
High-Density “Windowpane” Coordination Patterns of Water Clusters and Their NBO/NRT Characterization


Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin-Madison, Madison, WI 53706, USA
Molecules 2022, 27(13), 4218; https://doi.org/10.3390/molecules27134218
Submission received: 3 May 2022
/
Revised: 25 June 2022
/
Accepted: 27 June 2022
/
Published: 30 June 2022
(This article belongs to the Collection Hydrogen Bonds)
Abstract
:Cluster mixture models for liquid water at higher pressures suggest the need for water clusters of higher coordination and density than those commonly based on tetrahedral H-bonding motifs. We show here how proton-ordered water clusters of increased coordination and density can assemble from a starting cyclic tetramer or twisted bicyclic (Möbius-like) heptamer to form extended Aufbau sequences of stable two-, three-, and four-coordinate “windowpane” motifs. Such windowpane clusters exhibit sharply reduced (~90°) bond angles that differ appreciably from the tetrahedral angles of idealized crystalline ice Ih. Computed free energy and natural resonance theory (NRT) bond orders provide quantitative descriptors for the relative stabilities of clusters and strengths of individual coordinative linkages. The unity and consistency of NRT description is demonstrated to extend from familiar supra-integer bonds of the molecular regime to the near-zero bond orders of the weakest linkages in the present H-bond clusters. Our results serve to confirm that H-bonding exemplifies resonance–covalent (fractional) bonding in the sub-integer range and to further discount the dichotomous conceptions of “electrostatics” for intermolecular bonding vs. “covalency” for intramolecular bonding that still pervade much of freshman-level pedagogy and force-field methodology.
1. Introduction
The earliest applications of ab initio natural bond orbital (NBO) analysis [1,2,3,4] consistently revealed a “donor–acceptor” (resonance–covalency-type “charge transfer”) picture of hydrogen bonding that was sharply at odds with then-prevalent “electrostatic” conceptions of intermolecular interactions [5,6]. Although the IUPAC Gold Book definition of H-bonding was subsequently revised to acknowledge the importance of covalency in H-bonding [7], superficial “dipole–dipole” rationalizations of H bonding continue to survive in many freshman-level expositions [8]. Arguments against the charge-transfer picture or in support of classical-type long-range, multipole, or “electrostatically driven” conceptions of H-bonding continue to appear [9,10] (vs. replies in [11,12,13]) in the research literature, and similar simplifying approximations persist in the empirical force fields of popular molecular dynamics (MD) simulation methods [14] that are commonly adopted to describe H-bonding in condensed phases.
The daunting task of describing macroscopic phases of liquid water or other H-bonded fluids may seem to demand the drastic long-range approximations of intermolecular (“noncovalent”) interactions as compared to the exchange-type (“covalent”) interactions of the short-range molecular regime. However, a more practical and accurate approach to describing intermolecular H-bonding is achieved by adopting supramolecular clusters [15] {Cn} as the conceptual “building blocks” of the macroscopic liquid-phase description, based on the known continuity of high-density liquid and low-density gaseous phases around the fluid critical point [16]. More specifically, quantum cluster equilibrium (QCE) theory [17,18,19] provides a practical numerical implementation of such “cluster mixture” [20,21,22,23,24,25] modeling of macroscopic phase properties, based on accurate values of electronic and vibrational properties of H-bonded {Cn} clusters that can be obtained at any chosen ab initio or density functional theory (DFT) level. The key input for QCE-based thermodynamic modeling of an aqueous phase is the data set of supramolecular clusters whose self-consistent (T,P)-dependent equilibrium populations are determined from the computed partition functions for each cluster by the standard methods of quantum statistical thermodynamics [26].
Among the many H-bonded fluids of practical interest, water itself presents the most studied yet still most perplexing phase behavior of the terrestrial regime [27]. Even the microscopic structure and properties of “ordinary” liquid water under near-ambient conditions remain matters of controversy [28]. Further mysteries surround the phase behavior of water at higher temperatures and pressures, where both theory [29,30,31,32] and experiments [33,34,35,36] have suggested the existence of an alternative high-density phase of liquid water that could lead to a liquid–liquid critical point and an exotic new domain of thermodynamic behavior near 220 K and 1–2 kbar.
The primary goal of present work was to computationally search for a new class of water clusters {Wn} based on the quadrilateral (“windowpane”) coordination motif of the cyclic tetramer (Figure 1) that might contribute to equilibrium QCE populations in the neighborhood of the proposed high-density phase. In each case, we restricted attention to clusters that maintain maximal Grotthuss-type proton ordering for the powerful effects of cooperative stabilization [37,38], as exemplified by the clockwise ordering of in-ring OH bonds in the view of Figure 1. The near −90° coordination angles of the windowpane class correspond to reduced next-neighbor distances and increased mass/volume ratios compared to the characteristic tetrahedral angles and chair–hexagon coordination motifs of ice-I-like clusters. The search for cooperatively stabilized windowpane clusters is organized in Aufbau fashion toward increasing numbers of fully four-coordinate sites that more adequately sample the intermolecular interactions expected to dominate in the phase behavior of the low-temperature and high-pressure regime. The resulting windowpane clusters can serve as computational input for subsequent QCE studies to examine their possible role in the equilibrium cluster distributions of the water-phase diagram.
A secondary goal of this study was to characterize each computed cluster in deeper conceptual terms that can clarify distinctive features of the underlying H-bond interactions. Such characterization should include aspects of overall cluster stability, strengths of individual coordinative linkages, shifts in atomic charge distribution, and other orbital-level features of free vs. coordinated water molecules. For these purposes, we employed NBO analysis [39,40] to obtain localized descriptors of molecular and intermolecular bonding features. Of particular interest are natural resonance theory (NRT) bond orders [41], which are expected to exhibit useful correlations with bond lengths [42,43], bond energies [44,45], bond stretching frequencies [46,47,48], NMR 1J and 1hJ spin-coupling constants [49], and other experimentally measurable properties.
Although the present study of novel water clusters was primarily directed toward equilibrium thermodynamic properties, it is important to note that such studies can also yield information on the kinetics and mechanisms of water cluster reactions. This is particularly true when, as in the present case, each cluster of the class is created in a sequential Aufbau manner from a previous member, e.g., by successive dimer additions of the form
where Wk = (H2O)k is a k-mer of a chosen coordination pattern. Analogous to elementary A + B ⇌ C chemical reactions, one can compute the transition state (Wk···W2)‡ and other features of the intrinsic reaction coordinate [50] (IRC) for each such cluster reaction. Similarly, for other cluster species satisfying the simultaneous QCE equilibrium conditions,
standard quantum chemical methods can be employed to determine transition-state features and associated absolute rate constants along the associated IRC [51]. However, such deeper mechanistic aspects of cluster formation were not addressed in the present work.
Wk + W2 ⇌ Wk+2
Wj + Wk ⇌ Wj′ + Wk′ (j + k = j′ + k′)
2. Computational Methods
For direct comparisons with many previous chemical applications in the NBO/NRT literature [52,53], we employed the familiar B3LYP/6-311++G** level of hybrid density functional theory for all geometry optimizations and energy evaluations of the present work. As shown elsewhere [54,55], realistic treatment of thermodynamic properties requires balanced treatment of energetic (primarily electronic) and entropic (primarily vibrational) contributions to free energy. All species were fully optimized and checked for vibrational stability with standard options of the Gaussian-16 program [56]. NBO/NRT analyses were completed with the NBO7 program [57,58] in interactive G16/NBO7 configuration. Structural and orbital graphics were obtained with the NBOPro7@Jmol utility program [59]. For NRT analyses of larger clusters, keyword selections for enlarged dynamic memory and the number of resonance structures were required to obtain fully converged bond orders. Ready-to-run input files containing optimized cartesian coordinates and keyword input for each cluster are included in the Supporting Information (SI). As shown particularly in ref. [54], many DFT variants and additional “corrections” (for dispersion, counterpoise, etc.) give qualitatively similar results for individual cluster structures and relative energies, even if some choices prove “best” for a particular thermodynamic comparison. The provided SI files allow re-optimization of cluster structures for alternative method/basis levels of choice.
3. Sequential Aufbau of 2-, 3-, 4-Coordinate Windowpane Water Clusters
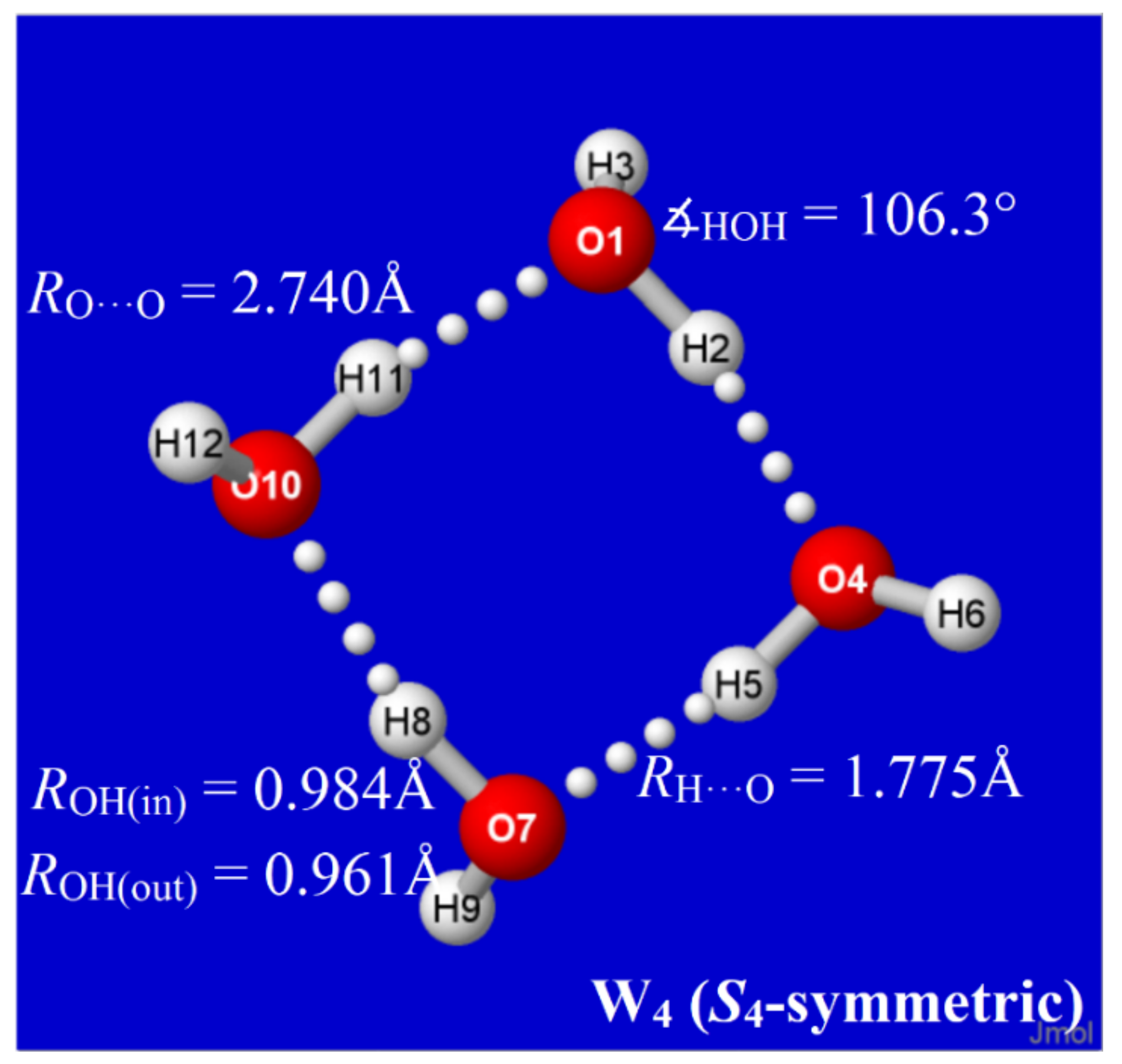
The properties of each water cluster Wk of an envisioned class are dictated by its specific H-bond coordination pattern. As primary descriptors of this pattern, we expect that each water molecule may generally be involved in two-, three-, or four-coordinate H-bonding to other molecules of the cluster (with singly coordinated “dangling” molecules excluded in leading clusters of the equilibrium thermodynamic distribution). For labeling purposes, the coordination pattern of each cluster may be usefully described by the number of quadruply (q), triply (t), or doubly (d) coordinated sites, appended as pre-superscripts (viz., q,t,dWn) to the cluster symbol. In this notation, the cyclic water tetramer of Figure 1 is labeled 0,0,4W4, with each monomer doubly coordinated in chain-like linkages to the substrate.
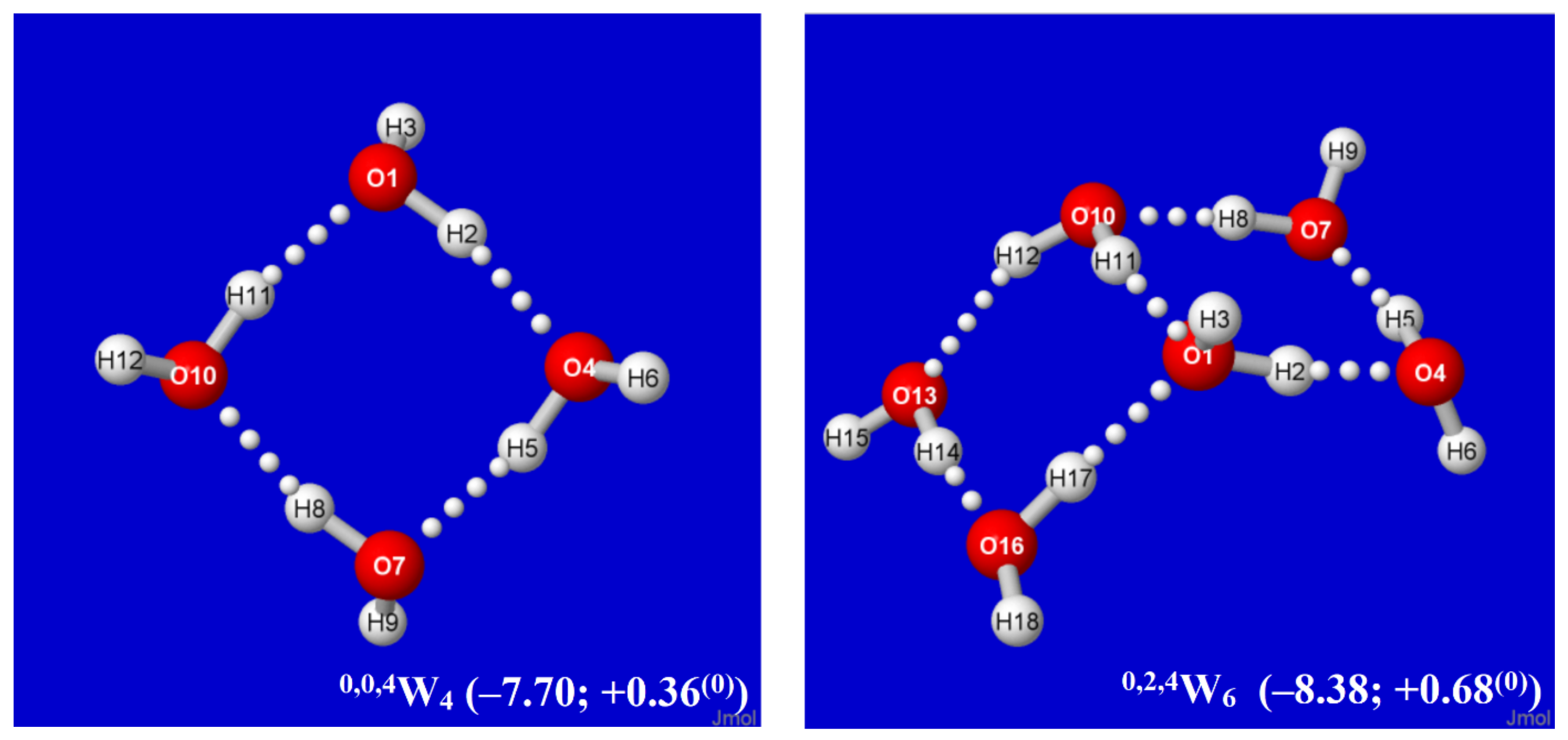
The structural logic for sequential Aufbau construction of windowpane clusters is straightforward. Starting from an existing cluster of this class, such as the cyclic water tetramer of Figure 1, one can choose any edge-type coordination (such as that between O(1) and O(10) in Figure 1) as a “base” for a new windowpane by attaching a water dimer in parallel fashion with two new H-bonds, as shown in the left panel of Figure 2. For maximum stabilization in forming this new H-bond attachment (e.g., from emanating H(12) at O(10)), the Grotthuss-type proton ordering should be continued around the edges of the newly formed windowpane that joins to O(1). The net result of this particular attachment is that sites O(10) and O(1) become tri-coordinate (t → t + 2), while other sites remain di-coordinate, leading to an overall 0,0,4W4 → 0,2,4W6 change in labeling. Some of these clusters, such as 0,0,4W4 itself or the cubane-like 0,8,0W8 described below, are featured in many previous cluster investigations, but the emphasis here is on hierarchical families of clusters that can be associated with a well-defined mechanistic Aufbau sequence of dimer additions, particularly leading to higher four-coordinate (q-type) motifs.
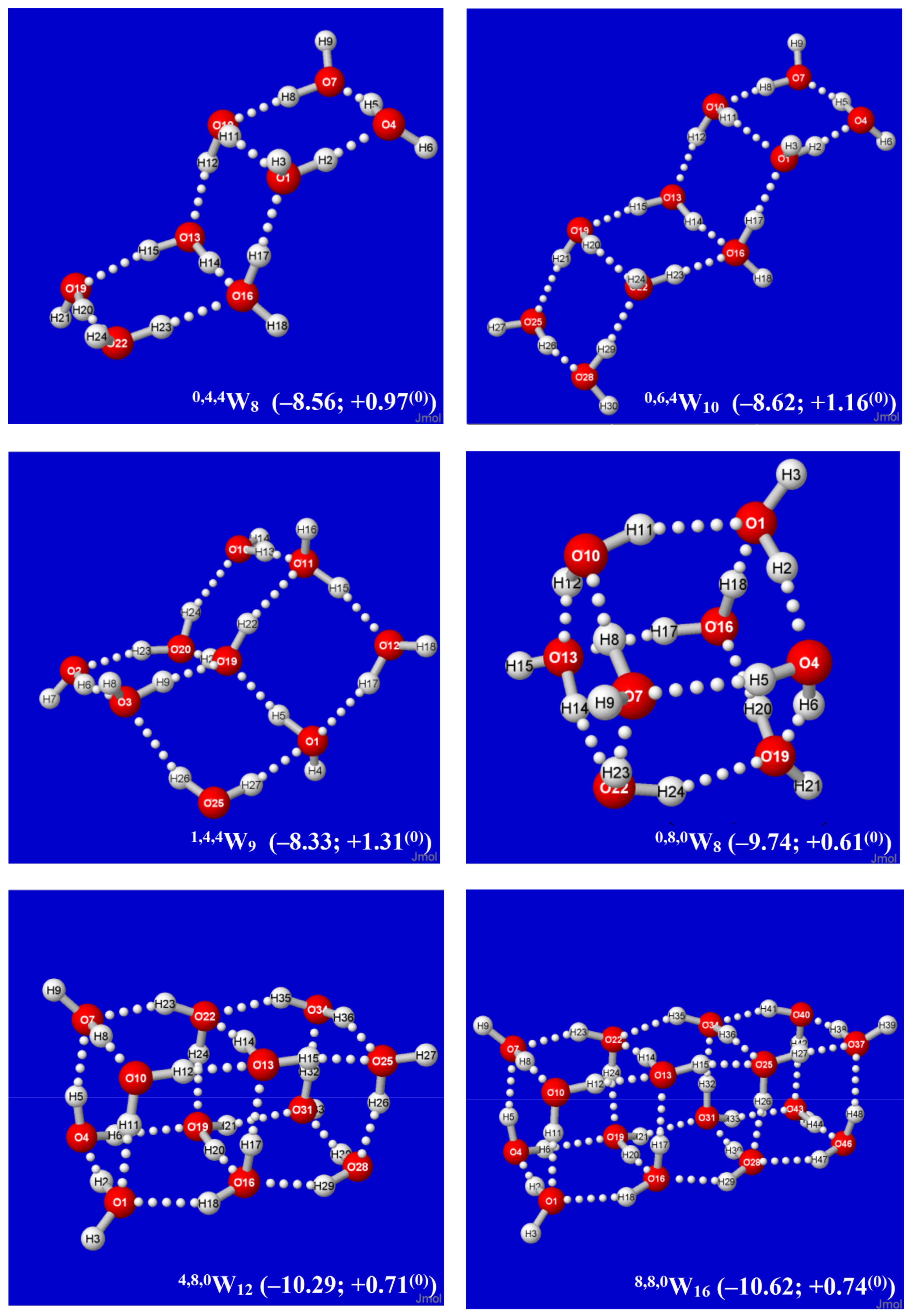
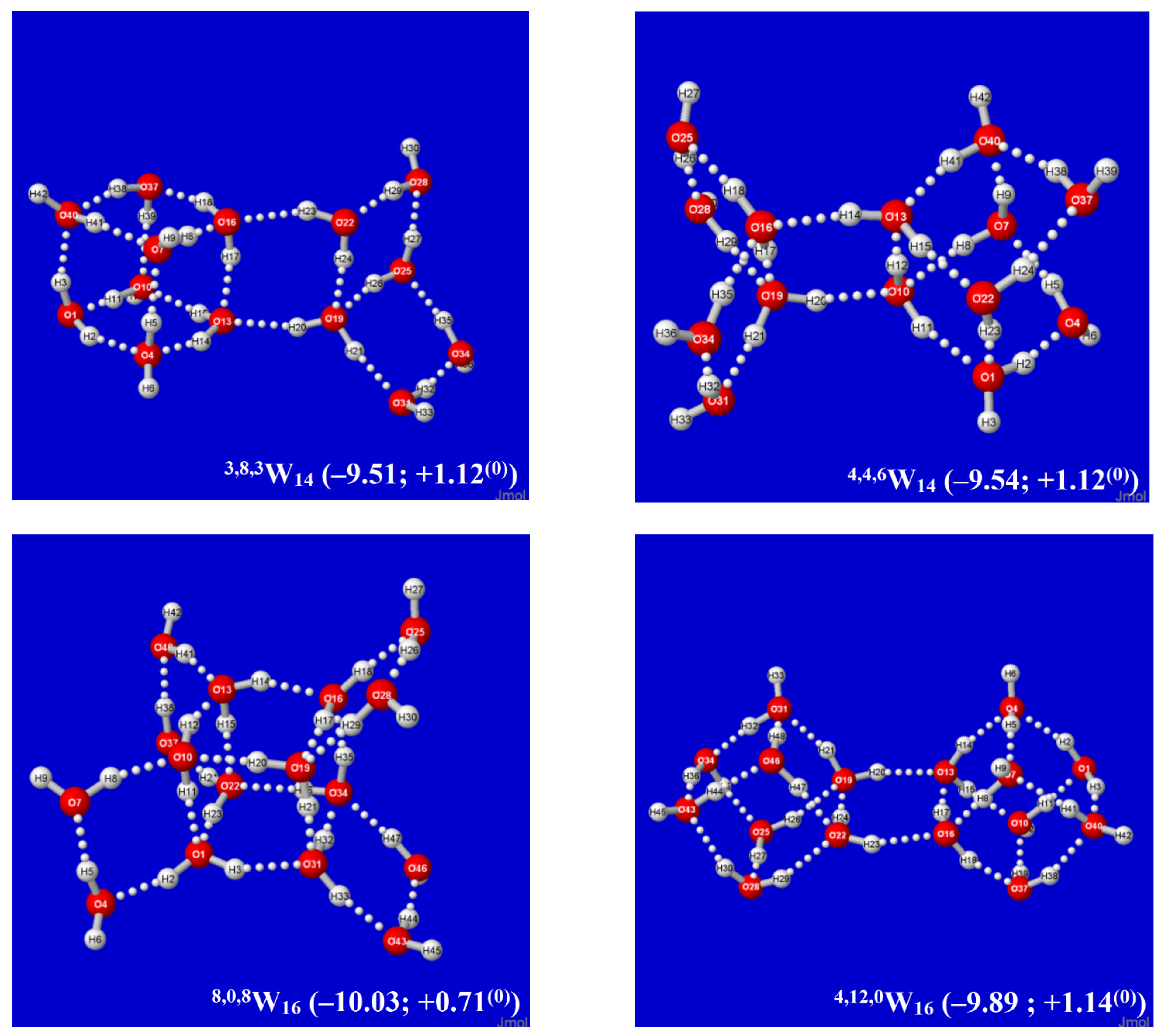
By alternating the sign of folding angles between panes, such additions can be continued indefinitely in “ladder-like” procession, as shown in successive panels of Figure 2. Each panel of Figure 2 includes (in parentheses) the per-monomer energy and standard-state Gibbs free energy change with respect to free water molecules, which serve to exhibit the important cooperative (nonadditive) effects of Grotthuss-ordered coordination patterns. The first four panels (0,0,4W4, 0,2,4W6, 0,4,4W8, 0,6,4W10) show the addition of successive rungs to the ladder pattern, up to the four-pane member. The ensuing 1,4,4W9 (row 3, left) is the alternative “2 × 2” four-pane cluster, which adopts a buckled saddle-shape deformation from planarity with a central four-coordinate monomer. From the starting two-pane ladder (0,2,4W6) at the upper right, one can also attempt to add another rung that curls backward (E-like) rather than forward (Z-like), but this optimizes to the cubane-like 0,8,0W8 cluster (row 3, right). The cubane motif becomes an evident building block for extensions to two-cube (4,8,0W12), three-cube (8,8,0W16), or longer rod-like clusters, as illustrated in the final row of the figure.
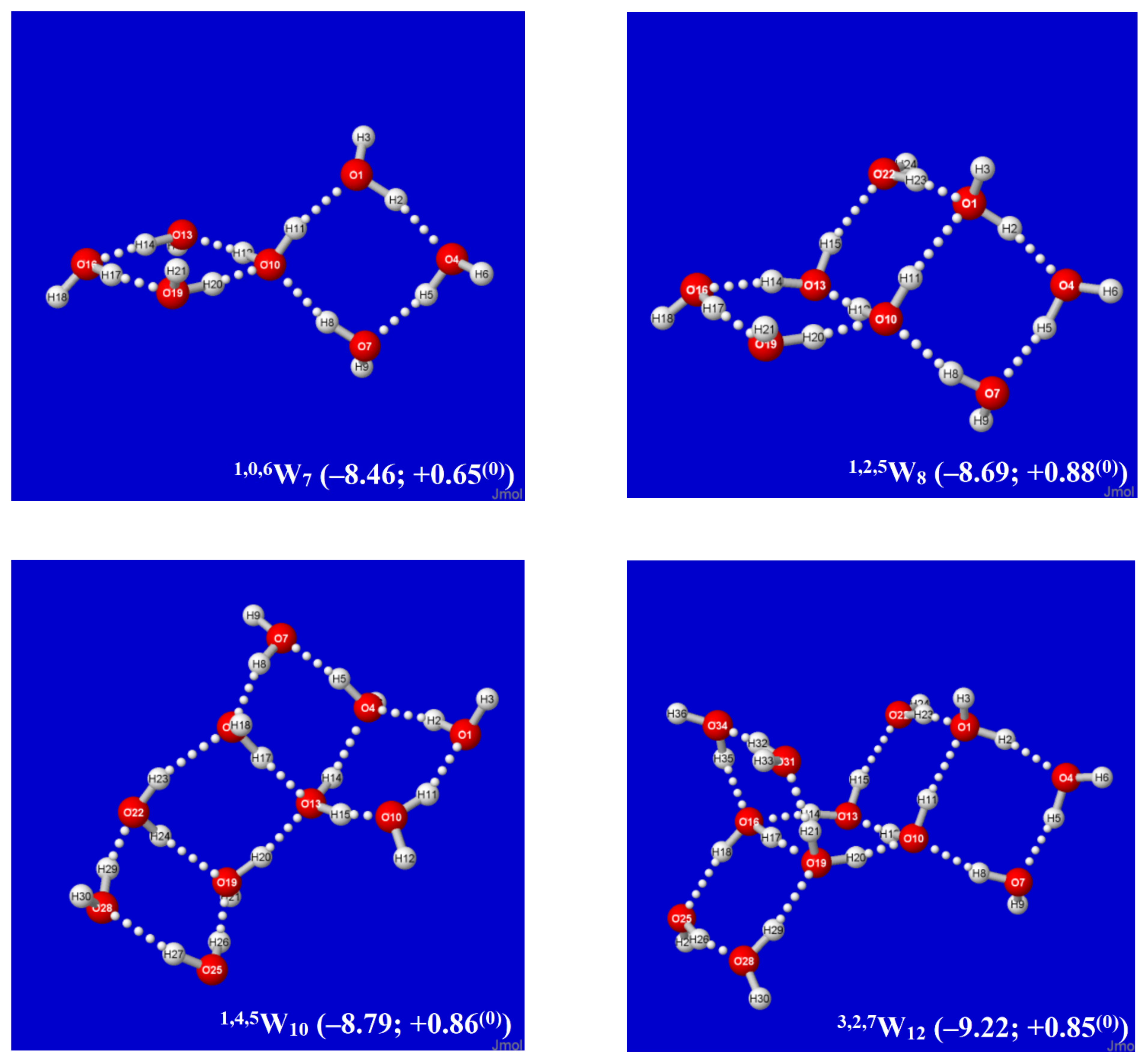
An alternative Aufbau starting point is provided by the twisted two-pane (1,0,6W7) cluster shown in the upper-left panel of Figure 3. This cluster features “Möbius-like” coordination with a continuous Grotthuss-ordered chain passing twice through the unique four-coordinate central monomer to form a closed loop. Remaining panels of Figure 3 show selected clusters that are obtained by successive Grotthuss-ordered dimer additions to 1,0,6W7, aimed at increasing q numbers of saturated four-coordinate sites. The resulting structures all incorporate the higher density coordination angles of the windowpane motif, but they exhibit irregular overall shapes that appear suitable as possible contributions to bulk liquid or amorphous solid phases. As seen in Figure 2 and Figure 3, the 8,8,0W16 cluster (Figure 2, lower right) achieves the largest number of three- and four-coordinate sites (q = t = 8) and the deepest per-monomer energy (−10.62 kcal/mol) in the depicted sequences. However, whether some or all of these clusters contribute significantly to known roots of the QCE equations, or whether (like the buckyball-type clathrate clusters previously studied [60]) they can serve as leading contributors to entirely new roots (phases) of the QCE phase diagram remains to be investigated.
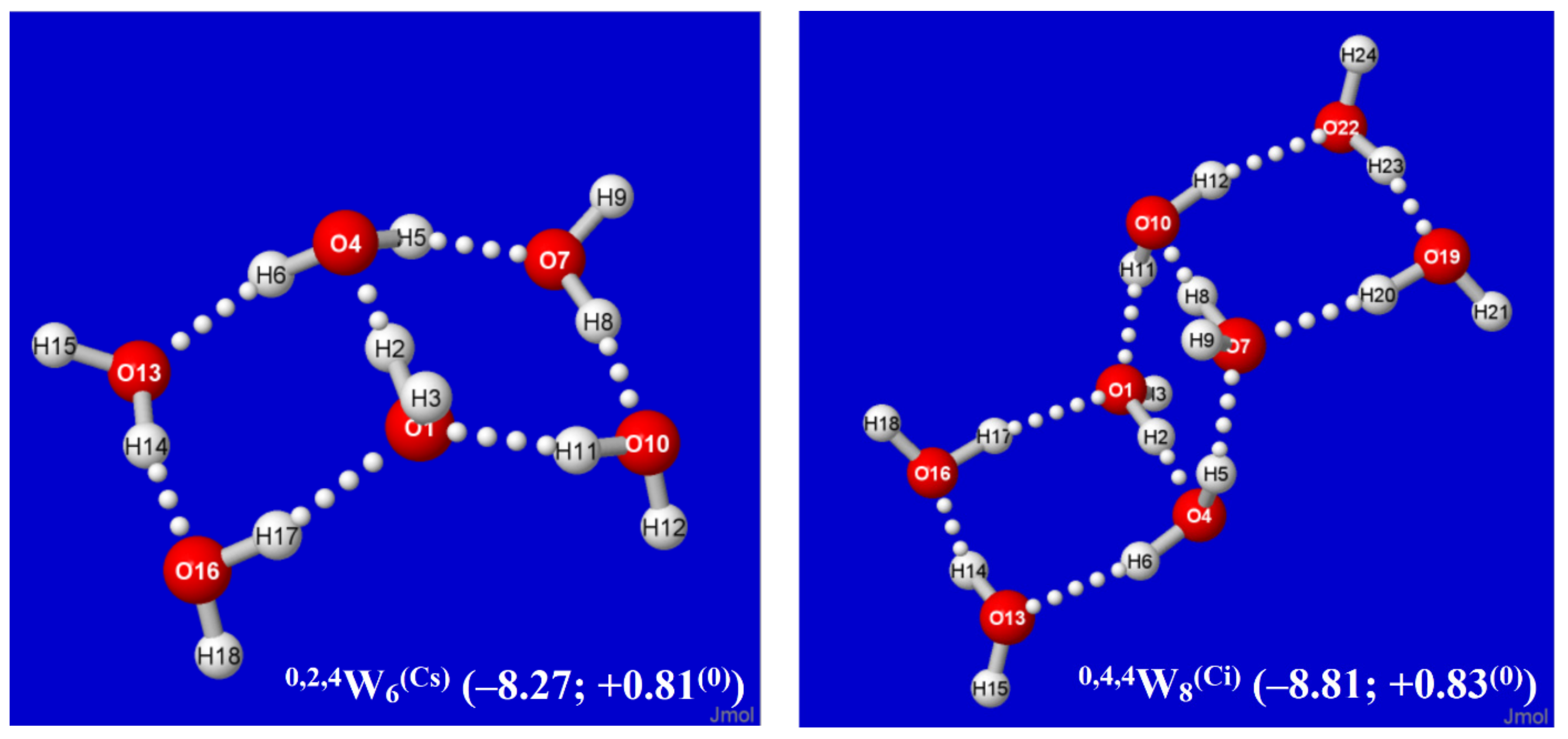
It is evident that each Aufbau cluster shown in Figure 2 and Figure 3 may have alternative isomeric rearrangements of the proton network without altering the q/t/d descriptors of O···(H)···O coordination linkages. Such alternative q,t,dWn(alt) isomers may have higher point group symmetry, different proton orderings (e.g., Grotthuss cycles around individual panes rather than overall periphery), and higher or lower energy than the Aufbau-derived clusters described above. Figure 4 displays two such alternative high-symmetry forms of the 0,2k,4W2k+4(sym) sequence (k = 1, 2), with respective Cs (k = 1), Ci (k = 2) symmetry. The Cs-symmetric 0,2,4W6(Cs) structure (Figure 4, left) is slightly higher in energy than 0,2,4W6 of Figure 2, but Ci-symmetric 0,4,4W8(Ci) (Figure 4, right) is slightly lower in energy than its low-symmetry counterpart in Figure 2. The inherent chirality of the coordination pattern about each O atom of higher-coordinated water clusters of Figure 2 and Figure 3 indicates that reduced symmetry (net chirality) is a high-probability feature of equilibrium water cluster distributions in any phase involving their participation.
Note that although H-bonds are considered weak noncovalent attractions, the cumulative energy release from larger cluster formation (viz., ΔE ≈ 170 kcal/mol for the 8,8,0W16 cluster) can readily exceed that necessary to dissociate a strong covalent bond, as in the ion pair clusters involved in self-dissociation (pH) of liquid water [54,55]. The per-monomer free energies of formation shown in Figure 2 and Figure 3 remain slightly positive under standard-state conditions, but the windowpane clusters are expected to gain increased stability relative to the ice-like clusters of the near-ambient regime as pressure increases. Full thermochemical and vibrational spectroscopic values for each cluster are included with the optimized coordinates in SI.
4. Natural Atomic Charge and Bond Order Characterizations
Among the many descriptors provided by NBO analysis, the natural atomic charges {QA} and interatomic bond orders {bAB} are most intimately associated with traditional empirical concepts of chemical bonding theory. Long-held perceptions of dichotomy between intra- vs. intermolecular forces (viz., “covalency” for chemical bond formation (bAB = 1, 2, 3,...) vs. “electrostatics” for H-bond formation (bH···O ≈ 0.1–0.2)) have long impeded true progress in the supramolecular domain. Demonstrations of how quantal QA, bAB descriptors extend seamlessly across the supposed divide can therefore serve to refute the obsolete dipole–dipole conceptions of H-bonding (and other so-called “non-covalent” interactions) that still pervade freshman-level pedagogy and classical force-field methodology. In the present section, we wish to test the usefulness of NBO/NRT-based QA, bAB descriptors when applied to the large data base of windowpane water clusters as described above.
4.1. General Features of Donor–Acceptor Interactions in Water Clusters
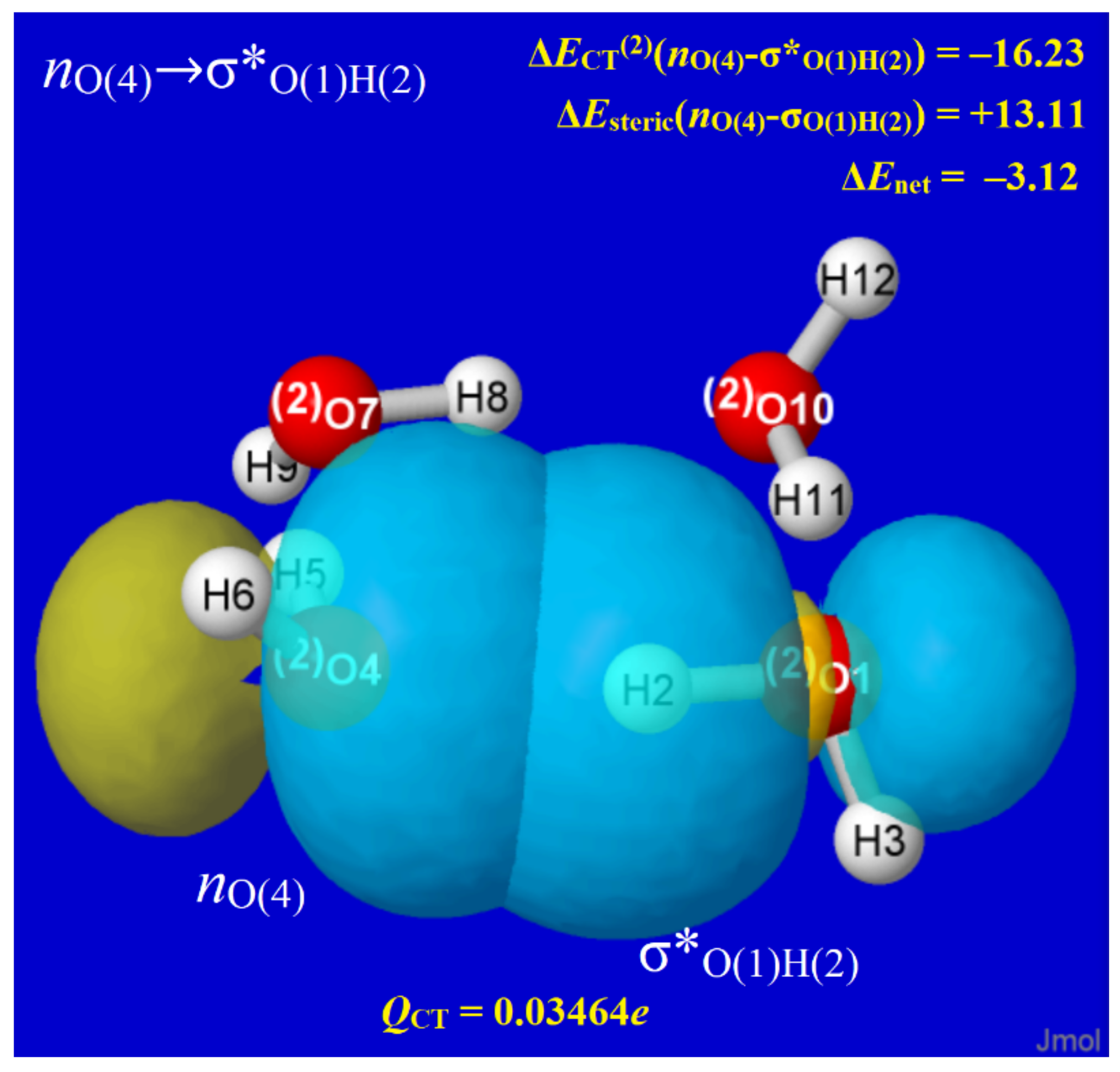
In every H-bond of every water cluster, NBO analysis reveals the characteristic nO→σ*OH donor–acceptor (“charge transfer”) interaction that transfers a slight electronic charge (QCT) from the oxygen lone pair (nO) of the Lewis base (LB) site into the valence antibond (σ*OH) of the proximal Lewis acid (LA) site. Figure 5 depicts the nO-σ*OH interaction for one of the H-bonds of W4c, showing the strongly overlapping forms of pre-orthogonal PNBOs deep inside van der Waals contact. The insets show details of the interaction that are routinely provided in NBO output, including (in kcal/mol; upper right) the second-order perturbative estimate of nO-σ*OH donor–acceptor attraction (ΔECT(2)), the corresponding steric opposition of nO-σOH donor–donor repulsion (ΔEsteric), and the net binding energy (ΔEnet). The known high transferability of NBOs [61] then assures that the individual nO, σ*OH orbitals are quite similar to those in water monomer and dimer as well as other windowpane clusters. However, one can also recognize the slight misalignments of ring strain that lower PNBO overlaps throughout the windowpane series and lead to the nuances in charge distribution, structure, and bond strength discussed below.
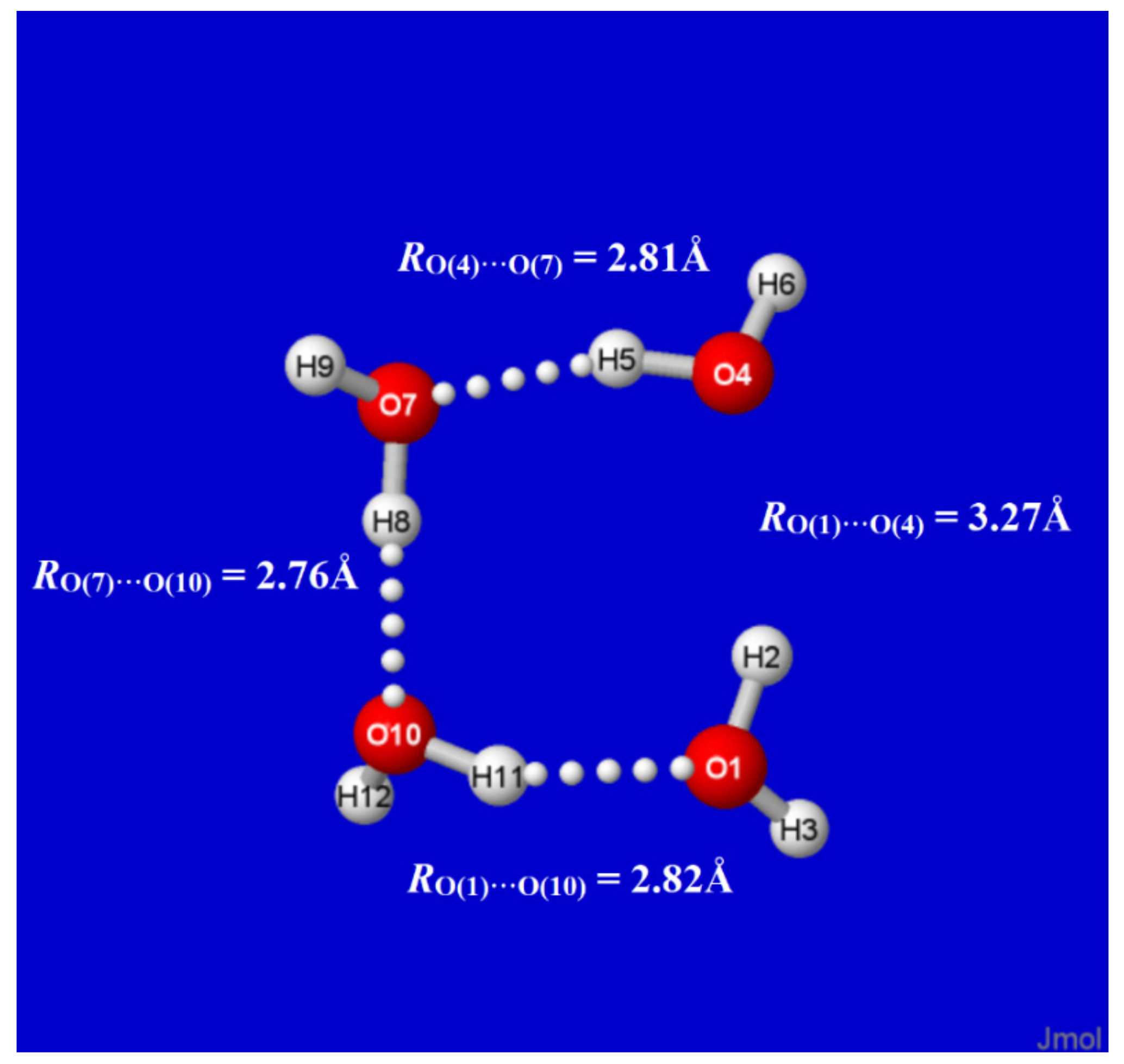
Alternatively, the effects of nO(4)→σ*O(1)H(2) interaction can be quantified by deleting this single specific matrix element from the DFT calculation (with standard $DEL keylist options [62]) and recalculating the energy and reoptimized geometry as though it were absent in nature. As shown in Figure 6, this single deletion “breaks” the O(4)···H(2)−O(1) hydrogen bond (and initial S4 symmetry) to give an open-chain structure with RO(1)···O(4) separation increased by ~0.5 Å to near-van der Waals contact distance. The monomers at each chain terminus also reorient to near coplanarity (contrary to the ~120° dihedral twisting of the two remaining monomers), thereby allowing partial re-gain of n(σ)O(4)→σ*O(1)H(2) attraction with the weaker in-plane n(σ)O(4) lone pair of O(4). By such $DEL deletion searches, one verifies that the specific nO(4)→σ*O(1)H(2) interaction is the unique “smoking gun” that is both necessary and sufficient for characteristic H-bonding between O(1) and O(4) monomers.
All such NBO-based energetic and $DEL deletion descriptors can be obtained for other windowpane clusters of Figure 2 and Figure 3. In the following, we focus instead on subtleties of the charge distributions and H-bond strengths that relate to the interesting cooperative effects of the highly ordered proton patterns (“water wires”) formed by the H-bond networks.
4.2. Natural Atomic Charge Distributions
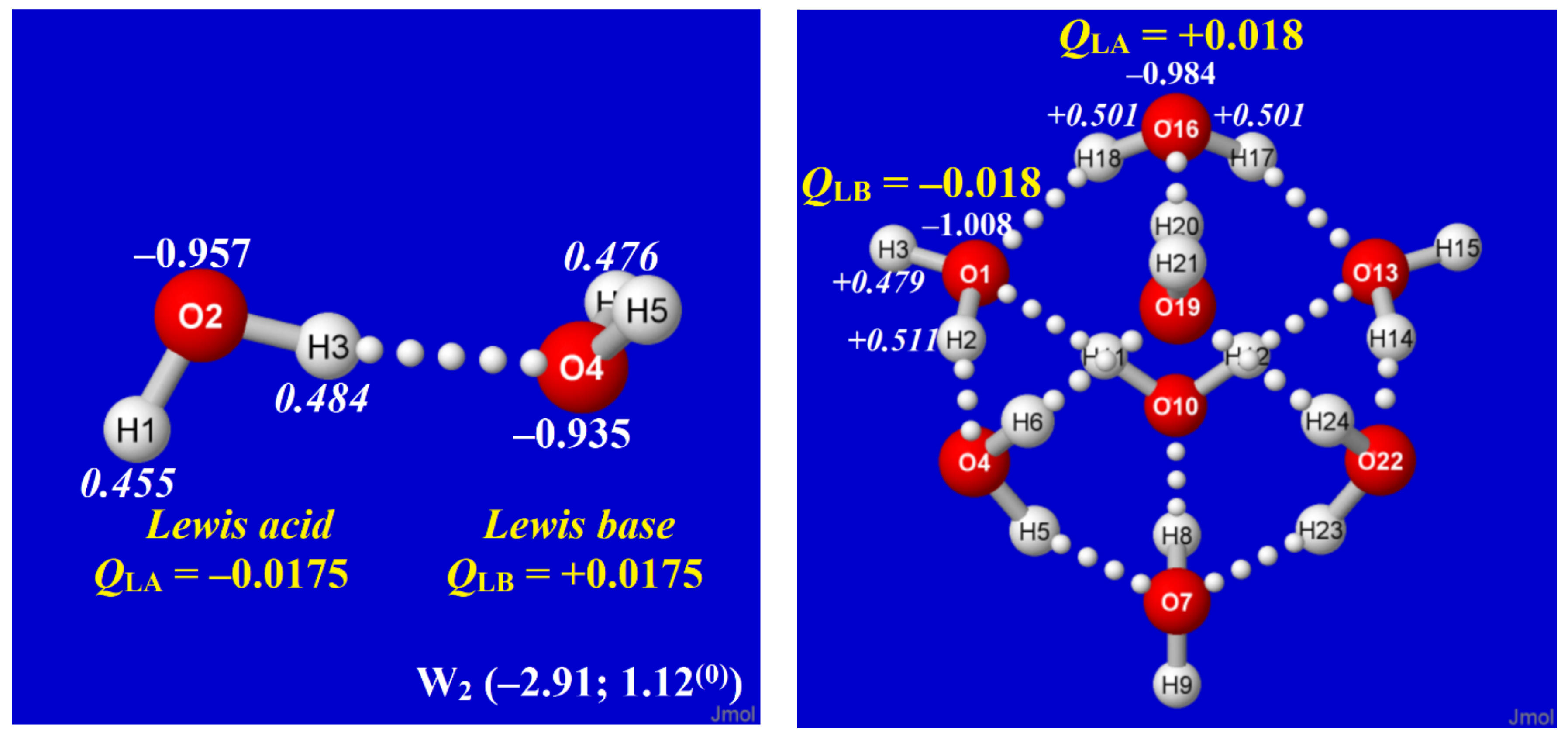
In principle, the simple water dimer (W2) might be seen as the fundamental conceptual building block for studies of electronic charge distribution and stability in clusters of higher complexity. However, Figure 7 exhibits the detailed comparisons of H atom (italic) and O atom (plain text) natural charges in W2 vs. cubane-like 0,8,0W8 to show the surprising contrasts between these species. In the two panels of Figure 7, the O(1) and O(16) monomers of the cubane cluster (right) are, respectively, the direct analogs of O(4) and O(2) monomers in the dimer (left), yet the net charges on the monomers of the dimer are directly opposite those in the cluster. Similar contrasts between charge distributions of the supposed “building block” dimer and those of higher coordination complexes are found throughout the clusters of Figure 2 and Figure 3.
How can the conflicting charge patterns of Figure 7 be rationalized? At the termini of each H-bond are two water monomers that can be identified as the LB (formal e-pair “donor”) and LA (formal σ*OH “acceptor” vacancy). In the simple water dimer, the nO→ σ*OH donor–acceptor interaction necessarily results in net charge transfer (ca. 0.017e) from LB to LA (Figure 7, left), resulting in the LBδ+···LAδ− charge pattern. However, in more complex water clusters, the surroundings of any chosen H-bond may be seen as a network of “water wires” that allow charge to redistribute as necessary to optimize overall cluster stability. Specifically, the multiple network connections allow electronic charge to be redistributed to achieve near neutrality at q- or d-coordinated sites, whose equal numbers of donor and acceptor interactions can be tuned to avoid capacitive build-up. However, at t-coordinated sites, which necessarily have an imbalance of donor (td, LB) vs. acceptor (ta, LA) connections, it becomes advantageous to confer excess anionic charge on td sites (increasing LB strength) and cationic charge on ta sites (increasing LA strength), thus leading to the commonly observed LBδ−···LAδ+ charge pattern.
To illustrate these propensities of cluster charge distribution, Figure 8 displays selected QO (plain text) and QH (italic) atomic charges of the 8,0,8W16 cluster for two q-type sites (centered at O(1), O(13)) and one d-type site (at O(46)), showing the significantly reduced net monomer charges compared to those of the water dimer. More complete listings of monomer charge values and coordination type at each O atom for all clusters of this study are included in SI, as illustrated for the 8,0,8W16 cluster in Table 1. The subtle variations in molecular charge indicate the extreme “feedback” sensitivity to every detail of the surrounding H-bond network, showing that overall network topology has taken precedence over characteristics of the water dimer (single H-bond) “building block” of which the network is composed.
4.3. Natural Bond Order Correlations
The distended shapes of windowpane clusters provide clear evidence of the severe effects of “ring strain” in altering the network O−H···O bonds from the idealized geometries of isolated H-bonds in binary complexes. Nevertheless, one expects that network H-bonds should continue to exhibit the robust correlations with NBO/NRT measures of bond order and charge transfer that were previously demonstrated for free binary H-bonded species [63]. We now turn to examining the supramolecular extension of such correlations for the classical bond order–bond length (BOBL) relationships that have long been fruitfully employed in the integer (single-, double-, triple-, etc., bond) range of covalent bonding in molecules [42,43].
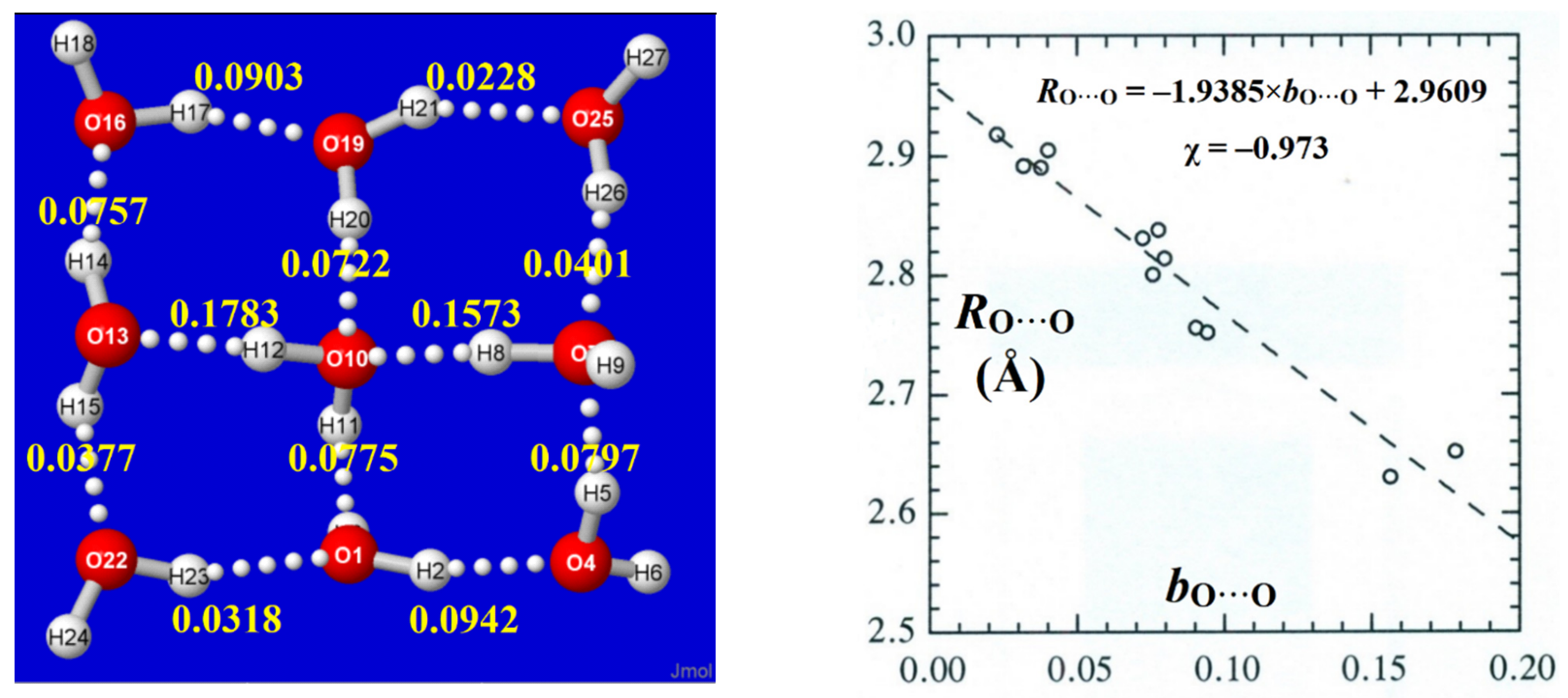
A simple example of such BOBL correlations is illustrated in Figure 9 for the 1,4,4W9 windowpane cluster of Figure 2. For each O···H−O linkage, the total bO···O bond order is obtained as the sum of bO···H (major) and “long-bond” [64] bO^O (minor) contributions,
with sub-integer values ranging from 0.02 to 0.18 in this simple cluster. As shown in the right panel, the BOBL correlation is of excellent quality (Pearson correlation coefficient χ ≈ −0.97), and the least-squares regression line (shown in the inset) allows close prediction of RO···O distances to near the 0.01Å level(!), despite the fact that NBO/NRT descriptors receive no input from real-space molecular geometry or spatial distribution of electron density. Thus, the resonance–covalency concepts underlying NRT bond order evaluations appear to extend seamlessly into this sub-integer range of weak H-bonding in clusters, practically as well as the familiar supra-integer range of strong covalent bonding and resonance in molecules.
bO···O = bO···H + bO^O
More complex three-dimensional structures of windowpane clusters obstruct clear visual representation of all relevant bO···O bond orders and tend to show additional effects of ring strain. Comprehensive listings of bO···O bond orders and RO···O distances (Å) for all H-bonds in all clusters (keyed to the atom numberings of Figure 2 and Figure 3) are presented as tables in SI, as exemplified for the 4,4,6W14 cluster in Table 2. In this case, the bO···O-RO···O correlation is found to be weaker, but still of reasonably high quality (χ ≈ −0.91), reflecting the heterogeneities of higher-coordination motifs.
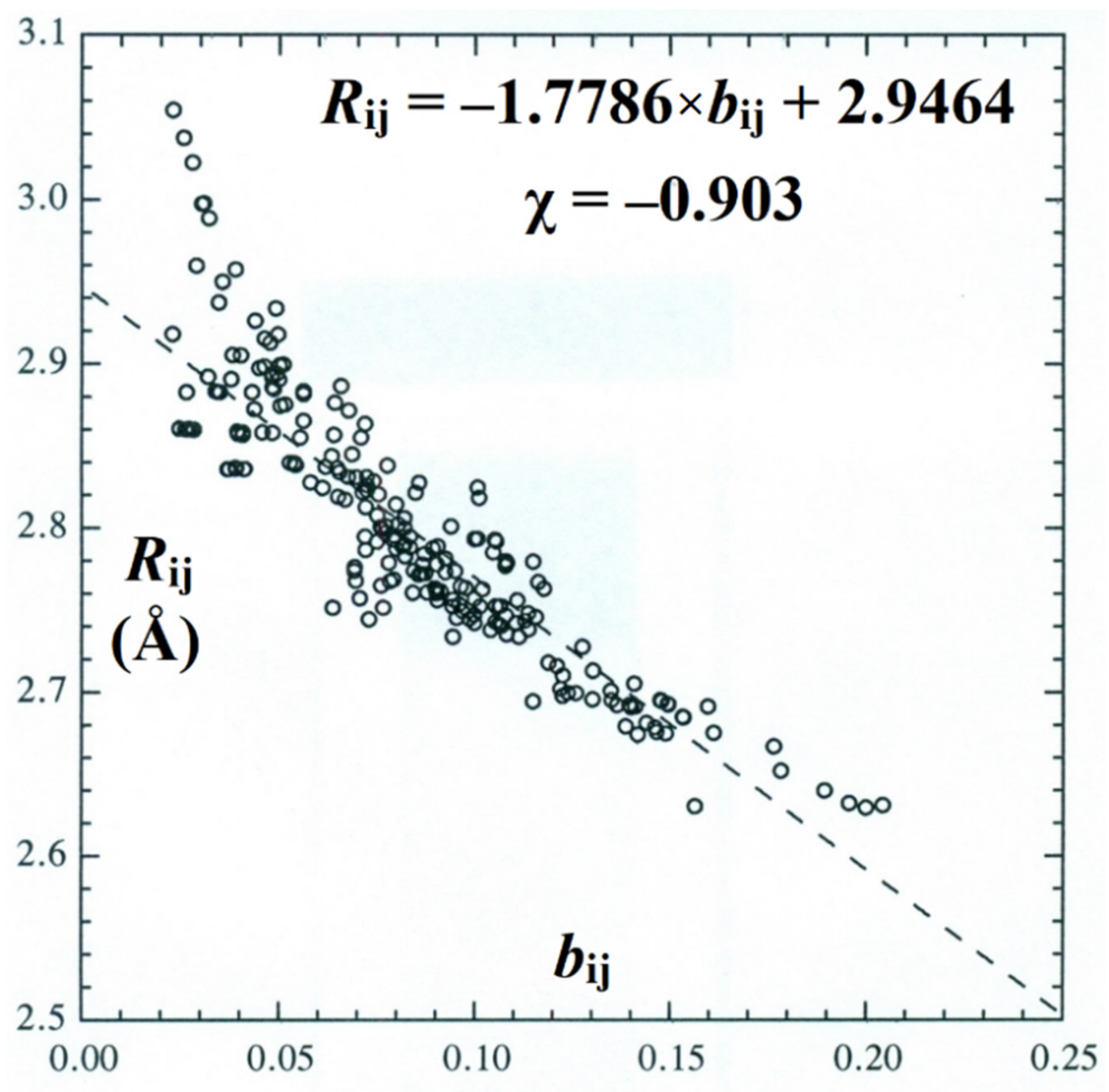
It is also of interest to examine the global BOBL correlations for all windowpane clusters of the present work, covering ca. 250 individual bO···O-RO···O H-bonded pairs in a broad variety of coupled coordination motifs. Figure 10 displays the BOBL scatter plot, least-squares regression line, and Pearson correlation coefficient for this entire data set of hydrogen bonds, showing the strong correlation (χ = −0.90) that persists in spite of increasingly heterogeneous cluster topologies.
The degraded accuracy of the linear least-squares regression fit in Figure 10 (compared, e.g., to that in Figure 9) can be primarily attributed to the upward deviations from linearity that are evident near bij → 0. However, it is important to recognize that these deviations are required on physical grounds, because intermonomer separation should asymptotically diverge (Rij → ∞) as bond order vanishes (bij → 0). Indeed, only the higher-order connectivity of the H-bond network prevents such asymptotic dissociation when any single H-bond is severed, so the proper appearance of such nonlinearity in the bij → 0 limit serves to further confirm the resonance–covalent nature of H-bonding even in this range of interaction strengths near the limit of chemical interest.
5. Conclusions
In the present work, we have employed standard density functional methods to computationally characterize a broad variety of unusual “windowpane” clusters that may play a role in the high-density fluid phase(s) of water. Despite their diverse topological forms and unusual angular features, we have demonstrated that these clusters are fully compliant with water’s known facility in forming doubly (d-type), triply (t-type), and quadruply (q-type) coordinative linkages to other water molecules, leading to multiply connected (“water-wired”) networks of increasing energetic stability when proper Grotthuss-type proton ordering is maintained. The Aufbau construction approach also suggests the mechanistic sequence by which such Grotthuss-ordered clusters can readily form from successive aggregation with water dimers.
We have also employed natural bond orbital (NBO) and natural resonance theory (NRT) analysis tools to demonstrate the consistency and accuracy with which H-bonding in these clusters conforms to the general conceptual picture of resonance–covalency (“charge transfer”) as the authentic origin of intermolecular O−H···O attractions. The charge flows and adaptive bond order and structural shifts in these clusters are shown to obey familiar bond order–bond length (BOBL) correlations with high accuracy (|χ| > 0.9). Moreover, the BOBL correlations also exhibit the expected deviations from linearity in the asymptotic limit of vanishing bond order where RO···O distance becomes divergent. Although connections can be shown between NBO and Bader-type descriptors [65], we believe that the NRT bond orders of the present work provide broader predictive utility and more nuanced inclusion of resonance effects than the topological descriptors as employed in previous studies of water clustering (e.g., [66]).
The reader is reminded that “correlation is not causation.” Nevertheless, the continuity of robust BOBL correlations that stretch across the broad extremes of supramolecular (sub-integer) vs. molecular (multi-integer) bond orders strongly implies their shared origin in unified “covalency” concepts, contrary to the dichotomous viewpoint that still dominates freshman-level teaching of chemical principles and many facets of force-field methodology.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27134218/s1. The Supporting Information (SI) file contains optimized geometrical coordinates, NBO/NRT keyword input, and other computational details in ready-to-run Gaussian input files for all equilibrium water clusters described in the paper. The file also contains tables of computed natural atomic charges and natural bond orders for all water clusters of the work.
Funding
Support for computational facilities was provided in part by the National Science Foundation Grant CHE-0840494.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Thanks are due to Eric Glendening (Indiana State University) for assistance in wrangling consistent NRT bond orders for the many challenging H-bonding interactions in this study.
Conflicts of Interest
The author declares no conflict of interest.
Sample Availability
Samples of the clusters are not available from the author.
References
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree-Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F.; Curtiss, L.A.; Pochatko, D.J. Natural bond orbital analysis of molecular interactions: Theoretical studies of binary complexes of HF, H2O, NH3, N2, O2, F2, CO, and CO2 with HF, H2O, and NH3. J. Chem. Phys. 1986, 84, 5687–5705. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Melendres, C.A.; Reed, A.E.; Weinhold, F. Theoretical studies of O2−:(H2O)n clusters. J. Comput. Chem. 1986, 7, 294–305. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Fowler, P.W.; Buckingham, A.D. The long-range model of intermolecular forces. Mol. Phys. 1983, 50, 1349–1361. [Google Scholar] [CrossRef]
- Buckingham, A.D.; Fowler, P.W.; Stone, A.J. Electrostatic predictions of shapes and properties of van der Waals molecules. Int. Rev. Phys. Chem. 1986, 5, 107–114. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Weinhold, F.; Klein, R.A. What is a hydrogen bond? Resonance covalency in the supramolecular domain. Chem. Educ. Res. Pract. 2014, 15, 276–285. [Google Scholar] [CrossRef]
- Stone, A.J. Natural bond orbitals and the nature of the hydrogen bond. J. Phys. Chem. A 2017, 121, 1531–1534. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. Sigma-holes, pi-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef]
- Weinhold, F.; Glendening, E.D. Comment on “Natural bond orbitals and the nature of the hydrogen bond”. J. Phys. Chem. A 2018, 122, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, F. Anti-electrostatic pi-hole bonding: How covalency conquers Coulombics. Molecules 2022, 27, 377. [Google Scholar] [CrossRef]
- Herbert, J.M. Neat, simple, and wrong: Debunking electrostatic fallacies regarding noncovalent interactions. J. Phys. Chem. A 2021, 125, 7125–7137. [Google Scholar] [CrossRef]
- Brooks, C.L., III; Case, D.A.; Plimpton, S.; Roux, B.; Van Der Spoel, D.; Tajkhorshid, E. Classical molecular dynamics. J. Chem. Phys. 2021, 154, 100401. [Google Scholar] [CrossRef]
- Ludwig, R. Water: From clusters to the bulk. Angew. Chem. Int. Ed. 2011, 40, 1808–1827. [Google Scholar] [CrossRef]
- Andrews, T. On the continuity of the gaseous and liquid states of matter. Philos. Trans. R. Soc. Lond. 1869, 159, 575–590. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, F. Quantum cluster equilibrium theory of liquids: General theory and computer implementation. J. Chem. Phys. 1998, 109, 367–372. [Google Scholar] [CrossRef]
- Weinhold, F. Quantum cluster equilibrium theory of liquids: Illustrative application to water. J. Chem. Phys. 1998, 109, 373–384. [Google Scholar] [CrossRef]
- Kirchner, B.; Weinhold, F.; Friedrich, J.; Perlt, E.; Lehmann, S.B.C. Quantum Cluster Equilibrium. In Many-Electron Approaches in Physics, Chemistry and Mathematics; Bach, V., Site, L.D., Eds.; Springer Mathematical Physics Studies: New York, NY, USA, 2014; pp. 77–96. [Google Scholar]
- Mayer, J.E.; Mayer, M.G. Statistical Mechanics; Wiley: New York, NY, USA, 1940. [Google Scholar]
- Frank, H.S.; Wen, W. Ion-solvent interaction. Structural aspects of ion-solvent interaction in aqueous solutions: A suggested picture of water structure. Discuss. Faraday Soc. 1957, 24, 133–140. [Google Scholar] [CrossRef]
- Nemethy, G.; Scheraga, H.A. Structure of water and hydrophobic bonding in proteins. I. A model for the thermodynamic properties of liquid water. J. Chem. Phys. 1962, 36, 3382–3400. [Google Scholar] [CrossRef]
- Walrafen, G.E. Raman and infrared spectral investigations of water structure. In Water: A Comprehensive Treatise; Franks, F., Ed.; Plenum: New York, NY, USA, 1972; Volume 1, pp. 151–214. [Google Scholar]
- Symons, M.C.R. Water structure and reactivity. Acc. Chem. Res. 1981, 14, 179–187. [Google Scholar] [CrossRef]
- Benson, S.W.; Siebert, E.D. A simple two-structure model for liquid water. J. Am. Chem. Soc. 1992, 114, 4269–4276. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Statistical Mechanics; Harper & Row: New York, NY, USA, 1976. [Google Scholar]
- Pettersson, L.G.M.; Henchman, R.H.; Nilsson, A. Water: The most anomalous liquid. Chem. Rev. 2016, 116, 7459–7462. [Google Scholar] [CrossRef] [Green Version]
- Wernet, P.; Nordlund, D.; Bergmann, U.; Cavalleri, M.; Odelius, M.; Ogasawara, H.; Näslund, L.Å.; Hirsch, T.K.; Ojamäe, L.; Glatzel, P.; et al. The structure of the first coordination shell in liquid water. Science 2004, 304, 995–999. [Google Scholar] [CrossRef] [Green Version]
- Speedy, R.; Angell, C. Isothermal compressibility of supercooled water and evidence for a thermodynamic singularity at −45 °C. J. Chem. Phys. 1976, 65, 851–858. [Google Scholar] [CrossRef]
- Mishima, O.; Stanley, H.E. The relationship between liquid, supercooled and glassy water. Nature 1998, 396, 329–335. [Google Scholar] [CrossRef]
- Gallo, P.; Amann-Winkel, K.; Angell, C.A.; Anisimov, M.A.; Caupin, F.; Chakravarty, C.; Lascaris, E.; Loerting, T.; Panagiotopoulos, A.Z.; Russo, J.; et al. Water: A tale of two liquids. Chem. Rev. 2016, 116, 7463–7500. [Google Scholar] [CrossRef]
- Hestand, N.J.; Skinner, J.L. Perspective: Crossing the Widom line in no man’s land: Experiments, simulations, and the location of the liquid-liquid critical point in supercooled water. J. Chem. Phys. 2018, 149, 140901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, A.; Pettersson, L.G.M. Perspective on the structure of liquid water. Chem. Phys. 2011, 389, 1–34. [Google Scholar] [CrossRef]
- Manka, A.; Pathak, H.; Tanimura, S.; Wölk, J.; Strey, R.; Wyslouzil, B.E. Freezing water in no-man’s land. Phys. Chem. Chem. Phys. 2012, 14, 4505–4516. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Petrik, N.G.; Smith, R.S.; Kay, B.D.; Kimmel, G.A. Growth rate of crystalline ice and the diffusivity of supercooled water from 126 to 262 K. Proc. Natl. Acad. Sci. USA 2016, 113, 14921–14925. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Späh, A.; Pathak, H.; Perakis, F.; Mariedahl, D.; Amann-Winkel, K.; Sellberg, J.A.; Lee, J.H.; Kim, S.; Park, J.; et al. Maxima in the thermodynamic response and correlation functions of deeply supercooled water. Science 2017, 358, 1589–1593. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, F. Nature of H-bonding in clusters, liquids and enzymes: An ab initio, natural bond orbital perspective. J. Mol. Struct. (THEOCHEM) 1997, 398–399, 181–197. [Google Scholar] [CrossRef]
- Weinhold, F. Resonance character of hydrogen-bonding interactions in water and other H-bonded species. In Peptide Solvation and H-Bonds: Advances in Protein Chemistry; Baldwin, R.L., Baker, D., Eds.; Elsevier: San Diego, CA, USA, 2006; Volume 72, pp. 121–155. [Google Scholar]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural Bond Orbital Theory: Discovering Chemistry with NBO7. In Complementary Bonding Analysis; Grabowsky, S., Ed.; Walter de Gruyter GmbH & Co KG: Amsterdam, The Netherlands, 2021; pp. 129–156. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Resonance theory reboot. J. Am. Chem. Soc. 2019, 141, 4156–4166. [Google Scholar] [CrossRef]
- Coulson, C.A.; Lennard-Jones, J.E. The electronic structure of some polyenes and aromatic molecules. VII. Bonds of fractional order by the molecular orbital method. Proc. R. Soc. Lond. 1939, A169, 413–428. [Google Scholar]
- Mishra, P.C.; Rai, D.K. Bond order-bond length relationship in all-valence-electron molecular orbital theory. Mol. Phys. 1972, 23, 631–634. [Google Scholar] [CrossRef]
- Johnston, H.S.; Parr, C. Activation energies from bond energies. I. Hydrogen transfer reactions. J. Am. Chem. Soc. 1963, 85, 2544–2551. [Google Scholar] [CrossRef]
- Johnstone, R.A.W.; Loureiro, R.M.S.; Lurdes, M.; Cristiano, S.; Labat, G. Bond energy/bond order relationships for NO linkages and a quantitative measure of ionicity: The role of nitro groups in hydrogen bonding. Arkivoc 2010, 2010, 142–169. [Google Scholar] [CrossRef]
- Badger, R.M. A relation between internuclear distances and bond force constants. J. Chem. Phys. 1934, 2, 128–131. [Google Scholar] [CrossRef] [Green Version]
- Boyer, M.A.; Marsalek, O.; Heindel, J.P.; Markland, T.E.; McCoy, A.B.; Xantheas, S.S. Beyond Badger’s rule: The origins and generality of the structure-spectra relationship of aqueous hydrogen bonds. J. Phys. Chem. Lett. 2019, 10, 918–924. [Google Scholar] [CrossRef]
- Bürgi, H.; Dunitz, J.D. Fractional bonds: Relations among their lengths, strengths, and stretching frequencies. J. Am. Chem. Soc. 1987, 109, 2924. [Google Scholar] [CrossRef]
- Gründemann, S.; Limbach, H.-H.; Buntkosky, G.; Sabo-Etienne, S.; Chaudret, B. Distance and scalar HH-coupling correlations in transition metal dihydrides and dihydrogen complexes. J. Phys. Chem. A 1999, 103, 4752–4754. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Weinhold, F. Kinetics and mechanism of water cluster equilibria. J. Phys. Chem. B 2014, 118, 7792–7798. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; John Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Perlt, E.; von Domaros, M.; Kirchner, B.; Ludwig, R.; Weinhold, F. Predicting the ionic product of water. Sci. Rep. 2017, 7, 10244. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, B.; Ingenmey, J.; von Domaros, M.; Perlt, E. The ionic product of water in the eye of the quantum cluster equilibrium. Molecules 2022, 27, 1286. [Google Scholar] [CrossRef]
- Frisch, M.E.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.P.; Petersson, G.A.; Nakatsuji, H.J.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018; Available online: https://nbo7.chem.wisc.edu/ (accessed on 2 May 2022).
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef]
- Weinhold, F.; Phillips, D.; Glendening, E.D.; Foo, Z.Y.; Hanson, R.M. NBOPro7@Jmol; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Ludwig, R.; Weinhold, F. Quantum cluster equilibrium theory of liquids: Freezing of QCE/3-21G water to tetrakaidecahedral “Bucky-ice”. J. Chem. Phys. 1999, 110, 508–515. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Weinhold, F. Transferability of natural bond orbitals. J. Am. Chem. Soc. 1988, 110, 368–372. [Google Scholar] [CrossRef]
- Weinhold, F.; Glendening, E.D. NBO 7.0 Program Manual, Sec. B-5. Available online: https://nbo7.chem.wisc.edu/nboman.pdf (accessed on 2 May 2022).
- Weinhold, F.; Klein, R.A. What is a hydrogen bond? Mutually consistent theoretical and experimental criteria for characterizing H-bonding interactions. Mol. Phys. 2012, 110, 565–579. [Google Scholar] [CrossRef]
- Landis, C.R.; Weinhold, F. 3c/4e -type long-bonding: A novel transitional motif toward the metallic delocalization limit. Inorg. Chem. 2013, 52, 5154–5166. [Google Scholar] [CrossRef]
- Weinhold, F. Natural bond critical point analysis: Quantitative relationships between NBO-based and QTAIM-based topological descriptors of chemical bonding. J. Comput. Chem. 2012, 33, 2440–2449. [Google Scholar] [CrossRef]
- Castor-Villegas, V.M.; Guevara-Vela, J.M.; Vallejo Narváez, W.E.; Pendása, A.M.; Rocha-Rinza, T.; Fernández-Alarcón, A. On the strength of hydrogen bonding within water clusters on the coordination limit. J. Comput. Chem. 2020, 41, 2266–2277. [Google Scholar] [CrossRef]
Figure 1.
Equilibrium structural properties of cyclic (H2O)4 “windowpane” cluster (B3LYP/6-311++G** level).
Figure 1.
Equilibrium structural properties of cyclic (H2O)4 “windowpane” cluster (B3LYP/6-311++G** level).

Figure 2.
Calculated Aufbau sequence of windowpane clusters q,t,dWk from starting cyclic tetramer 0,0,4W4 (upper left), showing parenthesized per-monomer changes (kcal/mol) in energy (ΔE) and Gibbs free energy (ΔG(0)) from free water molecules in each panel.
Figure 2.
Calculated Aufbau sequence of windowpane clusters q,t,dWk from starting cyclic tetramer 0,0,4W4 (upper left), showing parenthesized per-monomer changes (kcal/mol) in energy (ΔE) and Gibbs free energy (ΔG(0)) from free water molecules in each panel.


Figure 3.
Similar to Figure 2, for successive q,t,dWk windowpane clusters built from the Möbius-like 1,0,6W7 cluster (upper left).
Figure 3.
Similar to Figure 2, for successive q,t,dWk windowpane clusters built from the Möbius-like 1,0,6W7 cluster (upper left).


Figure 4.
Alternative higher-symmetry 0,2k,4W2k+4(sym) clusters (k = 1,2), one (Cs) of higher energy, the other (Ci) of lower energy than the corresponding low-symmetry structure of Figure 2.
Figure 4.
Alternative higher-symmetry 0,2k,4W2k+4(sym) clusters (k = 1,2), one (Cs) of higher energy, the other (Ci) of lower energy than the corresponding low-symmetry structure of Figure 2.

Figure 5.
Pre-orthogonal (PNBO) depiction of nO→σ*OH orbital interaction in one H-bond of W4c, with energetic (kcal/mol) and charge transfer (e) details as insets (see text).
Figure 5.
Pre-orthogonal (PNBO) depiction of nO→σ*OH orbital interaction in one H-bond of W4c, with energetic (kcal/mol) and charge transfer (e) details as insets (see text).

Figure 6.
$DEL (partially)-reoptimized structure of original W4c cluster (Figure 1), showing effects of deleting the single nO(4)→σ*O(1)H(2) interaction of Figure 5 (at the point where the maximum number of optimization steps was completed).

Figure 7.
Natural atomic charges for H (italics; white) and O (plain text; white) atoms of water dimer (left) and cubane-like 0,8,0W8 cluster (right), with corresponding net charges (yellow) of formal Lewis acid (e-acceptor) and Lewis base (e-donor) water molecules in each species, showing the reversal of apparent charge flow in the two cases. (Parenthesized per-monomer energy and free energy for W2 also allow direct stability comparisons with clusters of Figure 2, Figure 3 and Figure 4).
Figure 7.
Natural atomic charges for H (italics; white) and O (plain text; white) atoms of water dimer (left) and cubane-like 0,8,0W8 cluster (right), with corresponding net charges (yellow) of formal Lewis acid (e-acceptor) and Lewis base (e-donor) water molecules in each species, showing the reversal of apparent charge flow in the two cases. (Parenthesized per-monomer energy and free energy for W2 also allow direct stability comparisons with clusters of Figure 2, Figure 3 and Figure 4).

Figure 8.
Similar to Figure 7, for representative quadruply (q-type) coordinated O(1)H(2)H(3) and O(13)H(14)H(15) molecules of the cubane-like core, and doubly (d-type) coordinated O(46)H(47)H(48) molecule on a bridged wing of the 8,0,8W16 cluster.
Figure 8.
Similar to Figure 7, for representative quadruply (q-type) coordinated O(1)H(2)H(3) and O(13)H(14)H(15) molecules of the cubane-like core, and doubly (d-type) coordinated O(46)H(47)H(48) molecule on a bridged wing of the 8,0,8W16 cluster.

Figure 9.
Calculated NRT bond orders bO···O of the 1,4,4W9 windowpane cluster (left panel), showing their excellent BOBL correlation (Pearson χ = −0.973) with optimized RO···O bond lengths (right panel).
Figure 9.
Calculated NRT bond orders bO···O of the 1,4,4W9 windowpane cluster (left panel), showing their excellent BOBL correlation (Pearson χ = −0.973) with optimized RO···O bond lengths (right panel).

Figure 10.
Scatter plot, least-squares regression line, and Pearson correlation coefficient (χ) for bij-Rij BOBL correlations of all (~250) H-bonds in the clusters of Figure 2 and Figure 3.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Total natural charge Qi and q/t/d coordination type for each water monomer (centered on O(i)) of the 4,4,6W16 cluster. (Similar tables are found in SI for each q,t,dWn cluster of the present work).
Table 1.
Total natural charge Qi and q/t/d coordination type for each water monomer (centered on O(i)) of the 4,4,6W16 cluster. (Similar tables are found in SI for each q,t,dWn cluster of the present work).
| Cluster | Oi | Qi | q/t/d |
|---|---|---|---|
| 4,4,6W14 | 1 | −0.00662 | td |
| 4 | −0.00324 | d | |
| 7 | +0.00963 | ta | |
| 10 | −0.00636 | q | |
| 13 | −0.00107 | q | |
| 16 | −0.00414 | q | |
| 19 | +0.00264 | q | |
| 22 | +0.01427 | ta | |
| 25 | +0.00079 | d | |
| 28 | −0.00357 | d | |
| 31 | +0.00465 | d | |
| 34 | −0.00250 | d | |
| 37 | +0.00087 | d | |
| 40 | −0.00536 | td |
Table 2.
NRT bond orders bij and bond lengths Rij (Å) for all O(i)···O(j) H-bonds of the 4,4,6W14 cluster (with atom numberings as shown in Figure 3). (See SI for similar tables for all clusters of the present work).
Table 2.
NRT bond orders bij and bond lengths Rij (Å) for all O(i)···O(j) H-bonds of the 4,4,6W14 cluster (with atom numberings as shown in Figure 3). (See SI for similar tables for all clusters of the present work).
| 4,4,6W14 | i | j | bij | Rij |
|---|---|---|---|---|
| 1 | 4 | 0.1223 | 2.6972 | |
| 1 | 10 | 0.0638 | 2.8562 | |
| 1 | 22 | 0.0495 | 2.9173 | |
| 4 | 7 | 0.1301 | 2.7127 | |
| 7 | 10 | 0.0655 | 2.8861 | |
| 7 | 40 | 0.0490 | 2.9332 | |
| 10 | 13 | 0.1148 | 2.7789 | |
| 13 | 16 | 0.0923 | 2.7808 | |
| 13 | 40 | 0.1148 | 2.6940 | |
| 16 | 19 | 0.0937 | 2.8005 | |
| 16 | 25 | 0.0901 | 2.7772 | |
| 16 | 34 | 0.0923 | 2.7725 | |
| 19 | 28 | 0.0997 | 2.7415 | |
| 19 | 31 | 0.0984 | 2.7440 | |
| 22 | 37 | 0.0666 | 2.8167 | |
| 25 | 28 | 0.0839 | 2.7599 | |
| 31 | 34 | 0.0900 | 2.7594 | |
| 37 | 40 | 0.0616 | 2.8367 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Weinhold, F. High-Density “Windowpane” Coordination Patterns of Water Clusters and Their NBO/NRT Characterization. Molecules 2022, 27, 4218. https://doi.org/10.3390/molecules27134218
AMA Style
Weinhold F. High-Density “Windowpane” Coordination Patterns of Water Clusters and Their NBO/NRT Characterization. Molecules. 2022; 27(13):4218. https://doi.org/10.3390/molecules27134218
Chicago/Turabian StyleWeinhold, Frank. 2022. "High-Density “Windowpane” Coordination Patterns of Water Clusters and Their NBO/NRT Characterization" Molecules 27, no. 13: 4218. https://doi.org/10.3390/molecules27134218