
Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition
by
 , , and
, , and
Sushma Subedi
1,†, 
Santanu Sasidharan
2,† ,
,
Niharika Nag
1 ,
,
Prakash Saudagar
2,* and
and
Timir Tripathi
1,*
1
Molecular and Structural Biophysics Laboratory, Department of Biochemistry, North-Eastern Hill University, Shillong 793022, India
2
Department of Biotechnology, National Institute of Technology Warangal, Warangal 506004, India
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to the work.
Molecules 2022, 27(6), 1776; https://doi.org/10.3390/molecules27061776
Submission received: 30 November 2021
/
Revised: 4 March 2022
/
Accepted: 7 March 2022
/
Published: 8 March 2022
(This article belongs to the Special Issue Understanding Protein/Peptide Self-Assembly using Structural and Biophysical Chemistry)
Abstract
:Most neurodegenerative diseases such as Alzheimer’s disease, type 2 diabetes, Parkinson’s disease, etc. are caused by inclusions and plaques containing misfolded protein aggregates. These protein aggregates are essentially formed by the interactions of either the same (homologous) or different (heterologous) sequences. Several experimental pieces of evidence have revealed the presence of cross-seeding in amyloid proteins, which results in a multicomponent assembly; however, the molecular and structural details remain less explored. Here, we discuss the amyloid proteins and the cross-seeding phenomena in detail. Data suggest that targeting the common epitope of the interacting amyloid proteins may be a better therapeutic option than targeting only one species. We also examine the dual inhibitors that target the amyloid proteins participating in the cross-seeding events. The future scopes and major challenges in understanding the mechanism and developing therapeutics are also considered. Detailed knowledge of the amyloid cross-seeding will stimulate further research in the practical aspects and better designing anti-amyloid therapeutics.
1. Introduction
Amyloids are the aggregates of proteins that are insoluble and resistant to degradation. The formation of amyloids is generally associated with diseases collectively known as amyloidosis, though some amyloids do have functional roles Castellano and Shorter [1,2,3]. Several protein misfolding diseases (PMDs) are associated with the presence of amyloids, which are considered to be the hallmark of these diseases. For example, Parkinson’s disease (PD) is characterized by the presence of α-synuclein (α-syn) deposits, Alzheimer’s disease (AD) is characterized by the presence of amyloid-beta (Aβ) and tau plaques, and type-2 diabetes (T2D), apart from insulin resistance, is also characterized by the presence of human islet amyloid polypeptide (hIAPP) amyloid fibrils [4]. Structurally, significant conformational variability is seen in the amyloids formed by different proteins; however, they are predominantly composed of β-sheet secondary structures in a characteristic cross-β conformation stabilized by intermolecular hydrogen bonding [4,5]. The amyloid β-sheets can be arranged both in parallel [6,7] or antiparallel [8,9] orientations. Amyloids are also characterized by their physical features, such as the ability to bind with the dye Congo Red, resulting in apple-green birefringence in polarized light, and their ability to bind to fluorescent stains thioflavin-T and -S [10].
The rate-determining step in the formation of fibrils of misfolded proteins is the formation of “seeds”. Seeds are stable nuclei composed of polymerized proteins that can promote fibril formation by converting soluble proteins to fibrils [11]. The process of cross-seeding can either be homologous, i.e., seeds of the same protein, or they can be heterologous, i.e., seeds of one protein catalyzing the fibrillation of a different protein [12]. Cross-seeding aggregation between different amyloid proteins has been proposed to explain the presence of more than one misfolded protein in one disease and the coexistence of more than one PMD in the same individual [13,14]. It has also been observed that individuals diagnosed with one PMD are more susceptible to developing another [15,16]. The mechanism by which two different peptides form amyloids is not well understood. However, recent evidence suggests that heterotypic interactions between proteins via aggregation-prone homologous segments may contribute to it [17].
The terms cross-seeding and coaggregation are related but distinct. In the coaggregation process, two or more monomer protein influence the aggregation of each other independently of seeds, whereas in cross-seeding, the monomer or aggregate of one protein act as a seed for the aggregation of another protein. In the case of coaggregation, two proteins can polymerize together to form a mixed aggregate or fibrils or polymerize separately into distinct aggregates or fibrils.
2. Structure of Amyloids
Amyloids are the aggregates of protein that form long and unbranched fibers characterized by β-sheet structures in which the individual strands are arranged perpendicular to the axis of the fiber, forming a cross-β structure [18]. The cross-β pattern is considered to be the hallmark of the amyloid structure. The individual fibril, as visualized by transmission electron microscopy (TEM) and atomic force microscopy (AFM), are typically 7–13 nm in width and of the order of a few nanometers to micrometer in length [18,19]. X-ray diffraction studies have shown two characteristic scattering diffraction signals at 10 Å and 4.7 Å that correspond to the intersheet (stacking) and interstrand distance, respectively [18,19]. Amyloid fibrils are generally composed of substructures known as protofilaments [20,21]. These protofilaments vary in number and diameter and are often observed to twist around one another to form the 7–13 nm wide mature amyloid fibrils [4,5]. Each protofilament possesses the cross-β structure formed by variable numbers (typically 1–6) of sheets stacked on each other. The aggregation of a single amyloid protein can give rise to different structures. The amyloid polymorphism can arise due to differences in the arrangement, number, and packing of protofilaments. Solid-state nuclear magnetic resonance (ssNMR), a technique widely used for studying the structure of amyloids, was performed on amyloid fibrils of Aβ to show that β-sheets within the protofilaments can be arranged in parallel or antiparallel orientations [4]. Additionally, the ssNMR studies on Aβ and α-syn have revealed that the amyloid fibrils display in-register parallel β-sheet structures [22,23,24]. Cryo-electron microscope (cryo-EM) is another tool used to study high-resolution structures of amyloid fibrils. Cryo-EM studies of two types of amyloid fibrils- the paired helical filaments and straight filament, taken from the brains of patients with AD, revealed characteristic longitudinal cross-over distances ranging from 650–900 Å and filament widths ranging from 100–150 Å [25]. Another novel amyloid filament of tau, isolated from patients with chronic encephalopathy, revealed the presence of a more open conformation within the β-helix region than filaments observed in AD [26]. Recent cryo-EM studies on α-syn fibrils have shown the presence of two types of amyloid polymorphs—rod and twister structure [27]. The advancement in the use of multiple techniques to study the structure of amyloids associated with different diseases helps to understand the PMDs better and further the therapeutic and drugs development.
3. Intrinsically Disordered Proteins and Their Role in Amyloid Formation
Intrinsically disordered proteins (IDPs) lack a definite three-dimensional (3D) structure. The discovery of IDPs confronted the classical sequence–structure–function paradigm of proteins, which states that the 3D structure is responsible for its function. IDPs are flexible and exist in different conformational ensembles. They are rich in disorder-promoting amino acids such as Pro, Gly, Arg, Gln, Glu, Lys, Ser, and Ala and lack order-promoting amino acids such as Cys, Tyr, Trp, Val, Ile, Asn, Phe, and Leu [28,29]. IDPs have low hydrophobicity and high net charge, which promotes disorderedness due to strong electrostatic repulsion from the charged residues [30]. Many IDPs undergo a disorder-to-order structural transition upon binding to other biomolecules or can transform into “fuzzy complexes”, i.e., they remain partially disordered even when bound to another binding partner [31]. The amyloid-forming proteins such as Aβ, α-syn, tau, prion protein (PrP), etc. are partially or fully disordered in their native monomeric form [24,32,33,34,35,36]. The unique structural flexibility of these proteins allows for their aggregation through conformational polymorphism and oligomerization. The aggregation kinetics of amyloidogenic IDPs follows the typical nucleation-dependent polymerization. Many lines of evidence have shown that the soluble oligomeric species have disordered conformations and are more toxic than the matured fibrils [24,32,33,34,35,36]. Another feature of the amyloidogenesis and protein misfolding of almost all folded proteins is partial unfolding, which exposes the disordered region before the initiation of amyloid formation [37]. In globular proteins, the amyloidogenic sequence segments are buried within the core of the protein and, therefore, undergo partial unfolding before the formation of aggregates. For example, the conversion of α-helix to β-sheet in PrP is thought to occur due to helix-to-disorder transition. Additionally, the truncation of the C-terminal region of PrP, which is associated with diseases, yields a disordered variant that can readily undergo conversion into pathological amyloids [38,39]. However, besides their role in pathological amyloids formation, IDPs are primarily involved in a diverse range of biological functions such as cell division, cell cycle control, transcription and translational regulation, cell signaling, chromatin remodeling, and so forth [40,41]. Therefore, the aggregation of IDPs into amyloid fibrils has functional implications in a wide variety of organisms ranging from bacteria to humans [3,42,43].
4. Mechanism of Protein Aggregation and Amyloid Formation
The formation of amyloid fibrils from functional proteins involves a process of polymerization that is nucleation-dependent (NDP), resulting in the formation of β-sheet structures that are resistant to degradation and have a tendency to form larger aggregates [44,45,46]. The process starts with a slow nucleation phase, followed by an elongation phase, and ultimately ends in the saturation phase [44]. During polymerization, the rate-determining step is the formation of polymerized proteins, i.e., seeds. These seeds can then facilitate the further formation of amyloid fibrils [11]. A detailed description of the process is given in Section 6.4, with hIAPP as an example. In contrast to NDP, protein aggregation also occurs via isodesmic or linear polymerization, where the lag phase and critical monomer concentration are absent, which are the typical characteristics of NDP [47]. The kinetics of protein aggregation in linear polymerization does not require a separate nucleation and elongation rate, and therefore, the rate constants are identical for all the association steps.
A large number of studies suggest that the oligomeric intermediates that are formed during the aggregation process are toxic, and therefore, their interactions with cell membranes lead to cellular damages and cell death. The exposed hydrophobic regions in the oligomers seem to be responsible for their toxicity. Oligomers of the same size and morphology, but different exposed hydrophobic residues have variable toxicity levels [48]. The size of the oligomeric species is also responsible for toxicity, i.e., the smaller the size of the oligomers, the greater the toxicity it exhibits [48]. This could be due to the rapid diffusion of small oligomers within the cell, and hence, their interaction with different substrates could lead to cellular dysfunction. For instance, it has been shown that the toxic oligomers of Aβ and α-syn are responsible for the cellular damages in AD and PD, respectively [49,50]. Therefore, the toxicity of amyloids might be due to the interaction of oligomeric species with cellular compartments such as phospholipids, RNA, protein receptors, etc.
5. Seeding of Amyloid Proteins
The slow nucleation phase can be accelerated by the addition of preformed amyloid fibrils (seeds) (Figure 1). This phenomenon is known as seeding. Seeding can be either homologous or heterologous [12]. Homologous seeding occurs when the preformed amyloid fibrils catalyze the aggregation of the same protein. In contrast, heterologous seeding, also known as cross-seeding, occurs when the polymerization of one protein is catalyzed by a different protein [12]. Numerous studies suggest that the interactions between different amyloids are observed in many neurodegenerative diseases [51]. These studies support the idea that cross-seeding interactions between different amyloid proteins are perhaps the reason behind abnormal protein aggregations found in different PMDs. The process of cross-seeding polymerization is similar to that of homologous seeding. During the process, the unstructured monomers are converted into semistructured seeds and finally into amyloid fibrils that are composed of β-sheets [52]. However, cross-seeding polymerization is more complex than homologous-seeding due to the presence of different proteins. Additionally, unlike homologous seeding aggregation that always occurs, not every two different amyloid proteins can cross-seed each other, suggesting the existence of cross-seeding barriers. Several studies have shown that only a few pairs of amyloid protein can promote amyloid aggregation and fibrillization. These include the microtube-associated protein tau (MAPT, tau) and Aβ [53], Aβ and α-syn [54], tau and α-syn [55], Aβ and hIAPP [56], Aβ and scrapie-associated prion protein (PrPSC) [57], and rat islet amyloid polypeptide (rIAPP) and hIAPP [58].
6. Different Types of Amyloids Forming Proteins
Many proteins enter the amyloid formation stages and end up forming elongated fibers with the backbone spine composed of β-sheets. As of today, there are over 25 amyloid proteins that have been identified to form amyloid and are associated with diseases. The most common amyloid proteins are Aβ, tau, α-syn, hIAPP, and PrP, which can also cross-seed among themselves.
6.1. β-Amyloid
β-amyloid (Aβ) is a small peptide derived from a larger protein called amyloid precursor protein (APP). The human APP has two pathways for processing: non-amyloidogenic (α-secretase) and amyloidogenic (β-secretase and γ-secretase) (Figure 2). Even though the function of the APP protein remains elusive, the protein extends from the inside of a brain cell to the outside and passes through the fatty membrane that lies around the cell. In addition, the protein regulates synapsis formation and repair, anterograde neuronal transport, and iron export [59,60,61]. Aβ is a 4 kDa peptide and was first isolated from the amyloid deposits in the brain and cerebrovascular regions of the patients with AD and Down’s syndrome [62,63,64]. Aβ is formed when APP is proteolytically cleaved, and Aβ accumulates, resulting in the formation of senile plaques. The peptide is cleaved from the APP by two membrane-bound endoproteases: β-secretase and γ-secretase. Initially, the β-secretase cleaves the APP protein to release the soluble APP (sAPPβ), which is then sequentially cleaved by γ-secretase, to generate the Aβ. The γ-secretase cleavage is imprecise and, therefore, creates heterogeneity in the C-terminal end of the peptide. Structurally, the Aβ peptide contains a set of β-sheets that are parallel to the axis of the fibrils and extended strains perpendicular nearly to the fibril axis. The β-sheets are either parallel or antiparallel, and the sheets are aligned on top of one another in the fibril axis, i.e., they are “in register” (Figure 2) [34].
The multimeric assembly of the Aβ peptides is essential for their biological effects. Two phases of the assembly are known, and they vary in their characteristics and biological properties. Earlier, the amorphous and fibrillar deposits of Aβ were the point of focus but later shifted to the multimeric soluble forms of the peptide. The multimeric forms are much more toxic to cells than the fibrillar ones and can trigger several toxic events inside the cells [67]. Researchers argue that the soluble multimeric/oligomeric forms are active biologically and cause toxic effects in the cells; however, the mechanism of action remains elusive [68]. Structurally, both the phases are distinct to such an extent that the antibodies against multimeric forms do not recognize the monomer or fibrils and vice versa. The Aβ42 peptide readily undergoes oligomerization, while the Aβ40 forms, though abundant, do not oligomerize significantly [69]. The C-terminal region of the Aβ42 peptide is critical for the oligomerization process, and the ratio of Aβ42/40 is vital, as there lies a relation to the onset of the AD [70].
6.2. Tau Protein
Tau is another protein that contributes to the progress of AD and other neurodegenerative diseases such as dementia. The human tau protein is encoded by the microtubule-associated protein tau (MAPT) gene located on chromosome 17q21 [71,72]. The protein is found mainly in the axons of the central nervous system (CNS) and has six different isoforms generated by alternative splicing [73]. The proteins differ in the presence or absence of 29 amino acid repeats at the N terminal encoded by the exons 2 and 3 and by a 31 amino acid repeat in the C terminal. Structurally, tau protein has an N-terminal domain (1–165), followed by a proline-rich domain (166–242), a microtubule-binding region (MTBR) (243–367), and a C-terminal domain (368–441) (Figure 3) [74].
Tau is a vital microtubule-associated protein in the brain; however, several other MAPs have also been discovered [75]. The reason behind the limelight on tau is its association with AD. It is established that tau is required for the induction of Aβ toxicity [76]. The mechanism has been attributed to the hyperphosphorylation of the tau protein at several sites (eight or more phosphates per tau molecule) [77]. This can be due to several other physiological conditions and, therefore, should not be considered an indicator of AD onset [78]. Most of the phosphorylation sites in tau are in the proline-rich region and the C-terminal domain, except S262, S293, S324, and S356 [79,80,81]. The diseased condition might be either due to the upregulation of tau kinases or the downregulation of tau phosphatases [80,82]. The abnormally phosphorylated tau is incapable of binding to the tubulin and cannot promote microtubule assembly. On the other hand, it has also been known to inhibit assembly and disrupt the organization of microtubules [83,84]. Apart from hyperphosphorylation, another suggested mechanism is the acetylation of tau that leads to AD and other neurodegenerative disorders. The acetylation of K280 leads to the loss of capacity to bind to microtubules. This dysfunction causes paired helical filaments (PHF) aggregation and increased pools of cytosolic tau [85,86]. The typical arrangement of PHF is shown in Figure 3. It was also found that acetylation of tau was associated with hyperphosphorylation, indicating that tau dysfunction could be the cause of acetylation and hyperphosphorylation independently or in combination [87]. An alternative mechanism to the abnormal functioning of tau has been proposed to be due to proteolytic cleavage of tau [88]. The components of the PHF core are composed mainly of MTBR, which is truncated at E391 (C terminal), but the enzyme responsible remains unexplored [89]. Apart from AD, tau is also involved in other diseases such as Pick’s disease (straight filament (SF) aggregation), chronic traumatic encephalopathy (hyperphosphorylated tau with PHF and SF), and corticobasal degeneration (hyperphosphorylated tau) [90].
Figure 3.
Domain structure of tau protein. The N-terminal domain, proline-rich region (PRR), microtubule-binding region (MTBR), and C-terminal region (CTD) are shown. The PHF aggregate from 308–380 amino acids (in green) of tau protein solved by cryo-EM (PDB ID: 7NRQ) [91] shows the assembly of tau in the diseased fibril phase.
Figure 3.
Domain structure of tau protein. The N-terminal domain, proline-rich region (PRR), microtubule-binding region (MTBR), and C-terminal region (CTD) are shown. The PHF aggregate from 308–380 amino acids (in green) of tau protein solved by cryo-EM (PDB ID: 7NRQ) [91] shows the assembly of tau in the diseased fibril phase.

6.3. α-Synuclein
α-synuclein is a 140-amino-acid-long protein encoded by the SNCA gene. It is linked to diseases such as multiple system atrophy, AD, dementia, brain iron accumulation, neurodegeneration (type I), pure autonomic failure, and tremors [92]. The protein does not possess a defined structure in an aqueous solution and is, therefore, termed a natively unfolded protein. Interestingly, when α-syn binds to negatively charged lipids, it forms α-helix, and on prolonged incubation, it can also form β-sheet rich structures. There are two other members in the synuclein family of proteins: β-synuclein and γ-synuclein. They differ from each other in the central hydrophobic domain and localize preferentially. The protein comprises three unique domains—namely, (1) the N-terminal domain (NTD, 1–60) with lipid-binding motifs, which forms α-helices on membrane binding (amphiphilic helices); (2) a central hydrophobic domain (61–95), also called non-Aβ component (NAC), which confers β-sheet potential and contain minimal sequence required for aggregation; (3) a C-terminal domain (CTD, 96–140), which is highly unstructured, anti-amyloidogenic, and involved in Ca2+ binding (Figure 4) [93,94].
α-synuclein is capable of forming β-sheets under certain conditions, and this generated considerable interest, as it was similar to the β-sheets formed by Aβ. It was later found that the formation of β-sheets by α-syn is a result of the pathogenesis of neurodegenerative diseases. The mutations in α-syn induce the formation of amyloid-like fibrils, which has been observed only on prolonged incubation. The most common mutations are A53T and A30P, which forms protofibrils. The A53T mutant readily forms mature fibrils, and E46K formed fewer protofibrils when compared with wild-type α-syn [95,96]. Several other mutations are reported in the SNCA gene to cause PD, such as H50Q, G51D, A53T, A53E, A53V, and A30P. The mutations might increase the aggregation rate, a change in oligomerization and conformation, or a decrease in the tetramer/monomer ratio [97]. The α-syn undergoes multiple post-translational modifications (PTMs); the best-studied ones are phosphorylation. Earlier, it was believed that the soluble oligomer formation might be due to phosphorylation as observed in tau proteins [98,99], but later, it was shown that the effect of phosphorylation is contradictory. The phosphorylation of S129 occurs minimally in normal physiological conditions. The increased expression of α-syn phosphatases reduced the α-syn mediated neurotoxicity exhibited earlier [100].
6.4. hIAPP
Human islet amyloid polypeptide (hIAPP) is an amyloidogenic protein secreted as a randomly unstructured peptide. It plays a vital role in the progression of type 2 diabetes mellitus (T2DM). The autopsies of bodies with T2DM displayed hIAPP aggregates in the pancreatic islets. This was in line with the reduced pancreatic islets functioning and β-cell mass reduction, which, in turn, caused impaired insulin secretion [101]. Though the peptide is initially secreted with a random structure, it later assumes conformations such as helical, cross-β-sheets, and β-sheets, before it transforms into the amyloidogenic aggregated stage.
IAPP belongs to the calcitonin family comprising adrenomedullin, α- and β- calcitonin gene-related peptide (CGRP), calcitonin, and intermedin. All the peptides undergo extensive PTMs. Interestingly, IAPP shares sequence similarity with CGRP but shows a divergence in the 20–29 amino acid segment [102]. Moreover, hIAPP is amyloidogenic in nature, while CGRP does not form amyloid. Later, it was shown that the 10-residue region of hIAPP is responsible for the aggressive amyloidogenic property [102,103]. The residues V17, N22, and N23 in hIAPP were essential for the amyloid formation, and the 22–25 residue segment provided the nucleation template for the formation of fibrils [104]. The formation of fibrils is divided into three stages. Initially, the primary nucleation occurs, during which the monomeric peptides come together to form small oligomeric peptides that are soluble. These small oligomeric peptides form the critical nuclei that are energetically unstable, causing further amplification and growth [105]. This is followed by the elongation phase, in which the protofibrils propagate, and the monomer consumption and fibril growth occur [106]. This step is entropically favorable and results in the steady-state or plateau phase (Figure 5). In this phase, the concentration of fibrils dominates, along with equilibrium with the monomer concentration [107]. Apart from this, mutations have also been known to fasten the aggregation kinetics. Point mutations such as R18H, L23F, V26I in rat IAPP exhibited more rapid aggregation than the wild type [108]. The S20G mutation also displayed rapid aggregation, which was attributed to the smaller size of glycine when compared with serine, which might help in fibril packaging [109].
6.5. PrP Protein
Prion diseases cause some of the deadliest neurodegenerative diseases, affecting the motor and cognitive functioning of animals, including humans. They are caused by infectious proteinaceous particles called prions (PrP) [112,113]. PrP is a ubiquitous glycoprotein anchored to the plasma membrane of cells with the help of a glycosylphosphatidylinositol (GPI) anchor [114,115]. The protein is highly conserved in mammalian cells. The prion diseases include spongiform encephalopathy in cattle, scrapie in sheep and goats, chronic wasting in deer and elk, and kuru, fatal familial insomnia, Gerstmann–Straussler–Scheinker disease, and Creutzfeldt–Jakob disease (CJD) in humans [116]. Though the PrP protein has been studied, the precise function of the protein remains uncharacterized. The misfolding of the cellular prion protein (PrPc) results in the pathogenic isoform (PrPsc), which causes neurodegenerative prion diseases. Though PrPsc has been deciphered to be the causative agent in the related diseases, this is not always the case, as the absence of PrPsc has also displayed prion diseases. Therefore, other molecular species of PrPc, different from PrPsc, might also be the primary neurotoxic components. One such example is PrPctm, a transmembrane form of PrP, which is detected in the brains of human prion disease patients [117]. Therefore, it is imperative to understand the function, localization, and trafficking of the proteins that cause prion diseases. The function of the PrP protein has been studied to be the metabolism of copper, an essential metal cofactor required in various enzymes in the human nervous system [118]. The protein can reduce Cu2+ to Cu+ and decrease the formation of reactive oxygen species. In addition, the PrP protein is involved in cell–cell adhesion [119].
The PrP protein is synthesized from a single gene that codes for 250 amino acids. The N-terminal region of the protein houses five proline residues, followed by glycine-rich octapeptide repeats capable of binding copper. The hydrophobic domain (HD), which is the central segment of the PrP protein, is highly conserved and hydrophobic. The protein also contains a disulfide bridge and two glycosylation sites. The C-terminal region is hydrophobic and contains the signal peptide for GPI- anchor attachment. The important signal peptide in PrPc is the N-terminal signal peptide of 22 amino acids, which is recognized by signal recognizing particles. Therefore, the integrity of the N-terminal signal peptide is crucial for the import and export of PrPc [120]. The most important feature of PrPc is the endoproteolytic processing by two internal cleavage processes, i.e., α-cleavage and β-cleavage. In a healthy environment, PrPc is cleaved between amino acids 110/111 by an α-cleavage process resulting in a 17 kDa C-terminal fragment (C1) and 9 kDa N-terminal fragment (N1). These two fragments are the products of prion metabolism and increase PrPc dimerization. Under the diseased condition, β-cleavage of PrPc at amino acids 90/91 generates a 21 kDa C2 fragment, and this transition from C1 to C2 leads to the PrPsc propagation. The inefficient import of PrP protein to the ER results in its abnormal accumulation, thereby interfering with the cell viability [121]. Apart from the N-terminal region, the HD region (106–126) holds prominence in that it carries amyloidogenic properties and can form fibrils in vitro and produce toxic effects in cultured cells (Figure 6). Curiously, transgenic mice that expressed PrP lacking HD region could not develop transmissible prion infection, suggesting that the HD region was linked to the toxic effects exhibited by the PrP protein [122,123,124]. Furthermore, spectroscopic studies have identified that the PrPc and PrPsc differ conformationally in that the PrPc contains high α-helical content (~42%), with no β-sheets, while PrPsc comprises 30% of α-helix and 45% of β-sheets in its structure [125].
Transmissible spongiform encephalopathy (TSE) is another fatal neurodegenerative disorder caused by PrPc. The disorder results from the misfolding of PrPc to its scrapie isoform PrPsc. The PrPsc isoform is insoluble, partially protease-resistant, and can propagate by interacting with the normal isoform PrPc. PrPsc has a strong polymerizing tendency, and as a result, it forms amyloid aggregates. These aggregates are usually observed to be accumulated in the brain [120]. The accumulation is generally observed in the lymphoreticular region and brain, and the effect is typically neuronal vacuolation and death. Though the involvement of PrP protein in neurodegenerative disorders is known, the mechanism of conversion is still elusive. There are several hypothetical models, and they largely remain unexplored [127,128,129,130].
7. Cross-Seeding of Amyloid Proteins: Role and Mechanism
Amyloid-forming proteins aggregate independently to cause several neurodegenerative diseases. However, recently, these amyloids have been discovered to undergo cross-seeding. Cross-seeding is a biological event wherein the amyloid structures of one type of protein (homologous amyloids) can act as a seed and facilitate the aggregation of another amyloid protein, forming heterologous amyloids. Similar to homologous aggregation, the process of cross-seeding follows the same steps of nucleation, elongation, and the plateau phase (Section 6.4) [46,131,132]. Cross-seeding follows the nucleation-dependent aggregation pathway and requires a template to assist growth [133]. The nucleation phase is critical where the monomeric protein, in either mutated, denatured, or an oligomeric state, aggregates to overcome the high energy barrier. It is in the growth phase that various amyloid proteins come together in different forms to form deposits. The cross-seeding aggregation differs from the spontaneous aggregation in that the lag phase is reduced, and the aggregation kinetics is considerably faster in cross-seeding than in self-seeded aggregation [46,134]. Additionally, the aggregation in cross-seeding results from intermolecular interaction between different proteins, especially oppositely charged hetero-proteins [135], which contrasts to homologous amyloids, in which similarly charged proteins are electrostatically repulsed. The heterologous amyloids provide an electrostatically favorable environment and exposition of partially hydrophobic surfaces. These hydrophobic surfaces further trigger the nucleation and growth of aggregates [135]. However, not all two amyloid proteins can cross-seed each other, suggesting the existence of a cross-seeding barrier. Two models have been proposed for the mechanism of cross-seeding: template-assisted growth model and conformational and selection shift model. In the template-assisted growth model, depending on the aggregation and folding kinetics of different amyloids, the one that can form more populated seeds serves as a template and recruits the other amyloids for aggregation. In the other model, if both the amyloids have similarly populated seeds, the structural equilibrium lowers the barrier and selects those heterologous seeds with high conformational similarity, leading to cross-seeding [52]. A list of cross-seeding of various amyloid proteins deduced via multiple experimental and computational approaches is provided in Table 1 [52].
7.1. Cross-Seeding of Aβ and Tau
As discussed earlier, Aβ peptide found in plaques occurs in several forms, and studies have revealed that the different versions of Aβ interact with each other [160,161,162,163]. Apparently, cross-seeding has been observed between Aβ40 and Aβ42 peptides in in vitro conditions [161]. Apart from plaques, another hallmark of AD is the deposition of neurofibrillary tangles (NFTs), which mainly contain tau proteins [87]. Usually, Aβ plaques are deposited extracellularly, while the NFTs are formed intracellularly [164], but several pieces of evidence point toward the interaction between Aβ and tau protein. In vivo studies have shown that the Aβ species can accelerate the formation of NFTs [165] and form stable complexes with tau species [166]. The amyloid core sequence KLVFFA and the C-terminal residues of Aβ bind to tau [166]. The tau segments VQIINK and VQIVYK, located at the beginning of repeat 2 (R2) and repeat 3 (R3), respectively, of the four microtubule-binding repeats K18, can bind Aβ [167]. The peptides from regions of tau in exon 7 and 9 can also bind to Aβ [166]. Through the computational seeding model, it was predicted that the amyloid core of Aβ can form intermolecular β-sheet interactions with VQIINK or VQIVYK [143]. Recently, it has been reported that the peptide-based inhibitors of Aβ could reduce the aggregation and self-seeding of tau fibrils [168]. The ability of the inhibitors to interfere with the aggregation of both Aβ and tau suggested that both proteins share a common binding region. This supported the hypothesis that the interaction is through the cross-seeding mechanism [168,169].
7.2. Cross-Seeding of Aβ and α-Syn
The cross-seeding in α-syn has been observed between the isoforms of α-syn such as SNCA140, SNCA126, SNCA112, and SNCA95 [170,171]. Using in vitro experiments, it was shown that the C-terminal-truncated form of α-syn can seed the full-length form, leading to the formation of the Lewy body. Lewy body aggregates were also formed when the synphilin-1 protein interacted with α-syn [172,173]. The synphilin-1 A protein can interact with α-syn and synphilin-1 and cause aggregation [174]. The aggregation of α-syn into Lewy bodies (LBs) and Aβ into amyloid plaques are associated with PD and AD, respectively. LBs containing α-syn are usually found as aggregated intracellular vesicles [175], while Aβ is deposited extracellularly as senile plaques [176]; however, several studies have shown overlapping symptoms between the patients with AD and PD, suggesting a cross-talk between the two proteins. A study in transgenic mice demonstrated that Aβ could augment the aggregation of α-syn [177]. Conversely, α-syn has been shown to enhance the aggregation of Aβ both in vivo and in vitro. In addition, the non-amyloid component (NAC) region of α-syn was found in Aβ deposits in AD patients, indicating the interaction of Aβ with α-syn [178]. Moreover, employing nuclear magnetic resonance (NMR) spectroscopy in the membrane mimicking environment, it was shown that α-syn interacted more strongly with Aβ42 than with Aβ40 to produce more toxic oligomers [179].
7.3. Cross-Seeding of Aβ and IAPP
Several studies have shown that individuals with AD develop signs and symptoms of T2D or other glucose-related disorders, while individuals with T2D are at a higher risk of developing AD than healthy individuals. The exact mechanism behind the correlation of AD and T2D is still unknown, but multiple studies have indicated the cross-seeding interaction between Aβ and IAPP (amylin). A study on the interaction of Aβ and amylin had shown that hIAPP promoted Aβ42 oligomerization and the formation of larger aggregates [180]. It was also observed that Aβ42 and hIAPP interacted to form heterocomplex aggregates, which induced cell death in neuroblastoma cells [180]. In transgenic mice, intravenous injection with preformed Aβ fibrils triggered IAPP amyloid formation in the pancreas of the mice, suggesting that Aβ could enhance IAPP amyloid formation through cross-seeding [181].
7.4. Cross-Seeding of Tau and α-Syn
The coexistence of tau and α-syn proteins has been observed in many neurodegenerative diseases, indicating the interaction between these two proteins. Immunohistochemical examination of brains of Down’s syndrome patients has shown the coexistence of α-syn and tau in 50% of Down’s syndrome with AD patients [182]. In vitro studies using different cell models of synucleopathies have shown that tau can promote the aggregation and toxicity of α-syn [183]. In vivo studies using a mouse model have demonstrated that injecting α-syn oligomers derived from PD patients into Htau mice accelerated the formation of tau oligomers, along with neuronal cell loss [184]. It was observed that the coexpression of tau and α-syn in Dictyostelium discoideum had a positive effect on phagocytosis, growth, and respiration rate [185]. A study on the fruit fly model revealed the cross-seeding between tau and α-syn impaired the eyes and dopaminergic neurons [186], indicating a broad impact of cross-seeding. It has been reported that a simultaneous introduction of α-syn mouse preformed fibrils (mpffs) and AD lysate-derived tau seeds increased tau aggregation [187] Conversely, the absence of tau did not affect the aggregation of α-syn, showing that only α-syn can act as a seed for tau cross-seeding but not vice versa.
7.5. Cross-Seeding of α-Syn and IAPP
α-syn is the major aggregated peptide in substantia nigra neurons of patients with PD, while IAPP is the major peptide found in pancreatic beta cells; however, several studies have reported the presence of α-syn in the pancreatic beta cells [188,189,190]]. Recent studies on transgenic mice overexpressing hIAPP reported the colocalization of both α-syn and hIAPP in pancreatic beta cells of the transgenic mice, as well as in human pancreatic beta cells [191]. It was observed that α-syn promotes cross-seeding of hIAPP in a dose-dependent manner, both in vitro and in vivo [191]. It was also observed that injecting α-syn monomers exogenously in mice promoted faster aggregation of IAPP, whereas IAPP amyloid formation was reduced in mice lacking the gene encoding α-syn [191], further implying the cross-seeding interaction between α-syn and hIAPP. Recently, it was reported that the octapeptide TKEQVTNV from α-syn can cross-seed with hIAPP monomers and facilitate hIAPP fibrillation [192]. Moreover, the cross-seeding between the octapeptide from α-syn and hIAPP could increase cell viability and reduce cell apoptosis by reducing hIAPP induced cytotoxicity [192], suggesting a broader impact of cross-seeding.
7.6. Cross-Seeding in Prion Disease
Studies have revealed the coaggregation of prion proteins with other amyloid proteins. The Aβ42 extracted from the Alzheimer’s brain has been found to coaggregate with human prion proteins. The study suggested the presence of cross-interactions between the two proteins and also the diseases [193]. The coexistence of PD and prion proteins has also been shown in patients [194]. Curiously, it is claimed that the PrPc protein could facilitate the uptake of α-syn amyloid protein inside the cell, indicating the presence of possible intermolecular interactions between the two amyloid diseases [194]. It was reported that the peptide sequence “GNNQQNY” from a yeast protein (Sup35) could cross-seed with both hIAPP and Aβ to form β-structure aggregates, which accelerated amyloid fibrillization [191].
Although prions are neurogenerative diseases that affect humans and animals, evidence suggests that prions cross the species barrier. This results in cross-seeding of the prion proteins, and there is a consensus on the transmission of bovine spongiform encephalopathy from cattle to humans. There are key points for cross-species seeding that include (1) the difference in physiology between humans and animals under question, (2) the difference between the amino acid sequence of the humans and that of the animal PrPC, and (3) the prion strain of the animal coded in the conformation of PrPsc.
The prion cross-seeding has been studied using the protein misfolding cyclic amplification (PMCA) procedure. The prion strain variation and polymorphism in the codon 129 of the PRNP gene are the major factors responsible for the clinicopathological phenotype and the susceptibility of an individual to develop prion disease. In 2000, Parchi et al. showed that codon 129 is the primary determinant of the proteinase cleavage site in the PrPsc in CJD. A change in this codon leads to a change in the size of the PrP proteinase-resistant fragments [195]. The allelic variation percentage of this codon varies among different ethnic groups. One such case of M129V has been reported in the United Kingdom [196]. A recent study revealed the association between PRNP M129V polymorphism and mild cognitive impairment and dementia, including AD, in a Rotterdam-based population study [197]. Methionine homozygous individuals have a higher susceptibility to CJD, while codon 129 heterozygous individuals have a longer disease duration [198,199]. This is referred to as the variant CJD, predominantly found in the UK and affects younger people. On the other hand, the sporadic CJD is endemic throughout the world and affects patients of median age of 68 years. The two types have differences in symptoms, diagnosis, and incubation periods. Interestingly, Jones et al. studied the transmission properties of CJD using PMCA. The study used human brain tissues or transgenic mice models with three genotypes of codon 129 (MM, MV, and VV). The results showed that the MM genotype of codon 129 had strong PrPsc infectivity and conversion while the MV genotype and VV amplification had lesser and no amplification, respectively [200,201]. Therefore, the PRNP codon 129 genotype affects the susceptibility and phenotype of CJD.
7.7. Cross-Seeding in Other Proteins
Huntingtin (Htt), a hallmark protein of Huntington’s disease, has been observed to cross-seed and promote the fibrillation of TIA-1, an RNA-binding protein rich in glutamine and asparagine residues [202]. Another study found the occurrence of polyQ inclusions along with α-syn in samples of brain collected from HD rat models [203,204]. Aβ aggregation was influenced by cross-seeding with unrelated proteins that share a homologous aggregation-prone segment [205]. One clinical example for cross-seeding is the amyloidosis transthyretin (ATTR), which leads to a toxic function gain. TTR is a protein produced in the liver for the transport of thyroxin and retinol [206]. In the disease, the ATTR assembles into amyloid fibers and causes systematic organ dysfunction. Liver transplantation has been recommended as a treatment for ATTR variant (ATTRv) amyloidosis. However, due to the shortage of liver donors, there has been transplantation of livers from ATTRv patients. Fascinatingly, ATTR has been reported in patients who had received livers from ATTRv donors, specifically the V30M variant [207,208]. This is referred to as acquired ATTR following domino liver transplantation [207]. These recipients report the systemic amyloid deposition even before the appearance of amyloidosis symptoms in the patients with a mean time of 8 years [208,209]. In addition, liver transplant has led to the rapid and continuous deposition in cardiac tissues caused by the addition of the ATTR wild-type to the amyloid. This also has been observed in some V30M patients with cardiac amyloidosis [210,211,212,213].
8. Dual Inhibition
Cross-seeding of amyloid proteins has been observed to impact cell pathology [180,184,186], and therefore, targeting the involved amyloid proteins together might be a better therapeutic approach than individually targeting them [169]. Identification of the inhibitors having dual inhibition property is gaining the attention of many researchers. Many inhibitors, having dual inhibition effects against different pairs of amyloids aggregation, have been reported. The general mechanism of the working of dual inhibitors is depicted in Figure 7. We now discuss the available dual inhibitors of amyloid pairs involved in a few widely recognized PMDs.
8.1. Dual Inhibitors against Aβ and Tau
Several dual inhibitors against Aβ and tau have been reported. Recently, an extended in cellulo, in silico, and kinetic study was performed to test the inhibition efficiency of 1-benzylamino-2-hydroxyalkyl derivatives to identify a potent inhibitor against Aβ and tau [192]. It was observed that one of the compounds could inhibit 80% Aβ42 aggregation and 68.3% tau aggregation. The docking studies showed that the compound inhibited the aggregation process of Aβ by forming hydrophobic interactions, thereby stabilizing the α-helical structure of amyloid. The compound could inhibit tau fibrillization by binding to the central part of misfolded tau. The data from the docking studies also suggested the significant impact of chirality on the antiaggregation property of the inhibitor. The researchers suggested that S-isomers are favorable for Aβ and R-isomers for tau [192]. A series of peptide-based inhibitors have been designed that act as dual inhibitors against Aβ and tau [168]. The inhibitors could also reduce the efficiency of tau aggregation mediated by Aβ [168,169]. It has been reported that a curcumin derivative, PE859, could also act as a dual aggregation inhibitor against Aβ and tau in the mouse brains displaying the aging phenotype [214]. In addition, a furan coumarin (notopterol) has been identified to possess a dual inhibitory effect on β-secretase and GSK3β, the key enzymes responsible for Aβ production and tau phosphorylation, respectively [215]. The fact that these inhibitors could reduce the aggregation of both Aβ and tau further support the cross-seeding interaction between Aβ and tau. Recent in vivo studies on mice models have highlighted the neuroprotective properties of epigallocatechin-3-gallate (EGCG), a polyphenol constituent of green tea [216]. It was observed that treating the AD rats with EGCG decreased tau hyperphosphorylation in the hippocampus. Additionally, the expression and activity of Aβ42 and BACE1 were suppressed by EGCG, thereby improving the learning and memory function of AD rats. Methylene blue (MB) is another compound capable of reducing the aggregation of tau as well as Aβ. The ability of methylene blue to inhibit aggregated tau interaction with Aβ monomeric species in vitro through oxidation of cysteine residues serves it as a potent dual inhibitor against tau and Aβ [217]. Although MB reduces tau aggregation, it fails to act upon tau oligomers and thus showed poor performance in AD clinical trials [218].
8.2. Dual Inhibitors against Aβ and hIAPP
Numerous studies have reported that the potential link between AD and T2D could be due to the cross-seeding of Aβ and hIAPP [156,219,220,221]. Therefore, developing drugs targeting the cross-seeding between Aβ and hIAPP would be more effective than targeting the individual amyloids. Several inhibitors capable of inhibiting both the amyloids have been reported. Recently, bleomycin, a drug widely used as an antibiotic and antitumor, displayed a dual inhibitory effect on Aβ and hIAPP aggregation in vitro [222]. Genistein, a phytoestrogen in soybean, widely used as a cerebrovascular and anti-inflammatory drug, was also reported to have a dual inhibition effect on Aβ and hIAPP aggregation and increase cell viability and reduce cell apoptosis [223]. The polyphenol pentagalloyl glucose (PGG) has also been reported to inhibit the fibrillation of both Aβ and hIAPP [224,225]. PGG, at equal molar ratios to IAPP, was found to reduce fibril formation of IAPP [224]. In addition, tanshinones, the major component of the Chinese herb danshen (Salvia miltiorrhiza Bunge), could inhibit the aggregation of Aβ and hIAPP, disaggregate preformed hIAPP and Aβ fibrils, and also protect the cells from hIAPP and Aβ induced toxicity [226].
8.3. Dual Inhibitors against Aβ and α-Syn
Many AD patients develop signs and symptoms of PD and vice versa, indicating that the overlapping pathological pathways could be due to cross-seeding of Aβ and α-syn. Inhibitors have been reported to inhibit the aggregation of both Aβ and α-syn. Entacapone and tolcapone, anti-Parkinsonian drugs, could inhibit oligomerization and fibrillogenesis of both Aβ and α-syn in vitro and protect against the cytotoxicity induced by aggregation of both proteins as observed in the PC12 cell lines of rat adrenal gland pheochromocytoma [227]. Further, an in vitro study has shown that curcumin, the primary bioactive compound found in turmeric, has inhibitory effects against the aggregation of Aβ and α-syn [228].
8.4. Dual Inhibitors against Tau and α-Syn
The coexistence of aggregates of tau and α-syn in different pathologies has opened the doors to look for inhibitors that can act on both proteins, thereby preventing the formation of toxic aggregates. Recently, a small molecule (MG-2119) was identified as a potent dual inhibitor of monomeric tau and α-syn [229]. Using techniques such as cellular fluorescence resonance energy transfer, isothermal titration calorimetry, surface plasmon resonance, and microscale thermophoresis, the binding of the molecule to tau was investigated, and thioflavin T assay and dynamic light scattering results verified that it also inhibited the aggregation of α-syn. In SH-SY5Y neuroblastoma cells, the molecule reduced cytotoxicity in a dose-dependent manner.
9. Conclusions and Future Perspectives
Amyloid proteins can misfold and aggregate, causing disease conditions, and a few of them can interact homogenously and/or heterogeneously. Here, we attempt to shed light on the amyloid proteins, their cross-seeing behavior, and inhibition. Homologous aggregation of amyloid proteins has been extensively studied; however, the occurrence and mechanism of cross-seeding have not been extensively ventured. It is now accepted that cross-seeding is no longer an isolated but a well-established event in the growth of amyloid structures. Interestingly, the cross-seeding event is species-specific, and therefore, we discussed the aggregation of five hallmark amyloid proteins Aβ, αs, PrPc, tau, and hIAPP. It is hypothetically assumed that the cross-seeding between amyloids is dependent on conformations that lower the energy barrier for seeding. The mechanism of the formation of amyloid aggregates is still elusive for most proteins and requires a more profound understanding of anti-amyloid drug design and discovery. The knowledge of the protein components involved in the cross-seeding warrants further investigation to clarify the role and mechanism of cross-seeding and aggregation. The inhibitors targeting dually on amyloid proteins participating in the cross-seeding event inhibited heterologous aggregation but also caused disassembly of the aggregates. Understanding the molecular mechanism of interaction in cross-seeding will help develop better therapeutics against PMDs. Furthermore, researchers can strategize approaches to inhibit and disassemble both homologous and heterologous aggregates by designing site-specific inhibitors. Further exploration and in-depth studies on the amyloid cross-seeding will help gain more significant insights into understanding the amyloidogenesis mechanism, cross-talks of PMDs, and designing better anti-amyloid therapeutics.
Author Contributions
Methodology, formal analysis, investigation, data curation, writing—original draft preparation, S.S. (Sushma Subedi); methodology, formal analysis, investigation, data curation, writing—original draft preparation, S.S. (Santanu Sasidharan); data curation, formal analysis, writing—review and editing, N.N.; supervision, writing—review and editing, P.S.; conceptualization, supervision, writing—review and editing, T.T. All authors have read and agreed to the published version of the manuscript.
Funding
The review was prepared with the support of a project grant from the Indian Council of Medical Research (ICMR), Government of India, India (52/06/2020/BIO/BMS) to T.T.
Acknowledgments
N.N. would like to thank DST-INSPIRE for providing fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Castellano, L.M.; Shorter, J. The Surprising Role of Amyloid Fibrils in HIV Infection. Biology 2012, 1, 58–80. [Google Scholar] [CrossRef] [PubMed]
- Roan, N.R.; Sandi-Monroy, N.; Kohgadai, N.; Usmani, S.M.; Hamil, K.G.; Neidleman, J.; Montano, M.; Ständker, L.; Röcker, A.; Cavrois, M.; et al. Semen amyloids participate in spermatozoa selection and clearance. eLife 2017, 6, e24888. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabrizio, C.; Christopher, M.D. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C.F. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ã.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A structural model for Alzheimer’s beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [Green Version]
- Qiang, W.; Yau, W.-M.; Luo, Y.; Mattson, M.P.; Tycko, R. Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 4443–4448. [Google Scholar] [CrossRef] [Green Version]
- Tycko, R.; Sciarretta, K.L.; Orgel, J.P.R.O.; Meredith, S.C. Evidence for novel beta-sheet structures in Iowa mutant beta-amyloid fibrils. Biochemistry 2009, 48, 6072–6084. [Google Scholar] [CrossRef] [Green Version]
- Nizhnikov, A.A.; Antonets, K.S.; Inge-Vechtomov, S.G. Amyloids: From pathogenesis to function. Biochemistry 2015, 80, 1127–1144. [Google Scholar] [CrossRef]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid β-protein fibrillogenesis: Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, R.; Green, K.M.; Soto, C. Cross currents in protein misfolding disorders: Interactions and therapy. CNS Neurol. Disord. Drug Targets 2009, 8, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, D.J.; Lee, V.M.Y.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Reitz, C.; Patel, B.; Tang, M.-X.; Manly, J.J.; Mayeux, R. Relation of diabetes to mild cognitive impairment. Arch. Neurol. 2007, 64, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Konstantoulea, K.; Guerreiro, P.; Ramakers, M.; Louros, N.; Aubrey, L.D.; Houben, B.; Michiels, E.; De Vleeschouwer, M.; Lampi, Y.; Ribeiro, L.F.; et al. Heterotypic Amyloid β interactions facilitate amyloid assembly and modify amyloid structure. EMBO J. 2022, 41, e108591. [Google Scholar] [CrossRef]
- Sunde, M.; Blake, C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv. Protein Chem. 1997, 50, 123–159. [Google Scholar]
- Eisenberg, D.S.; Sawaya, M.R. Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [Green Version]
- Goldsbury, C.; Kistler, J.; Aebi, U.; Arvinte, T.; Cooper, G.J. Watching amyloid fibrils grow by time-lapse atomic force microscopy. J. Mol. Biol. 1999, 285, 33–39. [Google Scholar] [CrossRef]
- Jiménez, J.L.; Guijarro, J.I.; Orlova, E.; Zurdo, J.; Dobson, C.M.; Sunde, M.; Saibil, H.R. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J. 1999, 18, 815–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loquet, A.; El Mammeri, N.; Stanek, J.; Berbon, M.; Bardiaux, B.; Pintacuda, G.; Habenstein, B. 3D structure determination of amyloid fibrils using solid-state NMR spectroscopy. Methods 2018, 138, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, T. Solid-State NMR Studies of Amyloid Fibril Structure. Annu. Rev. Phys. Chem. 2011, 62, 279–299. [Google Scholar]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. eLife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Lyngdoh, D.L.; Shukla, H.; Sonkar, A.; Anupam, R.; Tripathi, T. Portrait of the Intrinsically Disordered Side of the HTLV-1 Proteome. ACS Omega 2019, 4, 10003–10018. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Riek, R. Biology of amyloid: Structure, function, and regulation. Structure 2010, 18, 1244–1260. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Knowles, T.P.; Klenerman, D. The molecular processes underpinning prion-like spreading and seed amplification in protein aggregation. Curr. Opin. Neurobiol. 2020, 61, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [Green Version]
- Kotler, S.M.; Walsh, P.; Brendera, J.R.; Ramamoorthy, A. Differences between amyloid-β aggregation in solution and on the membrane: Insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6692–6700. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, R.; Ranson, N.A.; Radford, S.E. Amyloid Structures: Much More than Just a Cross-β Fold. Curr. Opin. Struct. Biol. 2020, 60, 7–16. [Google Scholar] [CrossRef]
- Mukhopadhyay, S. The dynamism of intrinsically disordered proteins: Binding-induced folding, amyloid formation, and phase separation. J. Phys. Chem. 2020, 124, 11541–11560. [Google Scholar] [CrossRef]
- Theint, T.N.; Aucoin, D.; Helmus, J.J.; Pondaven, S.; Surewicz, K.S.; Jaroniec, C.P. Species-dependent structural polymorphism of Y145Stop prion protein amyloid revealed by solid-state NMR spectroscopy. Nat. Commun. 2017, 8, 753. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.M.; Surewicz, W.K. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell 2005, 121, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Fowler, D.M.K.; Balch, W.E.; Kelly, J.W. Functional Amyloid—From Bacteria to Humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef]
- Jain, N.; Chapman, M.R. Bacterial Functional Amyloids: Order from Disorder. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Otzen, D.; Riek, R. Functional Amyloids. Cold Spring Harb. Perspect. Biol. 2019, 11, a033860. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.; Cereghetti, G.; Feng, Y.; Picotti, P.; Peter, M.; Dechant, R. Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat. Cell Biol. 2017, 19, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Chatani, E.; Yamamoto, N. Recent progress on understanding the mechanisms of amyloid nucleation. Biophys. Rev. 2018, 10, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Morales, R.; Moreno-Gonzalez, I.; Soto, C. Cross-seeding of misfolded proteins: Implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog. 2013, 9, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Frieden, C. Protein aggregation processes: In search of the mechanism. Protein Sci. 2007, 16, 2334–2344. [Google Scholar] [CrossRef]
- Mannini, B.; Mulvihill, E.; Sgromo, C.; Cascella, R.; Khodarahmi, R.; Ramazzotti, M.; Dobson, C.M.; Cecchi, C.; Chiti, F. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 2014, 10, 2309–2317. [Google Scholar] [CrossRef]
- Benilova, I.; De Strooper, B. The toxic abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Roberts, H.L. Seeking a mechanism for the toxicity of oligomeric alphasynuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef] [Green Version]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Zhang, Y.; Zhang, M.; Liu, Y.; Zhang, D.; Gong, X.; Feng, Z.; Tang, J.; Chang, Y.; Zheng, J. Fundamentals of cross-seeding of amyloid proteins: An introduction. J. Mater. Chem. 2019, 7, 7267–7282. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, B.; Stancu, I.-C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.-N.L. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 2016, 131, 549–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid-β-protein and α-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Waxman, E.A.; Giasson, B.I. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Gonzalez, I.; Edwards, G., III; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- Morales, R.; Estrada, L.D.; Diaz-Espinoza, R.; Morales-Scheihing, D.; Jara, M.C.; Castilla, J.; Soto, C. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J. Neurosci. 2010, 30, 4528–4535. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Ren, B.; Zhang, M.; Chen, H.; Liu, Y.; Liu, L.; Gong, X.; Jiang, B.; Ma, J.; Zheng, J. Seed-induced heterogeneous cross-seeding self-assembly of human and rat islet polypeptides. ACS Omega 2017, 2, 784–792. [Google Scholar] [CrossRef]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 2006, 26, 7212–7221. [Google Scholar] [CrossRef]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Masters, C.L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985, 4, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerofolini, L.; Ravera, E.; Bologna, S.; Wiglenda, T.; Böddrich, A.; Purfürst, B.; Benilova, I.; Korsak, M.; Gallo, G.; Rizzo, D.; et al. Mixing Aβ(1–40) and Aβ(1–42) peptides generates unique amyloid fibrils. Chem. Commun. 2020, 56, 8830–8833. [Google Scholar] [CrossRef]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, A.; Mina, E.; Glabe, C.; Busciglio, J. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J. Neurosci. 2006, 26, 6011–6018. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; LeVine, H., III. Alzheimer’s disease and the amyloid-β peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Teplow, D.B.; Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B. Amyloid beta-protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar]
- Duering, M.; Grimm, M.O.; Grimm, H.S.; Schröder, J.; Hartmann, T. Mean age of onset in familial Alzheimer’s disease is determined by amyloid beta 42. Neurobiol. Aging 2005, 26, 785–788. [Google Scholar] [CrossRef]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol. Brain Res. 1986, 1, 271–280. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Spillantini, M.; Potier, M.; Ulrich, J.; Crowther, R. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of tau as a microtubule-associated protein: Structural and functional aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005, 6, 204. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Köpke, E.; Tung, Y.-C.; Shaikh, S.; Alonso, A.D.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau—Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef]
- Mandelkow, E.-M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Sergeant, N.; Bretteville, A.; Hamdane, M.; Caillet-Boudin, M.-L.; Grognet, P.; Bombois, S.; Blum, D.; Delacourte, A.; Pasquier, F.; Vanmechelen, E. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev. Proteom. 2008, 5, 207–224. [Google Scholar] [CrossRef]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Meyer, H.E.; Mandelkow, E.-M.; Drewes, G.; Trinczek, B.; Illenberger, S.; Biernat, J.; Schmitt-Ulms, G.; Mandelkow, E. Microtubule-associated Protein/Microtubule Affinity-regulating Kinase (p110mark): A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J. Biol. Chem. 1995, 270, 7679–7688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trojanowski, J.Q.; Lee, V.M.Y. Phosphorylation of paired helical filament tau in Alzheimer’s disease neurofibrillary lesions: Focusing on phosphatases. FASEB J. 1995, 9, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-Z.; Gong, C.-X.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer Paired Helical Filaments by Protein Phosphatase-2A and -2B. J. Biol. Chem. 1995, 270, 4854–4860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, B.L.; Feinstein, S.C. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J. Cell Biol. 1994, 124, 769–782. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer’s Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [Green Version]
- Wischik, C.; Novak, M.; Edwards, P.; Klug, A.; Tichelaar, W.; Crowther, R. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [Green Version]
- García-Sierra, F.; Wischik, C.M.; Harrington, C.R.; Luna-Muñoz, J.; Mena, R. Accumulation of C-terminally truncated tau protein associated with vulnerability of the perforant pathway in early stages of neurofibrillary pathology in Alzheimer’s disease. J. Chem. Neuroanat. 2001, 22, 65–77. [Google Scholar] [CrossRef]
- Oakley, S.S.; Maina, M.B.; Marshall, K.E.; Al-Hilaly, Y.K.; Harrington, C.R.; Wischik, C.M.; Serpell, L.C. Tau Filament Self-Assembly and Structure: Tau as a Therapeutic Target. Front. Neurol. 2020, 11, 1207. [Google Scholar] [CrossRef]
- Shi, Y.; Murzin, A.G.; Falcon, B.; Epstein, A.; Machin, J.; Tempest, P.; Newell, K.L.; Vidal, R.; Garringer, H.J.; Sahara, N.; et al. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol. 2021, 141, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.J. α-Synucleinopathy models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, T. A Master Regulator of α-Synuclein Aggregation. ACS Chem. Neurosci. 2020, 11, 1376–1378. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.-J.; Rochet, J.-C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Fredenburg, R.A.; Rospigliosi, C.; Meray, R.K.; Kessler, J.C.; Lashuel, H.A.; Eliezer, D.; Lansbury, P.T. The impact of the E46K mutation on the properties of α-synuclein in its monomeric and oligomeric states. Biochemistry 2007, 46, 7107–7118. [Google Scholar] [CrossRef] [Green Version]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease–lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Feany, M.B. α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of α-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Investig. 2009, 119, 3257–3265. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-W.; Chen, W.; Junn, E.; Im, J.-Y.; Grosso, H.; Sonsalla, P.K.; Feng, X.; Ray, N.; Fernandez, J.R.; Chao, Y. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J. Neurosci. 2011, 31, 6963–6971. [Google Scholar] [CrossRef]
- Zhao, J.; Luo, Y.; Jang, H.; Yu, X.; Wei, G.; Nussinov, R.; Zheng, J. Probing ion channel activity of human islet amyloid polypeptide (amylin). Biochim. Biophys. Acta (BBA)—Biomembr. 2012, 1818, 3121–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermark, P.; Engström, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburn, T.T.; Lansbury, P.T., Jr. Interspecies sequence variations affect the kinetics and thermodynamics of amyloid formation: Peptide models of pancreatic amyloid. J. Am. Chem. Soc. 1993, 115, 11012–11013. [Google Scholar] [CrossRef]
- Chakraborty, S.; Chatterjee, B.; Basu, S. A mechanistic insight into the amyloidogenic structure of hIAPP peptide revealed from sequence analysis and molecular dynamics simulation. Biophys. Chem. 2012, 168, 1–9. [Google Scholar] [CrossRef]
- Arosio, P.; Knowles, T.P.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of α-synuclein assembly by β-and γ-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef] [Green Version]
- Khatun, S.; Shikha, K.; Ganguly, A.; Pawar, N.; Gupta, A.N. Repulsive interaction induces fibril formation and their growth. Int. J. Biol. Macromol. 2019, 123, 20–25. [Google Scholar] [CrossRef]
- Bishoyi, A.K.; Roham, P.H.; Rachineni, K.; Save, S.; Hazari, M.A.; Sharma, S.; Kumar, A. Human islet amyloid polypeptide (hIAPP)-a curse in type II diabetes mellitus: Insights from structure and toxicity studies. Biol. Chem. 2021, 402, 133–153. [Google Scholar] [CrossRef]
- Kajava, A.V.; Aebi, U.; Steven, A.C. The parallel superpleated beta-structure as a model for amyloid fibrils of human amylin. J. Mol. Biol. 2005, 348, 247–252. [Google Scholar] [CrossRef]
- Nielsen, J.T.; Bjerring, M.; Jeppesen, M.D.; Pedersen, R.O.; Pedersen, J.M.; Hein, K.L.; Vosegaard, T.; Skrydstrup, T.; Otzen, D.E.; Nielsen, N.C. Unique identification of supramolecular structures in amyloid fibrils by solid-state NMR spectroscopy. Angew Chem. 2009, 48, 2118–2121. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structure and inhibitor design of human IAPP (amylin) fibrils. Nat. Struct. Mol. Biol. 2020, 27, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Calella, A.M. Prions: Protein aggregation and infectious diseases. Physiol. Rev. 2009, 89, 1105–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schätzl, H.M.; Da Costa, M.; Taylor, L.; Cohen, F.E.; Prusiner, S.B. Prion protein gene variation among primates. J. Mol. Biol. 1995, 245, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Wopfner, F.; Weidenhöfer, G.; Schneider, R.; von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schätzl, H.M. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef]
- Terry, C.; Wadsworth, J.D. Recent advances in understanding mammalian prion structure: A mini review. Front. Mol. Neurosci. 2019, 12, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, R.S.; Tremblay, P.; Groth, D.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature 1999, 402, 822–826. [Google Scholar] [CrossRef]
- Walter, E.D.; Stevens, D.J.; Spevacek, A.R.; Visconte, M.P.; Rossi, A.D.; Millhauser, G.L. Copper binding extrinsic to the octarepeat region in the prion protein. Curr. Protein Pept. Sci. 2009, 10, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Jiang, D.; McDonald, A.; Hao, Y.; Millhauser, G.L.; Zhou, F. Copper redox cycling in the prion protein depends critically on binding mode. J. Am. Chem. Soc. 2011, 133, 12229–12237. [Google Scholar] [CrossRef] [Green Version]
- Sarnataro, D.; Pepe, A.; Zurzolo, C. Cell biology of prion protein. Prog. Mol. Biol. Transl. Sci. 2017, 150, 57–82. [Google Scholar]
- Heller, U.; Winklhofer, K.F.; Heske, J.; Reintjes, A.; Tatzelt, J.R. Post-translational import of the prion protein into the endoplasmic reticulum interferes with cell viability: A critical role for the putative transmembrane domain. J. Biol. Chem. 2003, 278, 36139–36147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliavini, F.; Prelli, F.; Verga, L.; Giaccone, G.; Sarma, R.; Gorevic, P.; Ghetti, B.; Passerini, F.; Ghibaudi, E.; Forloni, G. Synthetic peptides homologous to prion protein residues 106-147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 9678–9682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, F.; Tolnay, M.; Brabeck, C.; Pahnke, J.; Kloz, U.; Niemann, H.H.; Heikenwalder, M.; Rülicke, T.; Bürkle, A.; Aguzzi, A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007, 26, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Miesbauer, M.; Rapaport, D.; Bartke, T.; Baier, M.; Winklhofer, K.F.; Tatzelt, J. Association of Bcl-2 with misfolded prion protein is linked to the toxic potential of cytosolic PrP. Mol. Biol. Cell 2006, 17, 3356–3368. [Google Scholar] [CrossRef] [Green Version]
- Pan, K.-M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Glynn, C.; Sawaya, M.R.; Ge, P.; Gallagher-Jones, M.; Short, C.W.; Bowman, R.; Apostol, M.; Zhou, Z.H.; Eisenberg, D.S.; Rodriguez, J.A. Cryo-EM structure of a human prion fibril with a hydrophobic, protease-resistant core. Nat. Struct. Mol. Biol. 2020, 27, 417–423. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Telling, G.C.; Scott, M.; Mastrianni, J.; Gabizon, R.; Torchia, M.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995, 83, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Goold, R.; Rabbanian, S.; Sutton, L.; Andre, R.; Arora, P.; Moonga, J.; Clarke, A.; Schiavo, G.; Jat, P.; Collinge, J. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2011, 2, 281. [Google Scholar] [CrossRef] [Green Version]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an intracellular site of prion conversion. PLoS Pathol. 2009, 5, e1000426. [Google Scholar] [CrossRef] [Green Version]
- Slepko, N.; Bhattacharyya, A.M.; Jackson, G.R.; Steffan, J.S.; Marsh, J.L.; Thompson, L.M.; Wetzel, R. Normal-repeat-length polyglutamine peptides accelerate aggregation nucleation and cytotoxicity of expanded polyglutamine proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 14367–14372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.-C.; Citron, M.; Biere, A.L. α-Synuclein Fibrillogenesis Is Nucleation-dependent: Implications for the pathogenesis of Parkinson’ s disease. J. Biol. Chem. 1999, 274, 19509–19512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, K.; Gerson, J.E.; Kayed, R. Oligomer Formation and Cross-Seeding: The New Frontier. Isr. J. Chem. 2017, 57, 665–673. [Google Scholar] [CrossRef]
- Dubey, K.; Anand, B.G.; Temgire, M.K.; Kar, K. Evidence of rapid coaggregation of globular proteins during amyloid formation. Biochemistry 2014, 53, 8001–8004. [Google Scholar] [CrossRef] [PubMed]
- Oki, S.; Iwashita, K.; Kimura, M.; Kano, H.; Shiraki, K. Mechanism of co-aggregation in a protein mixture with small additives. Int. J. Biol. Macromol. 2018, 107, 1428–1437. [Google Scholar] [CrossRef]
- Yoo, B.K.; Xiao, Y.; McElheny, D.; Ishii, Y. E22G pathogenic mutation of β-amyloid (Aβ) enhances misfolding of Aβ40 by unexpected prion-like cross talk between Aβ42 and Aβ40. J. Am. Chem. Soc. 2018, 140, 2781–2784. [Google Scholar] [CrossRef]
- Krotee, P.; Griner, S.L.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Shi, D.; Philipp, S.; Murray, K.; Saelices, L.; Lee, J. Common fibrillar spines of amyloid-β and human islet amyloid polypeptide revealed by microelectron diffraction and structure-based inhibitors. J. Biol. Chem. 2018, 293, 2888–2902. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Mizuguchi, M.; Hamaguchi, T.; Yamada, M. Exogenous amyloidogenic proteins function as seeds in amyloid β-protein aggregation. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2014, 1842, 646–653. [Google Scholar] [CrossRef] [Green Version]
- Qi, R.; Luo, Y.; Wei, G.; Nussinov, R.; Ma, B. Aβ “stretching-and-packing” cross-seeding mechanism can trigger tau protein aggregation. J. Phys. Chem. Lett. 2015, 6, 3276–3282. [Google Scholar] [CrossRef]
- Raz, Y.; Miller, Y. Interactions between Aβ and mutated Tau lead to polymorphism and induce aggregation of Aβ-mutated tau oligomeric complexes. PLoS ONE 2013, 8, e73303. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Chacko, A.R.; Belfort, G. Alzheimer’s Protective Cross-Interaction between Wild-Type and A2T Variants Alters Aβ42 Dimer Structure. ACS Chem. Neurosci. 2017, 8, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.; Ma, B.; Nussinov, R. Synergistic interactions between repeats in tau protein and Aβ amyloids may be responsible for accelerated aggregation via polymorphic states. Biochemistry 2011, 50, 5172–5181. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Luo, Y.; Dinkel, P.; Zheng, J.; Wei, G.; Margittai, M.; Nussinov, R.; Ma, B. Cross-seeding and conformational selection between three-and four-repeat human Tau proteins. J. Biol. Chem. 2012, 287, 14950–14959. [Google Scholar] [CrossRef] [Green Version]
- Berhanu, W.M.; Yaşar, F.; Hansmann, U.H. In silico cross seeding of Aβ and amylin fibril-like oligomers. ACS Chem. Neurosci. 2013, 4, 1488–1500. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Hu, R.; Chen, H.; Gong, X.; Zhou, F.; Zhang, L.; Zheng, J. Polymorphic associations and structures of the cross-seeding of Aβ1–42 and hIAPP1–37 polypeptides. J. Chem. Inf. Model. 2015, 55, 1628–1639. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, R.; Chen, H.; Chang, Y.; Ma, J.; Liang, G.; Mi, J.; Wang, Y.; Zheng, J. Polymorphic cross-seeding amyloid assemblies of amyloid-β and human islet amyloid polypeptide. Phys. Chem. Chem. Phys. 2015, 17, 23245–23256. [Google Scholar] [CrossRef]
- Luk, K.; Covell, D.; Kehm, V.; Zhang, B.; Song, I.; Byrne, M.; Pitkin, R.; Decker, S.; Trojanowski, J.; Lee, V. Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity. Cell Rep. 2016, 16, 3373–3387. [Google Scholar] [CrossRef] [Green Version]
- Terada, M.; Suzuki, G.; Nonaka, T.; Kametani, F.; Tamaoka, A.; Hasegawa, M. The effect of truncation on prion-like properties of α-synuclein. J. Biol. Chem. 2018, 293, 13910–13920. [Google Scholar] [CrossRef] [Green Version]
- Tavassoly, O.; Sade, D.; Bera, S.; Shaham-Niv, S.; Vocadlo, D.J.; Gazit, E. Quinolinic acid amyloid-like fibrillar assemblies seed α-synuclein aggregation. J. Mol. Biol. 2018, 430, 3847–3862. [Google Scholar] [CrossRef]
- Atsmon-Raz, Y.; Miller, Y. Non-amyloid-β component of human α-synuclein oligomers induces formation of new Aβ oligomers: Insight into the mechanisms that link Parkinson’s and Alzheimer’s diseases. ACS Chem. Neurosci. 2016, 7, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hall, C.K. Seeding and cross-seeding fibrillation of N-terminal prion protein peptides PrP (120–144). Protein Sci. 2018, 27, 1304–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katorcha, E.; Makarava, N.; Lee, Y.J.; Lindberg, I.; Monteiro, M.J.; Kovacs, G.G.; Baskakov, I.V. Cross-seeding of prions by aggregated α-synuclein leads to transmissible spongiform encephalopathy. PLoS Pathol. 2017, 13, e1006563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilitchev, A.I.; Giammona, M.J.; Olivas, C.; Claud, S.L.; Lazar Cantrell, K.L.; Wu, C.; Buratto, S.K.; Bowers, M.T. Hetero-oligomeric amyloid assembly and mechanism: Prion fragment PrP (106–126) catalyzes the islet amyloid polypeptide β-hairpin. J. Am. Chem. Soc. 2018, 140, 9685–9695. [Google Scholar] [CrossRef]
- Chua, K.P.; Chew, L.Y.; Mu, Y. Replica exchange molecular dynamics simulation of cross-fibrillation of IAPP and PrP106-126. Proteins Struct. Funct. Bioinform. 2016, 84, 1134–1146. [Google Scholar] [CrossRef]
- Hu, R.; Zhang, M.; Chen, H.; Jiang, B.; Zheng, J. Cross-seeding interaction between β-amyloid and human islet amyloid polypeptide. ACS Chem. Neurosci. 2015, 6, 1759–1768. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, R.; Chen, H.; Chang, Y.; Gong, X.; Liu, F.; Zheng, J. Interfacial interaction and lateral association of cross-seeding assemblies between hIAPP and rIAPP oligomers. Phys. Chem. Chem. Phys. 2015, 17, 10373–10382. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, R.; Liang, G.; Chang, Y.; Sun, Y.; Peng, Z.; Zheng, J. Structural and energetic insight into the cross-seeding amyloid assemblies of human IAPP and rat IAPP. J. Phys. Chem. B 2014, 118, 7026–7036. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, R.; Ren, B.; Chen, H.; Jiang, B.; Ma, J.; Zheng, J. Molecular understanding of Aβ-hIAPP cross-seeding assemblies on lipid membranes. ACS Chem. Neurosci. 2017, 8, 524–537. [Google Scholar] [CrossRef]
- Szczepankiewicz, O.; Linse, B.; Meisl, G.; Thulin, E.; Frohm, B.; Sala Frigerio, C.; Colvin, M.T.; Jacavone, A.C.; Griffin, R.G.; Knowles, T. N-terminal extensions retard Aβ42 fibril formation but allow cross-seeding and coaggregation with Aβ42. J. Am. Chem. Soc. 2015, 137, 14673–14685. [Google Scholar] [CrossRef] [Green Version]
- Tran, J.; Chang, D.; Hsu, F.; Wang, H.; Guo, Z. Cross-seeding between Aβ40 and Aβ42 in Alzheimer’s disease. FEBS Lett. 2017, 591, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Onstead, L.; Randle, S.; Price, R.; Smithson, L.; Zwizinski, C.; Dickson, D.W.; Golde, T.; McGowan, E. Aβ40 inhibits amyloid deposition in vivo. J. Neurosci. 2007, 27, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Sowade, R.; Jahn, T. Seed-induced acceleration of amyloid-β mediated neurotoxicity in vivo. Nat. Commun. 2017, 8, 512. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Chen, F.V.; Van Dorpe, J.; Nitsch, R. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-P.; Arai, T.; Miklossy, J.; McGeer, P.L. Aβ and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 1953–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidler, P.M.; Boyer, D.R.; Rodriguez, J.A.; Sawaya, M.R.; Cascio, D.; Murray, K.; Gonen, T.; Eisenberg, D.S. Structure-based inhibitors of tau aggregation. Nat. Chem. 2018, 10, 170–176. [Google Scholar] [CrossRef]
- Griner, S.L.; Seidler, P.; Bowler, J.; Murray, K.A.; Yang, T.P.; Sahay, S.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Philipp, S.; et al. Structure-based inhibitors of amyloid beta core suggest a common interface with tau. eLife 2019, 8, e46924. [Google Scholar] [CrossRef]
- Tripathi, T.; Khan, H. Direct Interaction between the β-Amyloid Core and Tau Facilitates Cross-Seeding: A Novel Target for Therapeutic Intervention. Biochemistry 2020, 59, 341–342. [Google Scholar] [CrossRef] [Green Version]
- Bungeroth, M.; Appenzeller, S.; Regulin, A.; Völker, W.; Lorenzen, I.; Grötzinger, J.; Pendziwiat, M.; Kuhlenbäumer, G. Differential aggregation properties of alpha-synuclein isoforms. Neurobiol. Aging 2014, 35, 1913–1919. [Google Scholar] [CrossRef]
- Cardo, L.F.; Coto, E.; de Mena, L.; Ribacoba, R.; Mata, I.F.; Menéndez, M.; Moris, G.; Alvarez, V. Alpha-synuclein transcript isoforms in three different brain regions from Parkinson’s disease and healthy subjects in relation to the SNCA rs356165/rs11931074 polymorphisms. Neurosci. Lett. 2014, 562, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; McLean, P.J.; Sharma, N.; Hyman, B.T. Interaction of α-synuclein and synphilin-1: Effect of Parkinson’s disease-associated mutations. J. Neurochem. 2001, 77, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S.; Kaminsky, Z.; Guo, X.; Sharp, A.H.; Amaravi, R.K.; Kleiderlein, J.J.; Margolis, R.L.; Troncoso, J.C.; Lanahan, A.A.; Worley, P.F. Synphilin-1 associates with α-synuclein and promotes the formation of cytosolic inclusions. Nat. Genet. 1999, 22, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Szargel, R.; Rott, R.; Engelender, S. Synphilin-1 isoforms in Parkinson’s disease: Regulation by phosphorylation and ubiquitylation. Cell. Mol. Life Sci. 2008, 65, 80–88. [Google Scholar] [CrossRef]
- Power, J.H.; Barnes, O.L.; Chegini, F. Lewy Bodies and the Mechanisms of Neuronal Cell Death in Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol 2017, 27, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Lendel, C. Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology. Mol. Neurodegener. 2021, 16, 59. [Google Scholar] [CrossRef]
- Bassil, F.; Brown, H.J.; Pattabhiraman, S.; Iwasyk, J.E.; Maghames, C.M.; Meymand, E.S.; Cox, T.O.; Riddle, D.M.; Zhang, B.; Trojanowski, J.Q.; et al. Amyloid-Beta (Aβ) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Aβ Pathology. Neuron 2020, 105, 260–275.e6. [Google Scholar] [CrossRef]
- Bodles, A.M.; Guthrie, D.J.S.; Greer, B.; Irvine, G.B. Identification of the region of non -Aβ component (NAC) of Alzheimer’s disease amyloid responsible for its aggregation and toxicity. J. Neurochem. 2001, 78, 384–395. [Google Scholar] [CrossRef]
- Mandal, P.K.; Pettegrew, J.W.; Masliah, E.; Hamilton, R.L.; Mandal, R. Interaction between Aβ Peptide and α-Synuclein: Molecular Mechanisms in Overlapping Pathology of Alzheimer’s and Parkinson’s in Dementia with Lewy Body Disease. Neurochem. Res. 2006, 31, 1153–1162. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Solomon, T.; Sahoo, B.R.; Ignasiak, K.; Gaskin, S.; Rowles, J.; Verdile, G.; Howard, M.J.; Bond, C.S.; Ramamoorthy, A.; et al. Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells. Sci. Rep. 2020, 10, 10356. [Google Scholar] [CrossRef]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In Vivo Seeding and Cross-Seeding of Localized Amyloidosis: A Molecular Link between Type 2 Diabetes and Alzheimer Disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Lippa, C.F.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q. Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann. Neurol. 1999, 45, 353–357. [Google Scholar] [CrossRef]
- Badiola, N.; de Oliveira, R.M.; Herrera, F.; Guardia-Laguarta, C.; Gonçalves, S.A.; Pera, M.; Suárez-Calvet, M.; Clarimon, J.; Outeiro, T.F.; Lleó, A. Tau enhances α-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS ONE 2011, 6, e26609. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Guerrero-Muñoz, M.J.; Sengupta, U.; Gerson, J.E.; Kayed, R. α-Synuclein Oligomers Induce a Unique Toxic Tau Strain. Biol. Psychiatry 2018, 84, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Mroczek, K.; Fernando, S.; Fisher, P.R.; Annesley, S.J. Interactions and Cytotoxicity of Human Neurodegeneration—Associated Proteins Tau and α-Synuclein in the Simple Model Dictyostelium discoideum. Front. Cell Dev. Biol. 2021, 9, 741662. [Google Scholar] [CrossRef]
- Ambegaokar, S.S.; Roy, B.; Jackson, G.R. Neurodegenerative models in Drosophila: Polyglutamine disorders, Parkinson disease, and amyotrophic lateral sclerosis. Neurobiol. Dis. 2010, 40, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Bassil, F.; Meymand, E.S.; Brown, H.J.; Xu, H.; Cox, T.O.; Pattabhiraman, S.; Maghames, C.M.; Wu, Q.; Zhang, B.; Trojanowski, J.Q.; et al. α-Synuclein modulates tau spreading in mouse brains. J. Exp. Med. 2021, 218, e20192193. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Amat-Villegas, I.; Valenti-Azcarate, R.; Carmona-Abellan, M.D.M.; Marcilla, I.; TuÃon, M.-T.; Luquin, M.-R. Interaction of amyloidogenic proteins in pancreatic β cells from subjects with synucleinopathies. Acta Neuropathol. 2018, 135, 877–886. [Google Scholar] [CrossRef]
- Mucibabic, M.; Steneberg, P.; Lidh, E.; Straseviciene, J.; Ziolkowska, A.; Dahl, U.; Lindahl, E.; Edlund, H. α-Synuclein promotes IAPP fibril formation in vitro and β-cell amyloid formation in vivo in mice. Sci. Rep. 2020, 10, 20438. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, D.; Liu, Y.; Zhang, Y.; Zhou, Y.; Chang, Y.; Zheng, B.; Xu, A.; Zheng, J. A new strategy to reconcile amyloid cross-seeding and amyloid prevention in a binary system of α-synuclein fragmental peptide and hIAPP. Protein Sci. 2022, 31, 485–497. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Liu, Y.; Zhang, D.; Tang, Y.; Ren, B.; Zheng, J. Dual amyloid cross-seeding reveals steric zipper-facilitated fibrillization and pathological links between protein misfolding diseases. J. Mater. Chem. B 2021, 9, 3300–3316. [Google Scholar] [CrossRef] [PubMed]
- Pasieka, A.; Panek, D.; Szałaj, N.; Espargaró, A.; Więckowska, A.; Malawska, B.; Sabaté, R.; Bajda, M. Dual Inhibitors of Amyloid-β and Tau Aggregation with Amyloid-β Disaggregating Properties: Extended In Cellulo, In Silico, and Kinetic Studies of Multifunctional Anti-Alzheimer’s Agents. ACS Chem. Neurosci. 2021, 12, 2057–2068. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.-Q.; Xiao, X.; Yuan, J.; Puoti, G.; Fujioka, H.; Wang, X.; Richardson, S.; Zhou, X.; Zou, R.; Li, S. Amyloid-β42 interacts mainly with insoluble prion protein in the Alzheimer brain. J. Biol. Chem. 2011, 286, 15095–15105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aulić, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P. α-Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci. Rep. 2017, 7, 10050. [Google Scholar] [CrossRef]
- Parchi, P.; Zou, W.; Wang, W.; Brown, P.; Capellari, S.; Ghetti, B.; Kopp, N.; Schulz-Schaeffer, W.J.; Kretzschmar, H.A.; Head, M.W. Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10168–10172. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.; Jaunmuktane, Z.; Joiner, S.; Campbell, T.; Morgan, C.; Wakerley, B.; Golestani, F.; Rudge, P.; Mead, S.; Jäger, H.R. Variant Creutzfeldt–Jakob disease in a patient with heterozygosity at PRNP codon 129. N. Engl. J. Med. 2017, 376, 292–294. [Google Scholar] [CrossRef]
- Karamujić-Čomić, H.; Ahmad, S.; Lysen, T.S.; Heshmatollah, A.; Roshchupkin, G.V.; Vernooij, M.W.; Rozemuller, A.J.; Ikram, M.A.; Amin, N.; van Duijn, C.M. Prion protein codon 129 polymorphism in mild cognitive impairment and dementia: The Rotterdam Study. Brain Commun. 2020, 2, fcaa030. [Google Scholar] [CrossRef]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous prion protein genotype predisposes to sporadic Creutzfeld-Jakob disease. Nature 1991, 352, 342. [Google Scholar]
- Jefferies, E.; Lambon Ralph, M.A. Semantic impairment in stroke aphasia versus semantic dementia: A case-series comparison. Brain 2006, 129, 2132–2147. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.; Peden, A.H.; Wight, D.; Prowse, C.; MacGregor, I.; Manson, J.; Turner, M.; Ironside, J.W.; Head, M.W. Effects of human PrPSc type and PRNP genotype in an in-vitro conversion assay. Neuroreport 2008, 19, 1783–1786. [Google Scholar] [CrossRef]
- Jones, M.; Wight, D.; Rona Barron, M.J.; Manson, J.; Prowse, C.; Ironside, J.W.; Head, M.W. Molecular model of prion transmission to humans. Emerg. Infect. Dis. 2009, 15, 2013. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kaneko, K.; Matsumoto, G.; Kurosawa, M.; Nukina, N. Cross-Seeding Fibrillation of Q/N-Rich Proteins Offers New Pathomechanism of Polyglutamine Diseases. J. Neurosci. 2009, 29, 5153. [Google Scholar] [CrossRef] [PubMed]
- Chánez-Cárdenas, M.E.; Vázquez-Contreras, E. The aggregation of huntingtin and α-synuclein. J. Biophys. 2012, 2012, 606172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomás-Zapico, C.; Díez-Zaera, M.; Ferrer, I.; Gomez-Ramos, P.; Morán, M.A.; Miras-Portugal, M.T.; Díaz-Hernández, M.; Lucas, J.J. α-Synuclein accumulates in huntingtin inclusions but forms independent filaments and its deficiency attenuates early phenotype in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2012, 21, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Nag, N.; Tripathi, T. Cross-Seeding with Homologous Sequences Alters Amyloid Aggregation Kinetics and Fibril Structure. ACS Chem. Neurosci. 2022, 13, 537–539. [Google Scholar] [CrossRef]
- Holmgren, G.; Steen, L.; Ekstedt, J.; Groth, C.G.; Ericzon, B.G.; Eriksson, S.; Andersen, O.; Karlberg, I.; Nordén, G.; Nakazato, M. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin. Genet. 1991, 40, 242–246. [Google Scholar] [CrossRef]
- Stangou, A.J.; Heaton, N.D.; Rela, M.; Pepys, M.B.; Hawkins, P.N.; Williams, R. Domino hepatic transplantation using the liver from a patient with familial amyloid polyneuropathy. Transplantation 1998, 65, 1496–1498. [Google Scholar] [CrossRef]
- Koike, H.; Kiuchi, T.; Iijima, M.; Ueda, M.; Ando, Y.; Morozumi, S.; Tomita, M.; Kawagashira, Y.; Watanabe, H.; Katsuno, M. Systemic but asymptomatic transthyretin amyloidosis 8 years after domino liver transplantation. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1287–1290. [Google Scholar] [CrossRef]
- Misumi, Y.; Ueda, M.; Masuda, T.; Tsuda, Y.; Nomura, T.; Okada, M.; Inoue, Y.; Tasaki, M.; Obayashi, K.; Yamashita, T. Characteristics of acquired transthyretin amyloidosis: A case series and review of the literature. Neurology 2019, 93, e1587–e1596. [Google Scholar] [CrossRef]
- Yazaki, M.; Mitsuhashi, S.; Tokuda, T.; Kametani, F.; Takei, Y.I.; Koyama, J.; Kawamorita, A.; Kanno, H.; Ikeda, S.I. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am. J. Transplant. 2007, 7, 235–242. [Google Scholar] [CrossRef]
- Liepnieks, J.J.; Benson, M.D. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid 2007, 14, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Yamashita, T.; Ando, Y.; Ueda, M.; Misumi, Y.; Obayashi, K.; Horibata, Y.; Uchino, M. Evaluation of myocardial changes in familial amyloid polyneuropathy after liver transplantation. Intern. Med. 2008, 47, 2133–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maetani, Y.; Itoh, K.; Egawa, H.; Shibata, T.; Ametani, F.; Kubo, T.; Kiuchi, T.; Tanaka, K.; Konishi, J. Factors influencing liver regeneration following living-donor liver transplantation of the right hepatic lobe. Transplantation 2003, 75, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, M.; Fujita, Y.; Hijikuro, I.; Wada, M.; Uemura, T.; Kobayashi, Y.; Waku, T.; Tanaka, N.; Nishimoto, T.; Izumi, Y. PE859, a novel curcumin derivative, inhibits amyloid-β and tau aggregation, and ameliorates cognitive dysfunction in senescence-accelerated mouse prone 8. J. Alzheimers Dis. 2017, 59, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lu, H.; Li, J.; Liu, W.; Wu, Q.; Xu, Z.; Qiao, Q.; Zhang, H.; Gao, H.; Zhao, Q. A natural BACE1 and GSK3β dual inhibitor Notopterol effectively ameliorates the cognitive deficits in APP/PS1 Alzheimer’s mice by attenuating amyloid-β and tau pathology. Clin. Transl. Med. 2020, 10, e50. [Google Scholar] [CrossRef] [PubMed]
- Nan, S.; Wang, P.; Zhang, Y.; Fan, J. Epigallocatechin-3-Gallate Provides Protection Against Alzheimer’s Disease-Induced Learning and Memory Impairments in Rats. Drug Des. Dev. Ther. 2021, 13, 2013–2024. [Google Scholar] [CrossRef]
- Crowe, A.; James, M.J.; Lee, V.M.Y.; Smith, A.B.; Trojanowski, J.Q.; Ballatore, C.; Brunden, K.R. Aminothienopyridazines and Methylene Blue Affect Tau Fibrillization via Cysteine Oxidation. J. Biol. Chem. 2013, 288, 11024–11037. [Google Scholar] [CrossRef] [Green Version]
- Soeda, Y.; Saito, M.; Maeda, S.; Ishida, K.; Nakamura, A.; Kojima, S.; Takashima, A. Methylene Blue Inhibits Formation of Tau Fibrils but not of Granular Tau Oligomers: A Plausible Key to Understanding Failure of a Clinical Trial for Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1677–1686. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, K.; Kenney, M.A.; Young, A.A.; Rink, T.J. High affinity amylin binding sites in rat brain. Mol. Pharmacol. 1993, 44, 493–497. [Google Scholar]
- Andreetto, E.; Yan, L.M.; Tatarek-Nossol, M.; Velkova, A.; Frank, R.; Kapurniotu, A. Identification of Hot Regions of the Aβ-IAPP Interaction Interface as High-Affinity Binding Sites in both Cross- and Self-Association. Angew. Chem. Int. Ed. 2011, 49, 3081–3085. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Sharma, R.; Shrivastava, N.; Somvanshi, P.; Grover, A. Bleomycin modulates amyloid aggregation in β-amyloid and hIAPP. RSC Adv. 2020, 10, 25929–25946. [Google Scholar] [CrossRef]
- Ren, B.; Liu, Y.; Zhang, Y.; Cai, Y.; Gong, X.; Chang, Y.; Xu, L.; Zheng, J. Genistein: A Dual Inhibitor of Both Amyloid β and Human Islet Amylin Peptides. ACS Chem. Neurosci. 2018, 9, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Bruno, E.; Pereira, C.; Roman, K.P.; Takiguchi, M.; Kao, P.-Y.; Nogaj, L.A.; Moffet, D.A. IAPP aggregation and cellular toxicity are inhibited by 1,2,3,4,6-penta-O-galloyl-β-d-glucose. Amyloid 2013, 20, 34–38. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, N.E.C.; Do, T.D.; LaPointe, N.E.; Tro, M.; Feinstein, S.C.; Shea, J.-E.; Bowers, M.T. 1,2,3,4,6-penta-O-galloyl-β-d-glucopyranose binds to the N-terminal metal binding region to inhibit amyloid β-protein oligomer and fibril formation. Int. J. Mass Spectrom. 2017, 420, 24–34. [Google Scholar] [CrossRef]
- Ren, B.; Liu, Y.; Zhang, Y.; Zhang, M.; Sun, Y.; Liang, G.; Xu, J.; Zheng, J. Tanshinones inhibit hIAPP aggregation, disaggregate preformed hIAPP fibrils, and protect cultured cells. J. Mater. Chem. B 2018, 6, 56–67. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Eleuteri, S.; Paleologou, K.E.; Yin, G.; Zweckstetter, M.; Carrupt, P.-A.; Lashuel, H.A. Entacapone and tolcapone, two catechol O-methyltransferase inhibitors, block fibril formation of alpha-synuclein and beta-amyloid and protect against amyloid-induced toxicity. J. Biol. Chem. 2010, 285, 14941–14954. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Yamada, M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for α-synuclein fibrils in vitro. J. Neurochem. 2006, 97, 105–115. [Google Scholar] [CrossRef]
- Gabr, M.T.; Peccati, F. Dual Targeting of Monomeric Tau and α-Synuclein Aggregation: A New Multitarget Therapeutic Strategy for Neurodegeneration. ACS Chem. Neurosci. 2020, 11, 2051–2057. [Google Scholar] [CrossRef]
Figure 1.
Amyloid seeding and aggregation. The addition of preformed seeds reduces the lag phase leading to faster aggregation. The seeds can be homologous or heterologous. Homologous seeds, which have the same nature as the existing nuclei, lead to homologous seeding, whereas the heterologous seeds differ from the initial nuclei and lead to heterologous or cross-seeding.
Figure 1.
Amyloid seeding and aggregation. The addition of preformed seeds reduces the lag phase leading to faster aggregation. The seeds can be homologous or heterologous. Homologous seeds, which have the same nature as the existing nuclei, lead to homologous seeding, whereas the heterologous seeds differ from the initial nuclei and lead to heterologous or cross-seeding.

Figure 2.
Human APP cleavage pathway. The human APP undergoes proteolytic cleavage in two different pathways: amyloidogenic and non-amyloidogenic. In the non-amyloidogenic pathway, the α-secretases cleave within the Aβ domain to form the α-C terminal fragments (CTFα), and the N-terminal soluble APP (sAPPα). The CTFα is subsequently cleaved by γ-secretase to form P3 and APP intracellular domain (AICD). In the amyloidogenic pathway, the β-secretase initially cleaves APP to form β-C-terminal fragments (CTFβ) and N-terminal-soluble APP (sAPPβ). The CTFβ is then cleaved by γ-secretase to form extracellular Aβ and AICD. The arrangement of Aβ40 (red) (PDB ID: 6TI5) [65] and Aβ42 (green) (PDB ID: 2BEG) [66] in the fibrillar phase are shown on the right. The β-sheet structure of the peptide and the parallel direction can be observed in the figure.
Figure 2.
Human APP cleavage pathway. The human APP undergoes proteolytic cleavage in two different pathways: amyloidogenic and non-amyloidogenic. In the non-amyloidogenic pathway, the α-secretases cleave within the Aβ domain to form the α-C terminal fragments (CTFα), and the N-terminal soluble APP (sAPPα). The CTFα is subsequently cleaved by γ-secretase to form P3 and APP intracellular domain (AICD). In the amyloidogenic pathway, the β-secretase initially cleaves APP to form β-C-terminal fragments (CTFβ) and N-terminal-soluble APP (sAPPβ). The CTFβ is then cleaved by γ-secretase to form extracellular Aβ and AICD. The arrangement of Aβ40 (red) (PDB ID: 6TI5) [65] and Aβ42 (green) (PDB ID: 2BEG) [66] in the fibrillar phase are shown on the right. The β-sheet structure of the peptide and the parallel direction can be observed in the figure.

Figure 4.
Domain structure of the α-syn protein. The lipid-binding N-terminal domain (NTD) is represented in orange, while the non-Aβ component (NAC) region necessary for the aggregation of α-syn protein is in blue. The C-terminal domain (CTD) of α-syn protein is represented in green. On top, the arrangement of α-syn protein in fibrillar form is shown (PDB ID: 6FLT) [27].
Figure 4.
Domain structure of the α-syn protein. The lipid-binding N-terminal domain (NTD) is represented in orange, while the non-Aβ component (NAC) region necessary for the aggregation of α-syn protein is in blue. The C-terminal domain (CTD) of α-syn protein is represented in green. On top, the arrangement of α-syn protein in fibrillar form is shown (PDB ID: 6FLT) [27].

Figure 5.
Schematic representation of hIAPP aggregation kinetics. The sigmoidal curves house the different forms: monomer, oligomer, protofibrils, and fibrils of hIAPP that are formed at various phases with time. The representation is shown for kinetics without seeding. The structures of different forms of hIAPP are shown (PDB IDs: 2KIB and 6VW2) [110,111].
Figure 5.
Schematic representation of hIAPP aggregation kinetics. The sigmoidal curves house the different forms: monomer, oligomer, protofibrils, and fibrils of hIAPP that are formed at various phases with time. The representation is shown for kinetics without seeding. The structures of different forms of hIAPP are shown (PDB IDs: 2KIB and 6VW2) [110,111].

Figure 6.
Domain structure of PrP protein. The signal peptide that guides the PrP protein is represented in red, with the octapeptide in orange. The globular C-terminal domain (CTD) with α-helices and β-sheets are shown in purple. The hydrophobic domain (HD) between the octapeptide and the CTD is colored in blue. The fibril form of human PrP is shown at the top (PDB ID: 6UUR) [126].
Figure 6.
Domain structure of PrP protein. The signal peptide that guides the PrP protein is represented in red, with the octapeptide in orange. The globular C-terminal domain (CTD) with α-helices and β-sheets are shown in purple. The hydrophobic domain (HD) between the octapeptide and the CTD is colored in blue. The fibril form of human PrP is shown at the top (PDB ID: 6UUR) [126].

Figure 7.
General mechanism of dual inhibitors of cross-seeding: (A) in the absence of an inhibitor, protein A acts as a seed and facilitates the cross-seeding of protein B; (B) in the presence of a dual inhibitor, the toxicity of the amyloid fibers of protein A is reduced, and the cross-seeding of protein B is also inhibited.
Figure 7.
General mechanism of dual inhibitors of cross-seeding: (A) in the absence of an inhibitor, protein A acts as a seed and facilitates the cross-seeding of protein B; (B) in the presence of a dual inhibitor, the toxicity of the amyloid fibers of protein A is reduced, and the cross-seeding of protein B is also inhibited.


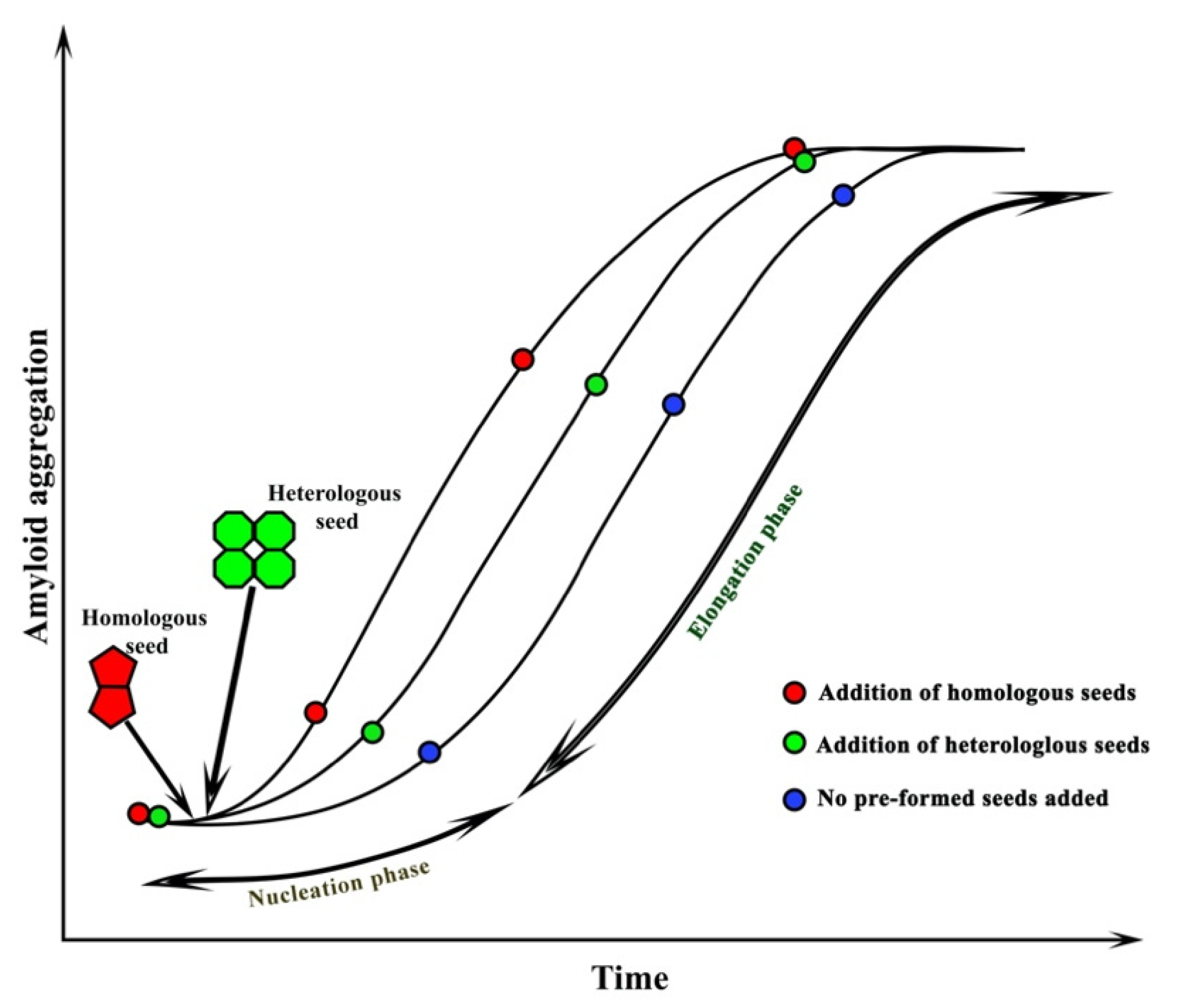{kind=link}
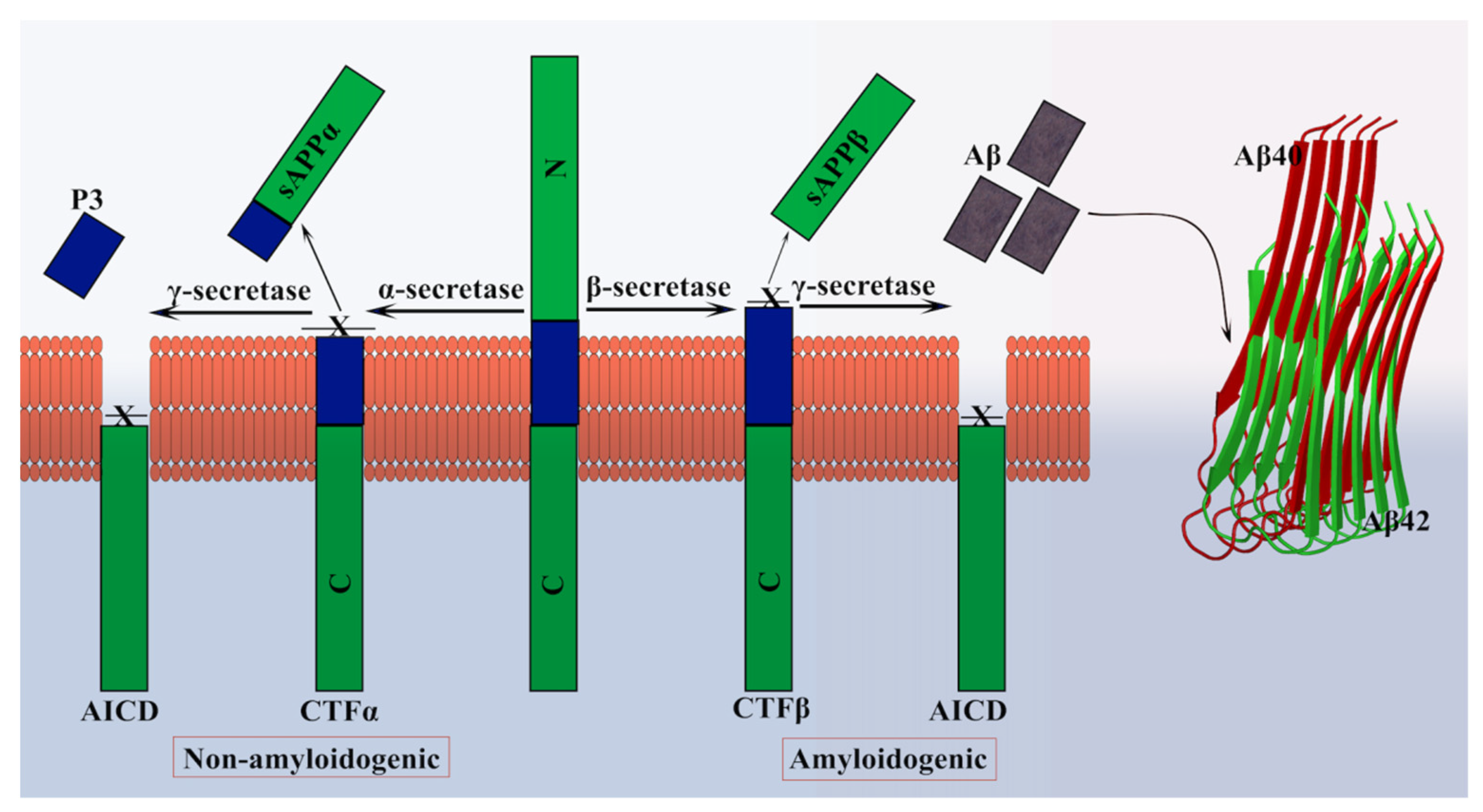{kind=link}
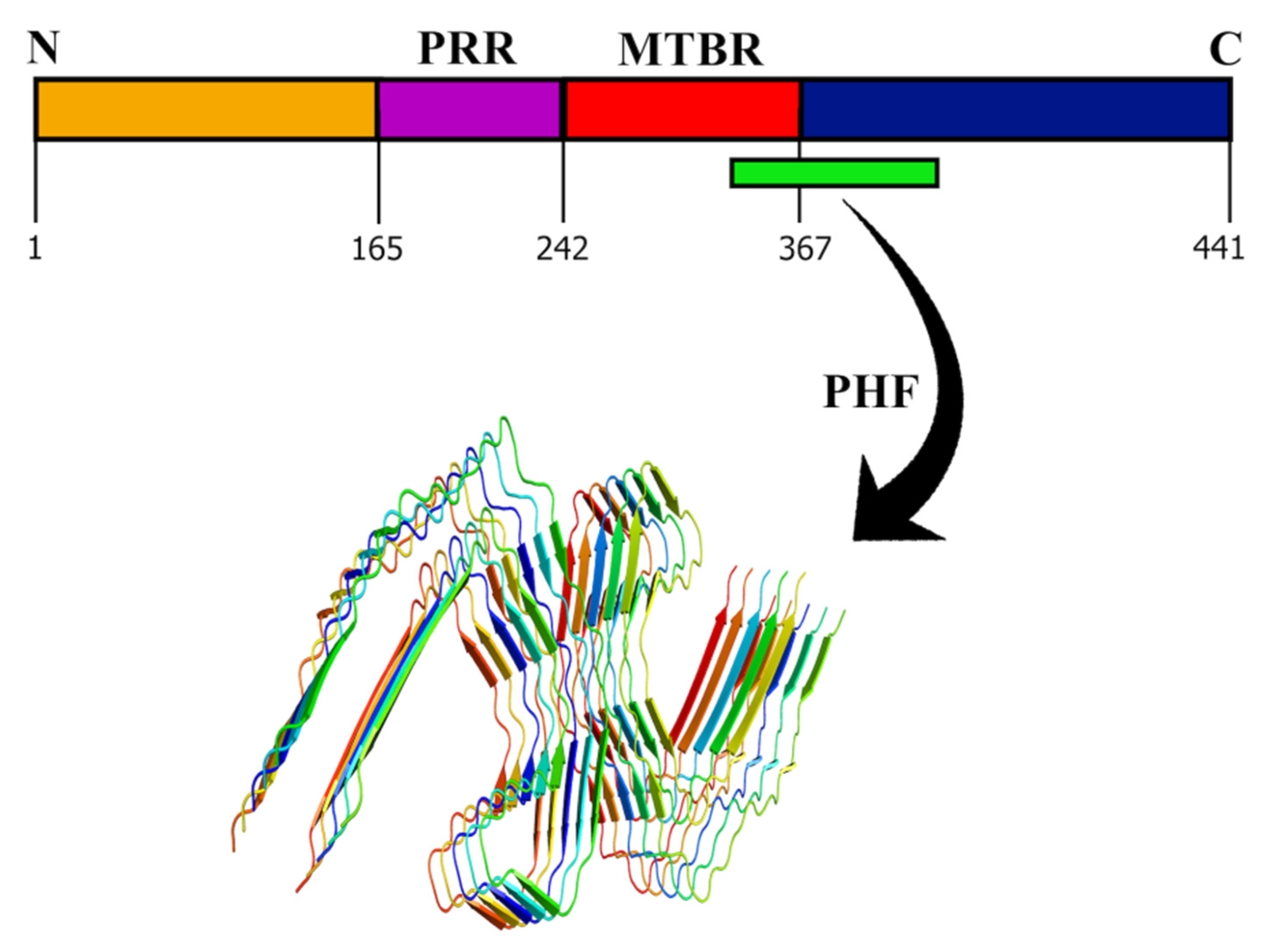{kind=link}
{kind=link}
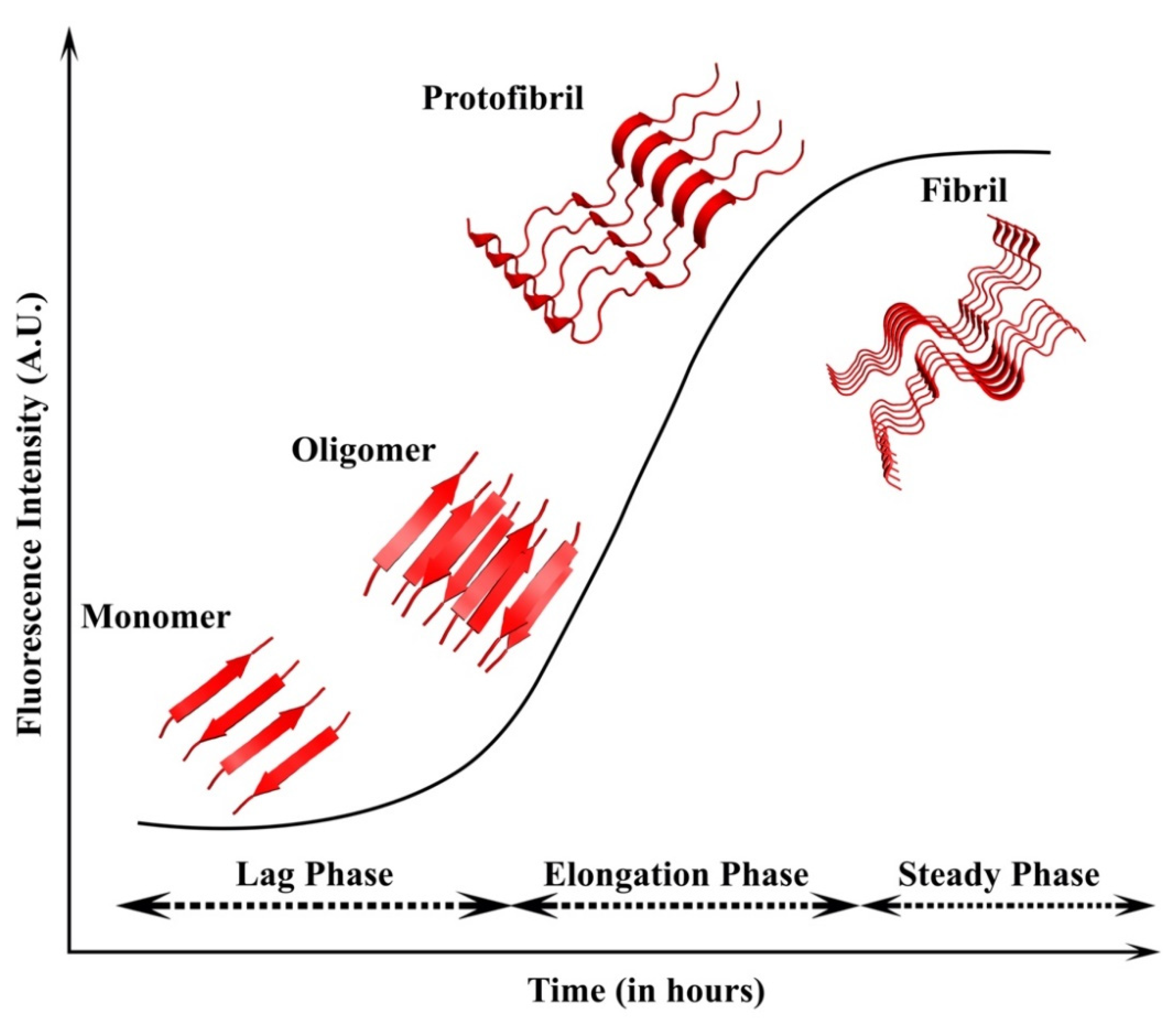{kind=link}
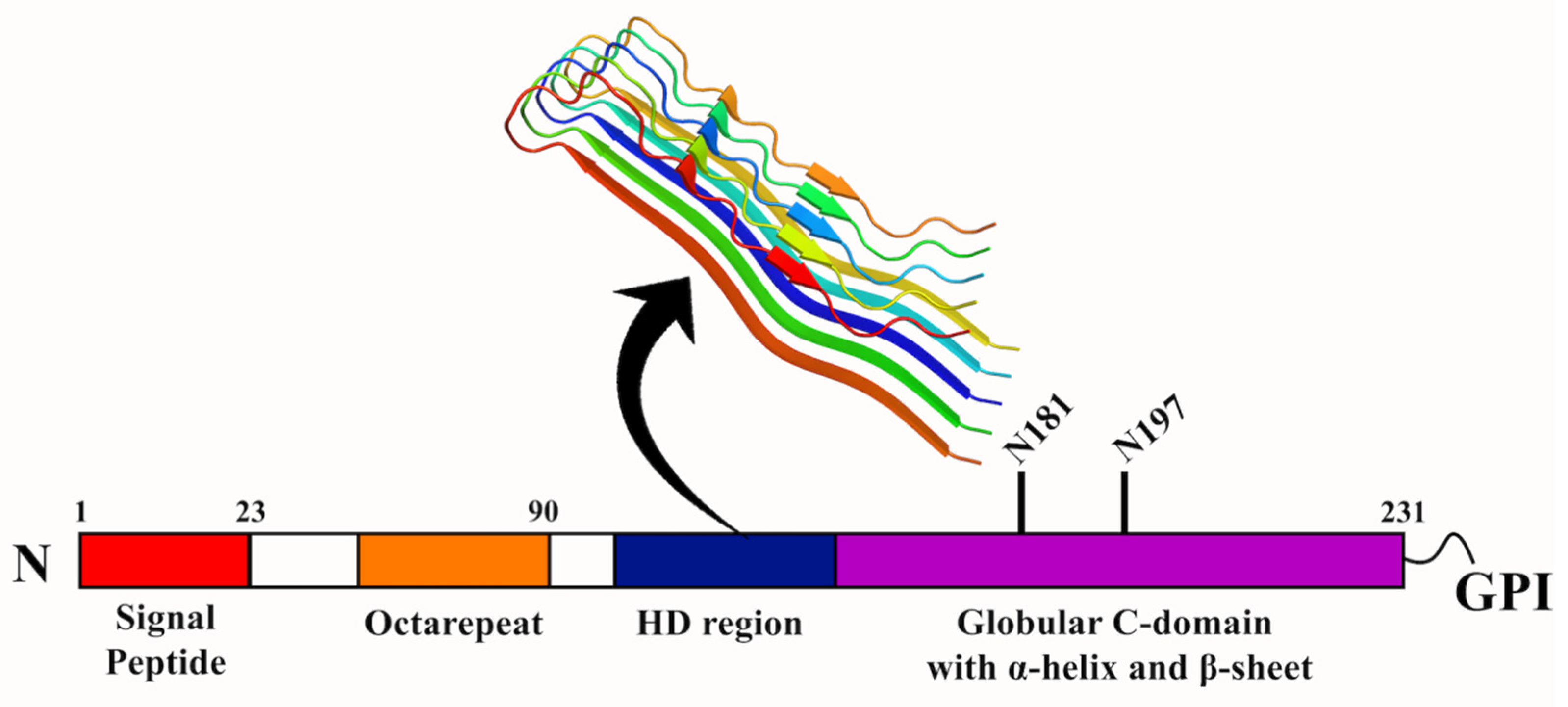{kind=link}
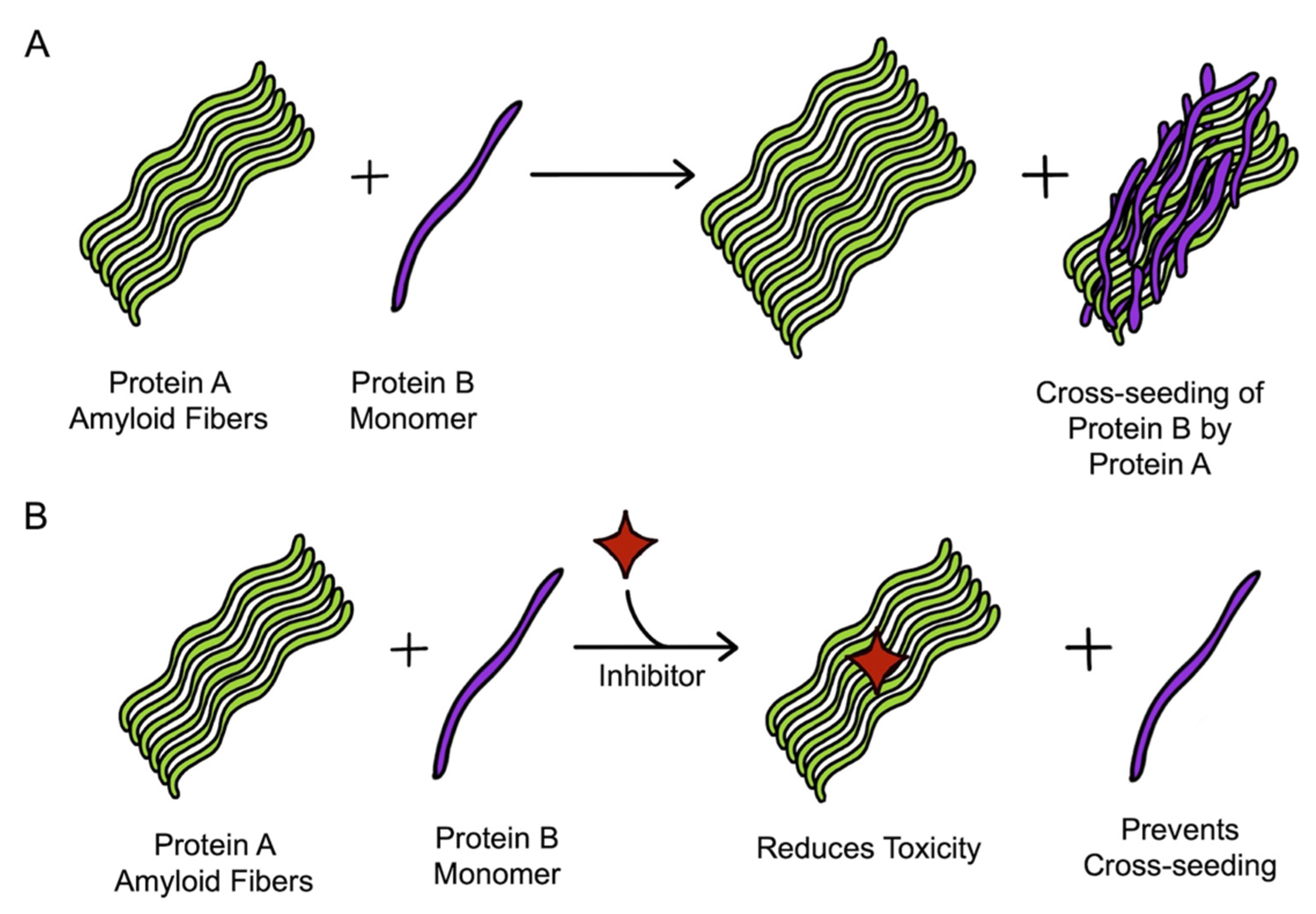{kind=link}
Table 1.
List of diseases and the cross-seeding amyloid proteins found by different experimental and computational approaches.
Table 1.
List of diseases and the cross-seeding amyloid proteins found by different experimental and computational approaches.
| Disease | Cross-Seeding Proteins | Ref. |
|---|---|---|
| Alzheimer’s disease | Aβ42-PrPsc | [57] |
| Aβ40-Aβ42 | [136] | |
| Aβ40-hIAPP37 | [56] | |
| Aβ24–34-hIAPP19–29S20G | [137] | |
| Aβ-ASC specks | [138] | |
| Aβ-casein, fibroin, sericin, actin, and IAPP | [139] | |
| Tau-Aβ42 | [53] | |
| Tau-αS | [55] | |
| Aβ42-Tau K18 | [140] | |
| Aβ42-Tau K19 | [140] | |
| Mutated Tau (R2)-Aβ17–42 | [141] | |
| Aβ42-A2T Aβ42 | [142] | |
| Aβ17–42-Tau (R2, R3 and R4) | [143] | |
| Tau K18-Tau K19 | [144] | |
| Aβ40-hIAPP37 | [145] | |
| Aβ42-hIAPP37 | [146] | |
| Aβ17–42-hIAPP37 | [147] | |
| Parkinson’s disease | αShuman-αSmouse | [148] |
| αSmouse-N-terminal truncated αShuman | [149] | |
| αSmouse-C-terminal truncated αShuman | [149] | |
| αShuman-Aβ | [54] | |
| αShuman-quinolinic acid | [150] | |
| Non-β-component of αS-Aβ42 | [151] | |
| Prion disease | PrP120–144 various species-PrP120–144 various species | [152] |
| PrP120–144 various species-PrP23–144 various species | [39] | |
| PrPc-αS | [153] | |
| PrP106–126-hIAPP | [154,155] | |
| Type 2 diabetes | hIAPP37-Aβ42 | [156] |
| rIAPP37-hIAPP37 | [58] | |
| hIAPP-Aβ | [156] | |
| hIAPP-rIAPP | [157,158] | |
| hIAPP37-Aβ17–42 | [159] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Subedi, S.; Sasidharan, S.; Nag, N.; Saudagar, P.; Tripathi, T. Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition. Molecules 2022, 27, 1776. https://doi.org/10.3390/molecules27061776
AMA Style
Subedi S, Sasidharan S, Nag N, Saudagar P, Tripathi T. Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition. Molecules. 2022; 27(6):1776. https://doi.org/10.3390/molecules27061776
Chicago/Turabian StyleSubedi, Sushma, Santanu Sasidharan, Niharika Nag, Prakash Saudagar, and Timir Tripathi. 2022. "Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition" Molecules 27, no. 6: 1776. https://doi.org/10.3390/molecules27061776