Computational Approaches for the Design of Novel Anticancer Compounds Based on Pyrazolo[3,4-d]pyrimidine Derivatives as TRAP1 Inhibitor

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Study of Pharmacophore Development
2.3. Pharmacophore Hypothesis Generation
2.4. Model Development by Three-Dimensional QSAR Study
2.4.1. Atom-Based QSAR
2.4.2. Generation of Contour Maps
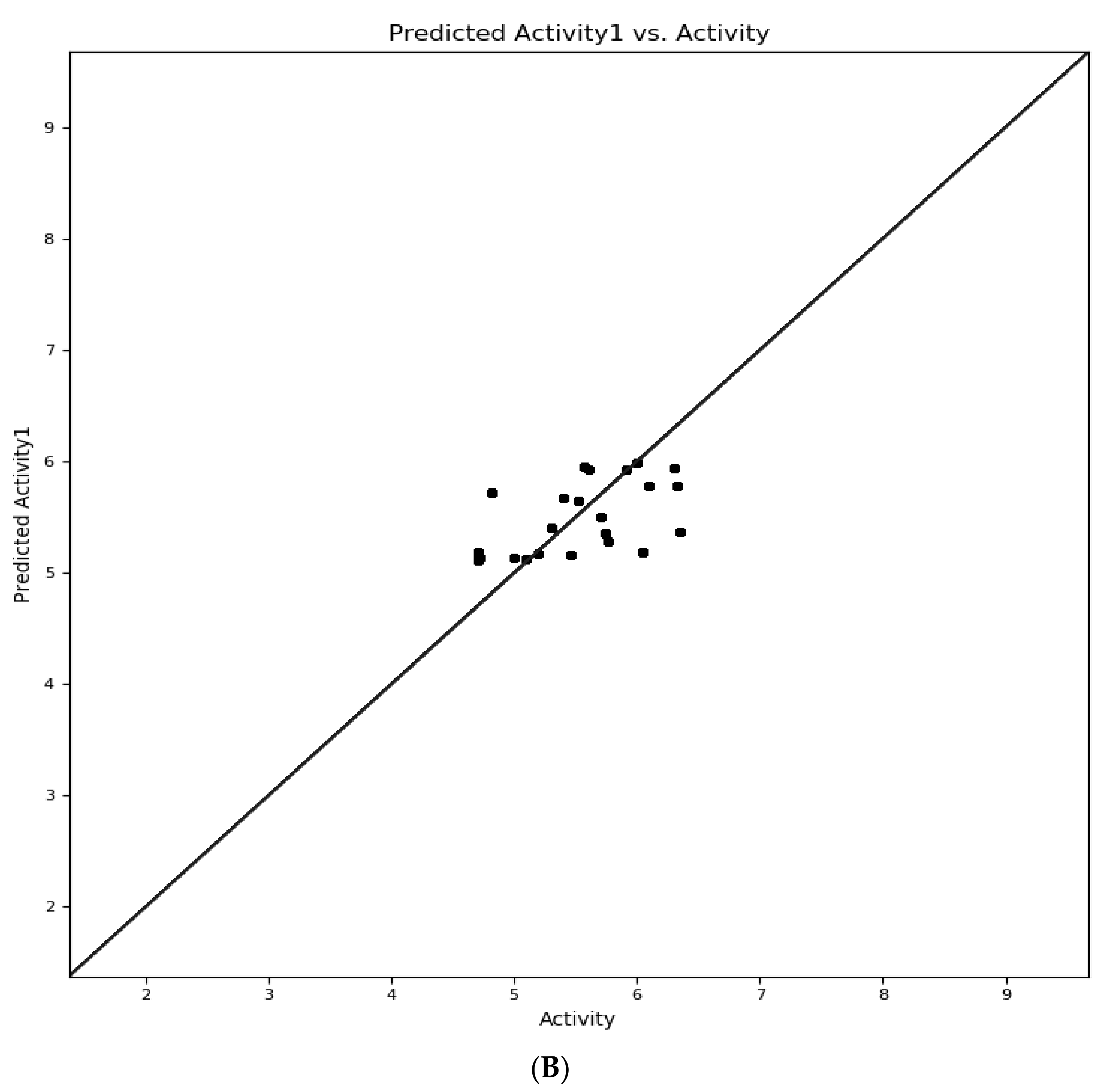
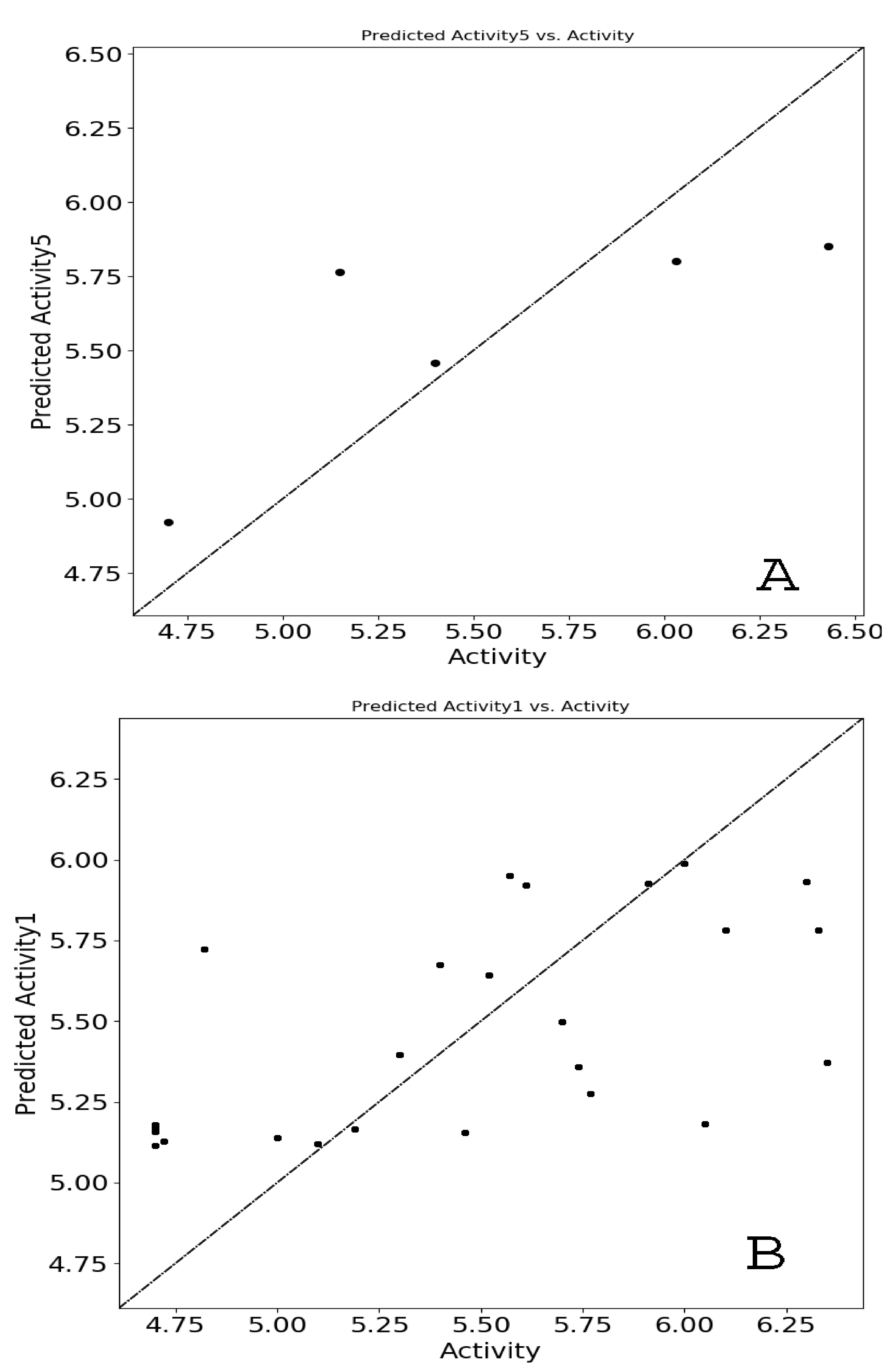
2.4.3. D-QSAR Model Evaluation
2.5. Virtual Screening Studies in the ZINC Database
2.6. Docking Study
2.7. Molecular Dynamics Simulations
2.8. Absorption, Distribution, Metabolism, and Excretion (ADME) Property Predictions
2.9. Enumeration Study
3. Results and Discussion
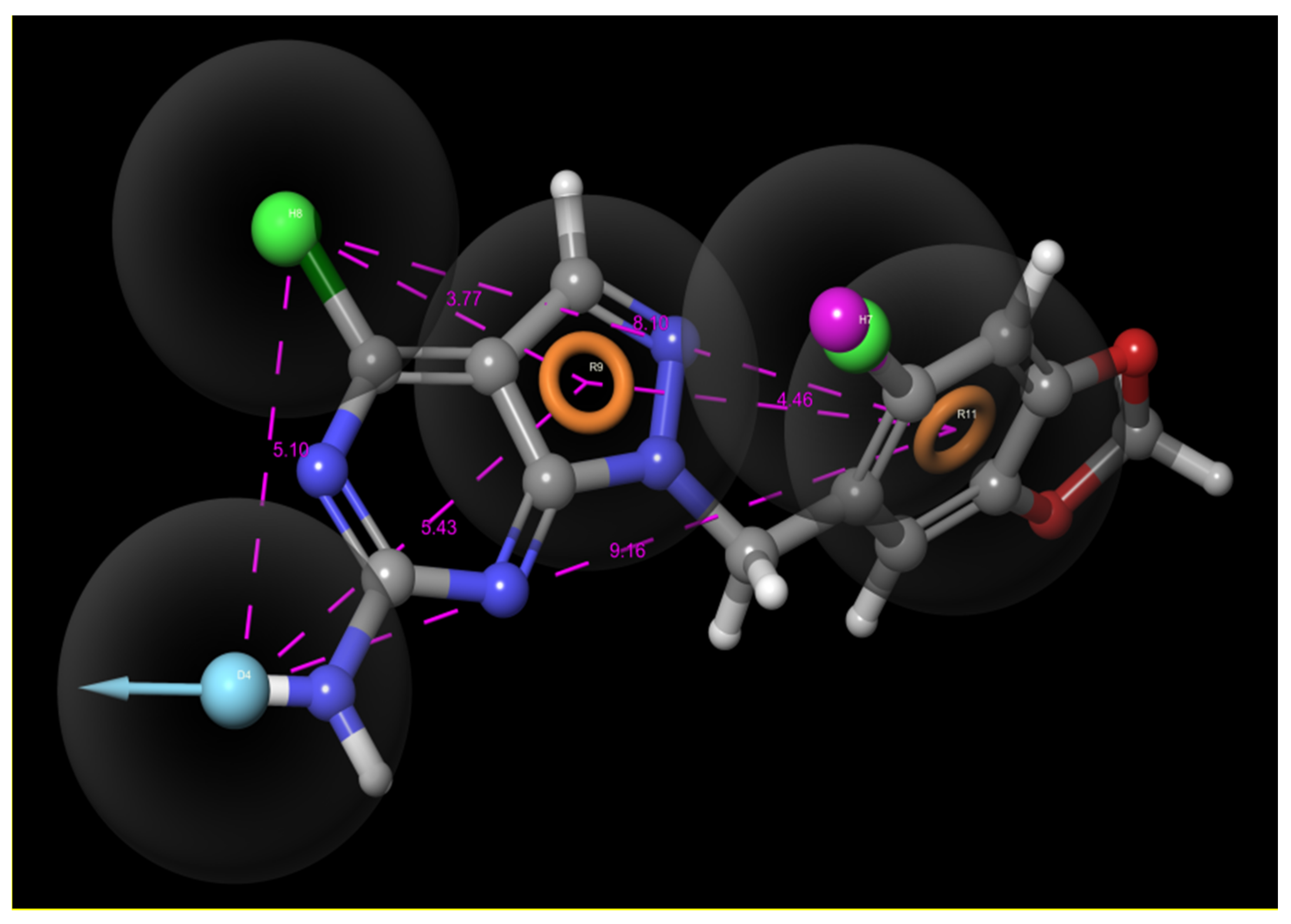
3.1. Selection of Best Pharmacophore Hypothesis
3.2. Pharmacophore Model Evaluation
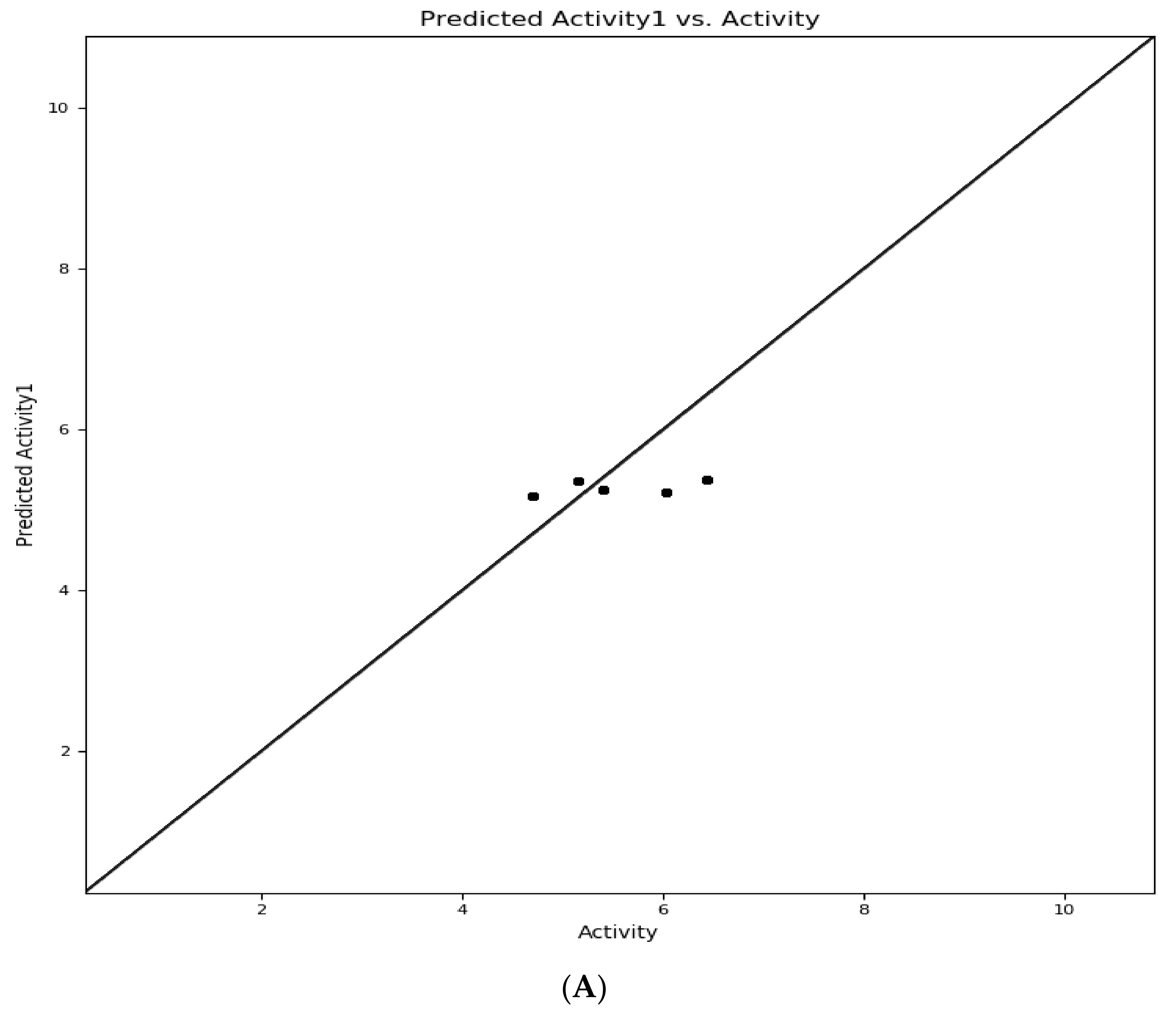
3.3. Selection of Atom-Based QSARmodel
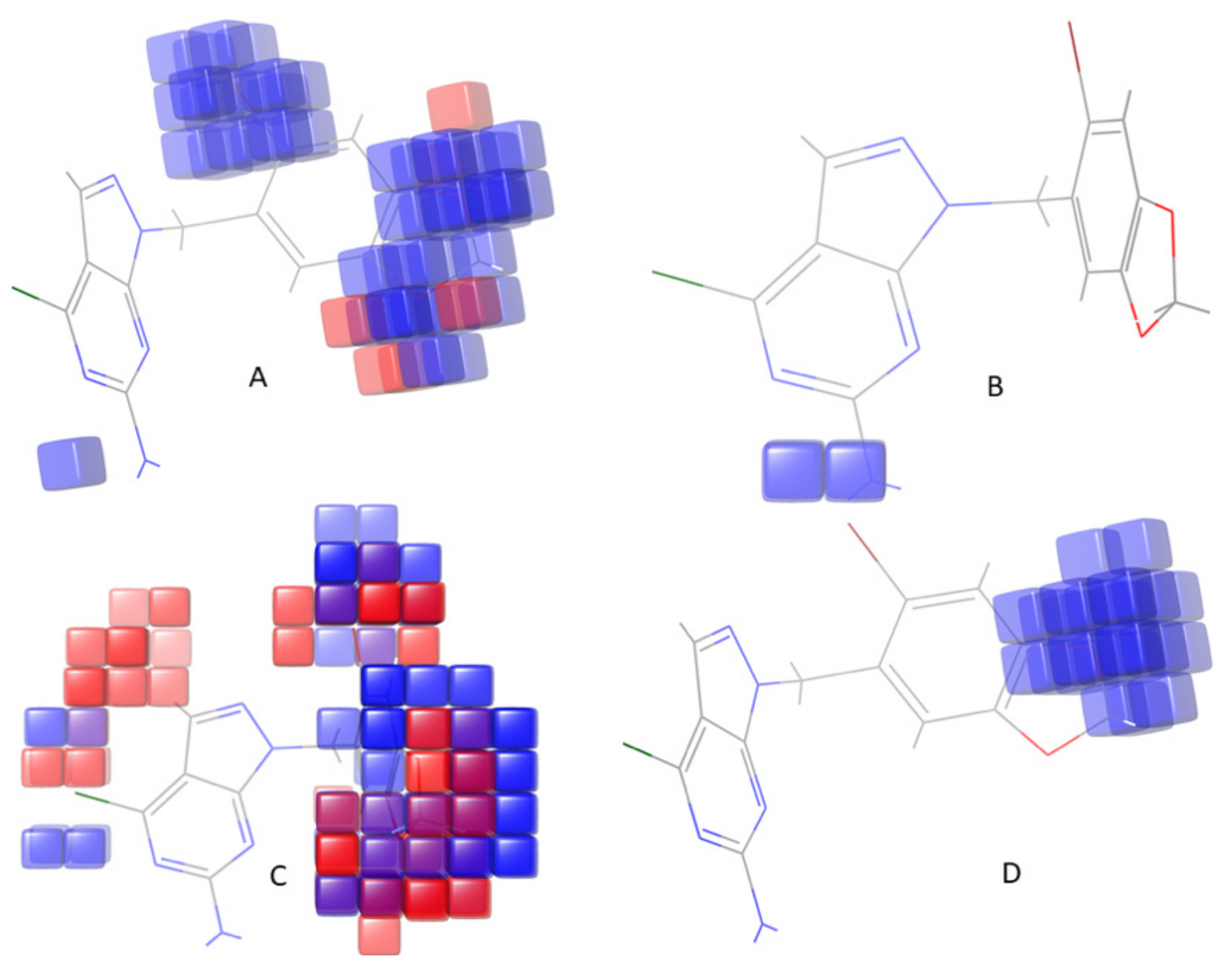
3.4. Contour Map Analysis
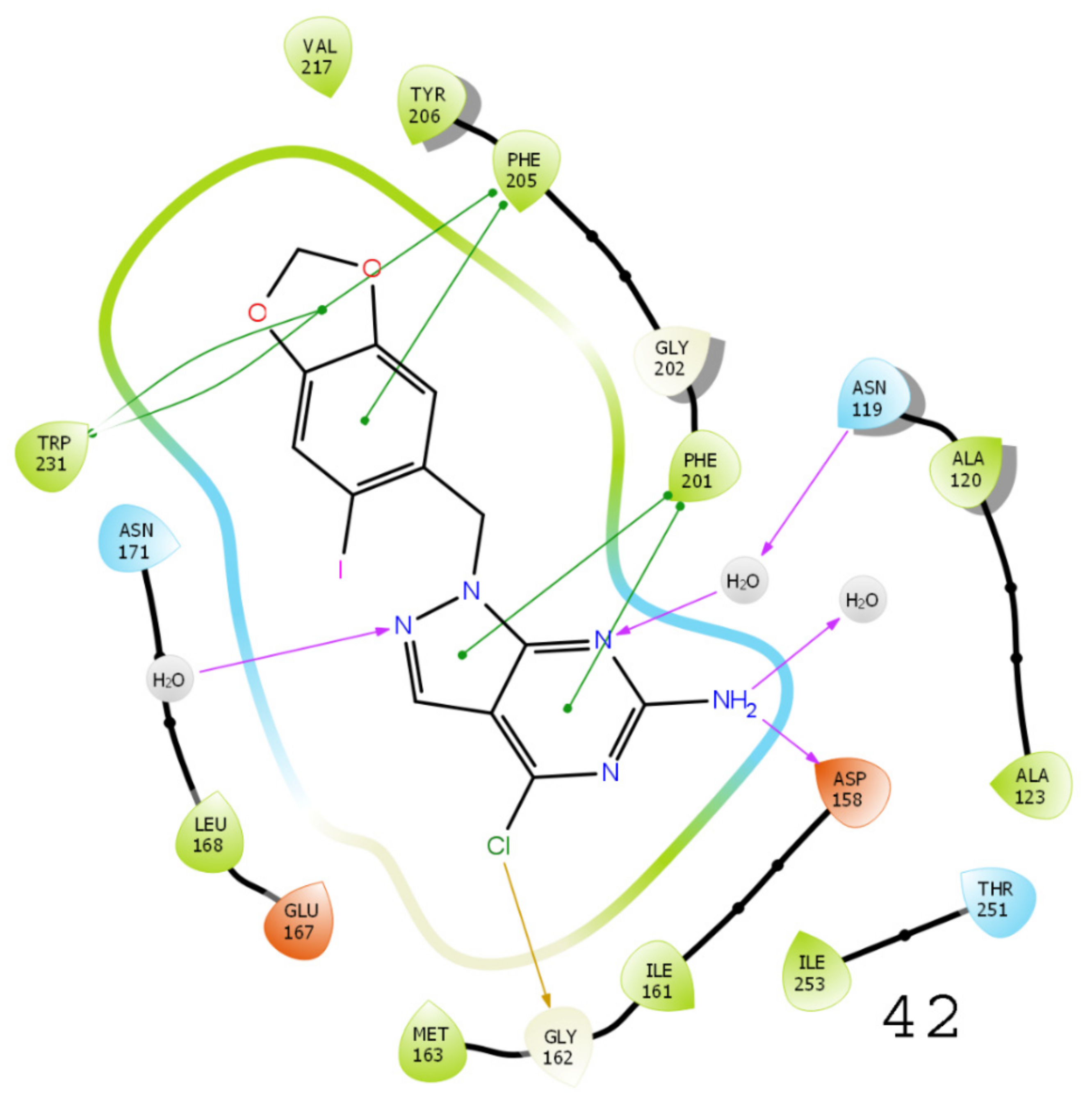
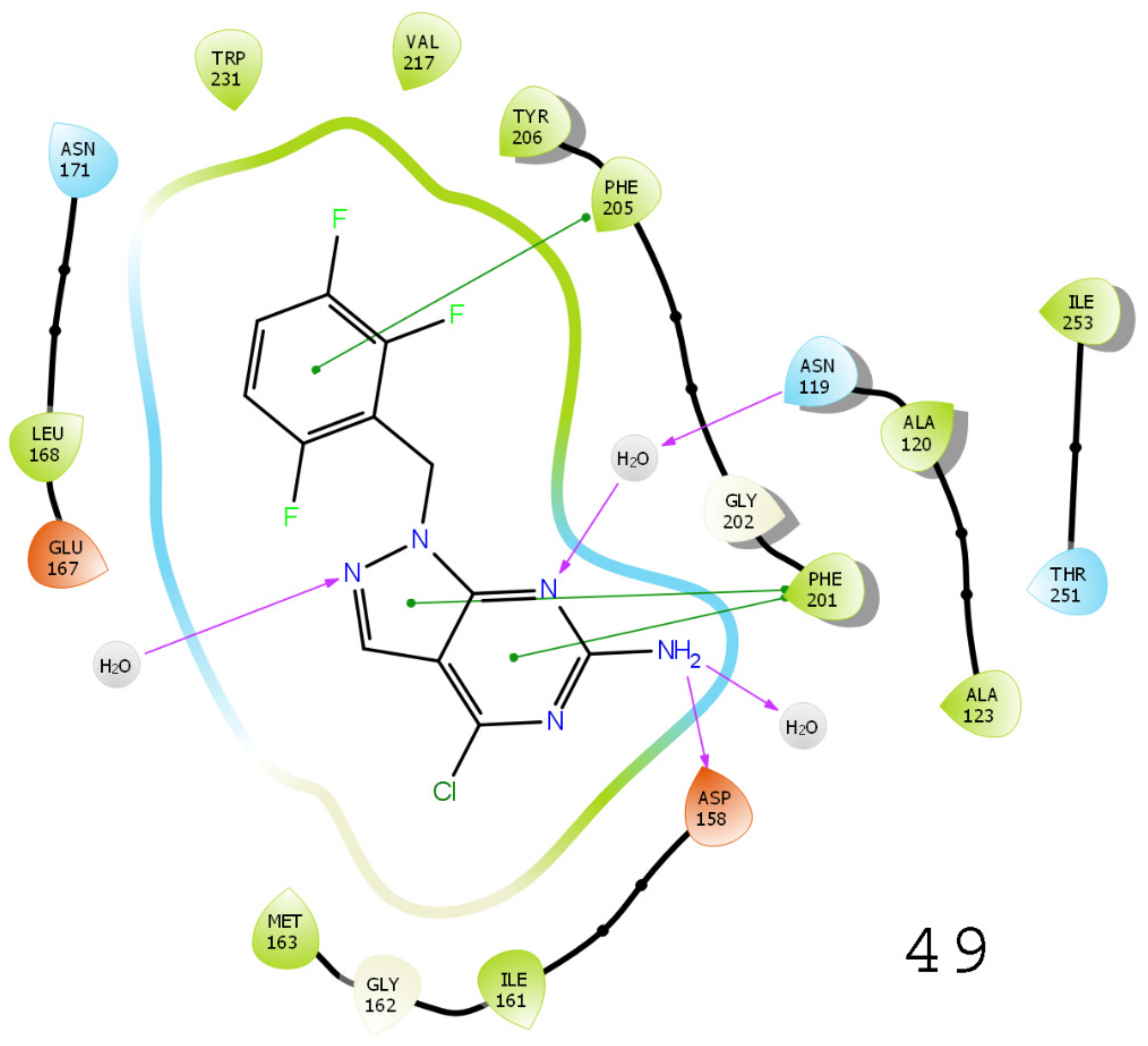
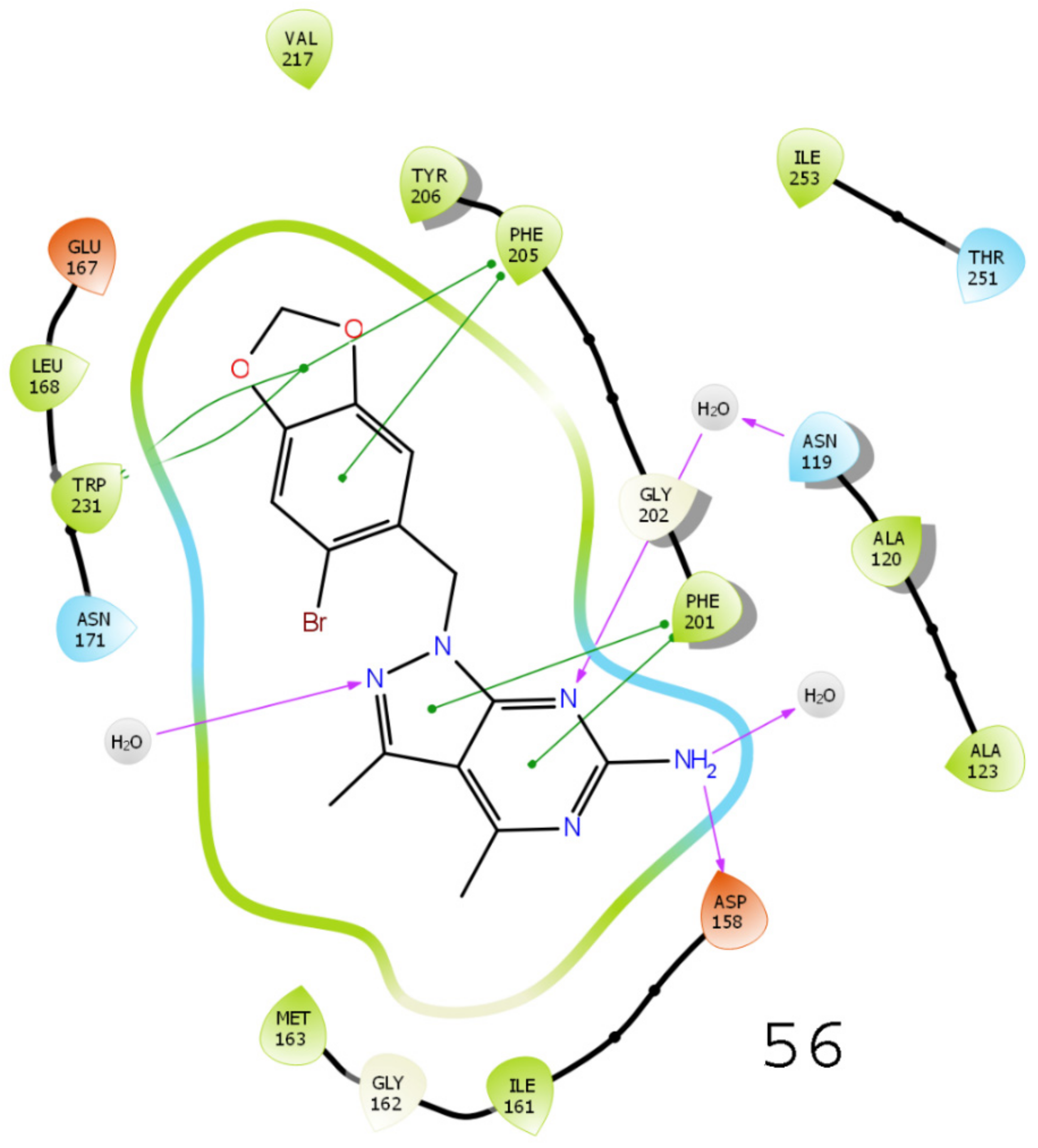
3.5. Results of Molecular Docking
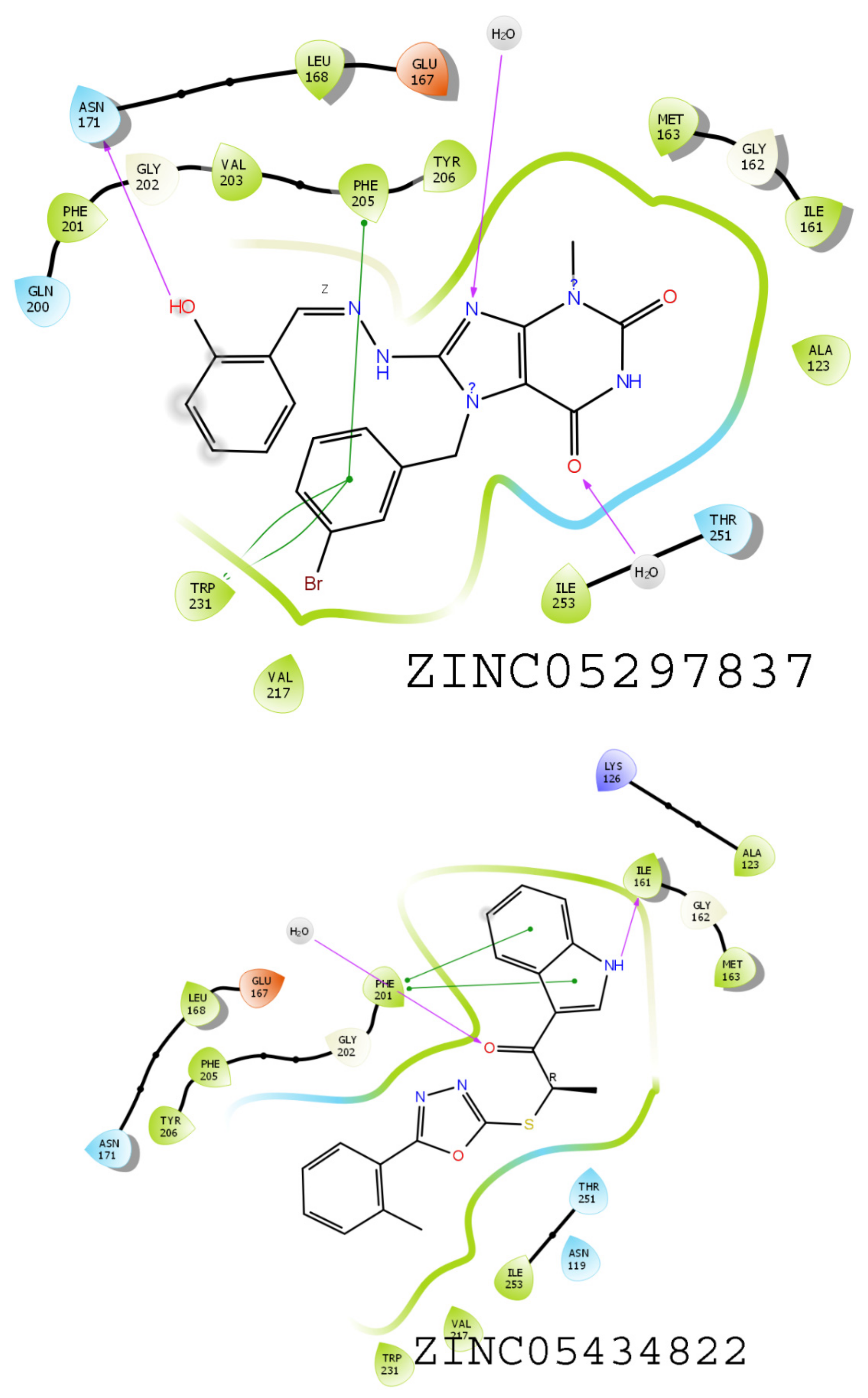
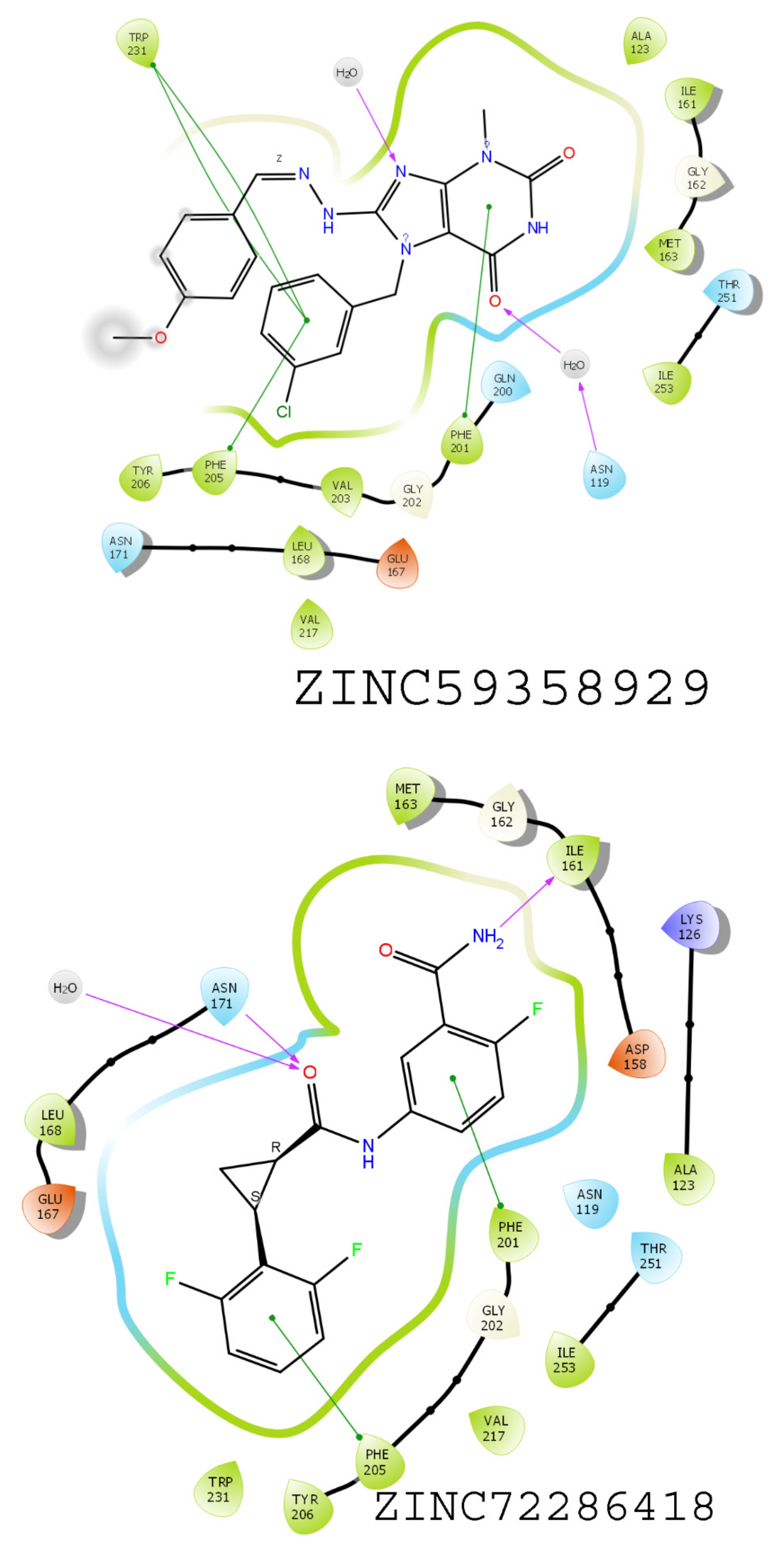
3.6. Results of Virtual Screening
3.7. MMGBSA-Based Rescoring
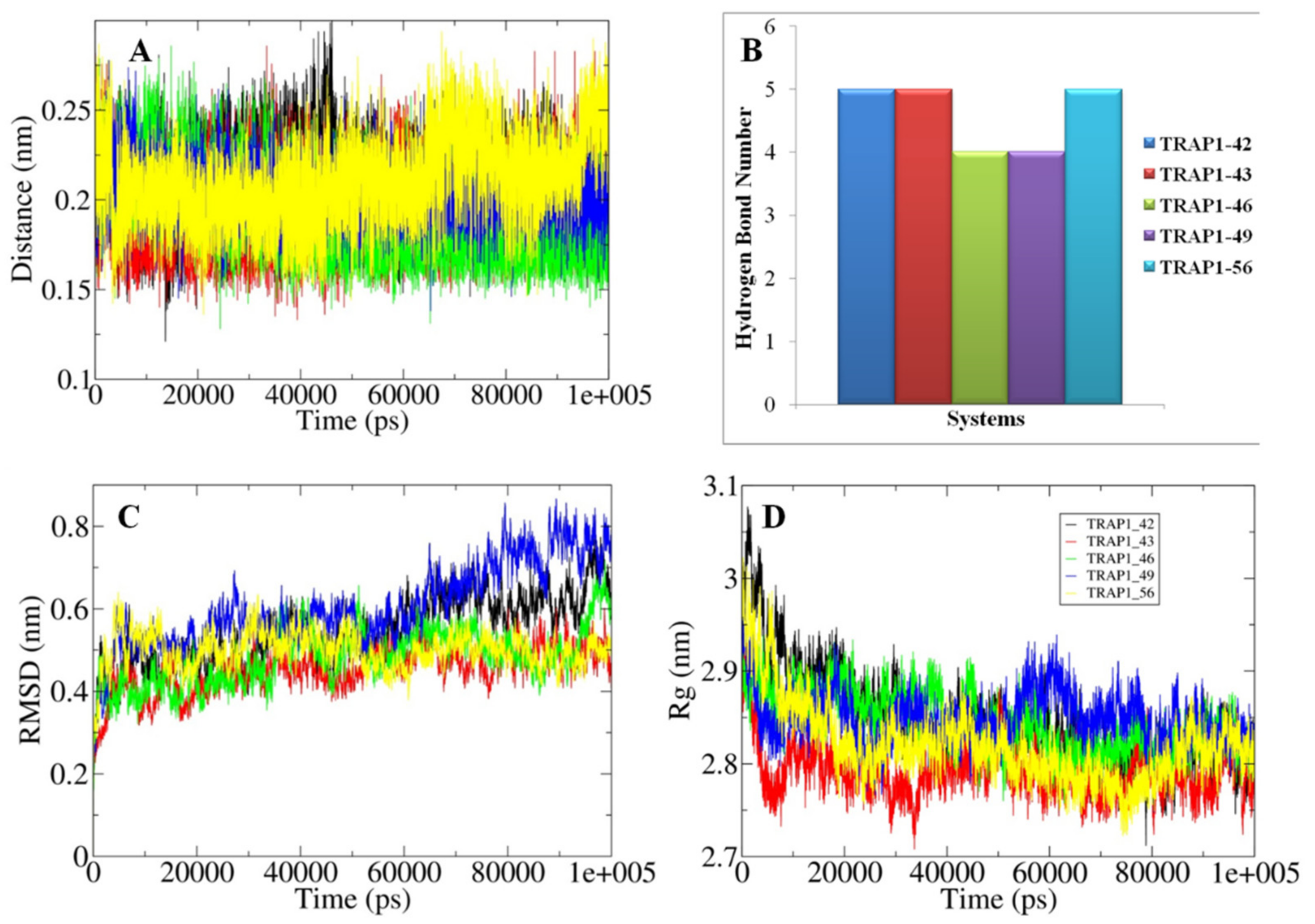
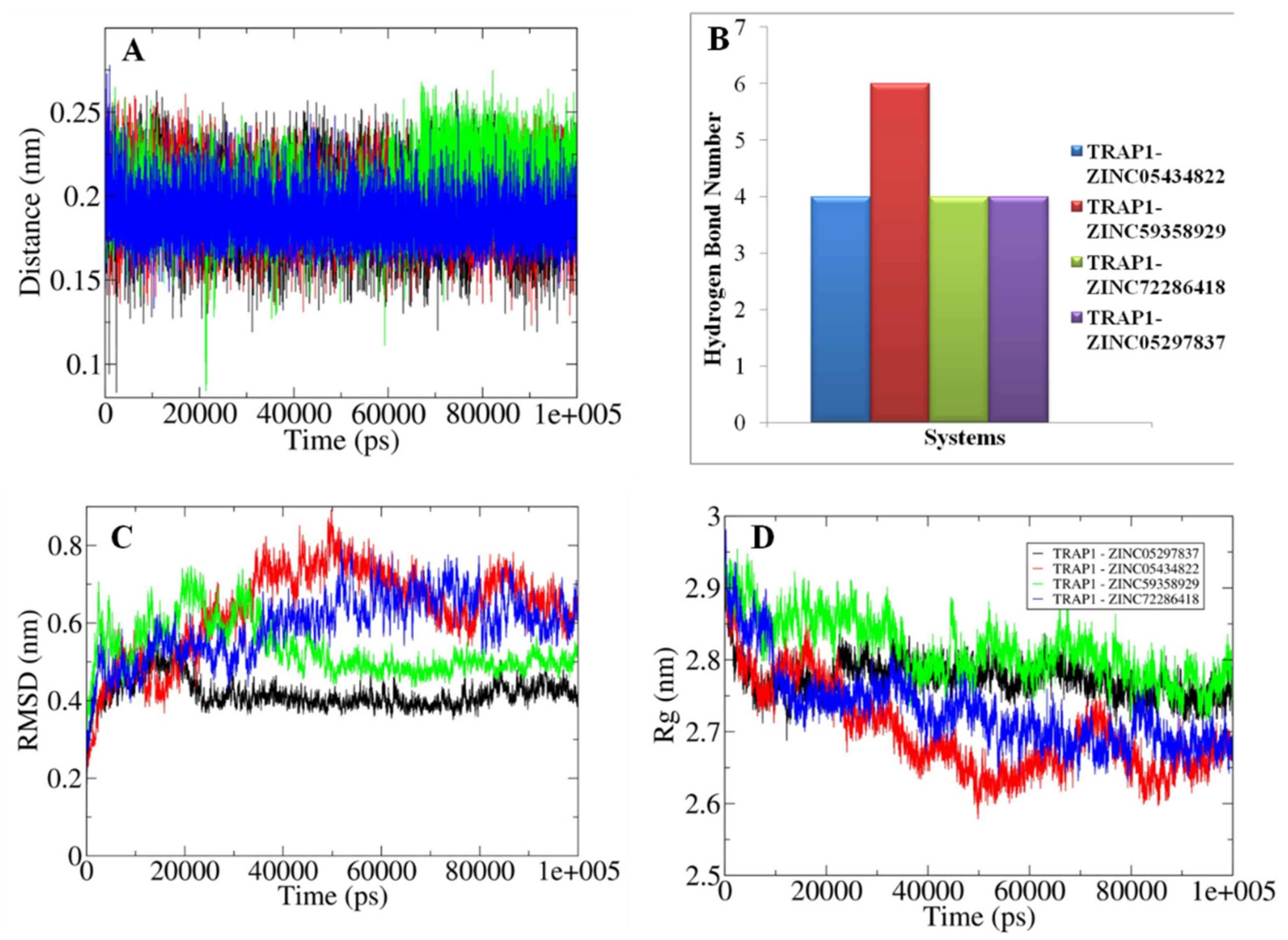
3.8. Analyses of the Conformations Obtained through MD Simulations
3.9. Prediction of ADME Properties
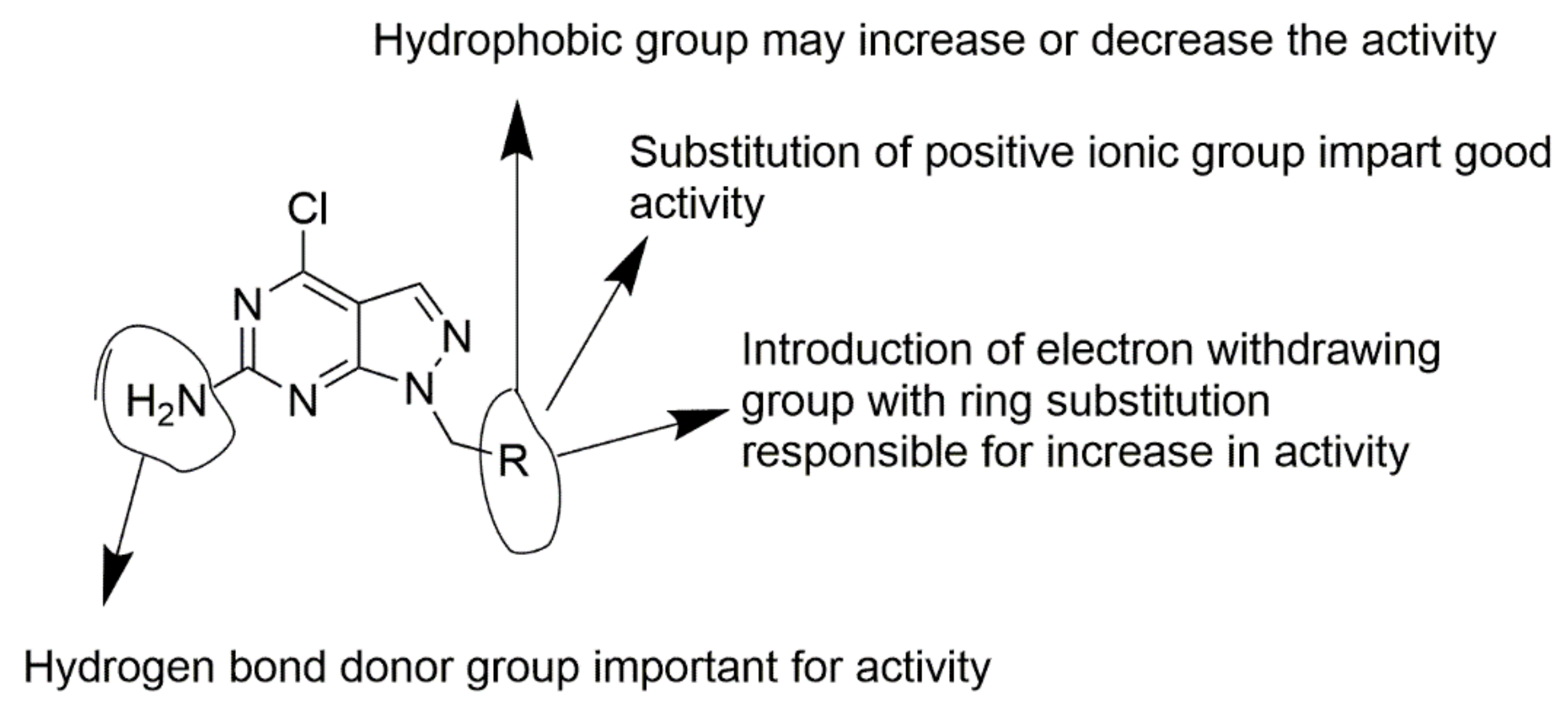
4. Optimization of Novel Ligands
5. Analysis of R Group Enumeration
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Felts, S.J.; Owen, B.A.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 2000, 275, 3305–3312. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Park, H.K.; Lee, J.E.; Lim, J.; Jo, D.E.; Park, S.A.; Suh, P.G.; Kang, B.H. Combination treatment with doxorubicin and gefitinib synergistically augments anticancer activity through enhanced activation of Bim. BMC Cancer 2014, 14, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.K.; Lee, J.E.; Lim, J.; Kang, B.H. Mitochondrial Hsp90s suppress calcium-mediated stress signals propagating from mitochondria to the ER in cancer cells. Mol. Cancer 2014, 13, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.K.; Hong, J.H.; Oh, Y.T.; Kim, S.S.; Yin, J.; Lee, A.J.; Chae, Y.C.; Kim, J.H.; Park, S.H.; Park, C.K.; et al. The interplay between TRAP1 and Sirtuin-3 Modulates Mitochondrial Respiration and Oxidative Stress to Maintain Stemness of Glioma Stem Cells. Cancer Res. 2019, 79, 1369–1382. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H. TRAP1 regulation of mitochondrial life or death decision in cancer cells and mitochondria-targeted TRAP1 inhibitors. BMB Rep. 2012, 45, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiesel, F.C.; James, E.D.; Hudec, R.; Springer, W. Mitochondrial targeted HSP90 inhibitor Gamitrinib-TPP (G-TPP) induces PINK1/Parkin-dependent mitophagy. Oncotarget 2017, 8, 106233–106248. [Google Scholar] [CrossRef] [Green Version]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Guzzo, G.; Sciacovelli, M.; Bernardi, P.; Rasola, A. Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti-oxidant and anti-apoptotic effects on tumor cells. Oncotarget 2014, 5, 11897–11908. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, S.Y.; Kim, D.; Yoon, N.G.; Yun, J.; Hong, K.B.; Lee, C.; Lee, J.H.; Kang, B.H.; Kang, S. Development of pyrazolo[3,4-d] pyrimidine-6-amine-based TRAP1 inhibitors that demonstrate in vivo anticancer activity in mouse xenograft models. Bioorganic Chem. 2020, 101, 103901. [Google Scholar] [CrossRef]
- Leonard, J.T.; Roy, K. On selection of training and test sets for the development of predictive QSAR models. QSAR Comb. Sci. 2006, 25, 235. [Google Scholar] [CrossRef]
- Schrödinger LLC. Ligprep; Version 2.5; Schrödinger LLC: New York, NY, USA, 2012. [Google Scholar]
- Asati, V.; Bharti, S.K.; Budhwani, A.K. 3D-QSAR and virtual screening studies of thiazolidine-2,4-dione analogs: Validation of experimental inhibitory potencies towards PIM-1 kinase. J. Mol. Struct. 2017, 1133, 278–293. [Google Scholar] [CrossRef]
- Schrödinger LLC. Phase; Version 4.4; Schrödinger LLC: New York, NY, USA, 2012. [Google Scholar]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Rajeswari, M.; Santhi, N.; Bhuvaneswari, V. Pharmacophore and virtual screening of JAK3 inhibitors. Bioinformation 2014, 10, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Crisan, L.; Borota, A.; Bora, A.; Pacureanu, L. Diarylthiazole and diarylimidazole selective COX-1 inhibitor analysis through pharmacophore modeling, virtual screening, and DFT-based approaches. Struct. Chem. 2019, 30, 2311–2326. [Google Scholar] [CrossRef]
- Schrödinger LLC. Phase 4.4 Quick Start Guide; Schrödinger LLC: New York, NY, USA, 2013. [Google Scholar]
- Sallam, A.A.; Houssen, W.E.; Gissendanner, C.R.; Orabi, K.Y.; Foudah, A.I.; El Sayed, K.A. Bioguided discovery and pharmacophore modeling of the mycotoxinindole diterpene alkaloids penitrems as breast cancer proliferation, migration, and invasion inhibitors. Med. Chem. Comm. 2013, 4, 1360–1369. [Google Scholar] [CrossRef]
- Hall, M.D.; Salam, N.K.; Hellawell, J.L.; Fales, H.M.; Kensler, C.B.; Ludwig, J.A.; Szakáes, G.; Hibbs, D.E.; Gottesman, M.M. Synthesis, activity, and pharmacophore development for isatin-β-thiosemicarbazones with selective activity toward multidrug-resistant cells. J. Med. Chem. 2009, 52, 3191–3204. [Google Scholar] [CrossRef]
- Teli, M.K.; Rajanikant, G.K. Pharmacophore generation and atom-based 3D-QSAR of Niso-propyl pyrrole-based derivatives as HMG-CoA reductase inhibitors. Org. Med. Chem. Lett. 2012, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Kamaria, P.; Kawathekar, N. Ligand-based 3D-QSAR analysis and virtual screening in an exploration of new scaffolds as Plasmodium falciparum glutathione reductase inhibitors. Med. Chem. Res. 2013, 23, 25–33. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Aid. Mol. Des. 2002, 16, 357–369. [Google Scholar] [CrossRef]
- Tanwar, O.P.; Saha, R.; Alam, M.M.; Akhtar, M. 3D-QSAR of amino-substituted pyrido [3,2B] pyrazinones as PDE-5 inhibitors. Med. Chem. Res. 2012, 21, 202–211. [Google Scholar] [CrossRef]
- Available online: http://gohom.win/ManualHom/Schrodinger_20152_docs/maestro/help_Maestro/phase/atom_based_qsar.html (accessed on 1 May 2021).
- Kirubakaran, P.; Muthusamy, K.; Singh, K.H.; Nagamani, S. Ligand-based pharmacophore modeling; atom-based 3D-QSAR analysis and molecular docking studies of phosphoinositide-dependent kinase-1 inhibitors. Indian J. Pharmaceut. Sci. 2012, 74, 141–151. [Google Scholar]
- Dixit, A.; Kashaw, S.K.; Gaur, S.; Saxena, A.K. Development of CoMFA, advance CoMFA and CoMSIA models in pyrroloquinazolines as thrombin receptor antagonist. Bioorganic Med. Chem. 2004, 12, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Shinde, M.G.; Modi, S.J.; Kulkarni, V.M. QSAR and molecular docking of phthalazine derivatives as epidermal growth factor receptor (EGFR) inhibitors. J. Appl. Pharm. Sci. 2017, 7, 181–191. [Google Scholar]
- Clark, M.; Cramer, R.D., III; Opdenbosch, N.V. Validation of the general-purpose Tripos 5.2 force field. J. Comp. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Kaushik, A.C.; Kumar, S.; Wei, D.Q.; Sahi, S. Structure-Based Virtual Screening Studies to Identify Novel Potential Compounds for GPR142 and Their Relative Dynamic Analysis for Study of Type 2 Diabetes. Front. Chem. 2018, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LLC. Protein Preparation Wizard; Schrödinger LLC: New York, NY, USA, 2012. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zielkiewicz, J. Structural properties of water: Comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J. Chem. Phys. 2005, 123, 104501. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The design of leadlike combinatorial libraries. Angew Chem. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Rondanin, R.; Lettini, G.; Oliva, P.; Baruchello, R.; Costantini, C.; Trapella, C.; Simoni, D.; Bernardi, T.; Sisinni, L.; Pietrafesa, M.; et al. New TRAP1 and Hsp90 chaperone inhibitors with cationic components: Preliminary studies on mitochondrial targeting. Bioorganic Med. Chem. Lett. 2018, 28, 2289–2293. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. Qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | Compounds | R | IC50 (µM) | pIC50 |
|---|---|---|---|---|
| 1 | 4 |  | 0.50 | 6.30 |
| 2 | 9 |  | 19.00 | 4.72 |
| 3 | 10 |  | 7.00 | 5.15 |
| 4 | 11 |  | 15.00 | 4.82 |
| 5 | 12 |  | 20.00 | 4.70 |
| 6 | 13 |  | 20.00 | 4.70 |
| 7 | 15 |  | 20.00 | 4.70 |
| 8 | 22 |  | 6.50 | 5.19 |
| 9 | 23 |  | 5.00 | 5.30 |
| 10 | 24 |  | 20.00 | 4.70 |
| 11 | 25 |  | 4.00 | 5.40 |
| 12 | 26 |  | 20.00 | 4.70 |
| 13 | 27 |  | 3.50 | 5.46 |
| 14 | 30 |  | 20.00 | 4.70 |
| 15 | 32 |  | 15.00 | 4.82 |
| 16 | 33 |  | 20.00 | 4.70 |
| 17 | 34 |  | 4.00 | 5.40 |
| 18 | 35 |  | 10.00 | 5.00 |
| 19 | 36 |  | 8.00 | 5.10 |
| 20 | 39 |  | 3.00 | 5.52 |
| 21 | 41 |  | 1.23 | 5.91 |
| 22 | 42 |  | 0.44 | 6.36 |
| 23 | 43 |  | 2.43 | 5.61 |
| 24 | 44 |  | 1.80 | 5.74 |
| 25 | 45 |  | 1.69 | 5.77 |
| 26 | 46 |  | 0.47 | 6.33 |
| 27 | 47 |  | 1.98 | 5.70 |
| 28 | 48 |  | 0.37 | 6.43 |
| 29 | 49 |  | 0.89 | 6.05 |
| 30 | 50 |  | 0.94 | 6.03 |
| 31 | 51 |  | 0.79 | 6.10 |
| 32 | 52 |  | 0.45 | 6.35 |
| 33 | 56 |  | 2.70 | 5.57 |
| 34 | 59 |  | 1.00 | 6.00 |
| Hypo ID | Survival | Site | Vector | Volume | Select | Matches | Inactive | Adjusted | BEDROC | Ref. Lig |
|---|---|---|---|---|---|---|---|---|---|---|
| DHHRR_1 | 5.94 | 0.93 | 1.00 | 0.91 | 2.15 | 9.00 | 2.66 | 3.28 | 1.00 | mol_32 |
| DHHRR_2 | 5.94 | 0.93 | 1.00 | 0.91 | 2.15 | 9.00 | 2.62 | 3.33 | 1.00 | mol_32 |
| DHHRR_3 | 5.94 | 0.93 | 1.00 | 0.91 | 2.15 | 9.00 | 2.68 | 3.26 | 1.00 | mol_32 |
| DHHRR_4 | 5.94 | 0.92 | 1.00 | 0.91 | 2.15 | 9.00 | 2.70 | 3.24 | 1.00 | mol_32 |
| DHHRR_5 | 5.94 | 0.93 | 1.00 | 0.91 | 2.15 | 9.00 | 2.63 | 3.30 | 1.00 | mol_32 |
| DHHRR_6 | 5.94 | 0.93 | 1.00 | 0.91 | 2.15 | 9.00 | 2.70 | 3.24 | 1.00 | mol_32 |
| DHHRR_7 | 5.93 | 0.92 | 1.00 | 0.91 | 2.15 | 9.00 | 2.75 | 3.19 | 1.00 | mol_32 |
| DHHRR_8 | 5.93 | 0.92 | 1.00 | 0.91 | 2.15 | 9.00 | 2.65 | 3.29 | 1.00 | mol_32 |
| DHHRR_9 | 5.93 | 0.93 | 1.00 | 0.91 | 2.14 | 9.00 | 2.64 | 3.29 | 1.00 | mol_32 |
| DHHRR_10 | 5.93 | 0.93 | 1.00 | 0.91 | 2.14 | 9.00 | 2.67 | 3.26 | 1.00 | mol_32 |
| DHRR_1 | 5.40 | 0.99 | 1.00 | 0.94 | 1.52 | 9.00 | 2.90 | 2.50 | 1.00 | mol_26 |
| DHRR_2 | 5.40 | 0.99 | 1.00 | 0.94 | 1.52 | 9.00 | 2.90 | 2.50 | 1.00 | mol_26 |
| DHRR_3 | 5.39 | 0.99 | 1.00 | 0.94 | 1.51 | 9.00 | 2.89 | 2.50 | 1.00 | mol_26 |
| DHRR_4 | 5.39 | 0.99 | 1.00 | 0.94 | 1.51 | 9.00 | 2.91 | 2.48 | 1.00 | mol_26 |
| DHRR_5 | 5.39 | 0.99 | 1.00 | 0.94 | 1.51 | 9.00 | 2.90 | 2.49 | 1.00 | mol_26 |
| DHRR_6 | 5.39 | 0.99 | 1.00 | 0.94 | 1.51 | 9.00 | 2.89 | 2.50 | 1.00 | mol_26 |
| DHRR_7 | 5.39 | 0.99 | 1.00 | 0.94 | 1.51 | 9.00 | 2.91 | 2.48 | 1.00 | mol_26 |
| DHRR_8 | 5.39 | 0.99 | 1.00 | 0.94 | 1.50 | 9.00 | 2.90 | 2.48 | 1.00 | mol_26 |
| DHRR_9 | 5.39 | 0.99 | 1.00 | 0.94 | 1.50 | 9.00 | 2.90 | 2.48 | 1.00 | mol_31 |
| DHRR_10 | 5.38 | 0.99 | 1.00 | 0.94 | 1.50 | 9.00 | 2.91 | 2.47 | 1.00 | mol_31 |
| # Factors | SD | R2 | R2 CV | R2 Scramble | Stability | F | RMSE | Q2 | Pearson’s r |
|---|---|---|---|---|---|---|---|---|---|
| 1.00 | 0.46 | 0.34 | 0.08 | 0.31 | 0.93 | 11.80 | 0.64 | 0.08 | 0.47 |
| 2.00 | 0.33 | 0.68 | 0.22 | 0.51 | 0.73 | 23.90 | 0.52 | 0.29 | 0.59 |
| 3.00 | 0.23 | 0.85 | 0.41 | 0.69 | 0.68 | 38.70 | 0.47 | 0.42 | 0.67 |
| 4.00 | 0.16 | 0.93 | 0.57 | 0.76 | 0.71 | 70.50 | 0.41 | 0.56 | 0.76 |
| 5.00 | 0.13 | 0.96 | 0.58 | 0.81 | 0.70 | 82.50 | 0.40 | 0.57 | 0.79 |
| # Factors | H-Bond Donor | Hydrophobic/Nonpolar | Electron-Withdrawing | Other |
|---|---|---|---|---|
| 1 | 0.011 | 0.577 | 0.391 | 0.021 |
| 2 | 0.007 | 0.741 | 0.225 | 0.027 |
| 3 | 0.035 | 0.703 | 0.216 | 0.046 |
| 4 | 0.043 | 0.738 | 0.198 | 0.021 |
| 5 | 0.045 | 0.755 | 0.192 | 0.008 |
| No r | Name | Actual pIC50 (X) | Atom-Based | |
|---|---|---|---|---|
| Predicted pIC50 (Ϋ) | Residuals (Ϋ−X) | |||
| 1 | 4 | 6.3 | 6.00 | −0.30 |
| 2 | 9 | 4.72 | 4.65 | −0.07 |
| 3 | 10 | 5.15 | 5.76 | 0.61 |
| 4 | 11 | 4.82 | 4.85 | 0.03 |
| 5 | 12 | 4.7 | 4.66 | −0.04 |
| 6 | 13 | 4.7 | 4.69 | −0.01 |
| 7 | 15 | 4.7 | 4.68 | −0.02 |
| 8 | 22 | 5.19 | 5.20 | 0.01 |
| 9 | 23 | 5.3 | 5.30 | 0.00 |
| 10 | 24 | 4.7 | 4.85 | 0.15 |
| 11 | 25 | 5.4 | 5.29 | −0.11 |
| 12 | 26 | 4.7 | 4.67 | −0.03 |
| 13 | 27 | 5.46 | 5.60 | 0.14 |
| 14 | 30 | 4.7 | 4.92 | 0.22 |
| 15 | 32 | 4.82 | 4.88 | 0.06 |
| 16 | 33 | 4.7 | 4.63 | −0.07 |
| 17 | 34 | 5.4 | 5.46 | 0.06 |
| 18 | 35 | 5 | 4.89 | −0.11 |
| 19 | 36 | 5.1 | 5.14 | 0.04 |
| 20 | 39 | 5.52 | 5.57 | 0.05 |
| 21 | 41 | 5.91 | 5.98 | 0.07 |
| 22 | 42 | 6.36 | 6.39 | 0.03 |
| 23 | 43 | 5.61 | 5.73 | 0.12 |
| 24 | 44 | 5.74 | 5.85 | 0.11 |
| 25 | 45 | 5.77 | 5.75 | −0.02 |
| 26 | 46 | 6.33 | 6.11 | −0.22 |
| 27 | 47 | 5.7 | 5.86 | 0.16 |
| 28 | 48 | 6.43 | 5.85 | −0.58 |
| 29 | 49 | 6.05 | 6.02 | −0.03 |
| 30 | 50 | 6.03 | 5.80 | −0.23 |
| 31 | 51 | 6.1 | 6.11 | 0.01 |
| 32 | 52 | 6.35 | 6.20 | −0.15 |
| 33 | 56 | 5.57 | 5.63 | 0.06 |
| 34 | 59 | 6 | 6.17 | 0.17 |
| Compound Name | PDB ID:5Y3N | ||
|---|---|---|---|
| Docking Score (Extra-Precision (XP)) kcal/mol | Docking Score (Standard Docking Precision (SP)) kcal/mol | MMGBSA ΔG Bind (XP Complex) kcal/mol | |
| 48 | −10.824 | −9.998 | 78.07 |
| 42 | −11.265 | −11.265 | −57.88 |
| 46 | −10.532 | −10.782 | −56.71 |
| 49 | −10.422 | −11.353 | −68.2 |
| 56 | −10.827 | −11.641 | −82.07 |
| 43 | −10.753 | −11.508 | −56.71 |
| ZINC05434822 | −11.63 | −11.641 | −68.2 |
| ZINC72286418 | −10.86 | −10.59 | −82.07 |
| ZINC05297837 | −10.42 | −6.25 | −59.752 |
| ZINC59358929 | −10.102 | −9.68 | −78.2 |
| S. No | Docked Complex | MM-PBSA Based Calculated Energies (kJ/mol) | |||
|---|---|---|---|---|---|
| ΔE (vdW) | ΔE (Elec) | ΔG (SASA) | ΔG (Binding) | ||
| 1 | NVP-AUY922 | −133.245 | −20.399 | −17.746 | −276.797 |
| 2 | TRAP1_42 | −174.918 | −6.766 | −16.056 | −213.795 |
| 3 | TRAP1_43 | −190.049 | −22.551 | −15.727 | −244.055 |
| 4 | TRAP1_46 | −201.654 | −12.756 | −16.761 | −247.931 |
| 5 | TRAP1_49 | −178.876 | −16.962 | −15.849 | −227.536 |
| 6 | TRAP1_56 | −184.313 | −16.979 | −16.403 | −234.098 |
| 7 | TRAP1_ZINC05297837 | −268.296 | −2.926 | −19.745 | −310.711 |
| 8 | TRAP1_ZINC05434822 | −277.273 | −13.936 | −18.528 | −328.265 |
| 9 | TRAP1_ZINC59358929 | −303.182 | −43.700 | −22.525 | −391.933 |
| 10 | TRAP1_ZINC72286418 | −219.998 | −12.829 | −17.952 | −268.731 |
| S. No. | Compound Name | QP log Po/w | QPP Caco | QP logBB | QPPMDCK | # Metab | QP logKhsa | Percent Human Oral Absorption |
|---|---|---|---|---|---|---|---|---|
| 1 | 48 | 3.156 | 920.499 | −0.145 | 4124.662 | 1 | 0.106 | 100 |
| 2 | 42 | 2.644 | 1011.799 | −0.193 | 2743.625 | 1 | −0.039 | 96.212 |
| 3 | 46 | 2.972 | 1006.302 | −0.295 | 2031.339 | 2 | 0.025 | 100 |
| 4 | 49 | 2.892 | 1037.573 | −0.101 | 3427.589 | 1 | 0.027 | 100 |
| 5 | 56 | 2.686 | 1271.458 | −0.292 | 1230.569 | 3 | 0.1 | 100 |
| 6 | 43 | 2.354 | 1053.419 | −0.305 | 1260.235 | 2 | −0.053 | 94.825 |
| 7 | ZINC05434822 | 4.687 | 1015.224 | −0.589 | 833.988 | 3 | 0.728 | 100 |
| 8 | ZINC72286418 | 2.653 | 431.141 | −0.802 | 581.552 | 3 | 0.1 | 89.635 |
| 9 | ZINC05297837 | 2.728 | 258.436 | −1.334 | 303.018 | 2 | 0.003 | 86.094 |
| S. No. | Name | Mol. Wt. (g/mol) | No. Rot. Bonds | No. H-Bond Acceptors | No. H-Bond Donors | Molar Refractivity |
|---|---|---|---|---|---|---|
| 1 | 48 | 356.58 | 2 | 4 | 1 | 78.14 |
| 2 | 42 | 429.6 | 2 | 5 | 1 | 89.27 |
| 3 | 46 | 325.7 | 3 | 6 | 1 | 76.89 |
| 4 | 49 | 313.67 | 2 | 6 | 1 | 70.36 |
| 5 | 56 | 376.21 | 2 | 5 | 1 | 89.17 |
| 6 | 43 | 317.73 | 2 | 5 | 1 | 81.52 |
| 7 | ZINC05434822 | 363.43 | 5 | 4 | 1 | 102.8 |
| 8 | ZINC72286418 | 334.29 | 5 | 5 | 2 | 80.71 |
| 9 | ZINC05297837 | 469.29 | 5 | 5 | 3 | 118.83 |
| Comp. Name | Structure | XP GS Score (PDB ID:5Y3N) | R1 s m Smiles | R2 s m Smiles | R3 s m Smiles |
|---|---|---|---|---|---|
| 1 | O=C(C)Nc(n1)nc([NH+](C)C)c(c12)cnn2Cc(c(c3)C(=O)N)cc(c34)OCO4 | −13.286 | [*][NH+](C)C | [*]NC(=O)C | [*]C(=O)N |
| 2 | n1c([NH3+])nc(O)c(c12)cnn2Cc(c(c3)C(=O)N)cc(c34)OCO4 | −13.286 | [*]O | [*][NH3+] | [*]C(=O)N |
| 3 | C1C[NH2+]CCC1c(nc(n2)C(=O)N)c(c23)cnn3Cc(c(c4)C(=O)N)cc(c45)OCO5 | −12.873 | [*]C1CC[NH2+]CC1 | [*]C(=O)N | [*]C(=O)N |
| 4 | NC(=O)c(n1)nc(O)c(c12)cnn2Cc(cc(c34)OCO4)c(c3)C(=O)Nc5ccccc5 | −12.73 | [*]O | [*]C(=O)N | [*]C(=O)Nc1ccccc1 |
| 5 | NC(=O)c(n1)nc(O)c(c12)cnn2Cc(cc(c34)OCO4)c(c3)-c5[nH]ccn5 | −12.674 | [*]O | [*]C(=O)N | [*]c1ncc[nH]1 |
| 6 | NC(=O)c(n1)nc(O)c(c12)cnn2Cc(cc(c34)OCO4)c(c3)-c5[nH]cnc5 | −12.622 | [*]O | [*]C(=O)N | [*]c1cnc[nH]1 |
| 7 | CNC(=O)Nc(n1)nc(O)c(c12)cnn2Cc(cc(c34)OCO4)c(c3)NC(=O)Nc5ccccc5 | −12.608 | [*]O | [*]NC(=O)NC | [*]NC(=O)Nc1ccccc1 |
| 8 | n1c([NH3+])nc(O)c(c12)cnn2Cc(c(c3)C(=O)N(C)C)cc(c34)OCO4 | −2.955 | from water 1 | 15173 | 15183 |
| 9 | c1nccn1-c(n2)nc(O)c(c23)cnn3Cc(cc(c45)OCO5)c(c4)C(=O)Nc6ccccc6 | −12.559 | [*]O | [*]n1ccnc1 | [*]C(=O)Nc1ccccc1 |
| 10 | n1c([NH3+])nc([NH2+]C)c(c12)cnn2Cc(cc(c34)OCO4)c(c3)C(=O)Nc5ccccc5 | −12.555 | [*][NH2+]C | [*][NH3+] | [*]C(=O)Nc1ccccc1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Abdellattif, M.H.; Ali, A.; AbuAli, O.; Shahbaaz, M.; Ahsan, M.J.; Hussien, M.A. Computational Approaches for the Design of Novel Anticancer Compounds Based on Pyrazolo[3,4-d]pyrimidine Derivatives as TRAP1 Inhibitor. Molecules 2021, 26, 5932. https://doi.org/10.3390/molecules26195932
Ali A, Abdellattif MH, Ali A, AbuAli O, Shahbaaz M, Ahsan MJ, Hussien MA. Computational Approaches for the Design of Novel Anticancer Compounds Based on Pyrazolo[3,4-d]pyrimidine Derivatives as TRAP1 Inhibitor. Molecules. 2021; 26(19):5932. https://doi.org/10.3390/molecules26195932
Chicago/Turabian StyleAli, Amena, Magda H. Abdellattif, Abuzer Ali, Ola AbuAli, Mohd Shahbaaz, Mohamed Jawed Ahsan, and Mostafa A. Hussien. 2021. "Computational Approaches for the Design of Novel Anticancer Compounds Based on Pyrazolo[3,4-d]pyrimidine Derivatives as TRAP1 Inhibitor" Molecules 26, no. 19: 5932. https://doi.org/10.3390/molecules26195932