (FMOs), i.e., Comp3. One can see that the substitution of -NO2 group lowered the EHOMO and ELUMO values of Comp3, resulting in reducing the Egap, i.e., 2.83 eV at S0.
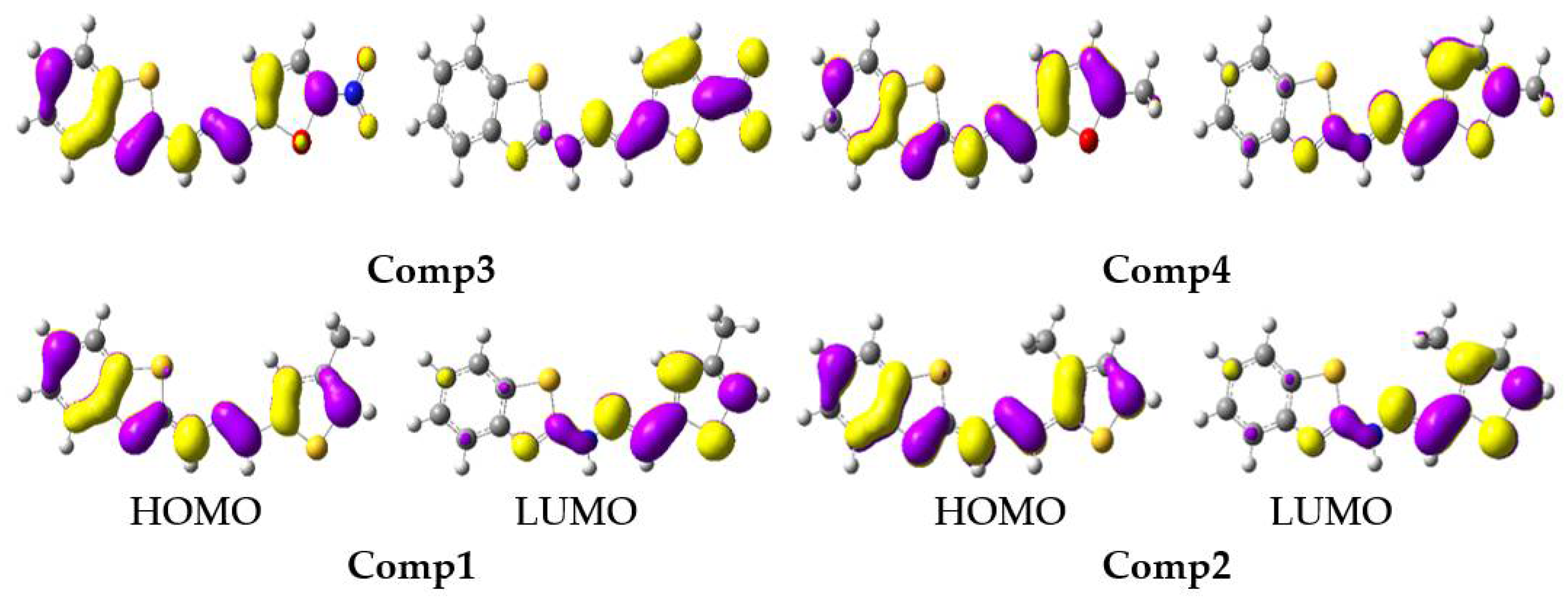
The tendency in the S1 EHOMO was: Comp4 (−4.73) > Comp2 (−4.76) > Comp1 (−4.77) > Comp3 (−5.13), while in the S1 ELUMO it was: Comp2 (−2.55) > Comp4 (−2.56) > Comp1 (−2.61) > Comp3 (−4.01). The substitution of -NO2 at position-5 on the furan ring led to lowering the energy levels of the HOMO and LUMO in Comp3, which reduced the Egap of S1, i.e., 1.12 eV, and was advantageous for charge transport and optoelectronic properties. The FMOs at S0 and S1 are illustrated in Figure 5 and Figure 6, respectively. The HOMO was delocalized on entire molecules in all studied systems, and the LUMO was localized toward the thiophene/furan moiety in Comp1, Comp2, and Comp4, while -NO2 in Comp3 was also involved in the formation of the LUMO showing an intra-molecular charge transfer (ICT) from H→L. At S1, the HOMO was delocalized on the benzothiazoles ring in all the derivatives, while the charge density at the LUMO was nearly the same as S0.
The work functions (ϕ) of Ag (Al) were 4.74 (4.08 eV) [57]. The hole/electron injection energy barricades (HIE/EIE) of benzothiazoles to the Al electrode were computed as (ϕ-EHOMO) and (ELUMO-ϕ), correspondingly. The EIE from Comp1–Comp4 to Al were 2.13, 2.20, 0.73, and 2.16 eV, respectively. The HIE for Comp1–Comp4 were 1.51, 1.50, 2.00, and 1.44 eV, correspondingly. The calculated EIE from Comp1–Comp4 to Ag were 2.79, 2.86, 1.39, and 1.82 eV, respectively. The HIE for Comp1–Comp4 were 0.85, 0.84, 1.44, and 0.78 eV, respectively. These outcomes suggested that the substitution of the -NO2 group at position 5 on the furan moiety in Comp3 would encourage the development of the electron injection, while the -CH3 group might top up the hole injection aptitude, see Table 2.
3.2. Absorption Spectra
Absorption (λmax), oscillator strengths (f), and the percentage contribution of the transitions of the FMOs to the studied derivatives of benzothiazole on the TD-B3LYP/6-31+G** level in gas-phase and various solvents are shown in Table 3. The solvent effect has attracted much attention because of many of the chemical processes that occur in the solution phase. In an effort to explore the effect of the solvent on the quantum–chemical calculations of the absorption maxima, the gas phase of the studies consisted of molecules (as a control), and then the data were compared with the λmax computed in a solvent such as ethanol, acetone, DMF, and DMSO. The effects of the substituents on the λmax at the TD-B3LYP/6-31+G** level in the gas phase and the solvent stage were compared and discussed. The effect of solvent polarity on the absorption spectra was measured in polar and non-polar solvents (ethanol, acetone, DMF, and DMSO), as demonstrated in Figures S5 and S6, and corresponding data are tabulated in Table S1.
Here, the first λmax band was noticed at 358 nm related to H→L (S0→S1), whereas the second was from H→L + 1 at 281 nm for Comp1 in the gas phase. The first λmax band was observed at 363 nm corresponding to H→ L (S0 → S1), while the second was from H→L + 1 at 281 nm for Comp1 in ethanol. Additionally, no significant effect was observed on λmax from the gas phase to solvent in Comp1, Comp2, and Comp4. The solvent polarity also had no significant effect on λmax. In the gas phase, the substitution of -NO2 at position 5 of furan in Comp3 led to a red shift, i.e., 93 nm in the first band and 42 nm in the second band compared with Comp1. We noted that a significant effect was observed in λmax from the gas phase to solvent in Comp3, i.e., 481 nm in ethanol vs. 451 nm in the gas phase. The computed data were in good agreement with the experimental findings, see Table S1. The substitution of -NO2 at position 5 of furan in Comp3 led to a redshift in λmax from the gas phase to ethanol, acetone, DMF, and DMSO, i.e., 30, 30, 33, and 33 nm for the first band and 12, 12 13, and 13 nm for the second. The effect of electron acceptor and electron donor moiety was mainly on the first λmax band, i.e., the introduction of -NO2 at position 5 of furan in Comp3 tuned the wavelength toward a longer wavelength.
All electronic transitions and corresponding oscillator strengths in the absorption spectra were π to π* type. A positive development in the oscillator strength was observed, i.e., it was largest from S0 to S1 in solvent compared with the gas phase. According to Table 3, the computed excitation energy for all studied compounds, except Comp3 in solvent, was almost the same. All of the compounds exhibited two absorption peaks related to the benzothiazole part, and the charge transfer was associated with the thiophene/furan substituted moiety (Figure 5). The HOMO was entirely located on the molecules and benzothiazole, while the LUMO was on the thiophene-furan part, which was an illuminating charge transfer-type transition (CT) of the π to π* in the first bands in the λmax.
The lowest energy gaps were the π to π* for Comp3 (2.8 eV), while the other benzothiazole derivatives showed larger band gaps as established from the absorption values (see Table 3). These results clearly showed that the interaction between the donor and the acceptor, either in an alternating manner or in a separate block in the molecule, played an important role in controlling the planarity and the photophysical properties [58].
3.3. Charge Transport Properties
Charge transports are sensitive to internal traps [59] as well as transport within p-type materials from low ionization potential levels, making the filling of deep traps less attractive. Likewise, materials of n-type benefit from a larger EA, where there are also fewer available traps. The hole injection from an electrode to a semiconductor HOMO is more effective when the electrode is closer, or even greater than a semiconductor IP. Similarly, for better electron injection, larger EA values would be suitable. For better stability of the device, it was certified that its charged and neutral states do not contribute to chemical reactions [60]. To preclude the thermodynamically efficient reaction (which contains oxygen and water), a neutral semiconductor is expected to require an ionization potential greater than 4.9 eV [60]. The shallow HOMO may diminish ambient O2 in the H2O existence to form OH−. The semiconductors having a deep HOMO can receive holes that oxidize the H2O in the atmosphere. Obstacles encountered during such an undesirable reaction by chance lead to overpotentials, which permits organic semiconductors to gently make redox to unveil the stabilities. Electron transport is most affected by the reaction with air, and it is important to prevent the electron polaron from degrading the material. To achieve this, the LUMO must be low to avert excited electrons from reducing the water-soluble O2 systems to O2− [61] or H2O to OH−. These undesirable electrochemical processes can reduce charge transfer and set about irreversible changes within the semiconductor. Defining the exact amount of EA that needs to be skipped to prevent this redox reaction requires consideration of the maximum response power and device morphology. It has been suggested that high EA can put down oxidation reactions [62].
The values of the ionization potential (IP) and the electron affinity (EA) are among the most important factors of organic compounds in xerography and electroluminescence. These parameters were interconnected to the amount of energy required to add or remove electrons and thus were considered as molecular stability with respect to donating or receiving electrons to create an exciton. The IPs and EAs for all systems are listed in Table 4. In addition, this entailed a positive correlation between the LUMO level and EAs. We set them as the injection of an electron is affected by the HOMO and LUMO levels. The π-conjugated organic materials with an electronic charge motion were created by a hopping mechanism. The reorganization energy (λ) as an important factor influencing the rate of the hopping charge because of its structural variation from neutral to ionic states (cation and/or anion). In order to have a high degree of material mobility, the λ must be reduced. The λ is a significant parameter for the estimation of the rate of charge transfer. Previously, it was discovered that the DFT can be a reliable way to replicate the experimental data [63,64,65,66]. The internal reorganization (λint) and external polarization (λext) are two components of λ [67]. Here, λint was projected for the hole (λhole) and electron (λelec). The λhole was estimated by Equations (1) and (2) [68]:
where E+ (B), E+ (B+), E (B+), and E (B) are the energies of the cation at neutral optimized geometry, neutral at the cationic optimized geometry, optimized cation, and optimized neutral geometry, respectively. Correspondingly, λelec was estimated by Equations (3) and (4):
where E− (B), E− (B−), and E (B−) are energies of the anion at neutral optimized geometry, neutral at anionic optimized geometry, and the anionic optimized geometry.
λ1 = E+ (B) − E+ (B+),
λ2 = E (B+) − E (B)
λ3 = E− (B) − E− (B−),
λ4 = E (B−) − E (B)
The vertical and adiabatic EA and IP were assessed by applying the following equations:
IPa = E(B+) − E(B),
EAa = E(B) − E(B−),
IPv = E+(B) − E(B),
IPv = E+(B) − E(B),
The computed (λhole and λelec) and λint of the benzothiazole derivatives at the level of B3LYP/6-31+G** are shown in Table 4. The λhole was calculated to be much smaller than the λelec, with the difference in the range of 0.071 to 0.271 eV. Hitherto, it was pointed out that lower λ values can increase the charge transfer rate [65,69]. The substitution of -NO2 at position 5 of the furan in Comp3 led to a decrease in the polarization of the neutral to cation state, which meant that the λhole values were lower compared with other compounds, suggesting that this compound was a good choice for the hole transfer. It may turn out that the -CH3 group at position 5 of furan in Comp4 led to a decrease in the polarization of the neutral to anion, which will result in a lower λelec value in comparison with other compounds, suggesting that this compound may be a good candidate for electron transport. Here we also compared the λhole value of the benzothiazole derivatives with TPD (λhole = 0.290 eV) [70]. The λhole values of the benzothiazole derivatives were much lower than those of the best practice TPD, which suggested that the compounds we studied may be better hole transfer contenders.
3.4. Molecular Electrostatic Potential
Earlier works reported that diffraction methods would experimentally determine the molecular electrostatic potential (MEP) [62,63]. In addition, it could be tested by simulation. The MEP measures the widespread electronic distribution across the board, which is a broad aspect of understanding and predicting the reactivity of different compounds [64]. Figure 7 contains the MEP map in color. Red represents regions with high negative potential and blue indicates regions with high positive potential. The high potentials of negative/positive regions are ideal for electrophilic/nucleophilic attacks. The MEP decreases in the order blue > green > yellow > orange > red; red indicates the greatest repulsion while blue indicates the most attractive phase of the nucleophilic attack.
Figure 7 reveals that the nitrogen atom in the benzothiazole moiety, in particular, had negative potential, while -NH on the bridge with the -CH3 group had good positive potential. In Comp3, the -NO2 group had a negative potential. These results clearly indicated that in the event of a nucleophilic attack, the repulsion will be in potentially negative MEP areas, i.e., the benzothiazole moiety, especially the nitrogen atom and the -NO2 group, while the main attraction will be on the -NH and -CH3 groups. In addition, in the event of an electrophilic attack, the attraction may be to the benzothiazole moiety, especially the nitrogen atom and the -NO2 group, while the greater repulsion may be to the atoms in the -NH and the -CH3 groups.
3.5. UV–vis Experiment, Thin Film Preparation and Characterization
3.5.1. UV–vis Experiment
A set of UV–vis experiments was performed in various polar solvents to gain insight into the effect of solvent polarity on Comp1–4. Comp1, Comp2, and Comp4 produced similar results because of the presence of a similar D-A system as well as methyl substituents. Comp3 produced entirely different results because of the presence of strong acceptor -NO2 groups, which facilitated charge transfer more effectively than Comp1, Comp2, and Comp4. The peaks at 350–360 nm and 250–270 nm corresponded to the n-p* and p-p* transitions for Comp1–4. The additional peak in Comp3 at 430–445 nm was due to the presence of charge transfer. In addition, upon increasing the polarity of the solvent, a sequential red shift was observed, signifying the effect of solvent polarity on the D-A-based materials, see Figure 8 and Figure 9. The experimental results supported the observation made from computational data.
3.5.2. Thin Film Preparation
The synthesized compounds Comp1, Comp4, Comp2, and Comp3 were separately dissolved in chloroform and DMSO to obtain 5 × 10−2 M solutions of the compounds. The prepared solutions were individually dropped onto a precleaned FTO glass substrate. The substrate was then placed on the spin coater (Holmarc, Model no-HO-TH-05) and rotated at 1000 rpm for 30 s. This process was repeated three times to achieve a uniform film on the substrate. The spin-coated film was then annealed at 70 °C in an oven. In addition, silver contact was provided by applying silver paste on top of the film to fabricate a device in a sandwich-like fashion (FTO-Comp-Ag). We also used Al contact for HIE and EIE measurements of the compounds.
3.5.3. Thin Film Characterization
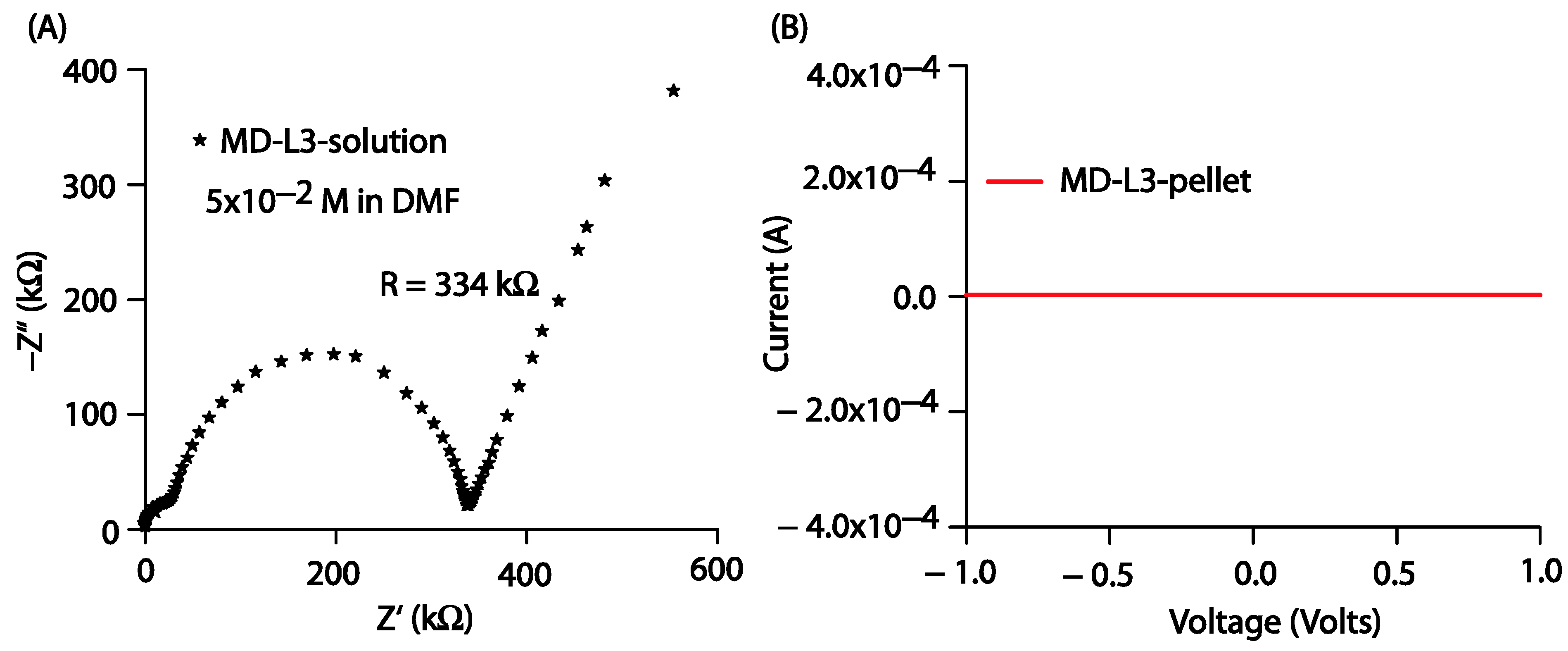
The current-voltage (I–V) characteristics of the prepared thin films were obtained using an electrochemical workstation (model CHI608E, CH Instruments) in linear sweep voltammetry mode at a scan rate of 100 mV/s. All the prepared thin films were subjected to the applied voltage; however, in response to the applied voltage, the prepared films failed to provide any output current, as shown in Figure S17. Furthermore, to gain insight into the conductance property of the prepared compounds in bulk, we performed electrochemical impedance spectroscopic (EIS) measurements using a 3-electrode system. The prepared solution of Comp2 in DMF was transferred into a cylindrical cell. The Nyquist impedance plot shown in Figure 10A provides the resistance of the Comp2 solution as 334 kW. We also tried I-V measurements on the Comp2 pellet, but there was no current in response to the applied voltage between −1 and 1 volt. We were likely unable to detect a significant current from the thin film because of its higher resistance value in bulk. However, further work related to molecular engineering to engage lone pairs with the objective of improving current response is under investigation.
4. Conclusions
The electronic, charge transfer, and optical properties of benzothiazole compounds were tuned. The absorption wavelengths were tested at the TD-B3LYP/6-31+G** level. Modification of chemical structures can greatly balance and improve electronic and charge transfer properties. In fact, the combination of benzothiazole with the furan unit and the -NO2 group resulted in a better separation of the absorption in the solar spectrum. The absorption maxima of this compound were red-shifted in the UV–vis region, expecting the blue region in the range of 481 nm in ethanol at the TD-B3LYP/6-31+G** level, which makes this material also suitable for efficient OFETs. The introduction of the -NO2 group into the furan at position 5 in Comp3 led to a red shift in the absorption spectra compared with other molecules. Comp1, Comp2, and Comp4 produced similar results because of the presence of a similar D-A system as well as the methyl substituents, while Comp3 produced entirely different results because of the presence of strong acceptor -NO2 groups, which facilitated charge transfer more effectively than Comp1, Comp2, and Comp4. Values of hole reorganization energies smaller than those of electrons would improve intrinsic hole mobility. The smaller electron reorganization energy value for Comp4 compared with other counterparts might lead to its better electron charge transfer ability. The insignificant current from the thin film was due to its higher resistance value in bulk. The reason we obtained insignificant hole/electron injection energy for the synthesized compounds was likely due to the presence of the free –NH group in the proposed molecules, resulting in the lowering of hole/electron mobility.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27248672/s1, Figures S1–S4: Characteristics of Comp1; Figures S5–S8: Characteristics of Comp2; Figures S9–S12: Characteristics of Comp3; Figures S13–S16: Characteristics of Comp4; Figure S17: I-V characteristics of (A) Comp1-film, (B) Comp2-film, (C) Comp3-film and (D) Comp4-film; Table S1: λmax for all the four compounds in acetone, ethanol, DMF and DMSO.
Author Contributions
Conceptualization, A.I.; methodology, A.G.A.-S. and A.I.; software, A.I.; validation, A.K., A.G.A.-S. and M.D.; formal analysis, A.K.; investigation, A.G.A.-S.; resources, A.I.; data curation, A.K.; writing—original draft preparation, M.D.; writing—review and editing, A.I.; visualization, A.G.A.-S.; supervision, A.I.; project administration, A.I.; funding acquisition, A.I. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful to King Abdulaziz City for Science and Technology (KACST), Riyadh, Saudi Arabia for funding this Project under Grant number: 12-ADV2976-07-R.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Reimers, J.R.; Picconnatto, C.A.; Ellenbogen, J.C.; Shashidhar, R. (Eds.) Molecular Electronics iii; New York Academy of Sciences: New York, NY, USA, 2003. [Google Scholar]
- Tour, J. Molecular Electronics; World Scientific: Singapore, 2003. [Google Scholar]
- Cunimberti, G.; Fagas, G.; Richter, K. (Eds.) Introducing Molecular Electronics. In Lecture Notes in Physics; Springer: Berlin, Germany, 2005. [Google Scholar]
- Habib, M.; Ghosh, N.N.; Sarkar, R.; Pramanik, A.; Sarkar, P.; Pal, S. Controlling the charge transfer and recombination dynamics in hollow zno qd based dye sensitized solar cell: An insight from ab initio simulation. Chem. Phys. Lett. 2018, 709, 21–25. [Google Scholar] [CrossRef]
- Pramanik, A.; Sarkar, S.; Pal, S.; Sarkar, P. Pentacene–fullerene bulk-heterojunction solar cell: A computational study. Phys. Lett. A 2015, 379, 1036–1042. [Google Scholar] [CrossRef]
- Bouit, P.-A.; Infantes, L.; Calbo, J.; Viruela, R.; Ortí, E.; Delgado, J.L.; Martín, N. Efficient light harvesters based on the 10-(1,3-dithiol-2-ylidene)anthracene core. Org. Lett. 2013, 15, 4166–4169. [Google Scholar] [CrossRef] [PubMed]
- Habib; Saha, S.; Sarkar, R.; Pramanik, A.; Sarkar, P.; Pal, S. Computational design of some TTF-substituted acene-based dyes for solar cell application using hollow ZnO quantum dot as acceptor. Comput. Theor. Chem. 2018, 1136–1137, 10–17. [Google Scholar] [CrossRef]
- Roy, P.; Biswas, S.; Pramanik, A.; Sarkar, P. Substitution induced carrier switching in s,n-heteroacene molecular junctions: A first principle analysis. Chem. Phys. Lett. 2018, 708, 87–93. [Google Scholar] [CrossRef]
- Irfan, A.; Muhammad, S.; Al-Sehemi, A.G.; Al-Assiri, M.S.; Kalam, A.; Chaudhry, A.R. The effect of anchoring groups on the electro-optical and charge injection in triphenylamine derivatives@ti6o12. J. Theor. Comput. Chem. 2015, 14, 1550027. [Google Scholar] [CrossRef]
- Irfan, A.; Al-Sehemi, A.G.; Al-Assiri, M.S. Push–pull effect on the electronic, optical and charge transport properties of the benzo [2,3-b]thiophene derivatives as efficient multifunctional materials. Comput. Theor. Chem. 2014, 1031, 76–82. [Google Scholar] [CrossRef]
- Şengez, B.; Doğruyol, Z.; San, S.E.; Kösemen, A.; Yılmaz, F.; Okutan, M.; Yerli, Y.; Demir, A.; Başaran, E. Use of side chain thiophene containing copolymer as a non-ionic gel-dielectric material for sandwich ofet assembly. Microelectron. Eng. 2013, 103, 111–117. [Google Scholar] [CrossRef]
- Szlachcic, P.; Danel, K.S.; Gryl, M.; Stadnicka, K.; Usatenko, Z.; Nosidlak, N.; Lewińska, G.; Sanetra, J.; Kuźnik, W. Organic light emitting diodes (oled) based on helical structures containing 7-membered fused rings. Dyes Pigm. 2015, 114, 184–195. [Google Scholar] [CrossRef]
- Jungsuttiwong, S.; Tarsang, R.; Surakhot, Y.; Khunchalee, J.; Sudyoadsuk, T.; Promarak, V.; Namuangruk, S. Theoretical study of α-fluorenyl oligothiophenes as color tunable emissive materials for highly efficient electroluminescent device. Org. Electron. 2012, 13, 1836–1843. [Google Scholar] [CrossRef]
- Marks, T.J.; Hersam, M.C. Materials science: Semiconductors grown large and thin. Nature 2015, 520, 631–632. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, A.; Prakasam, M.; Anbarasan, P.M. Influence of donor substitution at d-π-aarchitecture in efficient sensitizers for dye-sensitized solar cells: First-principle study. Bull. Mater. Sci. 2017, 40, 1389–1396. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.N.; Habib, M.; Pramanik, A.; Sarkar, P.; Pal, S. Tuning the bodipy core for its potential use in dssc: A quantum chemical approach. Bull. Mater. Sci. 2018, 41, 56. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.S.; Charanadhar, N.; Srikanth, V.V.; Rao, K.B.S.; Raj, B. Materials in harnessing solar power. Bull. Mater. Sci. 2018, 41, 62. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Huang, S.; Wang, C.; Yue, Y.; Zhang, Q. Computational prediction for oxidation and reduction potentials of organic molecules used in organic light-emitting diodes. Org. Electron. 2019, 64, 216–222. [Google Scholar] [CrossRef]
- Li, M.; Kou, L.; Diao, L.; Zhang, Q.; Li, Z.; Wu, Q.; Lu, W.; Pan, D. Theoretical Study of Acene-Bridged Dyes for Dye-Sensitized Solar Cells. J. Phys. Chem. A 2015, 119, 3299–3309. [Google Scholar] [CrossRef]
- Biswas, S.; Pramanik, A.; Pal, S.; Sarkar, P. A theoretical perspective on the photovoltaic performance of s,n-heteroacenes: An even–odd effect on the charge separation dynamics. J. Phys. Chem. C 2017, 121, 2574–2587. [Google Scholar] [CrossRef]
- Tarsang, R.; Promarak, V.; Sudyoadsuk, T.; Namuangruk, S.; Kungwan, N.; Khongpracha, P.; Jungsuttiwong, S. Triple bond-modified anthracene sensitizers for dye-sensitized solar cells: A computational study. RSC Adv. 2015, 5, 38130–38140. [Google Scholar] [CrossRef]
- Al-Horaibi, S.A.; Alrabie, A.A.; Alghamdi, M.T.; Al-Ostoot, F.H.; Garoon, E.M.; Rajbhoj, A.S. Novel hemicyanine sensitizers based on benzothiazole-indole for dye-sensitized solar cells: Synthesis, optoelectrical characterization and efficiency of solar cell. J. Mol. Struct. 2020, 1224, 128836. [Google Scholar] [CrossRef]
- Paczkowski, I.M.; Coelho, F.L.; Campo, L.F. 2,1,3-Benzothiadiazole dyes conjugated with benzothiazole and benzoxazole: Synthesis, solvatochromism and solid-state properties. J. Mol. Liq. 2020, 319, 114277. [Google Scholar] [CrossRef]
- Dutta, G.K.; Guha, S.; Patil, S. Synthesis of liquid crystalline benzothiazole based derivatives: A study of their optical and electrical properties. Org. Electron. 2010, 11, 1–9. [Google Scholar] [CrossRef]
- Al-Horaibi, S.A.; Asiri, A.M.; El-Shishtawy, R.M.; Gaikwad, S.T.; Rajbhoj, A.S. Indoline and benzothiazole-based squaraine dye-sensitized solar cells containing bis-pendent sulfonate groups: Synthesis, characterization and solar cell performance. J. Mol. Struct. 2019, 1195, 591–597. [Google Scholar] [CrossRef]
- Shehzad, R.A.; Iqbal, J.; Khan, M.U.; Hussain, R.; Javed, H.M.A.; Rehman, A.U.; Alvi, M.U.; Khalid, M. Designing of benzothiazole based non-fullerene acceptor (NFA) molecules for highly efficient organic solar cells. Comput. Theor. Chem. 2020, 1181, 112833. [Google Scholar] [CrossRef]
- Aziz, H.; Popovic, Z.D.; Hu, N.-X.; Hor, A.-M.; Xu, G. Degradation Mechanism of Small Molecule-Based Organic Light-Emitting Devices. Science 1999, 283, 1900–1902. [Google Scholar] [CrossRef]
- Grimsdale, A.C.; Chan, K.L.; Martin, R.E.; Jokisz, P.G.; Holmes, A.B. Synthesis of Light-Emitting Conjugated Polymers for Applications in Electroluminescent Devices. Chem. Rev. 2009, 109, 897–1091. [Google Scholar]
- Hughes, G.; Bryce, M.R. Electron-transporting materials for organic electroluminescent and electrophosphorescent devices. J. Mater. Chem. 2004, 15, 94–107. [Google Scholar] [CrossRef]
- Yoon, K.R.; Ko, S.-O.; Lee, S.M.; Lee, H. Synthesis and characterization of carbazole derived nonlinear optical dyes. Dye. Pigment. 2007, 75, 567–573. [Google Scholar] [CrossRef]
- Garnier, F. Organic-based electronics à la carte. Acc. Chem. Res. 1999, 32, 209–215. [Google Scholar] [CrossRef]
- Katz, H.E.; Bao, Z.; Gilat, S.L. Synthetic Chemistry for Ultrapure, Processable, and High-Mobility Organic Transistor Semiconductors. Acc. Chem. Res. 2001, 34, 359–369. [Google Scholar] [CrossRef]
- Shirota, Y.; Kageyama, H. Charge Carrier Transporting Molecular Materials and Their Applications in Devices. Chem. Rev. 2007, 107, 953–1010. [Google Scholar] [CrossRef]
- Izumi, T.; Kobashi, S.; Takimiya, K.; Aso, Y.; Otsubo, T. Synthesis and spectroscopic properties of a series of β-blocked long oligothiophenes up to the 96-mer: Revaluation of effective conjugation length. J. Am. Chem. Soc. 2003, 125, 5286–5287. [Google Scholar] [CrossRef] [PubMed]
- Meier, H.; Gerold, J.; Kolshorn, H.; Mühling, B. Extension of Conjugation Leading to Bathochromic or Hypsochromic Effects in OPV Series. Chem.-A Eur. J. 2004, 10, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.; Li, Z.H.; Tao, A.Y.; D’Iorio, M. Synthesis and Functional Properties of Donor−Acceptor π-Conjugated Oligomers. Chem. Mater. 2003, 15, 1198–1203. [Google Scholar] [CrossRef]
- Schweikart, K.-H.; Hanack, M.; Lüer, L.; Oelkrug, D. Synthesis, absorption and luminescence of a new series of soluble distyrylbenzenes featuring cyano substituents at the peripheral rings. Eur. J. Org. Chem. 2001, 2001, 293–302. [Google Scholar] [CrossRef]
- Lee, T.H.; Tong, K.L.; So, S.K.; Leung, L.M. Synthesis and electroluminescence of thiophene-based bipolar small molecules with different arylamine moieties. Synth. Met. 2005, 155, 116–124. [Google Scholar] [CrossRef]
- Cao, D.-X.; Fang, Q.; Wang, D.; Liu, Z.-Q.; Xue, G.; Xu, G.-B.; Yu, W.-T. Synthesis and two-photon-excited fluorescence of benzothiazole-based compounds with various π-electron donors. Eur. J. Org. Chem. 2003, 2003, 3628–3636. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Q.; Ren, H.; Yan, H.; Zhang, J.; Zhang, H.; Gu, J. Calculation of band gap in long alkyl-substituted heterocyclic-thiophene-conjugated polymers with electron donor–acceptor fragment. Sol. Energy Mater. Sol. Cells 2008, 92, 581–587. [Google Scholar] [CrossRef]
- Hutchison, G.R.; Ratner, M.A.; Marks, T.J. Accurate Prediction of Band Gaps in Neutral Heterocyclic Conjugated Polymers. J. Phys. Chem. A 2002, 106, 10596–10605. [Google Scholar] [CrossRef]
- Kwon, O.; McKee, M.L. Theoretical Calculations of Band Gaps in the Aromatic Structures of Polythieno[3,4-b]benzene and Polythieno[3,4-b]pyrazine. J. Phys. Chem. A 2000, 104, 7106–7112. [Google Scholar] [CrossRef]
- Hassanali, A.A.; Li, T.; Zhong, D.; Singer, S.J. A Molecular Dynamics Study of Lys-Trp-Lys: Structure and Dynamics in Solution Following Photoexcitation. J. Phys. Chem. B 2006, 110, 10497–10508. [Google Scholar] [CrossRef]
- Irfan, A.; Imran, M.; Thomas, R.; Basra, M.A.R.; Ullah, S.; Al-Sehemi, A.G.; Assiri, M.A. Exploring the effect of oligothiophene and acene cores on the optoelectronic properties and enhancing p- and n-type ability of semiconductor materials. J. Sulfur Chem. 2020, 42, 180–192. [Google Scholar] [CrossRef]
- Irfan, A. Push-pull effect on the charge transport characteristics in V-shaped organic semiconductor materials. Bull. Mater. Sci. 2021, 44, 43. [Google Scholar] [CrossRef]
- Wazzan, N.; Irfan, A. Promising architectures modifying the d-π-a architecture of 2,3-dipentyldithieno[3,2-f:2′,3′-h]quinoxaline-based dye as efficient sensitizers in dye-sensitized solar cells: A dft study. Mater. Sci. Semicond. Process. 2020, 120, 105260. [Google Scholar] [CrossRef]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M.; Ni, N.; Shaaban, M.I. Synthesis, antimicrobial, antiquorum-sensing and cytotoxic activities of new series of benzothiazole derivatives. Chin. Chem. Lett. 2015, 26, 1522–1528. [Google Scholar] [CrossRef]
- Soni, B.; Ranawat, M.S.; Sharma, R.; Bhandari, A.; Sharma, S. Synthesis and evaluation of some new benzothiazole derivatives as potential antimicrobial agents. Eur. J. Med. Chem. 2010, 45, 2938–2942. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. Iii. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Tang, S.-S.; Liu, J.-B.; Chen, G.; Jin, R.-F. Theoretical study on electronic and charge transfer properties of oligo[8]thiophene and its circular, hooped, and helical derivatives. Chin. J. Struct. Chem. 2014, 33, 104–114. [Google Scholar]
- Li, P.; Bu, Y.; Ai, H. Theoretical Determinations of Ionization Potential and Electron Affinity of Glycinamide Using Density Functional Theory. J. Phys. Chem. A 2004, 108, 1200–1207. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433–7447. [Google Scholar] [CrossRef]
- Frisch, G.W.T.M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Takano, Y.; Houk, K.N. Benchmarking the Conductor-like Polarizable Continuum Model (CPCM) for Aqueous Solvation Free Energies of Neutral and Ionic Organic Molecules. J. Chem. Theory Comput. 2004, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www2.chemistry.msu.edu/faculty/harrison/cem483/work_functions.pdf (accessed on 25 April 2021).
- Sonar, P.; Singh, S.P.; Leclère, P.; Surin, M.; Lazzaroni, R.; Lin, T.T.; Dodabalapur, A.; Sellinger, A. Synthesis, characterization and comparative study of thiophene–benzothiadiazole based donor–acceptor–donor (d–a–d) materials. J. Mater. Chem. 2009, 19, 3228–3237. [Google Scholar] [CrossRef]
- Nikolka, M.; Nasrallah, I.; Rose, B.; Ravva, M.K.; Broch, K.; Sadhanala, A.; Harkin, D.; Charmet, J.; Hurhangee, M.; Brown, A.; et al. High operational and environmental stability of high-mobility conjugated polymer field-effect transistors through the use of molecular additives. Nat. Mater. 2017, 16, 356–362. [Google Scholar] [CrossRef] [Green Version]
- De Leeuw, D.; Simenon, M.; Brown, A.; Einerhand, R. Stability of n-type doped conducting polymers and consequences for polymeric microelectronic devices. Synth. Met. 1997, 87, 53–59. [Google Scholar] [CrossRef]
- Abbaszadeh, D.; Kunz, A.; Kotadiya, N.B.; Mondal, A.; Andrienko, D.; Michels, J.J.; Wetzelaer, G.-J.A.H.; Blom, P.W.M. Electron trapping in conjugated polymers. Chem. Mater. 2019, 31, 6380–6386. [Google Scholar] [CrossRef] [Green Version]
- Usta, H.; Risko, C.; Wang, Z.; Huang, H.; Deliomeroglu, M.K.; Zhukhovitskiy, A.; Facchetti, A.; Marks, T.J. Design, synthesis, and characterization of ladder-type molecules and polymers. Air-stable, solution-processable n-channel and ambipolar semiconductors for thin-film transistors via experiment and theory. J. Am. Chem. Soc. 2009, 131, 5586–5608. [Google Scholar] [CrossRef]
- Felscia, U.R.; Rajkumar, B.J.M.; Mary, M.B. Charge transport properties of pyrene and its derivatives: Optoelectronic and nonlinear optical applications. J. Mater. Sci. 2018, 53, 15213–15225. [Google Scholar] [CrossRef]
- Yang, G.; Su, Z.; Qin, C. Theoretical study on the second-order nonlinear optical properties of asymmetric spirosilabifluorene derivatives. J. Phys. Chem. A 2006, 110, 4817–4821. [Google Scholar] [CrossRef]
- Wazzan, N.; El-Shishtawy, R.M.; Irfan, A. DFT and TD–DFT calculations of the electronic structures and photophysical properties of newly designed pyrene-core arylamine derivatives as hole-transporting materials for perovskite solar cells. Theor. Chim. Acta 2017, 137, 9. [Google Scholar] [CrossRef]
- Wazzan, N.; Irfan, A. Theoretical study of triphenylamine-based organic dyes with mono-, di-, and tri-anchoring groups for dye-sensitized solar cells. Org. Electron. 2018, 63, 328–342. [Google Scholar] [CrossRef]
- Brédas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-Transfer and Energy-Transfer Processes in π-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, W.; Ma, Y. Molecular design toward good hole transport materials based on anthra[2,3-c]thiophene: A theoretical investigation. Comput. Theor. Chem. 2013, 1010, 25–31. [Google Scholar] [CrossRef]
- Chai, W.; Jin, R. Theoretical investigations into optical and charge transfer properties of donor-acceptor 1,8-naphthalimide derivatives as possible organic light-emitting materials. J. Mol. Struct. 2016, 1103, 177–182. [Google Scholar] [CrossRef]
- Gruhn, N.E.; Filho, D.A.D.S.; Bill, T.G.; Malagoli, M.; Coropceanu, V.; Kahn, A.; Brédas, J.-L. The Vibrational Reorganization Energy in Pentacene: Molecular Influences on Charge Transport. J. Am. Chem. Soc. 2002, 124, 7918–7919. [Google Scholar] [CrossRef]
Figure 1.
Donor–acceptor (DA) model system diagram.

Figure 2.
The structures of benzothiazole derivatives.

Figure 3.
(A) Two adjacent molecules showing the π-π stacking interaction also displayed with the help of (B) the space fill model, (C) H-bonding interaction between two laterally close molecules (bond length shown in Å), (D) extended network of molecules though weak interactions (π-π stacking and H-bonding), and (E) labeling of each atom shown in ball stick model.
Figure 3.
(A) Two adjacent molecules showing the π-π stacking interaction also displayed with the help of (B) the space fill model, (C) H-bonding interaction between two laterally close molecules (bond length shown in Å), (D) extended network of molecules though weak interactions (π-π stacking and H-bonding), and (E) labeling of each atom shown in ball stick model.

Figure 4.
(A) View along the a* axis displaying the packing of molecules in monoclinic fashion, (B) Van der Waal interaction present between two molecules, and (C) ORTEP diagram of Comp2 (ellipsoids at 40% probability).
Figure 4.
(A) View along the a* axis displaying the packing of molecules in monoclinic fashion, (B) Van der Waal interaction present between two molecules, and (C) ORTEP diagram of Comp2 (ellipsoids at 40% probability).

Figure 5.
The frontier molecular orbitals distribution of benzothiazoles at a ground state (contour value = 0.035).
Figure 5.
The frontier molecular orbitals distribution of benzothiazoles at a ground state (contour value = 0.035).

Figure 6.
The frontier molecular orbitals distribution of benzothiazoles in an excited state (contour value = 0.035).
Figure 6.
The frontier molecular orbitals distribution of benzothiazoles in an excited state (contour value = 0.035).

Figure 7.
Molecular electrostatic potential surfaces views of benzothiazole derivatives.

Figure 8.
UV–vis spectrum of (A) Comp1 (0.5 × 10−5 M) in DMF and DMSO; (B) Comp2 (0.5 × 10−5 M) in acetone, ethanol, DMF, and DMSO.
Figure 8.
UV–vis spectrum of (A) Comp1 (0.5 × 10−5 M) in DMF and DMSO; (B) Comp2 (0.5 × 10−5 M) in acetone, ethanol, DMF, and DMSO.

Figure 9.
UV–vis spectrum of (A) Comp3 (0.5 × 10−5 M) in DMF and DMSO; (B) Comp4 (0.5 × 10−5 M) in acetone, ethanol, DMF, and DMSO.
Figure 9.
UV–vis spectrum of (A) Comp3 (0.5 × 10−5 M) in DMF and DMSO; (B) Comp4 (0.5 × 10−5 M) in acetone, ethanol, DMF, and DMSO.

Figure 10.
(A) Nyquist impedance plot for Comp2 solution in DMF (5 × 10−2 M), (B) I-V characteristics for Comp2 pellet.
Figure 10.
(A) Nyquist impedance plot for Comp2 solution in DMF (5 × 10−2 M), (B) I-V characteristics for Comp2 pellet.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The HOMO/LUMO energies (EHOMO/ELUMO) and energy gaps (Egap) of benzothiazoles at the B3LYP/6-31+G** and TD-B3LYP/6-31+G** levels for S0 and S1 (in eV), respectively.
Table 1.
The HOMO/LUMO energies (EHOMO/ELUMO) and energy gaps (Egap) of benzothiazoles at the B3LYP/6-31+G** and TD-B3LYP/6-31+G** levels for S0 and S1 (in eV), respectively.
| Compounds | EHOMO | ELUMO | Egap | EHOMO | ELUMO | Egap |
|---|---|---|---|---|---|---|
| Ground State | Excited State | |||||
| Comp1 | −5.59 | −1.95 | 3.64 | −4.77 | −2.61 | 2.16 |
| Comp2 | −5.58 | −1.88 | 3.70 | −4.76 | −2.55 | 2.21 |
| Comp3 | −6.18 | −3.35 | 2.83 | −5.13 | −4.01 | 1.12 |
| Comp4 | −5.52 | −1.92 | 3.60 | −4.73 | −2.56 | 2.17 |
Table 2.
The electron/hole injection energy (EIE/HIE) barriers in eV from benzothiazoles to silver (Ag)/aluminum (Al) electrodes calculated at the B3LYP/6-31+G** level.
Table 2.
The electron/hole injection energy (EIE/HIE) barriers in eV from benzothiazoles to silver (Ag)/aluminum (Al) electrodes calculated at the B3LYP/6-31+G** level.
| Compounds | HIE (Ag) | EIE (Ag) | HIE (Al) | EIE (Al) |
|---|---|---|---|---|
| Comp1 | 0.85 | 2.79 | 1.51 | 2.13 |
| Comp2 | 0.84 | 2.86 | 1.50 | 2.20 |
| Comp3 | 1.44 | 1.39 | 2.10 | 0.73 |
| Comp4 | 0.78 | 1.82 | 1.44 | 2.16 |
Table 3.
The absorption wavelengths (λmax, nm), oscillator strengths (f), percent contribution, and key transitions in the gas phase as well as in solvents (ethanol, acetone, DMF, and DMSO) of benzothiazole derivatives at the TD-B3LYP/6-31+G** level.
Table 3.
The absorption wavelengths (λmax, nm), oscillator strengths (f), percent contribution, and key transitions in the gas phase as well as in solvents (ethanol, acetone, DMF, and DMSO) of benzothiazole derivatives at the TD-B3LYP/6-31+G** level.
| Comp | λmax | f | Tran | %Con | λmax | f | Tran | %Con |
|---|---|---|---|---|---|---|---|---|
| Gas phase | In Ethanol | |||||||
| Comp1 | 358 | 0.8839 | H→L | 70% | 363 | 1.1034 | H→L | 70% |
| 281 | 0.2282 | H→L + 1 | 30% | 281 | 0.1313 | H→L + 1 | 29% | |
| Comp2 | 357 | 0.8301 | H→L | 70% | 359 | 1.0638 | H→L | 70% |
| 278 | 0.2280 | H→L + 1 | 35% | 277 | 0.1452 | H→L + 1 | 39% | |
| Comp3 | 451 | 0.6867 | H→L | 70% | 481 | 0.8210 | H→L | 70% |
| 323 | 0.4853 | H→L + 1 | 27% | 335 | 0.4279 | H→L + 1 | 23% | |
| Comp4 | 362 | 0.9571 | H→L | 70% | 367 | 1.1864 | H→L | 70% |
| 284 | 0.2341 | H→L + 1 | 29% | 283 | 0.1312 | H→L + 1 | 31% | |
| Comp | λmax | f | Tran | %Con | λmax | f | Tran | %Con |
| In acetone | In DMF | |||||||
| Comp1 | 363 | 1.1018 | H→L | 70% | 364 | 1.1213 | H→L | 70% |
| 281 | 0.1323 | H→L + 1 | 29% | 281 | 0.1280 | H→L + 1 | 29% | |
| Comp2 | 359 | 1.0620 | H→L | 70% | 360 | 1.0840 | H→L | 70% |
| 277 | 0.1461 | H→L + 1 | 39% | 277 | 0.1429 | H→L + 1 | 39% | |
| Comp3 | 481 | 0.8202 | H→L | 70% | 484 | 0.8388 | H→L | 70% |
| 335 | 0.4287 | H→L + 1 | 23% | 336 | 0.4224 | H→L + 1 | 23% | |
| Comp4 | 367 | 1.1847 | H→L | 70% | 368 | 1.2041 | H→L | 70% |
| 283 | 0.1322 | H→L + 1 | 31% | 283 | 0.1280 | H→L + 1 | 31% | |
| Comp | λmax | f | Tran | %Con | ||||
| In DMSO | ||||||||
| Comp1 | 364 | 1.1189 | H→L | 70% | ||||
| 281 | 0.1278 | H→L + 1 | 29% | |||||
| Comp2 | 360 | 1.0813 | H→L | 70% | ||||
| 277 | 0.1425 | H→L + 1 | 39% | |||||
| Comp3 | 484 | 0.8357 | H→L | 70% | ||||
| 336 | 0.4228 | H→L + 1 | 23% | |||||
| Comp4 | 368 | 1.2018 | H→L | 70% | ||||
| 283 | 0.1278 | H→L + 1 | 31% | |||||
Table 4.
Adiabatic/vertical ionization potential (IPa/IPv), adiabatic/vertical electron affinity (EAa/EAv), and electron/hole reorganization energies () in eV of benzothiazoles at the B3LYP/6-31+G** level.
Table 4.
Adiabatic/vertical ionization potential (IPa/IPv), adiabatic/vertical electron affinity (EAa/EAv), and electron/hole reorganization energies () in eV of benzothiazoles at the B3LYP/6-31+G** level.
| Compounds | IPa | EAa | IPv | EAv | |||
|---|---|---|---|---|---|---|---|
| Comp1 | 7.07 | 0.41 | 6.96 | 0.52 | 0.235 | 0.327 | 0.092 |
| Comp2 | 7.05 | 0.36 | 6.95 | 0.46 | 0.233 | 0.340 | 0.107 |
| Comp3 | 7.64 | 2.09 | 7.52 | 1.85 | 0.216 | 0.487 | 0.271 |
| Comp4 | 6.98 | 0.39 | 6.88 | 0.51 | 0.249 | 0.320 | 0.071 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Irfan, A.; Kalam, A.; Al-Sehemi, A.G.; Dubey, M. Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives. Molecules 2022, 27, 8672. https://doi.org/10.3390/molecules27248672
AMA Style
Irfan A, Kalam A, Al-Sehemi AG, Dubey M. Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives. Molecules. 2022; 27(24):8672. https://doi.org/10.3390/molecules27248672
Chicago/Turabian StyleIrfan, Ahmad, Abul Kalam, Abdullah G. Al-Sehemi, and Mrigendra Dubey. 2022. "Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives" Molecules 27, no. 24: 8672. https://doi.org/10.3390/molecules27248672