
Identification of Putative Vaccine and Drug Targets against the Methicillin-Resistant Staphylococcus aureus by Reverse Vaccinology and Subtractive Genomics Approaches

 ,
, 
 , and
, and
Abstract
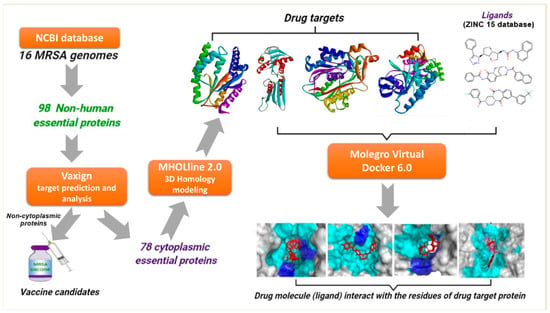
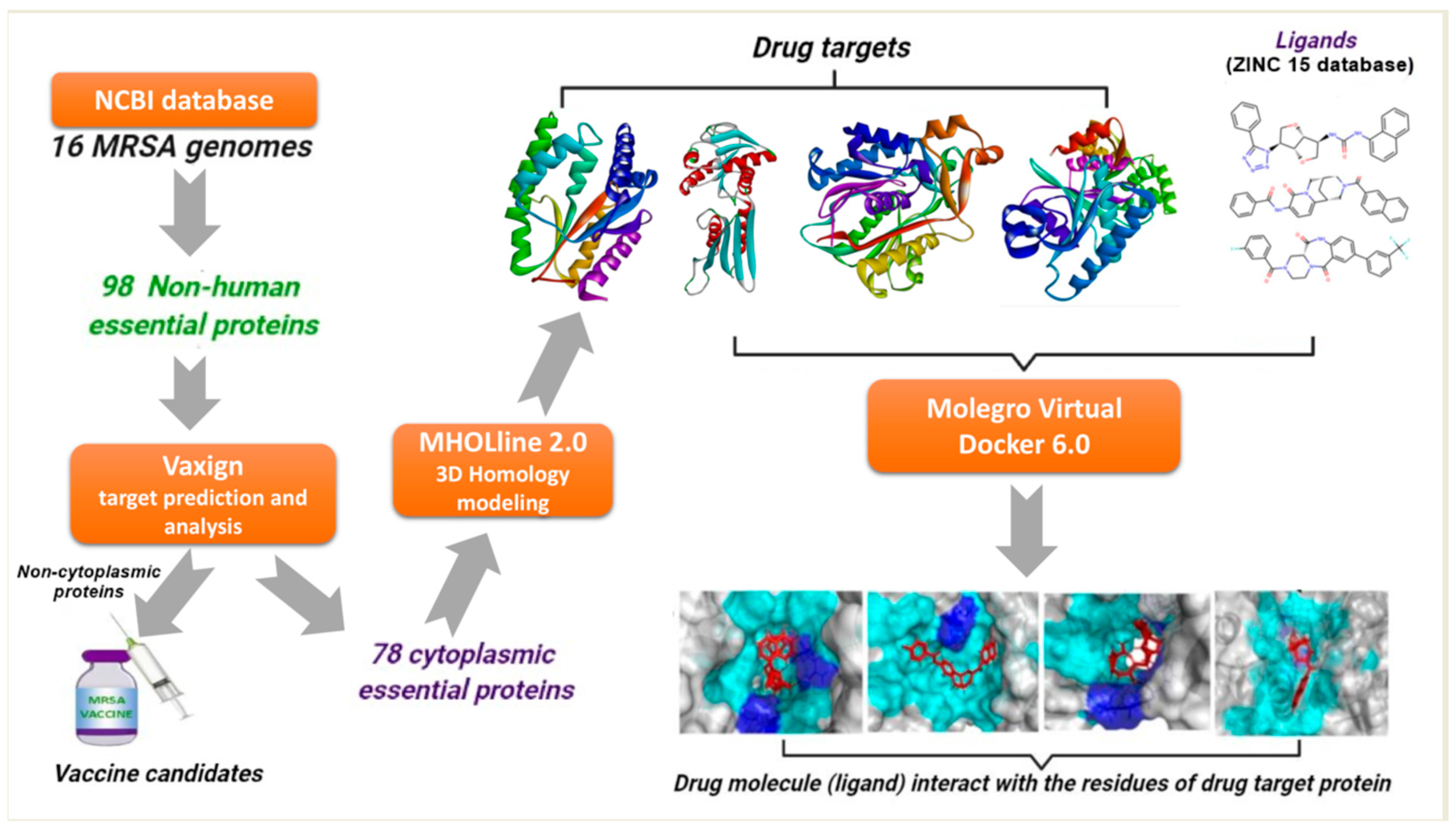
:
1. Introduction
2. Results and Discussion
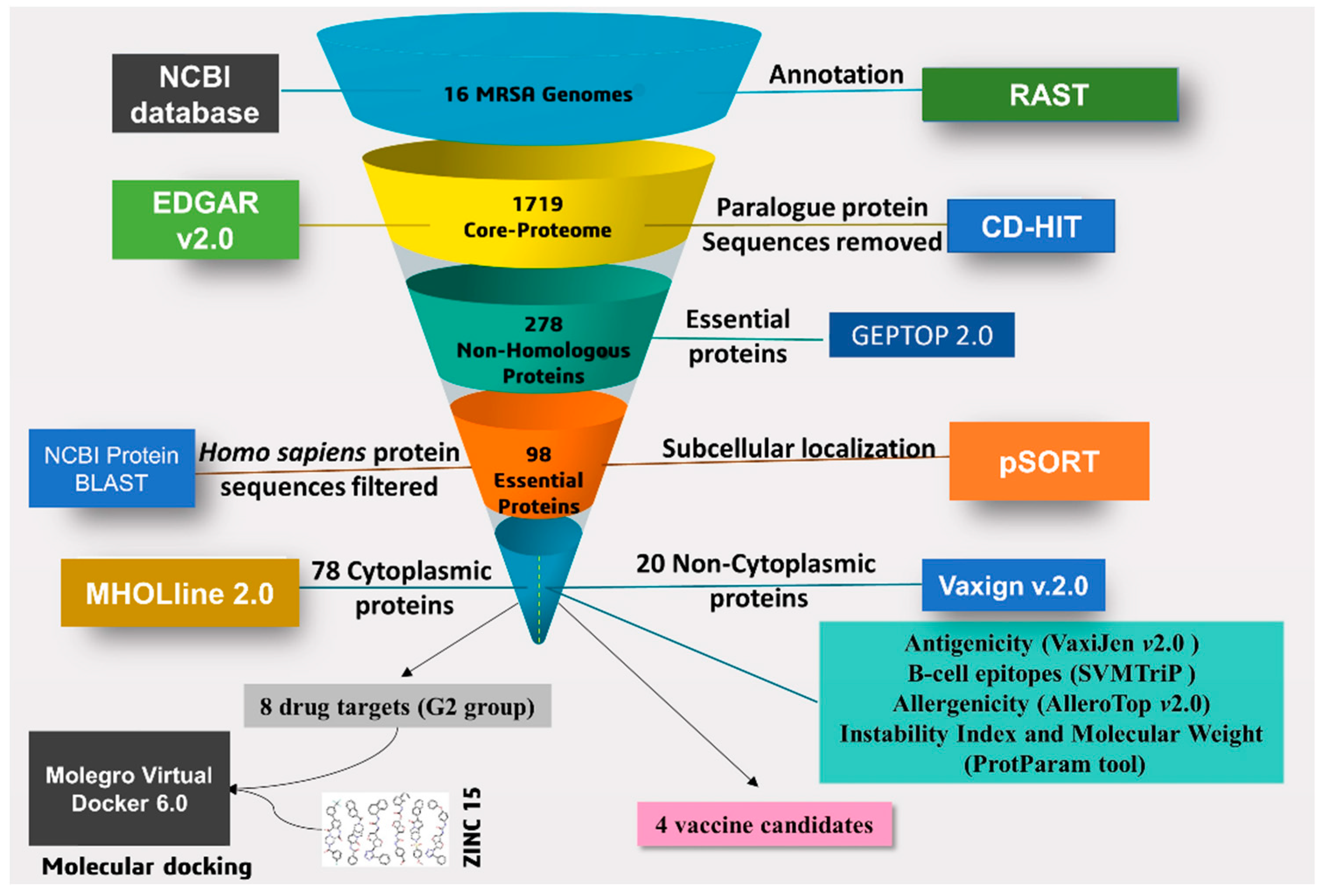
2.1. Prediction of Core-Genome
2.2. Identification of Essential Proteins and Non-Homologous Proteins
2.3. Characterization and Prediction of Subcellular Location of Proteins
2.4. Potential Vaccine Target Candidates
2.5. High-Throughput Structural Modelling and Druggability Analysis
2.6. Virtual Screening and Molecular Docking Analysis
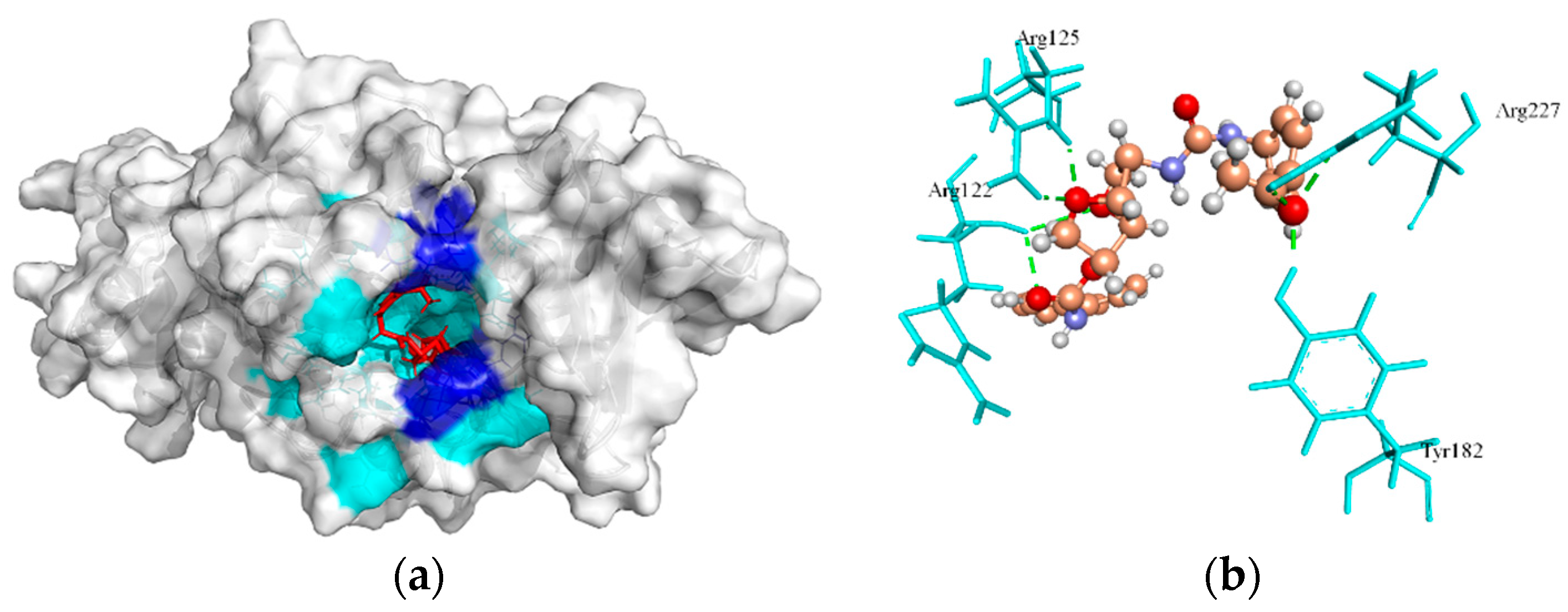
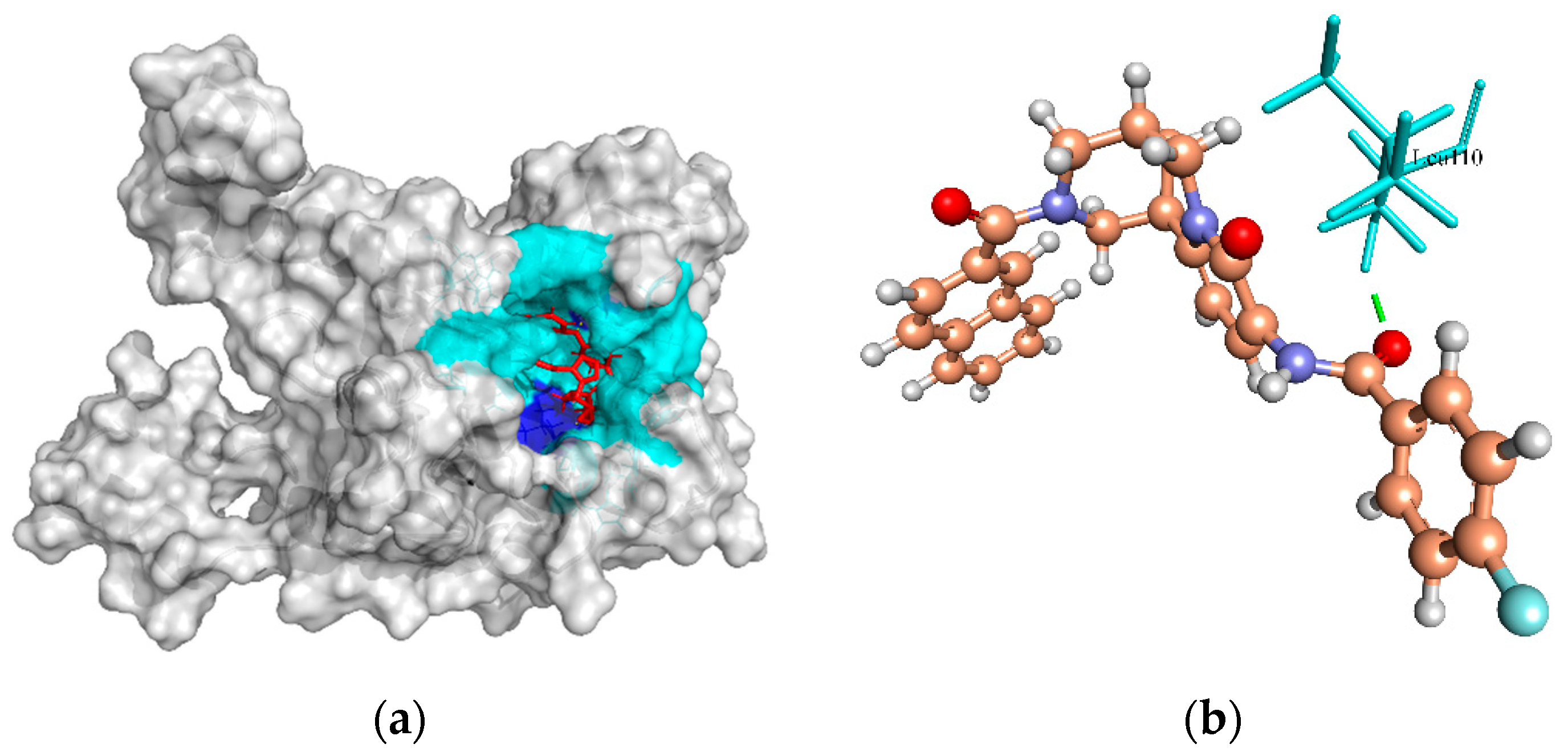
2.6.1. Biotin Protein Ligase
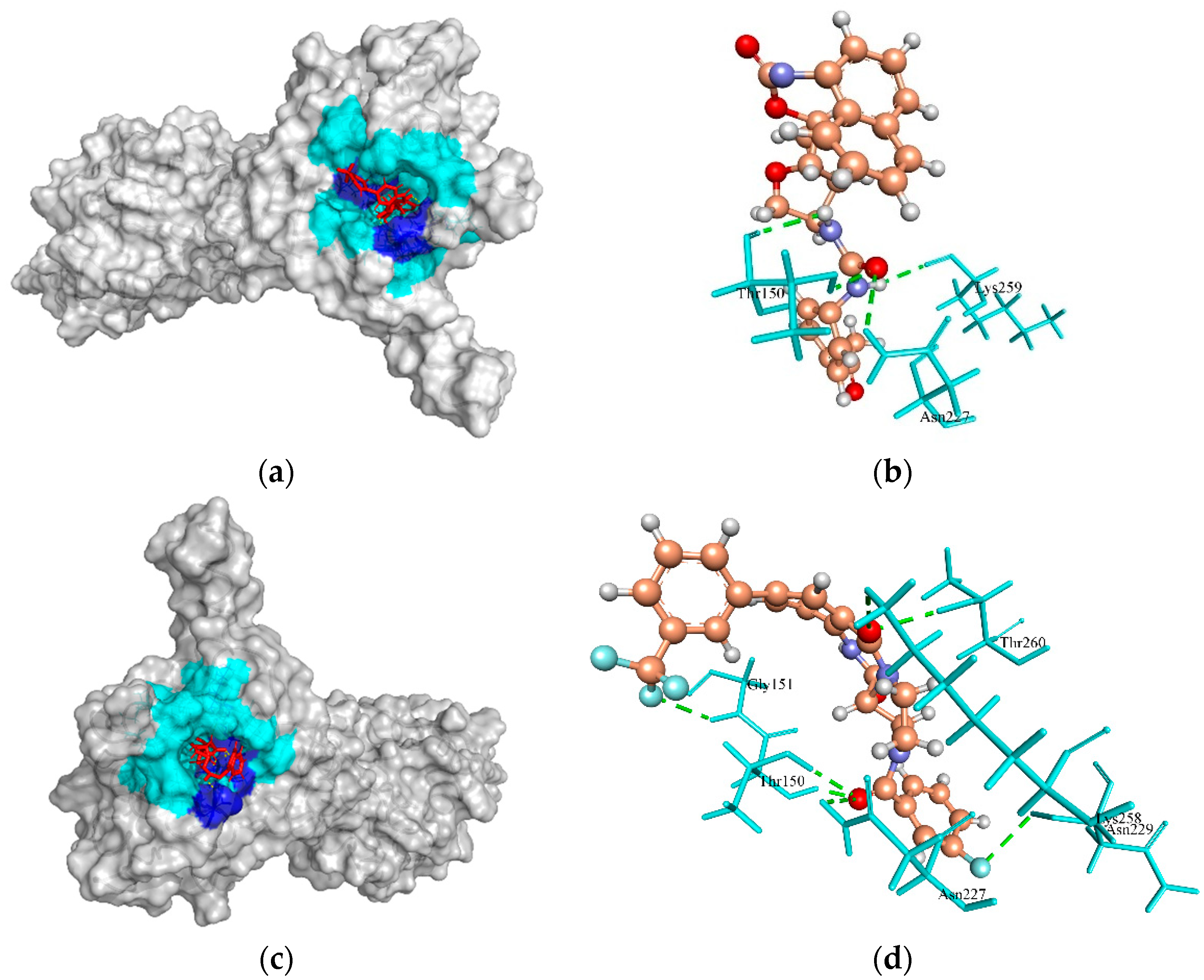
2.6.2. HPr Kinase/Phosphorylase
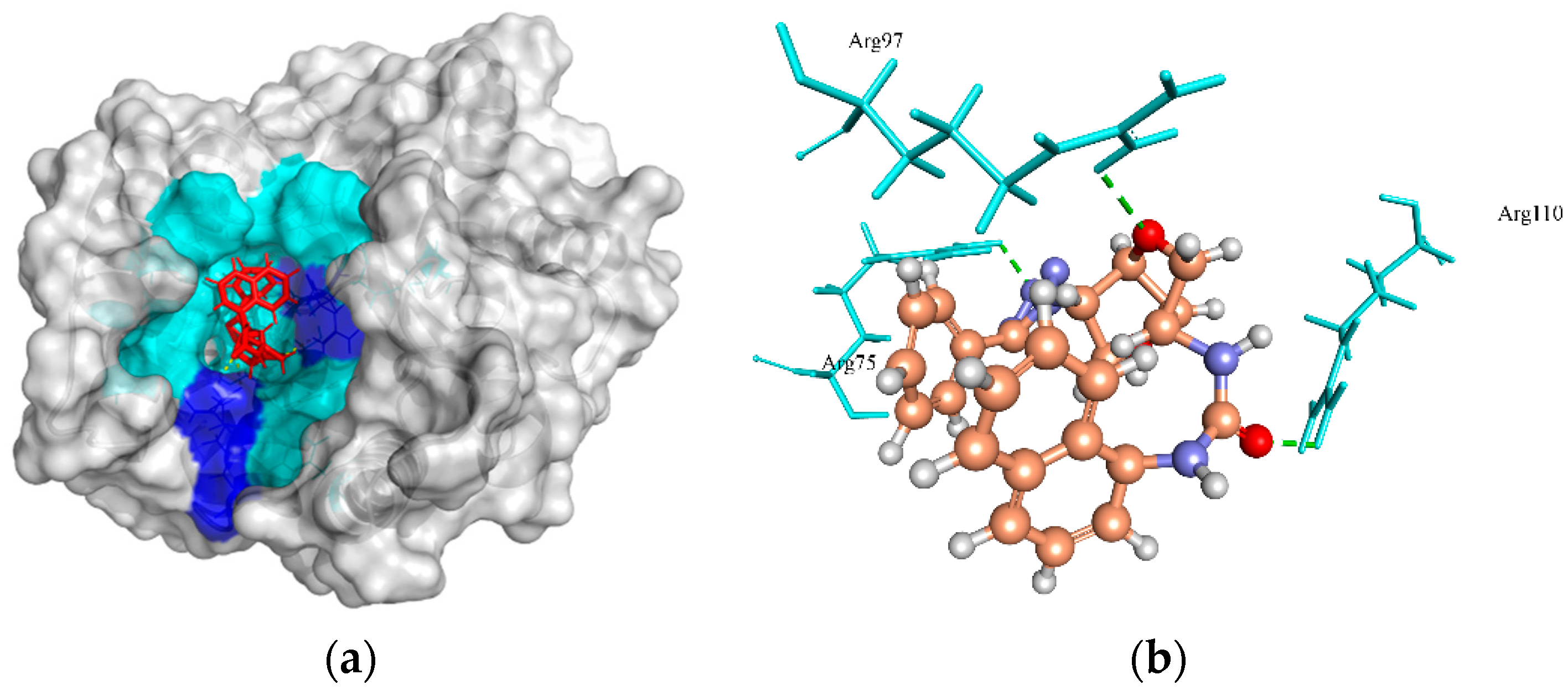
2.6.3. Thymidylate Kinase (TMK)
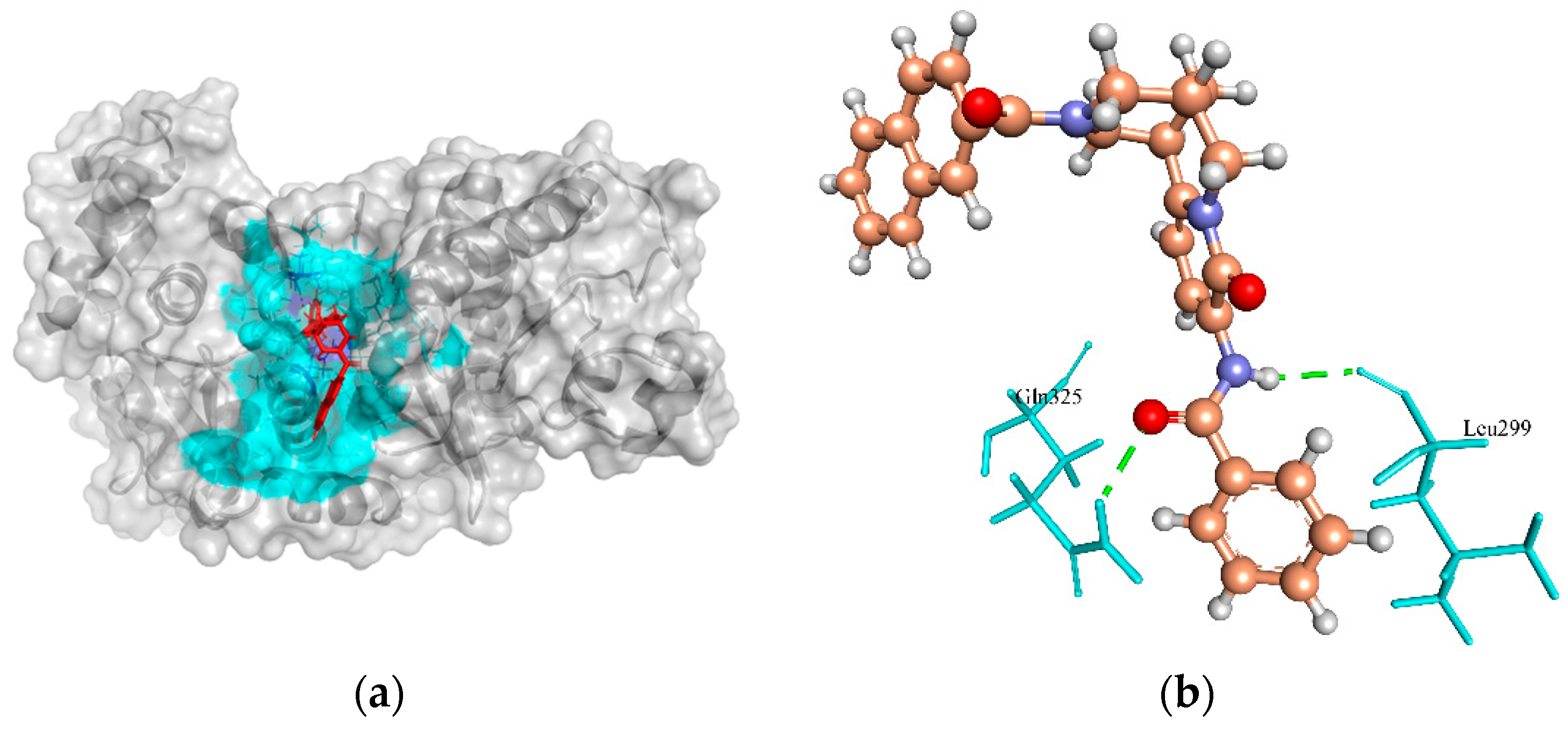
2.6.4. Phosphate Acetyltransferase (Pta)
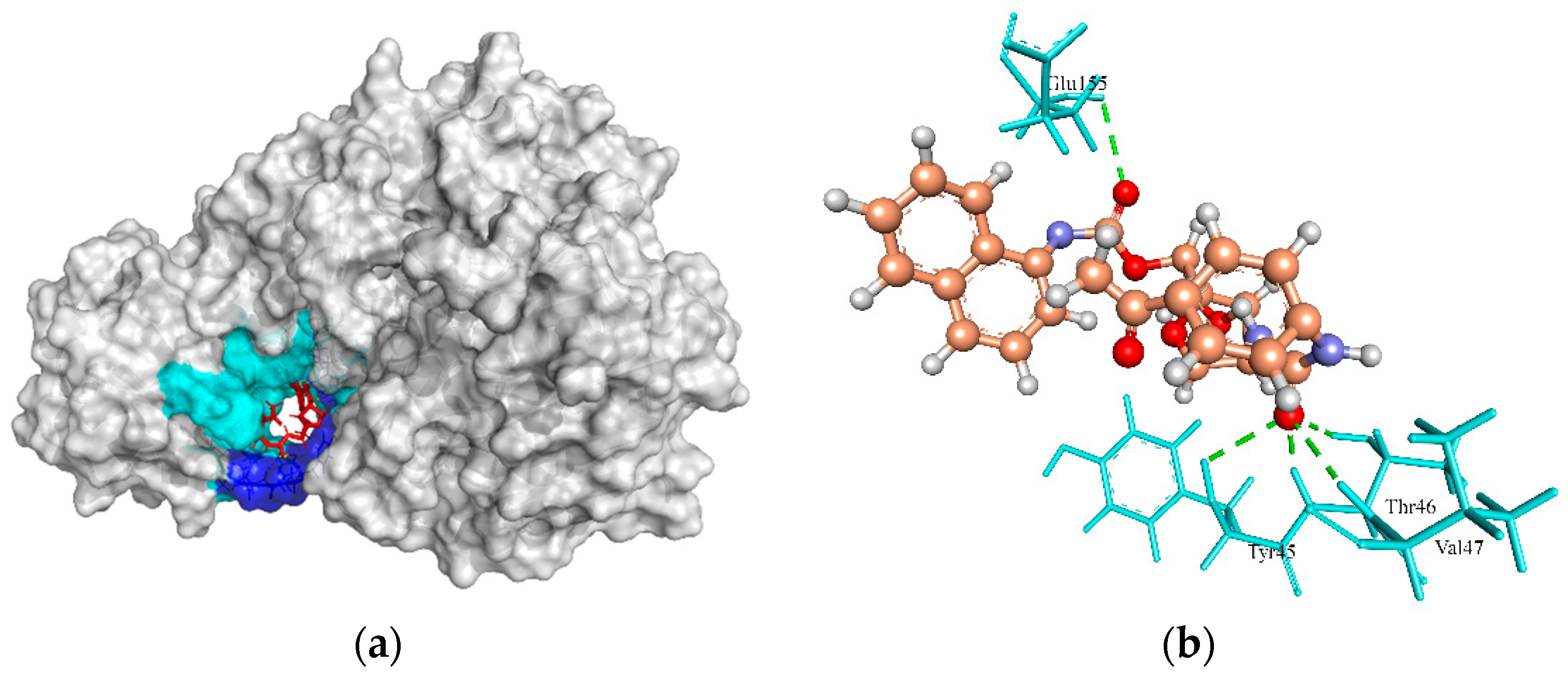
2.6.5. UDP-N-Acetylmuramoyl Alanyl-D-Glutamate-2,6-diaminopimelate Ligase (MurE)
2.6.6. UTP-Glucose-1-Phosphate Uridylyltransferase (UGPase)
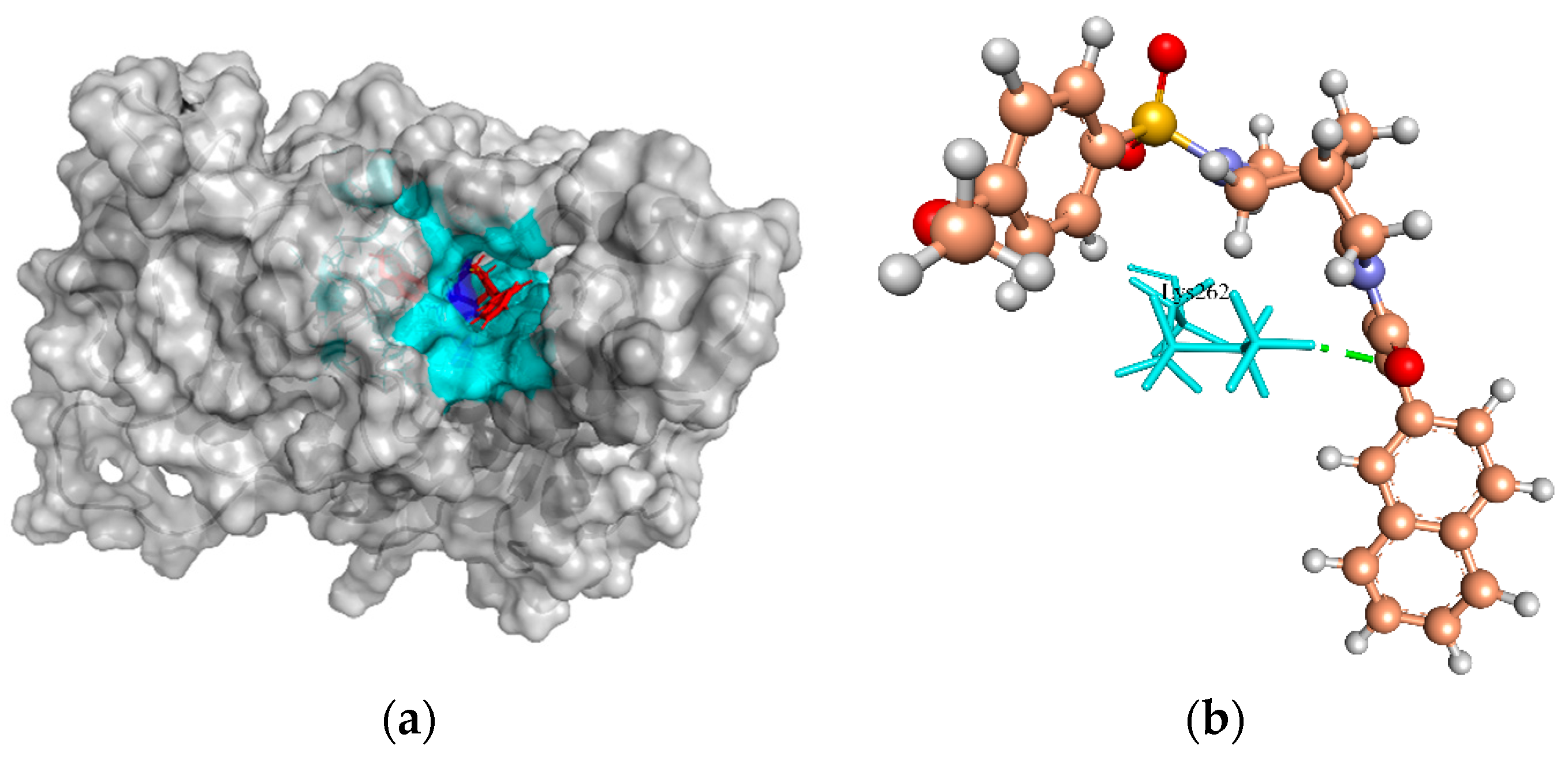
2.6.7. Putative Fatty Acid Synthesis Protein (PlsX)
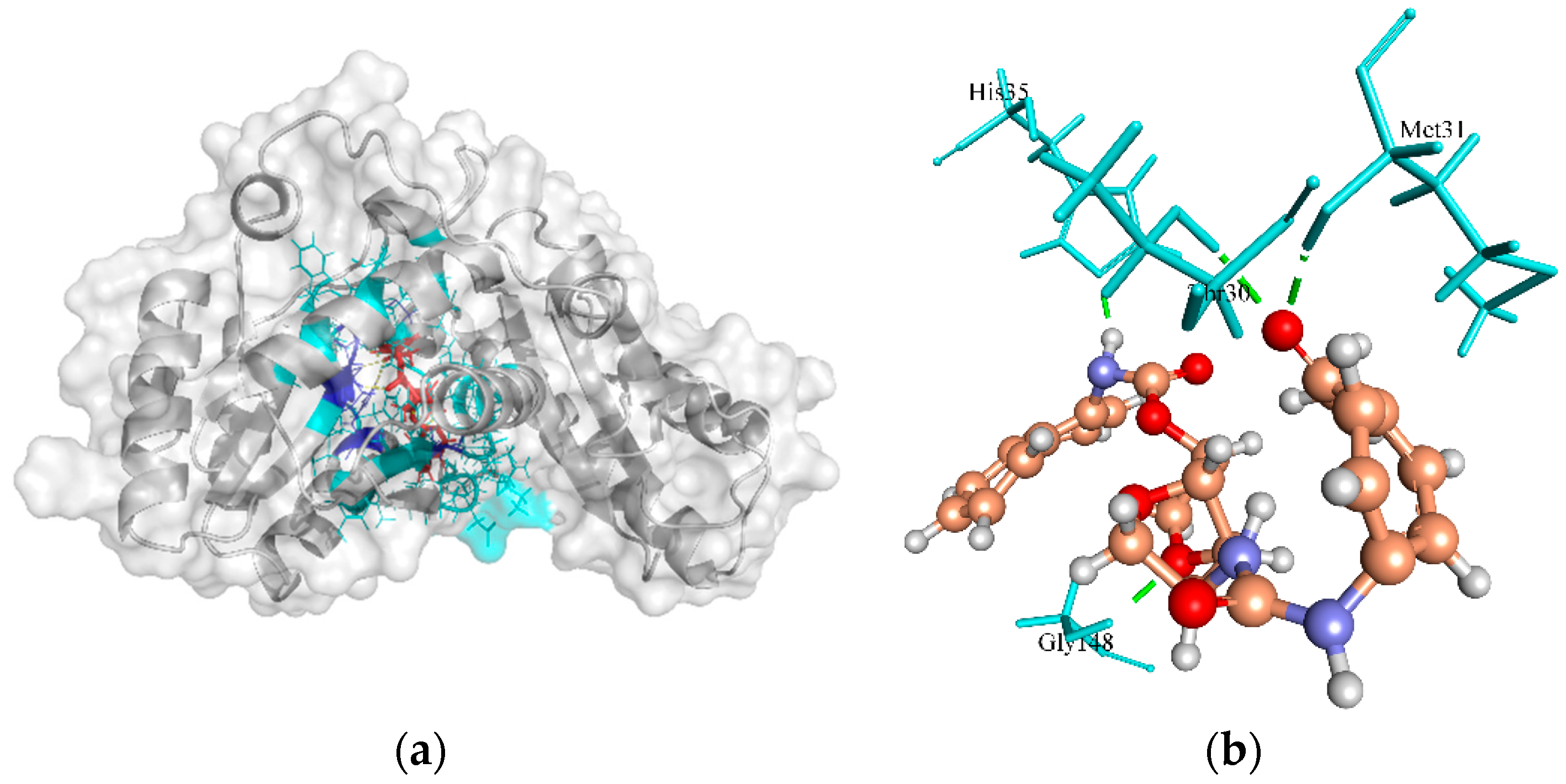
2.6.8. Pantoate Beta-Alanine Ligase (PanC)
3. Materials and Methods
3.1. Genome Sequences
3.2. Prediction of Core Proteome
3.3. Identification of Essential Non-Homologous Proteins
3.4. Characterization and Prediction of Subcellular Localization
3.5. Reverse Vaccinology Approach for Prediction of Putative Vaccine Candidates
3.6. High-Throughput Structural Modelling
3.7. Druggability Analysis of Drug Targets
3.8. Ligand Libraries and Docking Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chan, C.X.; Beiko, R.G.; Ragan, M.A. Lateral Transfer of Genes and Gene Fragments in Staphylococcus Extends beyond Mobile Elements. J. Bacteriol. 2011, 193, 3964–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Chen, C.-J.; Su, L.-H.; Hu, S.; Yu, J.; Chiu, C.-H. Evolution and Pathogenesis of Staphylococcus aureus: Lessons Learned from Genotyping and Comparative Genomics. FEMS Microbiol. Rev. 2008, 32, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenover, F.C.; Tickler, I.A.; Le, V.M.; Dewell, S.; Mendes, R.E.; Goering, R.V.; for the MRSA Consortium. Updating Molecular Diagnostics for Detecting Methicillin-Susceptible and Methicillin-Resistant Staphylococcus aureus Isolates in Blood Culture Bottles. J. Clin. Microbiol. 2019, 57, e01195-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, R.J.; Lowy, F.D. Pathogenesis of Methicillin-Resistant Staphylococcus aureus Infection. Clin. Infect. Dis. 2008, 46, S350–S359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G. Methicillin-Resistant Staphylococcus aureus: An Overview of Basic and Clinical Research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Denis, O. Route of Transmission of Staphylococcus aureus. Lancet Infect. Dis. 2017, 17, 124–125. [Google Scholar] [CrossRef] [Green Version]
- Rossolini, G.M.; Arena, F.; Pecile, P.; Pollini, S. Update on the Antibiotic Resistance Crisis. Curr. Opin. Pharmacol. 2014, 18, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Köck, R.; Becker, K.; Cookson, B.; van Gemert-Pijnen, J.E.; Harbarth, S.; Kluytmans, J.; Mielke, M.; Peters, G.; Skov, R.L.; Struelens, M.J.; et al. Methicillin-Resistant Staphylococcus aureus (MRSA): Burden of Disease and Control Challenges in Europe. Eurosurveillance 2010, 15, 19688. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.C. Livestock-Associated Staphylococcus aureus: The United States Experience. PLoS Pathog. 2015, 11, e1004564. [Google Scholar] [CrossRef]
- Adams, C.P.; Brantner, V.V. Estimating The Cost Of New Drug Development: Is It Really $802 Million? Health Aff. Millwood 2006, 25, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Kola, I.; Landis, J. Can the Pharmaceutical Industry Reduce Attrition Rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Perumal, D.; Lim, C.S.; Sakharkar, K.R.; Sakharkar, M.K. Differential Genome Analyses of Metabolic Enzymes in Pseudomonas aeruginosa for Drug Target Identification. In Silico Biol. 2007, 7, 453–465. [Google Scholar] [PubMed]
- Rappuoli, R. Reverse Vaccinology, a Genome-Based Approach to Vaccine Development. Vaccine 2001, 19, 2688–2691. [Google Scholar] [CrossRef]
- Kumar, H.; Frischknecht, F.; Mair, G.R.; Gomes, J. In Silico Identification of Genetically Attenuated Vaccine Candidate Genes for Plasmodium Liver Stage. Infect. Genet. Evol. 2015, 36, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Muzzi, A.; Masignani, V.; Rappuoli, R. The Pan-Genome: Towards a Knowledge-Based Discovery of Novel Targets for Vaccines and Antibacterials. Drug Discov. Today 2007, 12, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.S.; Tiwari, S.; Guimarães, L.C.; Jamal, S.B.; Folador, E.; Sharma, N.B.; de Castro Soares, S.; Almeida, S.; Ali, A.; Islam, A.; et al. Proteome Scale Comparative Modeling for Conserved Drug and Vaccine Targets Identification in Corynebacterium Pseudotuberculosis. BMC Genom. 2014, 15, S3. [Google Scholar] [CrossRef] [Green Version]
- Rappuoli, R. Reverse Vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Kumar, V.; Kancharla, S.; Kolli, P.; Jena, M. Reverse Vaccinology Approach towards the In-Silico Multiepitope Vaccine Development against SARS-CoV-2. F1000Research 2021, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Hoque, M.N.; Islam, M.R.; Akter, S.; Alam, A.S.M.R.U.; Siddique, M.A.; Saha, O.; Rahaman, M.M.; Sultana, M.; Crandall, K.A.; et al. Epitope-Based Chimeric Peptide Vaccine Design against S, M and E Proteins of SARS-CoV-2, the Etiologic Agent of COVID-19 Pandemic: An In Silico Approach. PeerJ 2020, 8, e9572. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Sarkar, B.; Islam, S.S. Exploiting the Reverse Vaccinology Approach to Design Novel Subunit Vaccines against Ebola Virus. Immunobiology 2020, 225, 151949. [Google Scholar] [CrossRef] [PubMed]
- Asif, S.M.; Asad, A.; Faizan, A.; Anjali, M.S.; Arvind, A.; Neelesh, K.; Hirdesh, K.; Sanjay, K. Dataset of Potential Targets for Mycobacterium Tuberculosis H37Rv through Comparative Genome Analysis. Bioinformation 2009, 4, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, A.; Singh, S.K.; Ghosh, P.; Mukherjee, R.; Mitter, S.; Bandyopadhyay, D. In Silico Identification of Potential Therapeutic Targets in the Human Pathogen Helicobacter pylori. In Silico Biol. 2006, 6, 43–47. [Google Scholar] [PubMed]
- Rathi, B.; Sarangi, A.N.; Trivedi, N. Genome Subtraction for Novel Target Definition in Salmonella typhi. Bioinformation 2009, 4, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serruto, D.; Bottomley, M.J.; Ram, S.; Giuliani, M.M.; Rappuoli, R. The New Multicomponent Vaccine against Meningococcal Serogroup B, 4CMenB: Immunological, Functional and Structural Characterization of the Antigens. Vaccine 2012, 30, B87–B97. [Google Scholar] [CrossRef] [Green Version]
- Maione, D.; Margarit, I.; Rinaudo, C.D.; Masignani, V.; Mora, M.; Scarselli, M.; Tettelin, H.; Brettoni, C.; Iacobini, E.T.; Rosini, R.; et al. Identification of a Universal Group B Streptococcus Vaccine by Multiple Genome Screen. Science 2005, 309, 148–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, V.; Tiwari, V. Subtractive Proteomics to Identify Novel Drug Targets and Reverse Vaccinology for the Development of Chimeric Vaccine against Acinetobacter baumannii. Sci. Rep. 2018, 8, 9044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, V.; Tiwari, M.; Tiwari, V. Prioritization of Potential Vaccine Targets Using Comparative Proteomics and Designing of the Chimeric Multi-Epitope Vaccine against Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 5240. [Google Scholar] [CrossRef]
- Amineni, U.; Pradhan, D.; Marisetty, H. In Silico Identification of Common Putative Drug Targets in Leptospira interrogans. J. Chem. Biol. 2010, 3, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalida, S.; Nadia, R.; Mohammad, S. Subtractive Genome Analysis for in Silico Identification and Characterization of Novel Drug Targets in C. trachomatis Strain d/Uw-3/Cx. Int. J. Curr. Res. 2012, 4, 017–021. [Google Scholar]
- Reddy, K.G.; Rao, N.K.; Rama, K.; Aravind, S. In Silico Identification of Potential Therapeutic Targets in Clostridium botulinum by the Approach Subtractive Genomics. Int. J. Pharm. Stud. Res. 2010, 2, 12. [Google Scholar]
- Gupta, E.; Mishra, R.K.; Niraj, R.R.K. Identification of Potential Vaccine Candidates against SARS-CoV-2, A Step Forward to Fight COVID-19: A Reverse Vaccinology Approach. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shahid, F.; Ashfaq, U.A.; Saeed, S.; Munir, S.; Almatroudi, A.; Khurshid, M. In Silico Subtractive Proteomics Approach for Identification of Potential Drug Targets in Staphylococcus saprophyticus. Int. J. Environ. Res. Public. Health 2020, 17, 3644. [Google Scholar] [CrossRef]
- Zhang, Z.; Ren, Q. Why Are Essential Genes Essential?—The Essentiality of Saccharomyces Genes. Microb. Cell 2015, 2, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barh, D.; Tiwari, S.; Jain, N.; Ali, A.; Santos, A.R.; Misra, A.N.; Azevedo, V.; Kumar, A. In Silico Subtractive Genomics for Target Identification in Human Bacterial Pathogens. Drug Dev. Res. 2011, 72, 162–177. [Google Scholar] [CrossRef]
- Uddin, R.; Sufian, M. Core Proteomic Analysis of Unique Metabolic Pathways of Salmonella enterica for the Identification of Potential Drug Targets. PLoS ONE 2016, 11, e0146796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, R.; Saeed, K. Identification and Characterization of Potential Drug Targets by Subtractive Genome Analyses of Methicillin Resistant Staphylococcus aureus. Comput. Biol. Chem. 2014, 48, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Goncearenco, A.; Li, M.; Simonetti, F.L.; Shoemaker, B.A.; Panchenko, A.R. Exploring Protein-Protein Interactions as Drug Targets for Anti-Cancer Therapy with In Silico Workflows. In Proteomics for Drug Discovery: Methods and Protocols; Methods in Molecular Biology; Lazar, I.M., Kontoyianni, M., Lazar, A.C., Eds.; Springer: New York, NY, USA, 2017; pp. 221–236. ISBN 978-1-4939-7201-2. [Google Scholar]
- Uddin, R.; Siddiqui, Q.N.; Sufian, M.; Azam, S.S.; Wadood, A. Proteome-Wide Subtractive Approach to Prioritize a Hypothetical Protein of XDR-Mycobacterium Tuberculosis as Potential Drug Target. Genes Genom. 2019, 41, 1281–1292. [Google Scholar] [CrossRef]
- Goyal, M.; Citu, C.; Singh, N. In Silico Identification of Novel Drug Targets in Acinetobacter baumannii by Subtractive Genomic Approach. Asian J. Pharm. Clin. Res. 2018, 11, 230. [Google Scholar] [CrossRef] [Green Version]
- Duffield, M.; Cooper, I.; McAlister, E.; Bayliss, M.; Ford, D.; Oyston, P. Predicting Conserved Essential Genes in Bacteria: In Silico Identification of Putative Drug Targets. Mol. Biosyst. 2010, 6, 2482–2489. [Google Scholar] [CrossRef]
- Mondal, S.I.; Ferdous, S.; Jewel, N.A.; Akter, A.; Mahmud, Z.; Islam, M.M.; Afrin, T.; Karim, N. Identification of Potential Drug Targets by Subtractive Genome Analysis of Escherichia coli O157:H7: An In Silico Approach. Adv. Appl. Bioinforma. Chem. 2015, 8, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdeva, G.; Kumar, K.; Jain, P.; Ramachandran, S. SPAAN: A Software Program for Prediction of Adhesins and Adhesin-like Proteins Using Neural Networks. Bioinformatics 2005, 21, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Vivona, S.; Bernante, F.; Filippini, F. NERVE: New Enhanced Reverse Vaccinology Environment. BMC Biotechnol. 2006, 6, 35. [Google Scholar] [CrossRef]
- Noori Goodarzi, N.; Bolourchi, N.; Fereshteh, S.; Soltani Shirazi, A.; Pourmand, M.R.; Badmasti, F. Investigation of Novel Putative Immunogenic Targets against Staphylococcus aureus Using a Reverse Vaccinology Strategy. Infect. Genet. Evol. 2021, 96, 105149. [Google Scholar] [CrossRef] [PubMed]
- Ünal, C.M.; Steinert, M. Microbial Peptidyl-Prolyl Cis/Trans Isomerases (PPIases): Virulence Factors and Potential Alternative Drug Targets. Microbiol. Mol. Biol. Rev. 2014, 78, 544–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, R.P.; Koch, J.R.; Burmann, B.M.; Schmidpeter, P.A.M.; Hunkeler, M.; Hiller, S.; Schmid, F.X.; Maier, T. Dimeric Structure of the Bacterial Extracellular Foldase PrsA. J. Biol. Chem. 2015, 290, 3278–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Yang, Y.; Zhou, J.; Liu, H.; Liao, X.; Luo, J.; Li, X.; Fang, W. Peptidyl Isomerase PrsA Is Surface-Associated on Streptococcus suis and Offers Cross-Protection against Serotype 9 Strain. FEMS Microbiol. Lett. 2019, 366, fnz002. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, B.; Shah, P.; Ramkumar, M.; Allen, E.B.; Swiatlo, E.; Burgess, S.C.; Lawrence, M.L. Quantitative Analysis of Streptococcus pneumoniae TIGR4 Response to in Vitro Iron Restriction by 2-D LC ESI MS/MS. Proteomics 2008, 8, 2104–2114. [Google Scholar] [CrossRef]
- Henningham, A.; Chiarot, E.; Gillen, C.M.; Cole, J.N.; Rohde, M.; Fulde, M.; Ramachandran, V.; Cork, A.J.; Hartas, J.; Magor, G.; et al. Conserved Anchorless Surface Proteins as Group A Streptococcal Vaccine Candidates. J. Mol. Med. 2012, 90, 1197–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Kitten, T.; Munro, C.L.; Conrad, D.H.; Xu, P. Pooled Protein Immunization for Identification of Cell Surface Antigens in Streptococcus sanguinis. PLoS ONE 2010, 5, e11666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.V.; Almonacid Mendoza, H.L.; Jackson, A.C.; Hung, M.-C.; Bielecka, M.K.; Heckels, J.E.; Christodoulides, M. Vaccine Potential of Bacterial Macrophage Infectivity Potentiator (MIP)-like Peptidyl Prolyl Cis/Trans Isomerase (PPIase) Proteins. Expert Rev. Vaccines 2015, 14, 1633–1649. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Du, H.; Zhang, H.; Shen, H.; Yan, R.; He, Y.; Wang, M.; Zhu, X. EsxA Might as a Virulence Factor Induce Antibodies in Patients with Staphylococcus aureus Infection. Braz. J. Microbiol. 2013, 44, 267–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gröschel, M.I.; Sayes, F.; Simeone, R.; Majlessi, L.; Brosch, R. ESX Secretion Systems: Mycobacterial Evolution to Counter Host Immunity. Nat. Rev. Microbiol. 2016, 14, 677–691. [Google Scholar] [CrossRef]
- Ma, Y.; Hao, L.; Liang, Z.; Ma, J.; Ke, H.; Kang, H.; Yang, H.; Wu, J.; Feng, G.; Liu, Z. Characterization of Novel Antigenic Vaccine Candidates for Nile Tilapia (Oreochromis niloticus) against Streptococcus agalactiae Infection. Fish Shellfish Immunol. 2020, 105, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Zarantonelli, M.L.; Antignac, A.; Lancellotti, M.; Guiyoule, A.; Alonso, J.-M.; Taha, M.-K. Immunogenicity of Meningococcal PBP2 during Natural Infection and Protective Activity of Anti-PBP2 Antibodies against Meningococcal bacteraemia in Mice. J. Antimicrob. Chemother. 2006, 57, 924–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, R.; Saeed, K.; Khan, W.; Azam, S.S.; Wadood, A. Metabolic Pathway Analysis Approach: Identification of Novel Therapeutic Target against Methicillin Resistant Staphylococcus aureus. Gene 2015, 556, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Das, A.K. Integrated Multi-Omics, Virtual Screening and Molecular Docking Analysis of Methicillin-Resistant Staphylococcus aureus USA300 for the Identification of Potential Therapeutic Targets: An In-Silico Approach. Int. J. Pept. Res. Ther. 2021, 27, 2735–2755. [Google Scholar] [CrossRef] [PubMed]
- Monterrubio-López, G.P.; González-Y-Merchand, J.A.; Ribas-Aparicio, R.M. Identification of Novel Potential Vaccine Candidates against Tuberculosis Based on Reverse Vaccinology. BioMed Res. Int. 2015, 2015, e483150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Aricò, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B.; et al. Identification of Vaccine Candidates Against Serogroup B Meningococcus by Whole-Genome Sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing Multi-Epitope Vaccine against Staphylococcus aureus by Employing Subtractive Proteomics, Reverse Vaccinology and Immuno-Informatics Approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef] [PubMed]
- Soltan, M.A.; Magdy, D.; Solyman, S.M.; Hanora, A. Design of Staphylococcus aureus New Vaccine Candidates with B and T Cell Epitope Mapping, Reverse Vaccinology, and Immunoinformatics. OMICS J. Integr. Biol. 2020, 24, 195–204. [Google Scholar] [CrossRef]
- Kim, J.; Gambhir, V.; Alatery, A.; Basta, S. Delivery of Exogenous Antigens to Induce Cytotoxic CD8+ T Lymphocyte Responses. J. Biomed. Biotechnol. 2010, 2010, e218752. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, V.; Tiwari, M.; Tiwari, V. Subtractive Proteomic Analysis of Antigenic Extracellular Proteins and Design a Multi-Epitope Vaccine against Staphylococcus aureus. Microbiol. Immunol. 2021, 65, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Abadio, A.K.R.; Kioshima, E.S.; Teixeira, M.M.; Martins, N.F.; Maigret, B.; Felipe, M.S.S. Comparative Genomics Allowed the Identification of Drug Targets against Human Fungal Pathogens. BMC Genom. 2011, 12, 75. [Google Scholar] [CrossRef] [Green Version]
- Abi Hussein, H.; Geneix, C.; Petitjean, M.; Borrel, A.; Flatters, D.; Camproux, A.-C. Global Vision of Druggability Issues: Applications and Perspectives. Drug Discov. Today 2017, 22, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Bitencourt-Ferreira, G.; de Azevedo, W.F. Molegro Virtual Docker for Docking. In Docking Screens for Drug Discovery; Methods in Molecular Biology; de Azevedo, W.F., Jr., Ed.; Springer: New York, NY, USA, 2019; pp. 149–167. ISBN 978-1-4939-9752-7. [Google Scholar]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Fu, H.; Fryar, K.L.; Landua, J.; Trevino, S.R.; Shirley, B.A.; Hendricks, M.M.; Iimura, S.; Gajiwala, K.; Scholtz, J.M.; et al. Contribution of Hydrophobic Interactions to Protein Stability. J. Mol. Biol. 2011, 408, 514–528. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares da Costa, T.P.; Tieu, W.; Yap, M.Y.; Zvarec, O.; Bell, J.M.; Turnidge, J.D.; Wallace, J.C.; Booker, G.W.; Wilce, M.C.J.; Abell, A.D.; et al. Biotin Analogues with Antibacterial Activity Are Potent Inhibitors of Biotin Protein Ligase. ACS Med. Chem. Lett. 2012, 3, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Paparella, A.S.; Booker, G.W.; Polyak, S.W.; Abell, A.D. Biotin Protein Ligase Is a Target for New Antibacterials. Antibiotics 2016, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Paparella, A.S.; da Costa, T.P.S.; Yap, M.Y.; Tieu, W.; Wilce, M.C.J.; Booker, G.W.; Abell, A.D.; Polyak, S.W. Structure Guided Design of Biotin Protein Ligase Inhibitors for Antibiotic Discovery. Curr. Top. Med. Chem. 2014, 14, 4–20. [Google Scholar] [CrossRef]
- Soares da Costa, T.P.; Tieu, W.; Yap, M.Y.; Pendini, N.R.; Polyak, S.W.; Sejer Pedersen, D.; Morona, R.; Turnidge, J.D.; Wallace, J.C.; Wilce, M.C.J.; et al. Selective Inhibition of Biotin Protein Ligase from Staphylococcus aureus. J. Biol. Chem. 2012, 287, 17823–17832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutscher, J.; Herro, R.; Bourand, A.; Mijakovic, I.; Poncet, S. P-Ser-HPr—A Link between Carbon Metabolism and the Virulence of Some Pathogenic Bacteria. Biochim. Biophys. Acta 2005, 1754, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Nessler, S. The Bacterial HPr Kinase/Phosphorylase: A New Type of Ser/Thr Kinase as Antimicrobial Target. Biochim. Biophys. Acta BBA Proteins Proteom. 2005, 1754, 126–131. [Google Scholar] [CrossRef]
- Martínez-Botella, G.; Loch, J.T.; Green, O.M.; Kawatkar, S.P.; Olivier, N.B.; Boriack-Sjodin, P.A.; Keating, T.A. Sulfonylpiperidines as Novel, Antibacterial Inhibitors of Gram-Positive Thymidylate Kinase (TMK). Bioorg. Med. Chem. Lett. 2013, 23, 169–173. [Google Scholar] [CrossRef]
- Keating, T.A.; Newman, J.V.; Olivier, N.B.; Otterson, L.G.; Andrews, B.; Boriack-Sjodin, P.A.; Breen, J.N.; Doig, P.; Dumas, J.; Gangl, E.; et al. In Vivo Validation of Thymidylate Kinase (TMK) with a Rationally Designed, Selective Antibacterial Compound. ACS Chem. Biol. 2012, 7, 1866–1872. [Google Scholar] [CrossRef]
- Morya, V.K.; Dewaker, V.; Kim, E.-K. In Silico Study and Validation of Phosphotransacetylase (PTA) as a Putative Drug Target for Staphylococcus aureus by Homology-Based Modelling and Virtual Screening. Appl. Biochem. Biotechnol. 2012, 168, 1792–1805. [Google Scholar] [CrossRef] [PubMed]
- Campos-Bermudez, V.A.; Bologna, F.P.; Andreo, C.S.; Drincovich, M.F. Functional Dissection of Escherichia coli Phosphotransacetylase Structural Domains and Analysis of Key Compounds Involved in Activity Regulation. FEBS J. 2010, 277, 1957–1966. [Google Scholar] [CrossRef]
- Sakinç, T.; Michalski, N.; Kleine, B.; Gatermann, S.G. The Uropathogenic Species Staphylococcus saprophyticus Tolerates a High Concentration of D-Serine. FEMS Microbiol. Lett. 2009, 299, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Amera, G.M.; Khan, R.J.; Jha, R.K.; Pathak, A.; Muthukumaran, J.; Singh, A.K. Prioritization of Mur Family Drug Targets against A. baumannii and Identification of Their Homologous Proteins through Molecular Phylogeny, Primary Sequence, and Structural Analysis. J. Genet. Eng. Biotechnol. 2020, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.; Flouret, B.; Chantalat, L.; van Heijenoort, J.; Mengin-Lecreulx, D.; Dideberg, O. Crystal Structure of UDP-N-Acetylmuramoyl-l-Alanyl-d-Glutamate:Meso-Diaminopimelate Ligase from Escherichia coli. J. Biol. Chem. 2001, 276, 10999–11006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lyne, P.D.; Giordanetto, F.; Lovell, T.; Li, J. On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors. J. Chem. Inf. Model. 2006, 46, 401–415. [Google Scholar] [CrossRef]
- Boittier, E.D.; Tang, Y.Y.; Buckley, M.E.; Schuurs, Z.P.; Richard, D.J.; Gandhi, N.S. Assessing Molecular Docking Tools to Guide Targeted Drug Discovery of CD38 Inhibitors. Int. J. Mol. Sci. 2020, 21, 5183. [Google Scholar] [CrossRef] [PubMed]
- Berbis, M.A.; Sanchez-Puelles, J.M.; Canada, F.J.; Jimenez-Barbero, J. Structure and Function of Prokaryotic UDP-Glucose Pyrophosphorylase, A Drug Target Candidate. Curr. Med. Chem. 2015, 22, 1687–1697. [Google Scholar] [CrossRef] [Green Version]
- Thoden, J.B.; Holden, H.M. The Molecular Architecture of Glucose-1-Phosphate Uridylyltransferase. Protein Sci. 2007, 16, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Bonofiglio, L.; García, E.; Mollerach, M. Biochemical Characterization of the Pneumococcal Glucose 1-Phosphate Uridylyltransferase (GalU) Essential for Capsule Biosynthesis. Curr. Microbiol. 2005, 51, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Genevaux, P.; Bauda, P.; DuBow, M.S.; Oudega, B. Identification of Tn10 Insertions in the RfaG, RfaP, and GalU Genes Involved in Lipopolysaccharide Core Biosynthesis That Affect Escherichia coli Adhesion. Arch. Microbiol. 1999, 172, 1–8. [Google Scholar] [CrossRef]
- Kim, Y.; Li, H.; Binkowski, T.A.; Holzle, D.; Joachimiak, A. Crystal Structure of Fatty Acid/Phospholipid Synthesis Protein PlsX from Enterococcus faecalis. J. Struct. Funct. Genom. 2009, 10, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-J.; Zhang, Y.-M.; Grimes, K.D.; Qi, J.; Lee, R.E.; Rock, C.O. Acyl-Phosphates Initiate Membrane Phospholipid Synthesis in Gram-Positive Pathogens. Mol. Cell 2006, 23, 765–772. [Google Scholar] [CrossRef]
- Yao, J.; Rock, C.O. Therapeutic Targets in Chlamydial Fatty Acid and Phospholipid Synthesis. Front. Microbiol. 2018, 9, 2291. [Google Scholar] [CrossRef] [PubMed]
- Von Delft, F.; Lewendon, A.; Dhanaraj, V.; Blundell, T.L.; Abell, C.; Smith, A.G. The Crystal Structure of E. coli Pantothenate Synthetase Confirms It as a Member of the Cytidylyltransferase Superfamily. Structure 2001, 9, 439–450. [Google Scholar] [CrossRef]
- Pradhan, S.; Sinha, C. High Throughput Screening against Pantothenate Synthetase Identifies Amide Inhibitors against Mycobacterium Tuberculosis and Staphylococcus aureus. In Silico Pharmacol. 2018, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An Enhanced Software Platform for Comparative Gene Content Analyses. Nucleic Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Wen, Q.-F.; Liu, S.; Dong, C.; Guo, H.-X.; Gao, Y.-Z.; Guo, F.-B. Geptop 2.0: An Updated, More Precise, and Faster Geptop Server for Identification of Prokaryotic Essential Genes. Front. Microbiol. 2019, 10, 1236. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved Protein Subcellular Localization Prediction with Refined Localization Subcategories and Predictive Capabilities for All Prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-S.; Chen, Y.-C.; Lu, C.-H.; Hwang, J.-K. Prediction of Protein Subcellular Localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, e2680160. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Xiang, Z.; Mobley, H.L.T. Vaxign: The First Web-Based Vaccine Design Program for Reverse Vaccinology and Applications for Vaccine Development. J. Biomed. Biotechnol. 2010, 2010, e297505. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A Method to Predict Antigenic Epitopes Using Support Vector Machine to Integrate Tri-Peptide Similarity and Propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-Throughput Prediction of Protein Antigenicity Using Protein Microarray Data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s Conserved Domain Database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T.K. A Novel Genomics Approach for the Identification of Drug Targets in Pathogens, with Special Reference to Pseudomonas aeruginosa. In Silico Biol. 2004, 4, 355–360. [Google Scholar]
- Rossi, A.D.; Oliveira, P.; Siqueira, D.G.; Reis, V.C.C.; Dardenne, L.; Goliatt, P.V.Z.C. MHOLline 2.0: Workflow for Automatic Large-Scale Modeling and Analysis of Proteins. Rev. Mund. Engen Tecnol. Gestão. 2020, 5, 1–14. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A Web Server for Automatic Binding Site Prediction, Analysis and Druggability Assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of Early Drug Discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef] [PubMed]
- Vilela Rodrigues, T.C.; Jaiswal, A.K.; de Sarom, A.; de Castro Oliveira, L.; Freire Oliveira, C.J.; Ghosh, P.; Tiwari, S.; Miranda, F.M.; de Jesus Benevides, L.; Ariston de Carvalho Azevedo, V.; et al. Reverse Vaccinology and Subtractive Genomics Reveal New Therapeutic Targets against Mycoplasma Pneumoniae: A Causative Agent of Pneumonia. R. Soc. Open Sci. 2019, 6, 190907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- PyMOL. Pymol.Org. Available online: https://pymol.org/2/ (accessed on 5 March 2022).
- BIOVIA Discovery Studio—BIOVIA—Dassault Systèmes®. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 5 March 2022).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine Target | Protein ID * | Length (bp) | Mol. Wt. (kDa) | Adherence Score | B-Cell Epitope Peptides | VaxiJen Score | Trans-Membrane Helix | Allergen |
|---|---|---|---|---|---|---|---|---|
| Foldase protein (PrsA) | WP_000782119.1 | 320 | 35.623 | 0.68 | 5 | 0.7662 | 0 | No |
| ESAT-6 machinery protein (EssA) | WP_000928935.1 | 152 | 17.392 | 0.58 | 1 | 0.7034 | 1 | No |
| Penicillin-binding protein 1 (PBP1) | WP_001118663.1 | 744 | 82.738 | 0.53 | 10 | 0.6351 | 1 | No |
| DD-transpeptidase (PBP2) | WP_000138351.1 | 727 | 80.356 | 0.82 | 10 | 0.6846 | 1 | No |
| Target Proteins | Position (x, y, z) | ZINC ID | MolDock Score | H-Bonds/Residues |
|---|---|---|---|---|
| Biotin protein ligase | 53.60, 18.59, 18.89 | ZINC4235426 | −176.846 | 7/Tyr182, Arg227, Arg125, and Arg122 |
| HPr kinase/phosphorylase | −60.93, 127.77, −83.27 | ZINC4235426 | −147.451 | 4/Lys259, Thr150, and Asn227 |
| ZINC4235924 | −137.549 | 7/Gly151, Thr150, Asn227, Lys258, Thr260, and Asn229 | ||
| Thymidylate kinase | 5.82, 10.91, 16.59 | ZINC4259578 | −139.656 | 3/Arg75, Arg97, and Arg110 |
| ZINC4235426 | −139.150 | 6/Arg75, Arg97, Glu106, Tyr105, and Glu42 | ||
| Phosphate acetyltransferase | 17.38, 20.45, 53.06 | ZINC4270981 | −134.847 | 2/Gln325, and Leu299 |
| UDP-N-acetylmuramoyl-L-alanyl-D-glutamate-L-lysine ligase | 99.73, 29.35, 28.72 | ZINC4235426 | −125.654 | 4/Tyr45, Thr46, Val47, and Glu155 |
| UTP-glucose-1-phosphate uridylyltransferase | −19.38, −1,2.96, −16.82 | ZINC428871 | −122.664 | 1/Leu110 |
| Fatty acid/phospholipid synthesis | 38.55, 51.23, 49.04 | ZINC4237105 | −130.756 | 1/Lys262 |
| Pantothenate synthetase | −6.64, −1.01, −43.92 | ZINC4235426 | −173.843 | 4/Met31, Gly148, His35, and Thr30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naorem, R.S.; Pangabam, B.D.; Bora, S.S.; Goswami, G.; Barooah, M.; Hazarika, D.J.; Fekete, C. Identification of Putative Vaccine and Drug Targets against the Methicillin-Resistant Staphylococcus aureus by Reverse Vaccinology and Subtractive Genomics Approaches. Molecules 2022, 27, 2083. https://doi.org/10.3390/molecules27072083
Naorem RS, Pangabam BD, Bora SS, Goswami G, Barooah M, Hazarika DJ, Fekete C. Identification of Putative Vaccine and Drug Targets against the Methicillin-Resistant Staphylococcus aureus by Reverse Vaccinology and Subtractive Genomics Approaches. Molecules. 2022; 27(7):2083. https://doi.org/10.3390/molecules27072083
Chicago/Turabian StyleNaorem, Romen Singh, Bandana Devi Pangabam, Sudipta Sankar Bora, Gunajit Goswami, Madhumita Barooah, Dibya Jyoti Hazarika, and Csaba Fekete. 2022. "Identification of Putative Vaccine and Drug Targets against the Methicillin-Resistant Staphylococcus aureus by Reverse Vaccinology and Subtractive Genomics Approaches" Molecules 27, no. 7: 2083. https://doi.org/10.3390/molecules27072083