
Synthesis of 2-Cyanobenzothiazoles via Pd-Catalyzed/Cu-Assisted C-H Functionalization/Intramolecular C-S Bond Formation from N-Arylcyanothioformamides †

Abstract
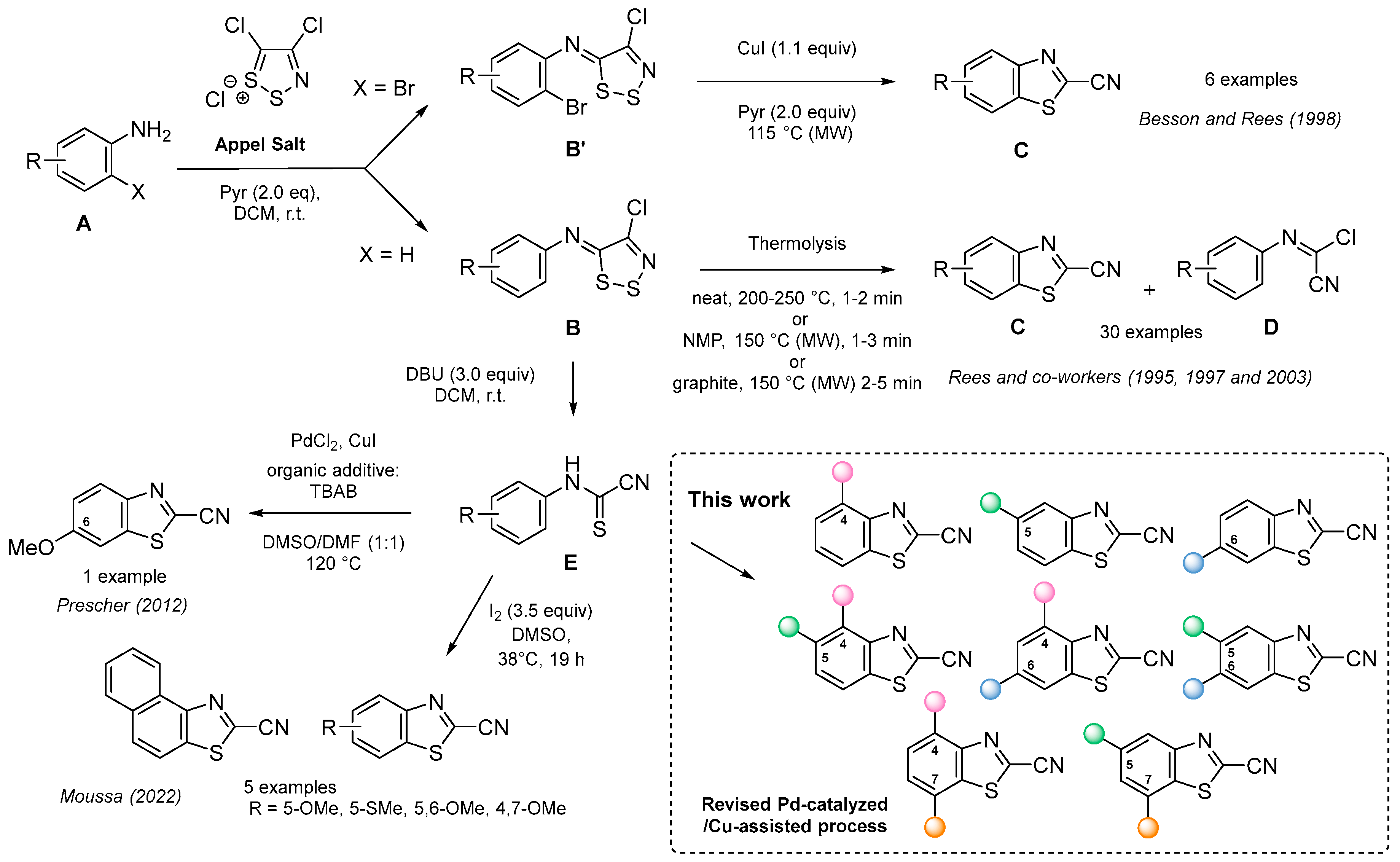
:1. Introduction
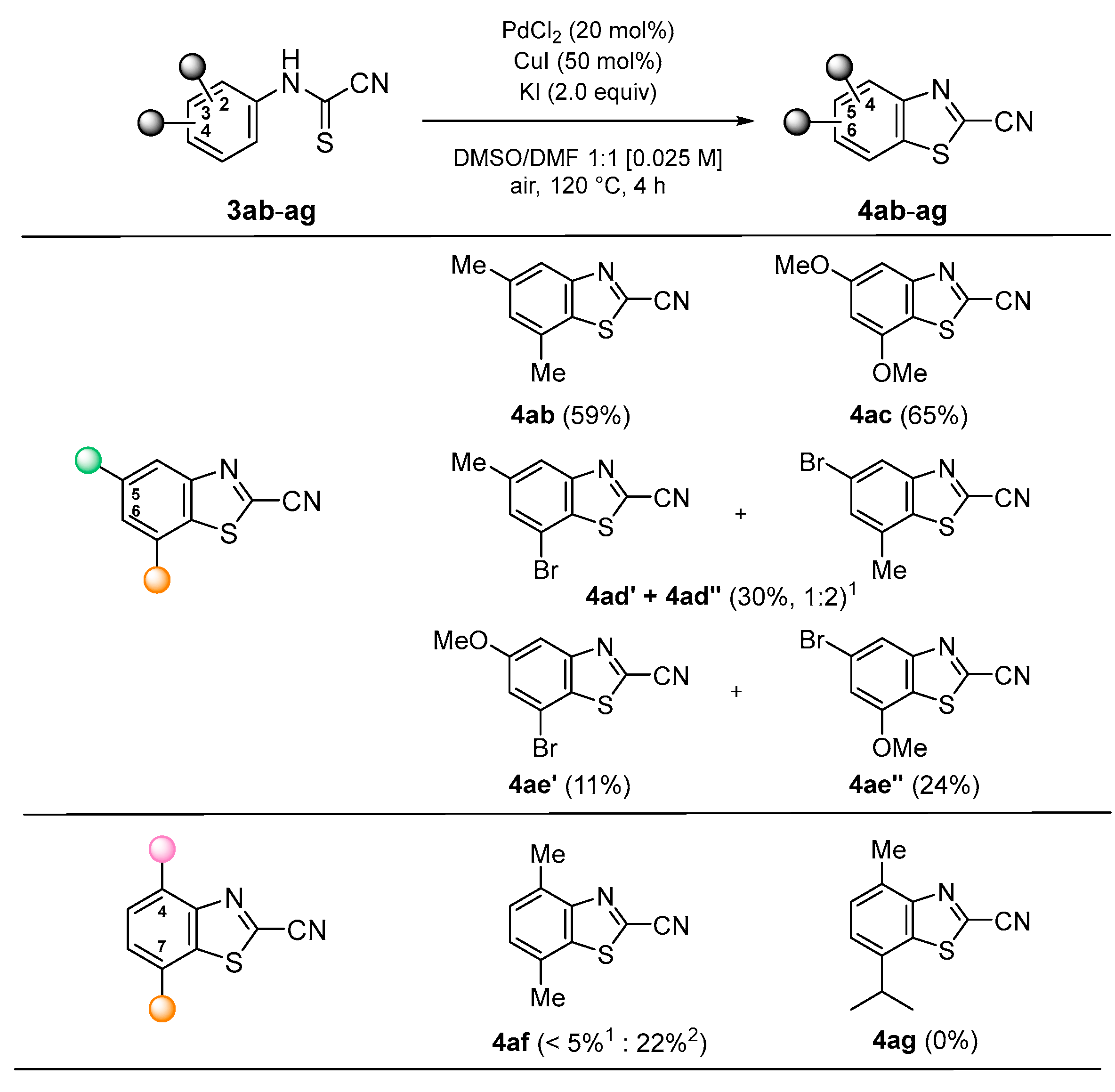
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Chemistry
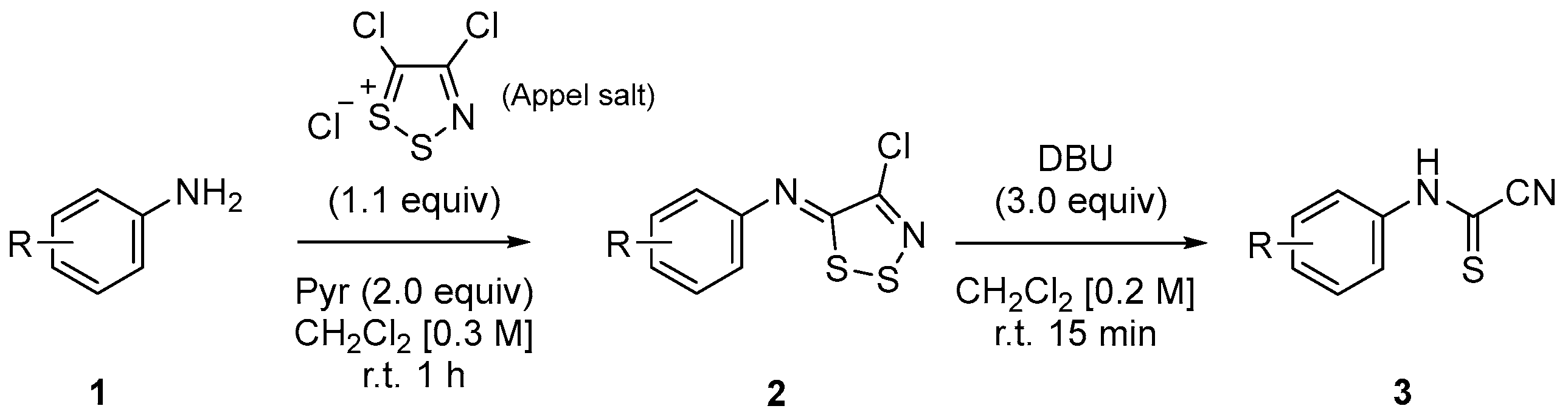
3.2.1. Synthesis of N-Arylimino-1,2,3-dithiazoles (2) and N-Arylcyanothioformamides (3)
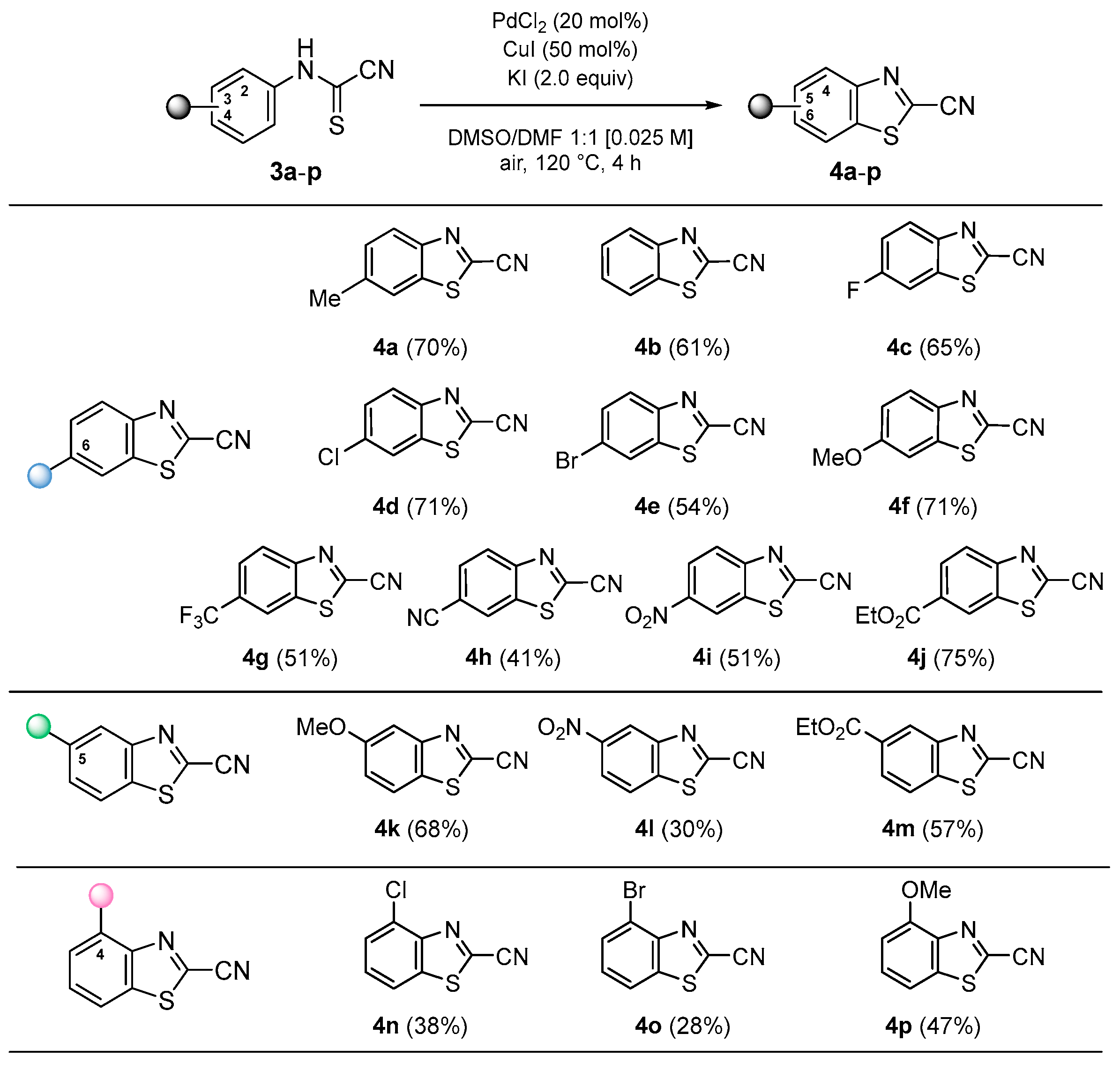
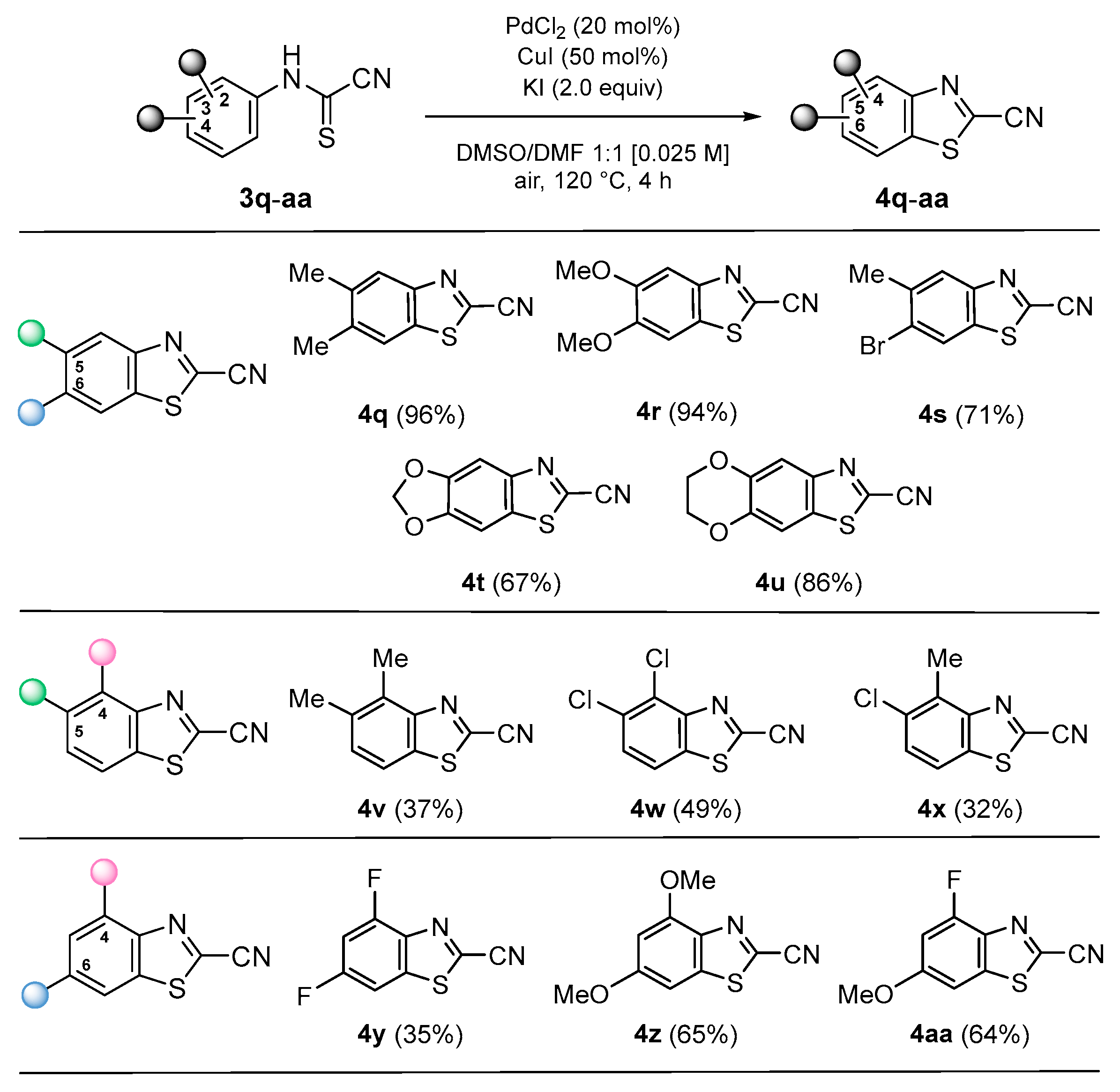
3.2.2. Synthesis of 2-Cyanobenzothiazoles (4)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kamal, A.; Syed, M.A.H.; Mohammed, S.M. Therapeutic potential of benzothiazoles: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Law, C.S.W.; Yeong, K.Y. Current trends of benzothiazoles in drug discovery: A patent review (2015–2020). Expert Opin. Ther. Pat. 2022, 32, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagumpi, S. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Gandhi, D.; Kalal, P. Benzothiazole: A versatile and multitargeted pharmacophore in the field of medicinal chemistry. Lett. Org. Chem. 2017, 14, 729–742. [Google Scholar] [CrossRef]
- Qadir, T.; Amin, A.; Salhotra, A.; Sharma, P.K.; Jeelani, I.; Abe, H. Recent advances in the synthesis of benzothiazole and its derivatives. Curr. Org. Chem. 2022, 26, 189–214. [Google Scholar] [CrossRef]
- Chanchal, S.; Rajnish, K.; Avijit, M.; Salahuddin, S.; Ajay, K.; Rakesh, S.; Shivali, M.; Mohd, A.M. Benzothiazole: Synthetic strategies, biological potential, and interactions with targets. Mini Rev. Org. Chem. 2022, 19, 242–256. [Google Scholar] [CrossRef]
- Noolvi, M.N.; Patel, H.M.; Kaur, M. Benzothiazoles: Search for anticancer agents. Eur. J. Med. Chem. 2012, 54, 447–462. [Google Scholar] [CrossRef]
- Hassan, A.Y.; Sarg, M.T.; Hussein, E.M. Design, synthesis, and anticancer activity of novel benzothiazole analogues. J. Heterocycl. Chem. 2019, 56, 1437–1457. [Google Scholar] [CrossRef]
- Irfan, A.; Batool, F.; Naqvi, S.A.Z.; Islam, A.; Osman, S.M.; Nocentini, A.; Alissa, S.A.; Supuran, C.T. Benzothiazole derivatives as anticancer agents, Journal of Enzyme Inhibition and Medicinal Chemistry. J. Enzym. Inhib. Med. Chem. 2020, 35, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.C.; Sharma, D.; Sharma, A.; Bansal, K.K.; Rajak, H.; Sharma, S.; Thakur, V.K. New horizons in benzothiazole scaffold for cancer therapy: Advances in bioactivity, functionality, and chemistry. Appl. Mater. Today 2020, 20, 100783. [Google Scholar] [CrossRef]
- Pathak, N.; Rathi, E.; Kumar, N.; Kini, S.G.; Rao, C.M. A Review on anticancer potentials of benzothiazole derivatives. Mini Rev. Med. Chem. 2020, 20, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Dhadda, S.; Raigar, A.K.; Saini, K.; Manju; Guleria, A. Benzothiazoles: From recent advances in green synthesis to anti-cancer potential. Sustain. Chem. Pharm. 2021, 24, 100521. [Google Scholar] [CrossRef]
- Prajapati, N.P.; Vekariya, R.H.; Borad, M.A.; Patel, H.D. Recent advances in the synthesis of 2-substituted benzothiazoles: A review. RSC Adv. 2014, 4, 60176–60208. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; Carradori, S.; Amoroso, R.; Fernández, I.F. 2-Substituted benzothiazoles as antiproliferative agents: Novel insights on structure-activity relationships. Eur. J. Med. Chem. 2020, 207, 112762. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liu, J.; Zuo, X.; Feng, X.; Gao, Y. Recent advances in synthesis of benzothiazole compounds related to green chemistry. Molecules 2020, 25, 1675. [Google Scholar] [CrossRef] [Green Version]
- Haider, K.; Rehman, S.; Pathak, A.; Naimi, A.K.; Yar, M.S. Advances in 2-substituted benzothiazole scaffold-based chemotherapeutic agents. Arch. Pharm. 2021, 354, 2100246. [Google Scholar] [CrossRef]
- Bénéteau, V.; Besson, T.; Guillard, J.; Leonce, S.; Pfeiffer, B. Synthesis and in vitro antitumour evaluation of benzothiazole-2-carbonitrile derivatives. Eur. J. Med. Chem. 1999, 34, 1053–1060. [Google Scholar] [CrossRef]
- Lamazzi, C.; Chabane, H.; Thiéry, V.; Pierré, A.; Léonce, S.; Pfeiffer, B.; Renard, P.; Guillaumet, G.; Besson, T. Synthesis and cytotoxic evaluation of novel thiazolocarbazoles. J. Enzym. Inhib. Med. Chem. 2002, 17, 397–401. [Google Scholar] [CrossRef]
- Testard, A.; Picot, L.; Fruitier-Arnaudin, I.; Piot, J.-M.; Chabane, H.; Domon, L.; Thiéry, V.; Besson, T. Microwave-assisted synthesis of novel thiazolocarbazoles and evaluation as potential anticancer agents. Part III. J. Enzym. Inhib. Med. Chem. 2004, 19, 467–473. [Google Scholar] [CrossRef]
- Amess, R.; Baggett, N.; Darby, P.R.; Goode, A.R.; Vickers, E.E. Synthesis of luciferin glycosides as substrates for novel ultrasensitive enzyme assays. Carbohydr. Res. 1990, 205, 225–233. [Google Scholar] [CrossRef]
- Testard, A.; Picot, L.; Lozach, O.; Blairvac, M.; Meijer, L.; Murillo, L.; Piot, J.-M.; Thiéry, V.; Besson, T. Synthesis and evaluation of the antiproliferative activity of novel thiazoloquinazolinone kinases inhibitors. J. Enzym. Inhib. Med. Chem. 2005, 20, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Logé, C.; Testard, A.; Thiéry, V.; Lozach, O.; Blairvacq, M.; Robert, J.-M.; Meijer, L.; Besson, T. Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1 (CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2008, 43, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Meronni, G.; Ciana, P.; Meda, C.; Maggi, A.; Santaniello, E. 2,6-Disubstituted benzothiazoles, analogues of the aromatic core of D-luciferin: Synthesis and evaluation of the affinity for Photinus pyralis luciferase. ARKIVOC 2009, xi, 22–30. [Google Scholar] [CrossRef]
- Couly, F.; Dubouilh-Benard, C.; Besson, T.; Fruit, C. Late-stage C–H arylation of thiazolo[5,4-f]quinazolin-9(8H)-one backbone: Synthesis of an array of potential kinase inhibitors. Synthesis 2017, 49, 4615–4622. [Google Scholar] [CrossRef]
- Pacheco-Benichou, A.; Ivendengani, E.; Kostakis, I.K.; Besson, T.; Fruit, C. Copper-catalyzed C–H arylation of fused-pyrimidinone derivatives using diaryliodonium salts. Catalysts 2021, 11, 28. [Google Scholar] [CrossRef]
- Liang, G.; Ren, H.; Rao, J. A biocompatible condensation reaction for controlled assembly of nanostructures in living cells. Nat. Chem. 2010, 2, 54–60. [Google Scholar] [CrossRef]
- Yuan, Y.; Liang, G. A biocompatible, highly efficient click reaction and its applications. Org. Biomol. Chem. 2014, 12, 865–871. [Google Scholar] [CrossRef]
- Dragulescu-Andrasi, A.; Kothapalli, S.-R.; Tikhomirov, G.A.; Rao, J. Activatable oligomerizable imaging agents for photoacoustic imaging of furin-like activity in living subjects. J. Am. Chem. Soc. 2013, 135, 11015–11022. [Google Scholar] [CrossRef] [Green Version]
- Inkster, J.A.H.; Colin, D.J.; Seimbille, Y. A novel 2-cyanobenzothiazole-based 18F prosthetic group for conjugation to 1,2-aminothiol-bearing targeting vectors. Org. Biomol. Chem. 2015, 13, 3667–3676. [Google Scholar] [CrossRef]
- Jones, K.A.; Portefield, W.B.; Rathbun, C.M.; McCutcheon, D.C.; Paley, M.A.; Prescher, J.A. Orthogonal luciferase-luciferin pairs for bioluminescence imaging. J. Am. Chem. Soc. 2017, 139, 2351–2358. [Google Scholar] [CrossRef]
- Wang, W.; Gao, J. N,S-Double labeling of N-terminal cysteines via an alternative conjugation pathway with 2-cyanobenzothiazole. J. Org. Chem. 2020, 85, 1756–1763. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.J.; Hwang, C.S.; Prescher, J.A. Orthogonal bioluminescent probes from disubstituted luciferins. Biochemistry 2021, 60, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Li, D.; Zhao, L.; Liu, M.; Ma, P.; Luo, Y.; Yang, X. Bioorthogonal in situ assembly of nanomedicines as drug depots for extracellular drug delivery. Nat. Commun. 2022, 13, 2038. [Google Scholar] [CrossRef] [PubMed]
- White, E.H.; McCapra, F.; Field, G.H.; McElroy, W.D. The structure and synthesis of firefly luciferin. J. Am. Chem. Soc. 1961, 83, 2402–2403. [Google Scholar] [CrossRef]
- White, E.H.; McCapra, F.; Field, G.H. The structure and synthesis of firefly luciferin. J. Am. Chem. Soc. 1963, 85, 337–343. [Google Scholar] [CrossRef]
- Ioka, S.; Saitoh, T.; Iwano, S.; Suzuki, K.; Maki, S.A.; Miyawaki, A.; Imoto, M.; Nishiyama, S. Synthesis of firefly luciferin analogues and evaluation of the luminescent properties. Chem. Eur. J. 2016, 22, 9330–9337. [Google Scholar] [CrossRef]
- Barbero, M.; Cadamuro, S.; Dughera, S. Copper-free Sandmeyer cyanation of arenediazonium o-benzenedisulfonimides. Org. Biomol. Chem. 2016, 14, 1437–1441. [Google Scholar] [CrossRef]
- White, E.H.; Wörther, H.; Seliger, H.H.; McElroy, W.D. Amino analogs of firefly luciferin and biological activity thereof. J. Am. Chem. Soc. 1966, 88, 2015–2019. [Google Scholar] [CrossRef]
- White, E.H.; Wörther, H. Analogs of firefly luciferin. Ill. J. Org. Chem. 1966, 31, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Klement, I.; Lennick, K.; Tucker, C.E.; Knochel, P. Preparation of polyfunctional nitriles by the cyanation of functionalized organozinc halides with p-toluenesulfonyl cyanide. Tetrahedron Lett. 1993, 34, 4623–4626. [Google Scholar] [CrossRef]
- Do, H.-Q.; Daugulis, O. Copper-catalyzed cyanation of heterocycle carbon−hydrogen bonds. Org. Lett. 2010, 12, 2517–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Würfel, H.; Weiss, D.; Beckert, R.; Güther, A. A new application of the “mild thiolation” concept for an efficient three-step synthesis of 2-cyanobenzothiazoles: A new approach to firefly-luciferin precursors. J. Sulfur Chem. 2012, 33, 9–16. [Google Scholar] [CrossRef]
- Hauser, J.R.; Beard, H.A.; Bayana, M.E.; Jolley, K.E.; Warriner, S.L.; Bon, R.S. Economical and scalable synthesis of 6-amino-2-cyanobenzothiazole. Beilstein J. Org. Chem. 2016, 12, 2019–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.-L.; Sun, K.-K.; Cai, C. Copper-catalyzed cyanation of heterocycle C–H bonds with ethyl(ethoxymethylene)-cyanoacetate as a cyanating agent and its mechanism. Org. Chem. Front. 2018, 5, 1848–1853. [Google Scholar] [CrossRef]
- Shahmoradi, G.; Amani, S. Convenient cyanation of diazonium tetrafluoroborate salt toward firefly luciferin precursors. Polycycl. Aromat. Compd. 2020, 40, 660–669. [Google Scholar] [CrossRef]
- Rees, C.W. Polysulfur-nitrogen heterocyclic chemistry. J. Heterocycl. Chem. 1992, 29, 639–651. [Google Scholar] [CrossRef]
- Besson, T.; Rees, C.W. Some chemistry of 4,5-dichloro-l,2,3-dithiazolium chloride and its Derivatives. J. Chem. Soc. Perkin Trans. 1 1995, 1659–1662. [Google Scholar] [CrossRef]
- English, R.F.; Rakitin, O.A.; Rees, C.W.; Vlasova, O.G. Conversion of imino-1,2,3-dithiazoles into 2-cyanobenzothiazoles, cyanoimidoyl chlorides and diatomic sulfur. J. Chem. Soc. Perkin Trans. 1 1997, 201–205. [Google Scholar] [CrossRef]
- Appel, R.; Janssen, H.; Siray, M.; Knoch, F. Synthese und reaktionen des 4,5-dichlor-1,2,3-dithiazolium-chlorids. Chem. Ber. 1985, 118, 1632–1643. [Google Scholar] [CrossRef]
- Bénéteau, V.; Besson, T.; Rees, C.W. Rapid Synthesis of 2-cyanobenzothiazoles from N-aryliminodithiazoles under microwave irradiation. Synth. Commun. 1997, 27, 2275–2280. [Google Scholar] [CrossRef]
- Frère, S.; Thiéry, V.; Besson, T. Scaleable Synthesis of 2-cyanobenzothiazoles via N-arylimino-1,2,3-dithiazoles. Synth. Commun. 2003, 33, 3789–3804. [Google Scholar] [CrossRef]
- Besson, T.; Dozias, M.-J.; Guillard, J.; Rees, C.W. New route to 2-cyanobenzothiazoles via N-arylimino-1,2,3-dithiazoles. J. Chem. Soc. Perkin Trans. 1 1998, 3925–3926. [Google Scholar] [CrossRef]
- Inamoto, K.; Hasegawa, C.; Hiroya, K.; Doi, T. Palladium-catalyzed synthesis of 2-substituted senzothiazoles via a C-H Functionalization/intramolecular C-S Bond formation process. Org. Lett. 2008, 10, 5147–5150. [Google Scholar] [CrossRef]
- Inamoto, K.; Hasegawa, C.; Kawasaki, J.; Hiroya, K.; Doi, T. Use of molecular oxygen as a reoxidant in the Synthesis of 2-substituted benzothiazoles via palladium-catalyzed C-H Functionalization/Intramolecular C-S Bond Formation. Adv. Synth. Catal. 2010, 352, 2643–2655. [Google Scholar] [CrossRef]
- McCutcheon, D.C.; Paley, M.A.; Steinhardt, R.C.; Prescher, J.A. Expedient synthesis of electronically modified luciferins for bioluminescence imaging. J. Am. Chem. Soc. 2012, 134, 7604–7607. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, D.C.; Porterfield, W.B.; Prescher, J.A. Rapid and scalable assembly of firefly luciferasesubstrates. Org. Biomol. Chem. 2015, 13, 2117–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussa, Z.; Judeh, Z.M.A.; Alzamly, A.; Ahmed, S.A.; Al-Masri, H.T.; Al-Hindawi, B.; Rasool, F.; Saada, S. Iodine-DMSO mediated conversion of N-arylcyanothioformamides to N-arylcyanoformamides and the unexpected formation of 2-cyanobenzothiazoles. RSC Adv. 2022, 12, 6133–6148. [Google Scholar] [CrossRef] [PubMed]
- Foucourt, A.; Hédou, D.; Dubouilh-Benard, C.; Désiré, L.; Casagrande, A.-S.; Leblond, B.; Loäec, N.; Meijer, L.; Besson, T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part I. Molecules 2014, 19, 15546–15571. [Google Scholar] [CrossRef] [Green Version]
- Foucourt, A.; Hédou, D.; Dubouilh-Benard, C.; Girard, A.; Taverne, T.; Casagrande, A.-S.; Désiré, L.; Leblond, B.; Besson, T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part II. Molecules 2014, 19, 15411–15439. [Google Scholar] [CrossRef] [Green Version]
- Hédou, D.; Godeau, J.; Loaëc, N.; Meijer, L.; Fruit, C.; Besson, T. Synthesis of thiazolo[5,4-f]quinazolin-9(8H)-ones as multi-target directed ligands of Ser/Thr kinases. Molecules 2016, 21, 578. [Google Scholar] [CrossRef]
- Hédou, D.; Dubouilh-Benard, C.; Loaëc, N.; Meijer, L.; Fruit, C.; Besson, T. Synthesis of bioactive 2-(arylamino)thiazolo[5,4-f]-quinazolin-9-ones via the Hügershoff reaction or Cu-catalyzed intramolecular C-S bond formation. Molecules 2016, 21, 794. [Google Scholar] [CrossRef] [Green Version]
- Michaelidou, S.S.; Koutentis, P.A. The synthesis of 2-cyano-cyanothioformanilides from 2-(4-chloro-5H-1,2,3-dithiazol-5-ylideneamino)benzonitriles using DBU. Synthesis 2009, 24, 4167–4174. [Google Scholar] [CrossRef]
- Strieter, E.R.; Blackmond, D.G.; Buchwald, S.L. The role of chelating diamine ligands in the goldberg reaction: A kinetic study on the copper-catalyzed amidation of aryl Iodides. J. Am. Chem. Soc. 2005, 127, 4120–4121. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, K.; Kim, K. Novel Synthesis of 5-(Arylimino)-4-(dialkylamino)-5H-1,2,3-dithiazoles and the Mechanism of Their Formation. J. Org. Chem. 1994, 59, 6179–6183. [Google Scholar] [CrossRef]
- Besson, T.; Emayan, K.; Rees, C.W. 1,2,3-Dithiazoles and new routes to 3,l-benzoxazin-4-ones, 3,l-benzothiazin-4-ones and N-arylcyanothioformamides. J. Chem. Soc. Perkin Trans. 1 1995, 2097–2102. [Google Scholar] [CrossRef]
- Oppedisano, F.; Catto, M.; Koutentis, P.A.; Nicolotti, O.; Pochini, L.; Koyioni, M.; Introcaso, A.; Michaelidou, S.S.; Carotti, A.; Indiveri, C. 1,2,3-Dithiazoles—New reversible melanin synthesis inhibitors: A chemical genomics study. Toxicol. Appl. Pharmacol. 2012, 265, 93–102. [Google Scholar] [CrossRef]
- Besson, T.; Guillard, J.; Rees, C.W.; Thiéry, V. New syntheses of aryl isothiocyanates. J. Chem. Soc. Perkin Trans. 1 1998, 889–892. [Google Scholar] [CrossRef]
- Soares do Rêgo Barros, O.; Nogueira, C.W.; Stangherlin, E.C.; Menezes, P.H.; Zeni, G. Copper-promoted carbon-nitrogen bond formation with 2-iodo-selenophene and amides. J. Org. Chem. 2006, 71, 1552–1557. [Google Scholar] [CrossRef]
- Deau, E.; Dubouilh-Benard, C.; Levacher, V.; Besson, T. Microwave-assisted synthesis of novel N-(4-phenylthiazol-2-yl)-benzo[d]thiazole-, thiazolo[4,5-b]pyridine, thiazolo[5,4-b]pyridine- and benzo[d]oxazole-2-carboximidamides inspired by marine topsentines and nortopsentines. Tetrahedron 2014, 70, 5532–5540. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | PdCl2 (x mol%) | Solvent [c] (M) | Yield (%) 1 |
| 1 | 10 | 0.050 | 40 |
| 2 | 10 | 0.025 | 44 |
| 3 | 20 | 0.050 | 46 |
| 4 | 20 | 0.025 | 49 2 |
 | |||||
|---|---|---|---|---|---|
| Entry | Additive | Yield (%) 1 | Entry | Additive | Yield (%) 1 |
| 1 | CsF | 15 | 6 | KF | 16 |
| 2 | CsI | 32 | 7 | LiBr | 53 |
| 3 | NaI | 53 | 8 | LiCl | 33 |
| 4 | KBr | 66 | 9 | LiI | 55 |
| 5 | KI | 70 2,3,4,5 | 10 | none | 37 |
 | ||||
|---|---|---|---|---|
| Entry | [Pd] | [Cu] | Solvent/Co-Solvent | Yield (%) 1 |
| 1 | PdCl2 | CuI | DMSO/DMF | 70 |
| 2 | PdBr2 | CuI | DMSO/DMF | 67 |
| 3 | Pd(OAc)2 | CuI | DMSO/DMF | 22 |
| 4 | Pd2dba3 | CuI | DMSO/DMF | 7 |
| 5 | none | CuI | DMSO/DMF | 0 |
| 6 | PdCl2 | CuBr | DMSO/DMF | 53 |
| 7 | PdCl2 | CuCl2 | DMSO/DMF | 57 |
| 8 | PdCl2 | Cu(OAc)2 | DMSO/DMF | 39 |
| 9 | PdCl2 | none | DMSO/DMF | 41 |
| 10 | PdCl2 | CuI | DMSO/NMP | 18 |
| 11 | PdCl2 | CuI | DMSO/- | 34 |
| 12 | PdCl2 | CuI | DMF/- | 14 |
 | ||||
|---|---|---|---|---|
| Entry | PdCl2 (x mol%) | CuI (y mol%) | [c] M | Yield (%) 1 |
| 1 | 20 | 50 | 0.025 | 70 |
| 2 | 20 | 50 | 0.050 | 53 |
| 3 | 20 | 20 | 0.025 | 51 |
| 4 | 10 | 50 | 0.025 | 21 |
| 5 | 10 | 20 | 0.025 | 14 |
| Aniline | R | Yield of 2 (%) 1 | Yield of 3 (%) 1 | Aniline | R | Yield of 2 (%) 1 | Yield of 3 (%) 1 |
|---|---|---|---|---|---|---|---|
| 1a | 4-Me | 67 2 | 46 6 | 1r | 3,4-diOMe | 61 8 | 71 6 |
| 1b | H | 59 2 | 41 6 | 1s | 3-Me, 4-Br | 84 | 63 |
| 1c | 4-F | 86 3 | 47 6 | 1t | 3,4(-OCH2O-) | 26 9 | 55 |
| 1d | 4-Cl | 82 2 | 45 6 | 1u | 3,4(-OCH2CH2O-) | 65 10 | 71 |
| 1e | 4-Br | 86 4 | 47 | 1v | 2,3-diMe | 64 | 79 |
| 1f | 4-OMe | 48 2 | 33 6 | 1w | 2,3-diCl | 87 | 78 6 |
| 1g | 4-CF3 | 72 | 56 6 | 1x | 2-Me, 3-Cl | 77 | 85 |
| 1h | 4-CN | 69 5 | 71 | 1y | 2,4-diF | 94 3 | 64 6 |
| 1i | 4-NO2 | 85 | 32 6 | 1z | 2,4-diOMe | 75 5 | 75 6 |
| 1j | 4-CO2Et | 78 | 82 6 | 1aa | 2-F, 4-OMe | 92 | 75 |
| 1k | 3-OMe | 74 2 | 68 6 | 1ab | 3,5-diMe | 91 | 77 |
| 1l | 3-NO2 | 90 | 81 6 | 1ac | 3,5-diOMe | 45 | 65 |
| 1m | 3-CO2Et | 85 | 75 | 1ad | 3-Br, 5-Me | 63 | 44 |
| 1n | 2-Cl | 89 | 83 | 1ae | 3-Br, 5-OMe | 95 | 85 |
| 1o | 2-Br | 85 2 | 80 6 | 1af | 2,5-diMe | 69 4 | 77 |
| 1p | 2-OMe | 90 7 | 79 | 1ag | 2-Me, 5-iPr | 76 5 | 78 |
| 1q | 3,4-diMe | 88 | 52 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broudic, N.; Pacheco-Benichou, A.; Fruit, C.; Besson, T. Synthesis of 2-Cyanobenzothiazoles via Pd-Catalyzed/Cu-Assisted C-H Functionalization/Intramolecular C-S Bond Formation from N-Arylcyanothioformamides. Molecules 2022, 27, 8426. https://doi.org/10.3390/molecules27238426
Broudic N, Pacheco-Benichou A, Fruit C, Besson T. Synthesis of 2-Cyanobenzothiazoles via Pd-Catalyzed/Cu-Assisted C-H Functionalization/Intramolecular C-S Bond Formation from N-Arylcyanothioformamides. Molecules. 2022; 27(23):8426. https://doi.org/10.3390/molecules27238426
Chicago/Turabian StyleBroudic, Nathan, Alexandra Pacheco-Benichou, Corinne Fruit, and Thierry Besson. 2022. "Synthesis of 2-Cyanobenzothiazoles via Pd-Catalyzed/Cu-Assisted C-H Functionalization/Intramolecular C-S Bond Formation from N-Arylcyanothioformamides" Molecules 27, no. 23: 8426. https://doi.org/10.3390/molecules27238426