

Preparation of Chiral Enantioenriched Densely Substituted Cyclopropyl Azoles, Amines, and Ethers via Formal SN2′ Substitution of Bromocylopropanes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
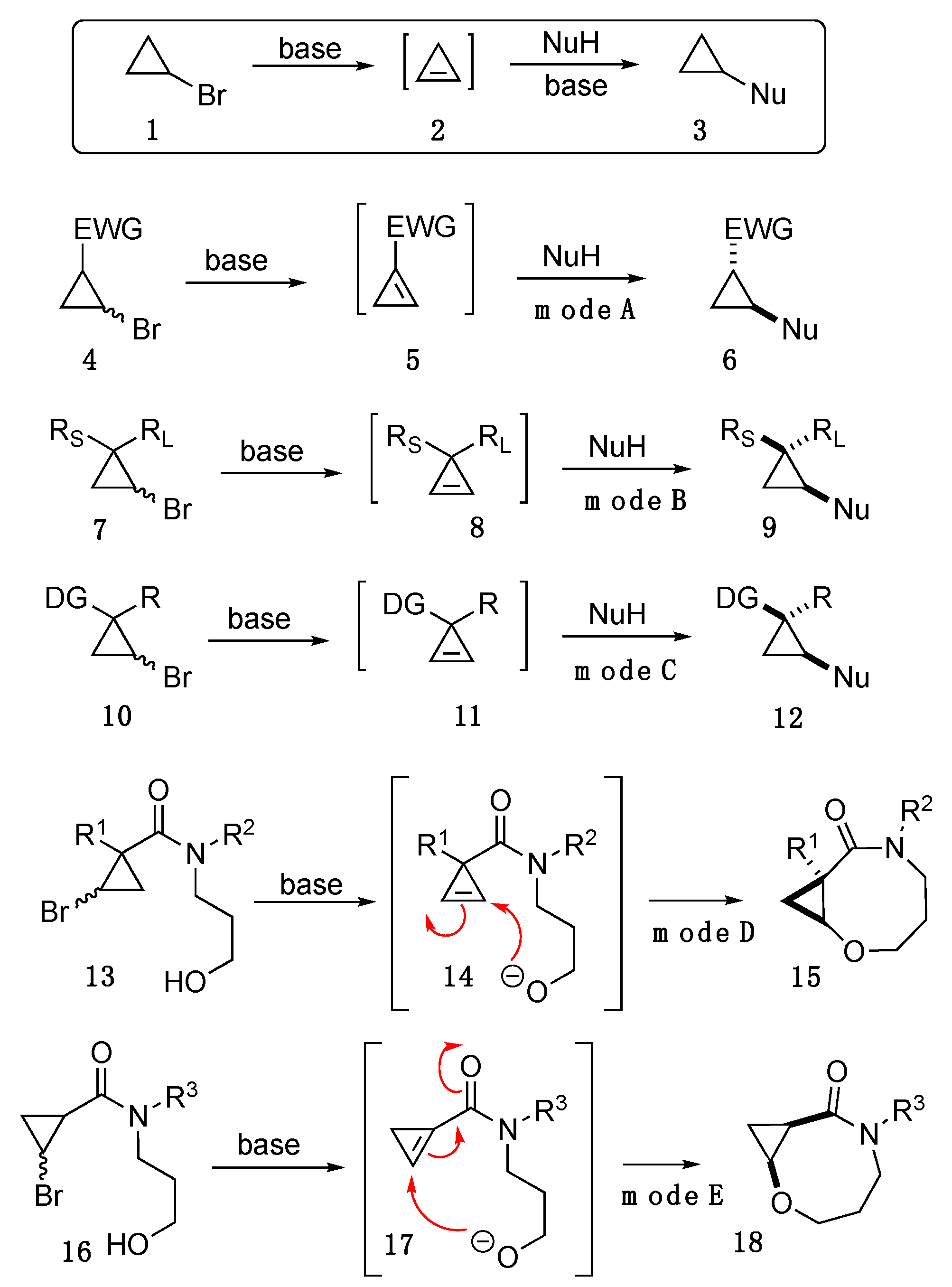
:1. Introduction
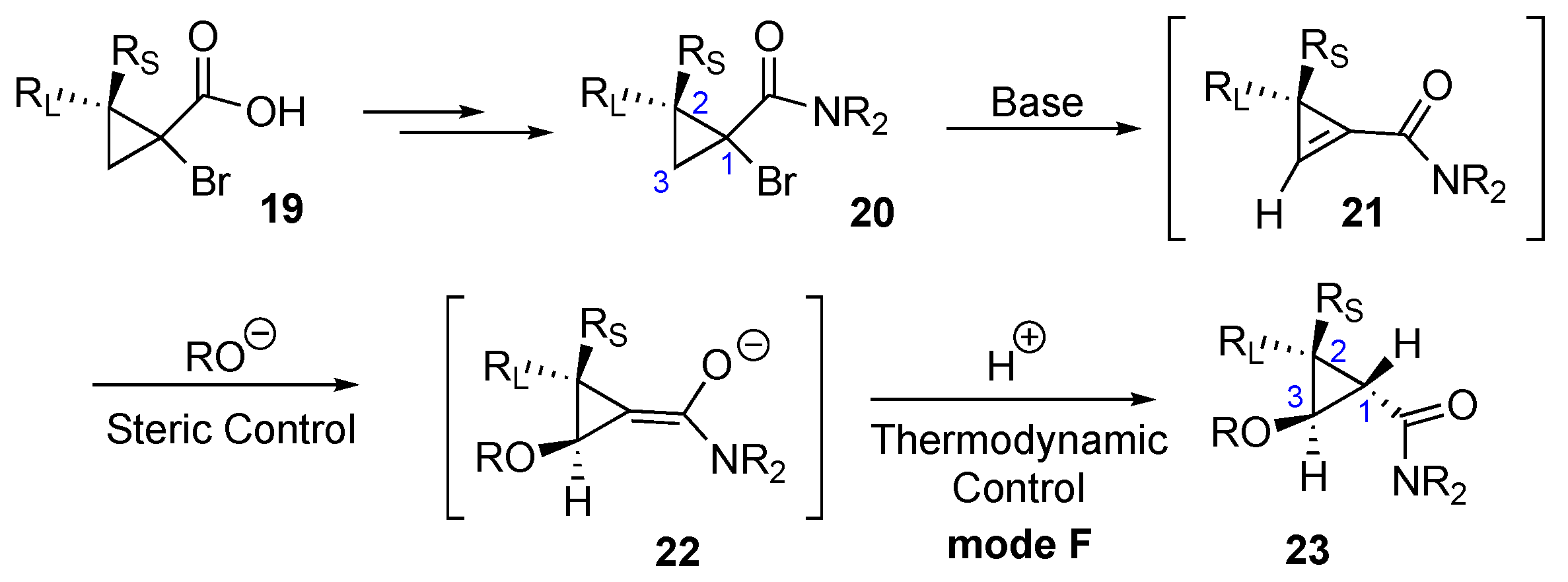
2. Results and Discussion
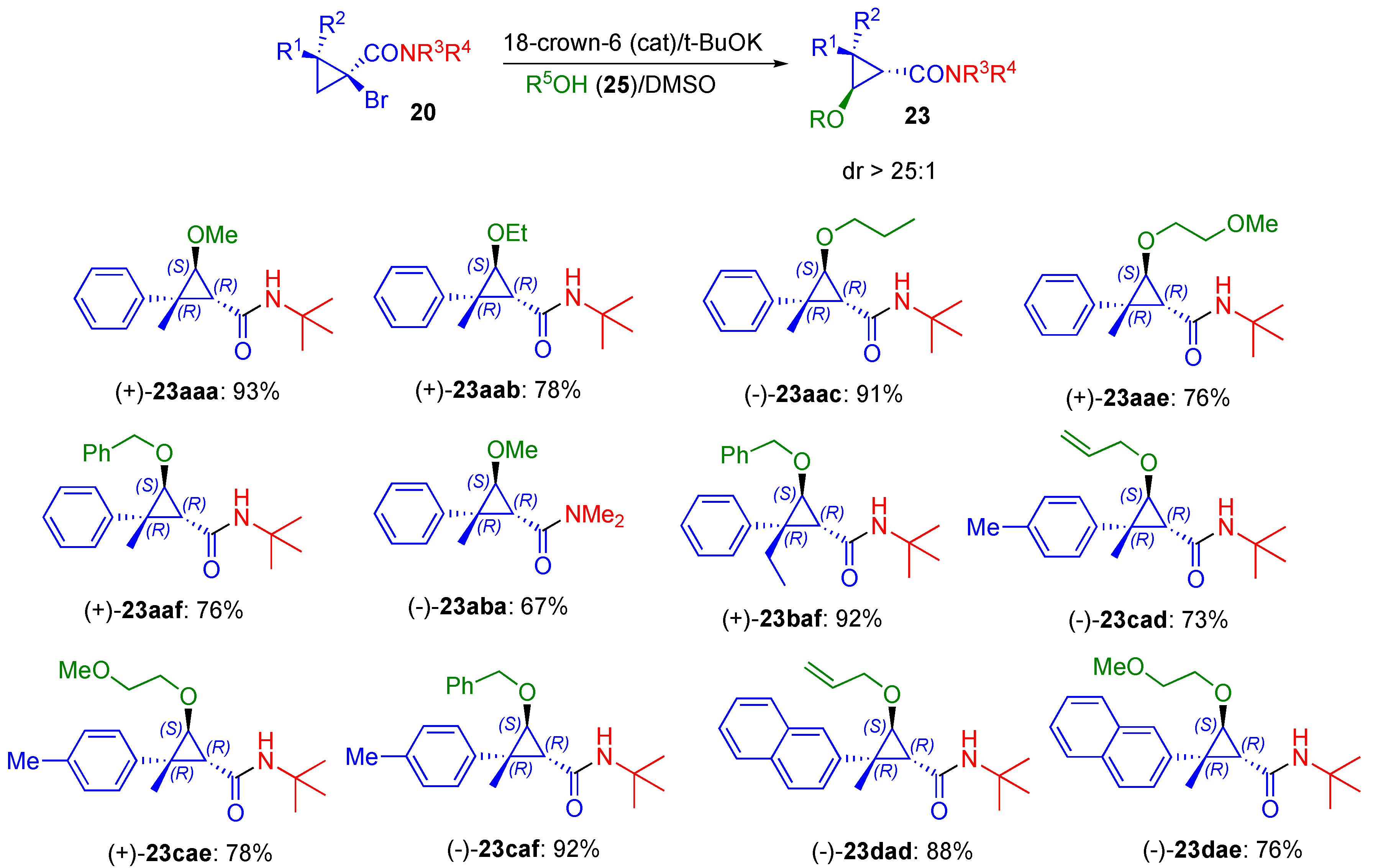
2.1. Alcohol Nucleophiles
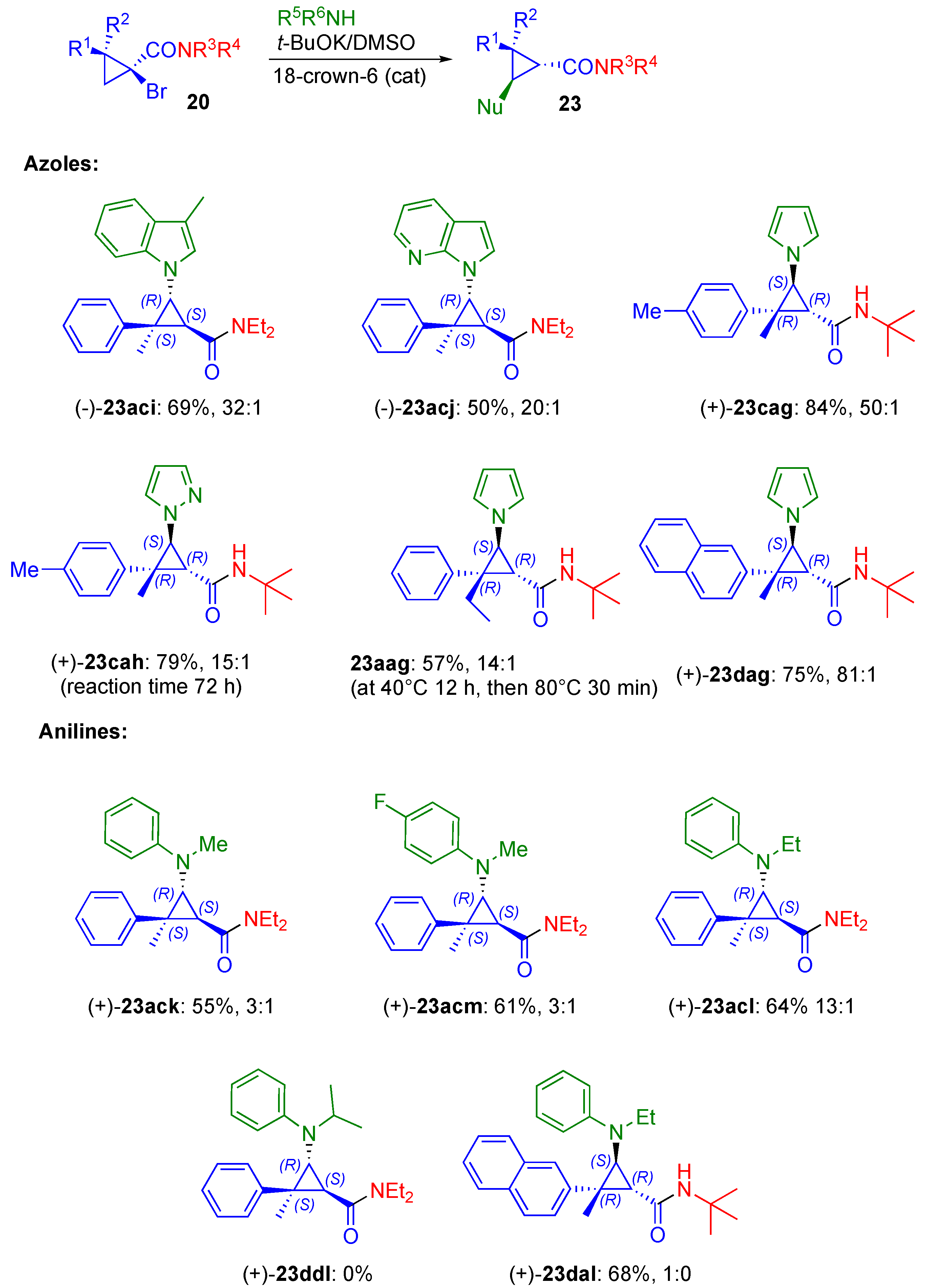
2.2. Azole Nucleophiles
2.3. Aniline Nucleophiles
3. Materials and Methods
3.1. General
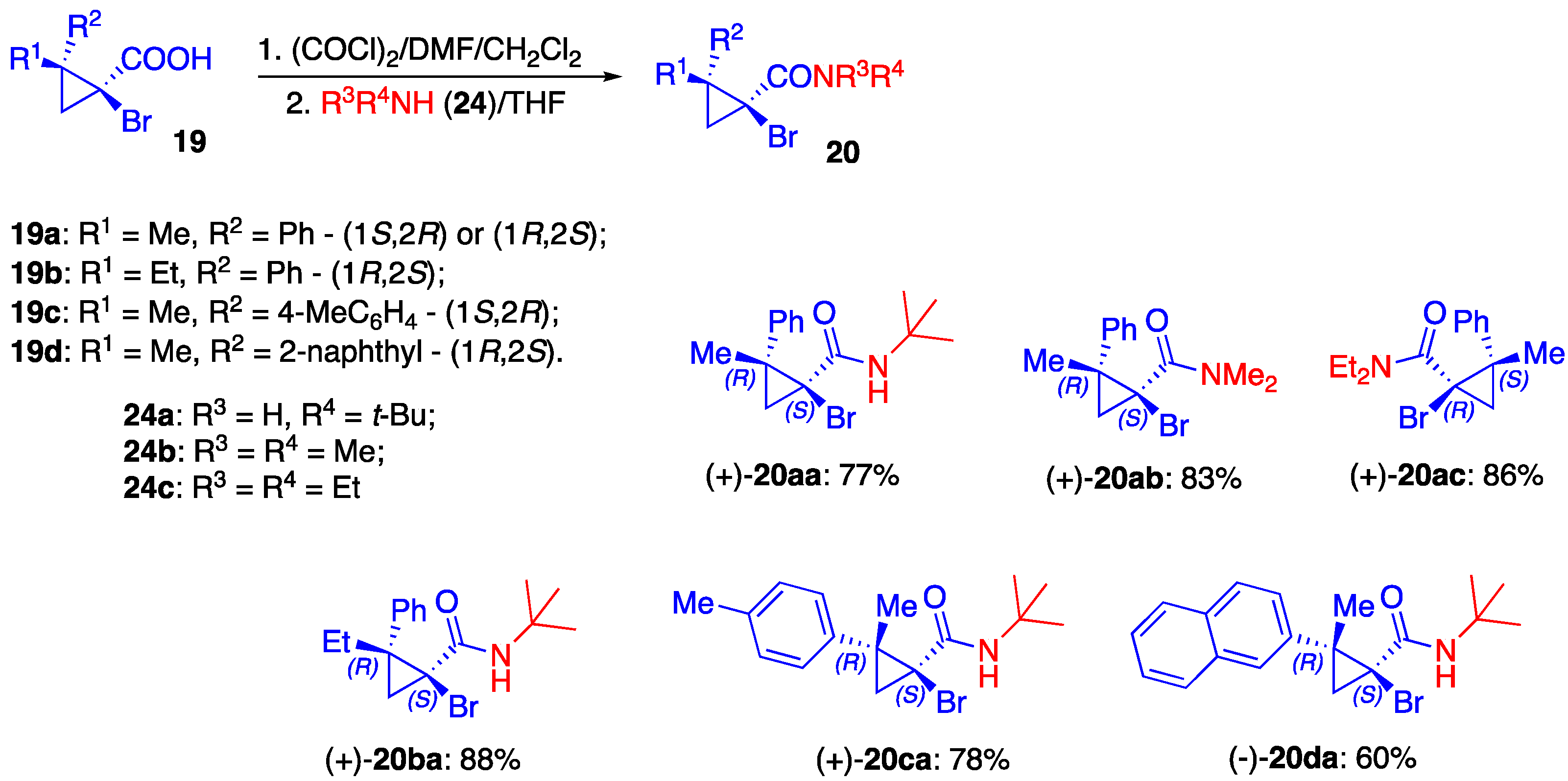
3.2. Preparation of Starting Materials
3.3. Nucleophilic Addition Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, D.Y.K.; Pouwer, R.H.; Richard, J.-A. Recent advances in the total synthesis of cyclopropane-containing natural products. Chem. Soc. Rev. 2012, 41, 4631–4642. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.; Steck, V.; Potenzino, R.J.; Fasan, R. A diverse library of chiral cyclopropane scaffolds via chemoenzymic assembly and diversification of cyclopropyl ketones. J. Am. Chem. Soc. 2021, 143, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, W.A. Synthesis of cyclopropane containing natural products. Tetrahedron 2001, 57, 8589–8627. [Google Scholar] [CrossRef]
- Keglevich, P.; Keglevich, A.; Hazai, L.; Kalaus, G.; Szantay, C. Natural Compounds Containing a Condensed Cyclopropane Ring. Natural and Synthetic Aspects. Curr. Org. Chem. 2014, 18, 2037–2042. [Google Scholar] [CrossRef]
- Shim, S.Y.; Kim, J.Y.; Nam, M.; Hwang, G.-S.; Ryu, D.H. Enantioselective Cyclopropanation with α-Alkyl-α-diazoesters Catalyzed by Chiral Oxazaborolidinium Ion: Total Synthesis of (+)-Hamavellone B. Org. Lett. 2016, 18, 160–163. [Google Scholar] [CrossRef]
- Williams, J.D.; Yazarians, J.A.; Almeyda, C.C.; Anderson, K.; Boyce, G.R. Detection of the Previously Unobserved Stereoisomers of Thujone in the Essential Oil and Consumable Products of Sage (Salvia officinalis L.) Using Headspace Solid-Phase Microextraction-Gas Chromatography-Mass Spectrometry. J. Agric. Food Chem. 2016, 64, 4319–4326. [Google Scholar] [CrossRef]
- Green, R.; Cheeseman, M.; Duffill, S.; Merritt, A.; Bull, S.D. An efficient asymmetric synthesis of grenadamide. Tetrahedron Lett. 2005, 46, 7931–7934. [Google Scholar] [CrossRef]
- Ren, X.; Chandgude, A.L.; Fasan, R. Highly Stereoselective Synthesis of Fused Cyclopropane-γ-Lactams via Biocatalytic Iron-Catalyzed Intramolecular Cyclopropanation. ACS Catal. 2020, 10, 2308–2313. [Google Scholar] [CrossRef]
- Wang, L.; Tang, Y. Asymmetric Ring-Opening Reactions of Donor-Acceptor Cyclopropanes and Cyclobutanes. Isr. J. Chem. 2016, 56, 463–475. [Google Scholar] [CrossRef]
- Sanchez-Diez, E.; Vesga, D.L.; Reyes, E.; Uria, U.; Carrillo, L.; Vicario, J.L. Organocatalytically Generated Donor-Acceptor Cyclopropanes in Domino Reactions. One-Step Enantioselective Synthesis of Pyrrolo[1,2-a]quinolines. Org. Lett. 2016, 18, 1270–1273. [Google Scholar] [CrossRef]
- Cao, B.; Mei, L.-Y.; Li, X.-G.; Shi, M. Palladium-catalyzed asymmetric [3+2] cycloaddition to construct 1,3-indandione and oxindole-fused spiropyrazolidine scaffolds. RSC Adv. 2015, 5, 92545–92548. [Google Scholar] [CrossRef]
- Gharpure, S.J.; Nanda, L.N.; Shukla, M.K. Donor-Acceptor Substituted Cyclopropane to Butanolide and Butenolide Natural Products: Enantiospecific First Total Synthesis of (+)-Hydroxyancepsenolide. Org. Lett. 2014, 16, 6424–6427. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.; Cohen, A.; Marek, I. Creating Stereocenters Within Acyclic Systems by C-C Bond Cleavage of Cyclopropanes. Chem. Rev. 2021, 121, 140–161. [Google Scholar] [CrossRef] [PubMed]
- Rubina, M.; Sherrill, W.M.; Rubin, M. Dramatic Stereo- and Enantiodivergency in the Intermolecular Asymmetric Heck Reaction Catalyzed by Palladium Complexes with Cyclopropane-Based PHOX Ligands. Organometallics 2008, 27, 6393–6395. [Google Scholar] [CrossRef]
- Rubina, M.; Sherrill, W.M.; Barkov, A.Y.; Rubin, M. Rational design of cyclopropane-based chiral PHOX ligands for intermolecular asymmetric Heck reaction. Beilstein J. Org. Chem. 2014, 10, 1536–1548. [Google Scholar] [CrossRef]
- Khlebnikov, A.F.; Kozhushkov, S.I.; Yufit, D.S.; Schill, H.; Reggelin, M.; Spohr, V.; de Meijere, A. A Novel Type of Chiral Triangulane-Based Diphosphane Ligands for Transition Metals. Eur. J. Org. Chem. 2012, 2012, 1530–1545. [Google Scholar] [CrossRef]
- Molander, G.A.; Burke, J.P.; Carroll, P.J. Synthesis and Application of Chiral Cyclopropane-Based Ligands in Palladium-Catalyzed Allylic Alkylation. J. Org. Chem. 2004, 69, 8062–8069. [Google Scholar] [CrossRef]
- Onajole, O.K.; Vallerini, G.P.; Eaton, J.B.; Lukas, R.J.; Brunner, D.; Caldarone, B.J.; Kozikowski, A.P. Synthesis and Behavioral Studies of Chiral Cyclopropanes as Selective α4β2-Nicotinic Acetylcholine Receptor Partial Agonists Exhibiting an Antidepressant Profile. Part III. ACS Chem. Neurosci. 2016, 7, 811–822. [Google Scholar] [CrossRef]
- Reddy, C.N.; Nayak, V.L.; Mani, G.S.; Kapure, J.S.; Adiyala, P.R.; Maurya, R.A.; Kamal, A. Synthesis and biological evaluation of spiro[cyclopropane-1,3’-indolin]-2’-ones as potential anticancer agents. Bioorg. Med. Chem. Lett. 2015, 25, 4580–4586. [Google Scholar] [CrossRef]
- Mei, H.; Pan, G.; Zhang, X.; Lin, L.; Liu, X.; Feng, X. Catalytic Asymmetric Ring-Opening/Cyclopropanation of Cyclic Sulfur Ylides: Construction of Sulfur-Containing Spirocyclopropyloxindoles with Three Vicinal Stereocenters. Org. Lett. 2018, 20, 7794–7797. [Google Scholar] [CrossRef]
- Rodriguez, K.X.; Howe, E.N.; Bacher, E.P.; Burnette, M.; Meloche, J.L.; Meisel, J.; Schnepp, P.; Tan, X.; Chang, M.; Zartman, J.; et al. Combined Scaffold Evaluation and Systems-Level Transcriptome-Based Analysis for Accelerated Lead Optimization Reveals Ribosomal Targeting Spirooxindole Cyclopropanes. ChemMedChem 2019, 14, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Riss, P.J.; Roesch, F. A convenient chemo-enzymatic synthesis and 18F-labelling of both enantiomers of trans-1-toluenesulfonyloxymethyl-2-fluoromethyl-cyclopropane. Org. Biomol. Chem. 2008, 6, 4567–4574. [Google Scholar] [CrossRef] [PubMed]
- Unzner, T.A.; Grossmann, A.S.; Magauer, T. Rapid Access to Orthogonally Functionalized Naphthalenes: Application to the Total Synthesis of the Anticancer Agent Chartarin. Angew. Chem. Int. Ed. 2016, 55, 9763–9767. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.D.; Schmitz, J.C.; Chu, E.; Wipf, P. Total Synthesis of (-)-CP2-Disorazole C1. Org. Lett. 2011, 13, 4088–4091. [Google Scholar] [CrossRef] [Green Version]
- Casar, Z. Synthetic Approaches to Contemporary Drugs that Contain the Cyclopropyl Moiety. Synthesis 2020, 52, 1315–1345. [Google Scholar] [CrossRef]
- Bender, T.A.; Dabrowski, J.A.; Zhong, H.; Gagne, M.R. Diastereoselective B(C6F5)3-Catalyzed Reductive Carbocyclization of Unsaturated Carbohydrates. Org. Lett. 2016, 18, 4120–4123. [Google Scholar] [CrossRef]
- Wu, W.; Lin, Z.; Jiang, H. Recent advances in the synthesis of cyclopropanes. Org. Biomol. Chem. 2018, 16, 7315–7329. [Google Scholar] [CrossRef]
- Li, J.-H.; Feng, T.-F.; Du, D.-M. Construction of Spirocyclopropane-Linked Heterocycles Containing Both Pyrazolones and Oxindoles through Michael/Alkylation Cascade Reactions. J. Org. Chem. 2015, 80, 11369–11377. [Google Scholar] [CrossRef]
- Tollefson, E.J.; Erickson, L.W.; Jarvo, E.R. Stereospecific Intramolecular Reductive Cross-Electrophile Coupling Reactions for Cyclopropane Synthesis. J. Am. Chem. Soc. 2015, 137, 9760–9763. [Google Scholar] [CrossRef]
- Munnuri, S.; Falck, J.R. Directed, Remote Dirhodium C(sp3)-H Functionalization, Desaturative Annulation, and Desaturation. J. Am. Chem. Soc. 2022, 144, 17989–17998. [Google Scholar] [CrossRef]
- Calo, F.P.; Fuerstner, A. A Heteroleptic Dirhodium Catalyst for Asymmetric Cyclopropanation with α-Stannyl α-Diazoacetate. “Stereoretentive” Stille Coupling with Formation of Chiral Quarternary Carbon Centers. Angew. Chem. 2020, 59, 13900–13907. [Google Scholar] [CrossRef] [PubMed]
- Chanthamath, S.; Nguyen, D.T.; Shibatomi, K.; Iwasa, S. Highly Enantioselective Synthesis of Cyclopropylamine Derivatives via Ru(II)-Pheox-Catalyzed Direct Asymmetric Cyclopropanation of Vinylcarbamates. Org. Lett. 2013, 15, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-Q.; Liu, W.; Zheng, B.-H.; Liu, X.Y.; Yang, Z.; Ding, C.-H.; Li, H.; Peng, Q.; Hou, X.-L. Pd-Catalyzed Asymmetric Cyclopropanation Reaction of Acyclic Amides with Allyl and Polyenyl Carbonates. Experimental and Computational Studies for the Origin of Cyclopropane Formation. ACS Catal. 2018, 8, 1964–1972. [Google Scholar] [CrossRef]
- Inoue, H.; Nga, P.T.T.; Fujisawa, I.; Iwasa, S. Synthesis of Forms of a Chiral Ruthenium Complex Containing a Ru-Colefin(sp2) Bond and Their Application to Catalytic Asymmetric Cyclopropanation Reactions. Org. Lett. 2020, 22, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Roesler, L.; Wissel, T.; Breitzke, H.; Hofmann, K.; Limbach, H.-H.; Gutmann, T.; Buntkowsky, G. Design and characterization of novel dirhodium coordination polymers-the impact of ligand size on selectivity in asymmetric cyclopropanation. Catal. Sci. Technol. 2021, 11, 3481–3492. [Google Scholar] [CrossRef]
- Purins, M.; Waser, J. Asymmetric Cyclopropanation and Epoxidation via a Catalytically Formed Chiral Auxiliary. Angew. Chem. Int. Ed. 2022, 61, e202113925. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Luo, Y.; Wang, X.-L.; Lu, S.; Zhao, Y.-L.; Zhang, J. Theoretical Studies on the Mechanism, Enantioselectivity, and Axial Ligand Effect of a Ru(salen)-Catalyzed Asymmetric Cyclopropanation Reaction. Organometallics 2014, 33, 3673–3682. [Google Scholar] [CrossRef]
- Wang, H.-X.; Li, W.-P.; Zhang, M.-M.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M. Synthesis of chiral pyrimidine-substituted diester D-A cyclopropanes via asymmetric cyclopropanation of ylides. Chem. Commun. 2020, 56, 11649–11652. [Google Scholar] [CrossRef]
- Wei, B.; Sharland, J.C.; Lin, P.; Wilkerson-Hill, S.M.; Fullilove, F.A.; McKinnon, S.; Blackmond, D.G.; Davies, H.M.L. In Situ Kinetic Studies of Rh(II)-Catalyzed Asymmetric Cyclopropanation with Low Catalyst Loadings. ACS Catal. 2020, 10, 1161–1170. [Google Scholar] [CrossRef]
- White, J.D.; Shaw, S. A New Cobalt-Salen Catalyst for Asymmetric Cyclopropanation. Synthesis of the Serotonin-Norepinephrine Reuptake Inhibitor (+)-Synosutine. Org. Lett. 2014, 16, 3880–3883. [Google Scholar] [CrossRef]
- Yuan, W.-C.; Lei, C.-W.; Zhao, J.-Q.; Wang, Z.-H.; You, Y. Organocatalytic Asymmetric Cyclopropanation of 3-Acylcoumarins with 3-Halooxindoles: Access to Spirooxindole-cyclopropa[c]coumarin Compounds. J. Org. Chem. 2021, 86, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Hoshiya, N.; Takenaka, K.; Shuto, S.; Uenishi, J.-I. Pd(II)-Catalyzed Alkylation of Tertiary Carbon via Directing-Group-Mediated C(sp3)-H Activation: Synthesis of Chiral 1,1,2-Trialkyl Substituted Cyclopropanes. Org. Lett. 2016, 18, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Hoshiya, N.; Kondo, M.; Fukuda, H.; Arisawa, M.; Uenishi, J.I.; Shuto, S. Entry to Chiral 1,1,2,3-Tetrasubstituted Arylcyclopropanes by Pd(II)-Catalyzed Arylation via Directing Group-Mediated C(sp3)-H Activation. J. Org. Chem. 2017, 82, 2535–2544. [Google Scholar] [CrossRef]
- Jerhaoui, S.; Poutrel, P.; Djukic, J.P.; Wencel-Delord, J.; Colobert, F. Stereospecific C-H activation as a key step for the asymmetric synthesis of various biologically active cyclopropanes. Org. Chem. Front. 2018, 5, 409–414. [Google Scholar] [CrossRef]
- Minami, T.; Fukuda, K.; Hoshiya, N.; Fukuda, H.; Watanabe, M.; Shuto, S. Synthesis of Enantiomerically Pure 1,2,3-Trisubstituted Cyclopropane Nucleosides Using Pd-Catalyzed Substitution via Directing Group-Mediated C(sp3)-H Activation as a Key Step. Org. Lett. 2019, 21, 656–659. [Google Scholar] [CrossRef]
- Vicente, R. Recent Progresses towards the Strengthening of Cyclopropene Chemistry. Synthesis 2016, 48, 2343–2360. [Google Scholar] [CrossRef]
- Li, P.; Zhang, X.; Shi, M. Recent developments in cyclopropene chemistry. Chem. Commun. 2020, 56, 5457–5471. [Google Scholar] [CrossRef]
- Muller, D.S.; Marek, I. Copper mediated carbometalation reactions. Chem. Soc. Rev. 2016, 45, 4552–4566. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.-B.; Wei, Y.; Shi, M. Recent developments of cyclopropene chemistry. Chem. Soc. Rev. 2011, 40, 5534–5563. [Google Scholar] [CrossRef]
- Dian, L.; Marek, I. Asymmetric Preparation of Polysubstituted Cyclopropanes Based on Direct Functionalization of Achiral Three-Membered Carbocycles. Chem. Rev. 2018, 118, 8415–8434. [Google Scholar] [CrossRef]
- Edwards, A.; Rubina, M.; Rubin, M. Nucleophilic Addition of Cyclopropenes. Curr. Org. Chem. 2016, 20, 1862–1877. [Google Scholar] [CrossRef]
- Fumagalli, G.; Stanton, S.; Bower, J.F. Recent Methodologies That Exploit C-C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes. Chem. Rev. 2017, 117, 9404–9432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, M.; Rubina, M.; Gevorgyan, V. Transition Metal Chemistry of Cyclopropenes and Cyclopropanes. Chem. Rev. 2007, 107, 3117–3179. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Lu, A.; Kuker, E.L.; Dong, V.M. Enantioselective Hydrothiolation: Diverging Cyclopropenes through Ligand Control. J. Am. Chem. Soc. 2021, 143, 6176–6184. [Google Scholar] [CrossRef]
- Yamanushkin, P.M.; Rubina, M.; Rubin, M. Amide-Directed Reactions of Small Carbocycles. Curr. Org. Chem. 2021, 25, 1686–1703. [Google Scholar] [CrossRef]
- Liu, F.; Bugaut, X.; Schedler, M.; Froehlich, R.; Glorius, F. Designing N-Heterocyclic Carbenes: Simultaneous Enhancement of Reactivity and Enantioselectivity in the Asymmetric Hydroacylation of Cyclopropenes. Angew. Chem., Int. Ed. 2011, 50, 12626–12630. [Google Scholar] [CrossRef]
- Liu, X.; Fox, J.M. Enantioselective, Facially Selective Carbomagnesation of Cyclopropenes. J. Am. Chem. Soc. 2006, 128, 5600–5601. [Google Scholar] [CrossRef]
- Alnasleh, B.K.; Sherrill, W.M.; Rubina, M.; Banning, J.; Rubin, M. Highly Diastereoselective Formal Nucleophilic Substitution of Bromocyclopropanes. J. Am. Chem. Soc. 2009, 131, 6906–6907. [Google Scholar] [CrossRef]
- Banning, J.E.; Prosser, A.R.; Rubin, M. Thermodynamic Control of Diastereoselectivity in the Formal Nucleophilic Substitution of Bromocyclopropanes. Org. Lett. 2010, 12, 1488–1491. [Google Scholar] [CrossRef]
- Prosser, A.R.; Banning, J.E.; Rubina, M.; Rubin, M. Formal Nucleophilic Substitution of Bromocyclopropanes with Amides en route to Conformationally Constrained β-Amino Acid Derivatives. Org. Lett. 2010, 12, 3968–3971. [Google Scholar] [CrossRef]
- Ryabchuk, P.; Rubina, M.; Xu, J.; Rubin, M. Formal Nucleophilic Substitution of Bromocyclopropanes with Azoles. Org. Lett. 2012, 14, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Banning, J.E.; Gentillon, J.; Ryabchuk, P.G.; Prosser, A.R.; Rogers, A.; Edwards, A.; Holtzen, A.; Babkov, I.A.; Rubina, M.; Rubin, M. Formal Substitution of Bromocyclopropanes with Nitrogen Nucleophiles. J. Org. Chem. 2013, 78, 7601–7616. [Google Scholar] [CrossRef] [PubMed]
- Banning, J.E.; Prosser, A.R.; Alnasleh, B.K.; Smarker, J.; Rubina, M.; Rubin, M. Diastereoselectivity Control in Formal Nucleophilic Substitution of Bromocyclopropanes with Oxygen- and Sulfur-Based Nucleophiles. J. Org. Chem. 2011, 76, 3968–3986. [Google Scholar] [CrossRef]
- Alnasleh, B.K.; Rubina, M.; Rubin, M. Templated assembly of medium cyclic ethers via exo-trig nucleophilic cyclization of cyclopropenes. Chem. Commun. 2016, 52, 7494–7496. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Bennin, T.; Rubina, M.; Rubin, M. Synthesis of 1,5-dioxocanes via the two-fold C-O bond forming nucleophilic 4 + 4-cyclodimerization of cycloprop-2-en-1-ylmethanols. RSC Adv. 2015, 5, 71849–71853. [Google Scholar] [CrossRef] [Green Version]
- Ryabchuk, P.; Matheny, J.P.; Rubina, M.; Rubin, M. Templated Assembly of Chiral Medium-Sized Cyclic Ethers via 8-endo-trig Nucleophilic Cyclization of Cyclopropenes. Org. Lett. 2016, 18, 6272–6275. [Google Scholar] [CrossRef]
- Ryabchuk, P.; Edwards, A.; Gerasimchuk, N.; Rubina, M.; Rubin, M. Dual Control of the Selectivity in the Formal Nucleophilic Substitution of Bromocyclopropanes en Route to Densely Functionalized, Chirally Rich Cyclopropyl Derivatives. Org. Lett. 2013, 15, 6010–6013. [Google Scholar] [CrossRef]
- Edwards, A.; Ryabchuk, P.; Barkov, A.; Rubina, M.; Rubin, M. Preparative resolution of bromocyclopropylcarboxylic acids. Tetrahedron Asymmetry 2014, 25, 1537–1549. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Straub, H.; Ryabchuk, P.; Rubina, M.; Rubin, M. Preparation of Chiral Enantioenriched Densely Substituted Cyclopropyl Azoles, Amines, and Ethers via Formal SN2′ Substitution of Bromocylopropanes. Molecules 2022, 27, 7069. https://doi.org/10.3390/molecules27207069
Straub H, Ryabchuk P, Rubina M, Rubin M. Preparation of Chiral Enantioenriched Densely Substituted Cyclopropyl Azoles, Amines, and Ethers via Formal SN2′ Substitution of Bromocylopropanes. Molecules. 2022; 27(20):7069. https://doi.org/10.3390/molecules27207069
Chicago/Turabian StyleStraub, Hillary, Pavel Ryabchuk, Marina Rubina, and Michael Rubin. 2022. "Preparation of Chiral Enantioenriched Densely Substituted Cyclopropyl Azoles, Amines, and Ethers via Formal SN2′ Substitution of Bromocylopropanes" Molecules 27, no. 20: 7069. https://doi.org/10.3390/molecules27207069