
In Silico Evaluation of Iranian Medicinal Plant Phytoconstituents as Inhibitors against Main Protease and the Receptor-Binding Domain of SARS-CoV-2


 , ,
, ,  , , ,
, , ,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Molecular Docking Analysis
2.2. ADMET Analysis
2.3. In Silico Inhibition Constant
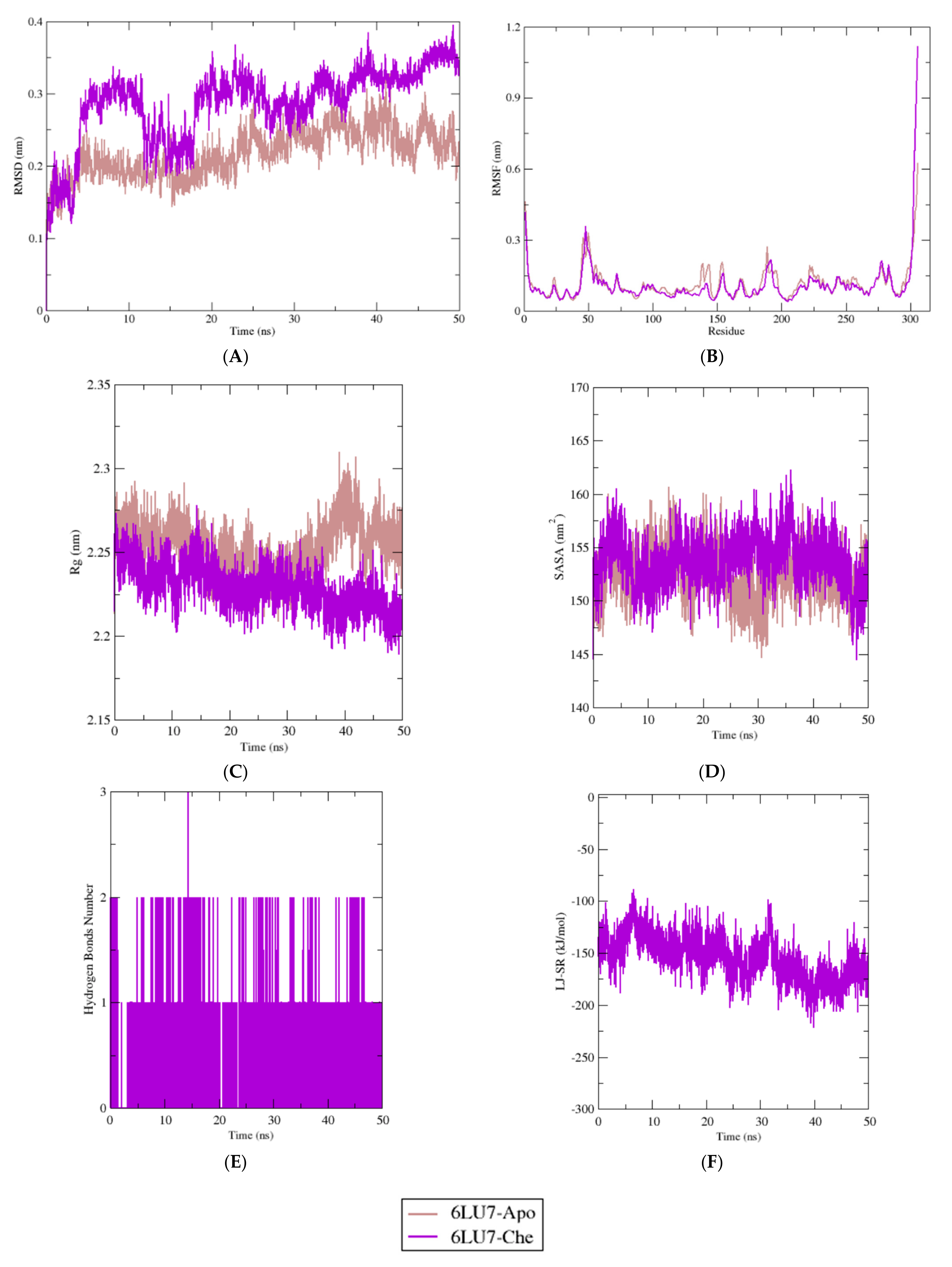
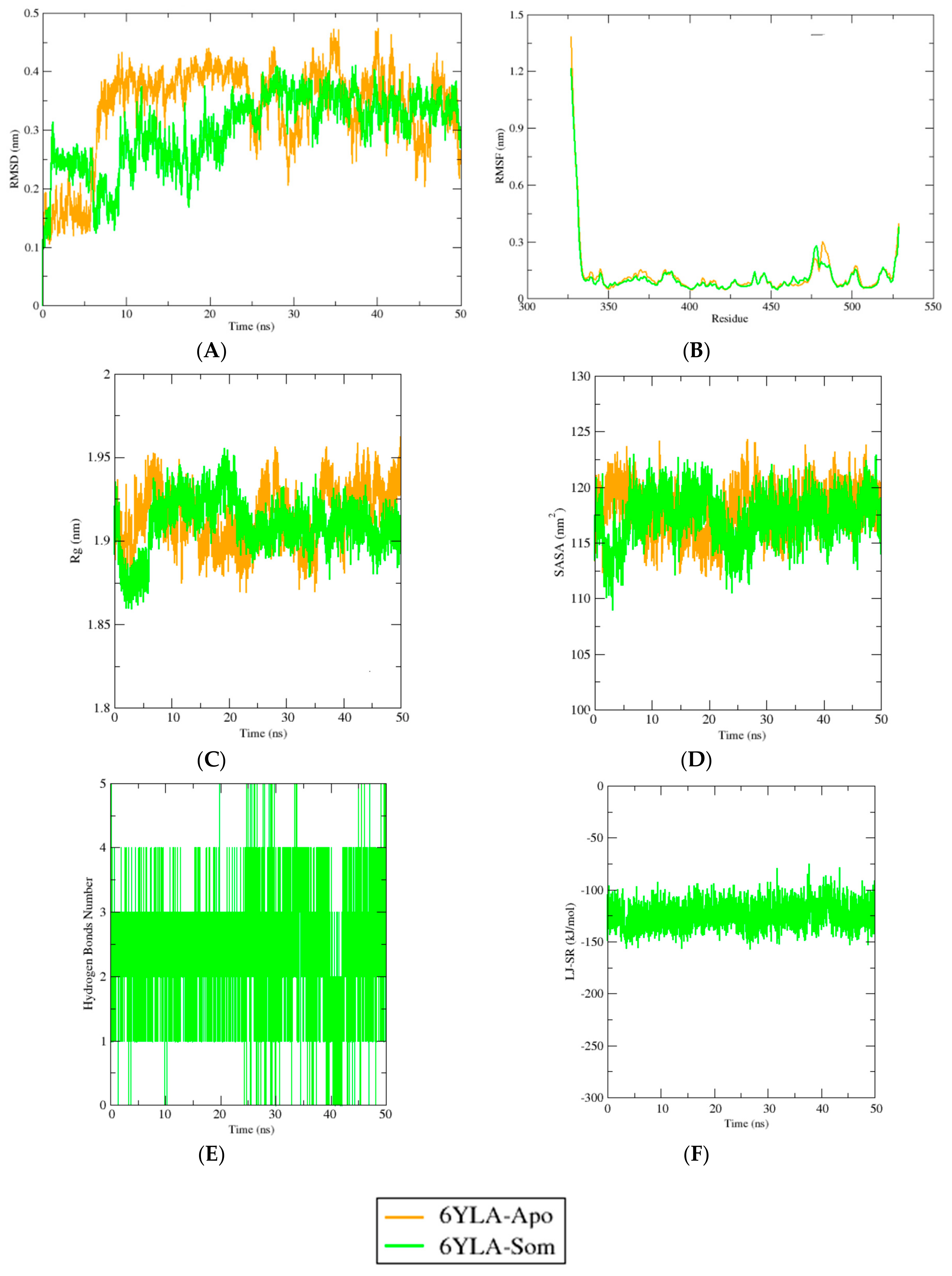
2.4. Molecular Dynamics Simulation Study
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Molecular Docking Analysis
4.2.1. Ligand Preparation
4.2.2. Receptor Preparation
4.2.3. Determination of Active Sites
4.2.4. Molecular Docking and Visualization of Ligand–Receptor Interactions
4.3. ADMET Analysis
4.4. Calculation of Predicted IC50
4.5. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- MacKenzie, J.S.; Smith, D.W. COVID-19-A Novel Zoonotic Disease: A Review of the Disease, the Virus, and Public Health Measures. Asia-Pac. J. Public Health 2020, 32, 145–153. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, X.; Qu, J. Coronavirus disease 2019 (COVID-19): A clinical update. Front. Med. 2020, 14, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Ayerdi, O.; Puerta, T.; Clavo, P.; Vera, M.; Ballesteros, J.; Fuentes, M.E.; Estrada, V.; Rodríguez, C.; Del Romero, J. Preventive Efficacy of Tenofovir/Emtricitabine Against Severe Acute Respiratory Syndrome Coronavirus 2 Among Pre-Exposure Prophylaxis Users. Open Forum Infect. Dis. 2020, 7, ofaa455. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Kumar, S.; Li, X.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Gong, X. A Review of Protein Inter-residue Distance Prediction. Curr. Bioinform. 2020, 15, 821–830. [Google Scholar] [CrossRef]
- Tan, Q.; Duan, L.; Ma, Y.; Wu, F.; Huang, Q.; Mao, K.; Xiao, W.; Xia, H.; Zhang, S.; Zhou, E.; et al. Is oseltamivir suitable for fighting against COVID-19: In silico assessment, in vitro and retrospective study. Bioorg. Chem. 2020, 104, 104257. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Tallei, T.E.; Tumilaar, S.G.; Lombogia, L.T.; Adam, A.A.; Sakib, S.A.; Emran, T.B.; Idroes, R. Potential of betacyanin as inhibitor of SARS-CoV-2 revealed by molecular docking study. IOP Conf. Ser. Earth Environ. Sci. 2021, 711, 12028. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Du, H.; Hou, X.-Y.; Miao, Y.-H.; Huang, B.-S.; Liu, D.-H. Traditional Chinese Medicine: An effective treatment for 2019 novel coronavirus pneumonia (NCP). Chin. J. Nat. Med. 2020, 18, 206–210. [Google Scholar] [CrossRef]
- Yang, Y.; Islam, M.S.; Wang, J.; Li, Y.; Chen, X. Traditional Chinese Medicine in the Treatment of Patients Infected with 2019-New Coronavirus (SARS-CoV-2): A Review and Perspective. Int. J. Biol. Sci. 2020, 16, 1708–1717. [Google Scholar] [CrossRef]
- Benarba, B.; Pandiella, A. Medicinal Plants as Sources of Active Molecules Against COVID-19. Front. Pharmacol. 2020, 11, 1189. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Dutta, M.; Tareq, A.M.; Rakib, A.; Mahmud, S.; Sami, S.A.; Mallick, J.; Islam, M.N.; Majumder, M.; Uddin, M.Z.; Alsubaie, A.; et al. Phytochemicals from Leucas zeylanica targeting main protease of SARS−CoV−2: Chemical profiles, molecular docking, and molecular dynamics simulations. Biology 2021, 10, 789. [Google Scholar] [CrossRef]
- Mahmud, S.; Biswas, S.; Paul, G.K.; Mita, M.A.; Promi, M.M.; Afrose, S.; Hasan, R.; Zaman, S.; Uddin, M.S.; Dhama, K.; et al. Plant-based phytochemical screening by targeting main protease of SARS-CoV-2 to design effective potent inhibitors. Biology 2021, 10, 589. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- David, A.B.; Diamant, E.; Dor, E.; Barnea, A.; Natan, N.; Levin, L.; Chapman, S.; Mimran, L.C.; Epstein, E.; Zichel, R.; et al. Identification of SARS-CoV-2 Receptor Binding Inhibitors by In Vitro Screening of Drug Libraries. Molecules 2021, 26, 3213. [Google Scholar] [CrossRef]
- Bojadzic, D.; Alcazar, O.; Chen, J.; Chuang, S.T.; Condor Capcha, J.M.; Shehadeh, L.A.; Buchwald, P. Small-Molecule Inhibitors of the Coronavirus Spike: ACE2 Protein–Protein Interaction as Blockers of Viral Attachment and Entry for SARS-CoV-2. ACS Infect. Dis. 2021, 7, 1519–1534. [Google Scholar] [CrossRef]
- Mahmud, S.; Mita, M.A.; Biswas, S.; Paul, G.K.; Promi, M.M.; Afrose, S.; Hasan, R.; Shimu, S.S.; Zaman, S.; Uddin, S.; et al. Molecular docking and dynamics study to explore phytochemical ligand molecules against the main protease of SARS-CoV-2 from extensive phytochemical datasets. Expert Rev. Clin. Pharmacol. 2021, 1–11. [Google Scholar] [CrossRef]
- Talevi, A. Computer-Aided Drug Design: An Overview. Methods Mol. Biol. 2018, 1762, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S. Computational Approaches for Drug Discovery. Molecules 2019, 24, 3061. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.-L.; Chen, C.-C. Computational approaches for drug discovery. Drug Dev. Res. 2014, 75, 412–418. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef] [Green Version]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Çubuk, H.; Özbİl, M. Comparison of clinically approved molecules on SARS-CoV-2 drug target proteins: A molecular docking study. Turk. J. Chem. 2021, 45, 35–41. [Google Scholar] [CrossRef]
- Sailah, I.; Tumilaar, S.G.; Lombogia, L.T. Molecular Docking and Dynamics Simulations Study of Selected Phytoconstituents of ‘Pangi’ (Pangium edule Reinw) Leaf as Anti-SARS-CoV-2. Philipp. J. Sci. 2021, 150, 925–937. [Google Scholar]
- Fatimawali; Maulana, R.R.; Windah, A.; Wahongan, I.F.; Tumilaar, S.G.; Adam, A.A.; Kepel, B.J.; Bodhi, W.; Tallei, T.E. Data on the docking of phytoconstituents of betel plant and matcha green tea on SARS-CoV-2. Data Br. 2021, 36, 107049. [Google Scholar] [CrossRef]
- Aljofan, M.; Netter, H.J.; Aljarbou, A.N.; Hadda, T.B.; Orhan, I.E.; Sener, B.; Mungall, B.A. Anti-hepatitis B activity of isoquinoline alkaloids of plant origin. Arch. Virol. 2014, 159, 1119–1128. [Google Scholar] [CrossRef]
- Tallei, T.E.; Tumilaar, S.G.; Niode, N.J.; Fatimawali; Kepel, B.J.; Idroes, R.; Effendi, Y.; Sakib, S.A.; Emran, T.B. Potential of Plant Bioactive Compounds as SARS-CoV-2 Main Protease (Mpro) and Spike (S) Glycoprotein Inhibitors: A Molecular Docking Study. Scientifica 2020, 1–18. [Google Scholar] [CrossRef]
- Luthar, Z.; Germ, M.; Likar, M.; Golob, A.; Vogel-Mikuš, K.; Pongrac, P.; Kušar, A.; Pravst, I.; Kreft, I. Breeding Buckwheat for Increased Levels of Rutin, Quercetin and Other Bioactive Compounds with Potential Antiviral Effects. Plants 2020, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.; Tabrez, S.; Ali, R.; Alqahtani, A.S.; Ahmed, M.Z.; Rub, A. Molecular docking analysis of rutin reveals possible inhibition of SARS-CoV-2 vital proteins. J. Tradit. Complement. Med. 2021, 11, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Calzada, F.; Correa-Basurto, J.; Barbosa, E.; Mendez-Luna, D.; Yepez-Mulia, L. Antiprotozoal Constituents from Annona cherimola Miller, a Plant Used in Mexican Traditional Medicine for the Treatment of Diarrhea and Dysentery. Pharmacogn. Mag. 2017, 13, 148–152. [Google Scholar] [CrossRef]
- Bharathi, D.; Bhuvaneshwari, V. Synthesis of zinc oxide nanoparticles (ZnO NPs) using pure bioflavonoid rutin and their biomedical applications: Antibacterial, antioxidant and cytotoxic activities. Res. Chem. Intermed. 2019, 45, 2065–2078. [Google Scholar] [CrossRef]
- Rizzuti, B.; Grande, F.; Conforti, F.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Vega, S.; Reyburn, H.T.; Abian, O.; Velazquez-Campoy, A. Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs. Biomedicines 2021, 9, 375. [Google Scholar] [CrossRef]
- Rakib, A.; Paul, A.; Chy, M.; Sami, S.A.; Baral, S.K.; Majumder, M.; Tareq, A.M.; Amin, M.N.; Shahriar, A.; Uddin, M.Z.; et al. Biochemical and Computational Approach of Selected Phytocompounds from Tinospora crispa in the Management of COVID-19. Molecules 2020, 25, 3936. [Google Scholar] [CrossRef]
- Shang, X.-F.; Yang, C.J.; Morris-Natschke, S.L.; Li, J.C.; Yin, X.D.; Liu, Y.Q.; Guo, X.; Peng, J.W.; Goto, M.; Zhang, J.Y.; et al. Biologically active isoquinoline alkaloids covering 2014-2018. Med. Res. Rev. 2020, 40, 2212–2289. [Google Scholar] [CrossRef]
- Garg, S.; Roy, A. In silico analysis of selected alkaloids against main protease (M(pro)) of SARS-CoV-2. Chem. Biol. Interact. 2020, 332, 109309. [Google Scholar] [CrossRef]
- Gyebi, G.A.; Adegunloye, A.P.; Ibrahim, I.M.; Ogunyemi, O.M.; Afolabi, S.O.; Ogunro, O.B. Prevention of SARS-CoV-2 cell entry: Insight from in silico interaction of drug-like alkaloids with spike glycoprotein, human ACE2, and TMPRSS2. J. Biomol. Struct. Dyn. 2020, 1–25. [Google Scholar] [CrossRef]
- Ghoran, S.H.; Espadinha, M.; Machado, M.; Gut, J.; Gonçalves, L.M.; Rosenthal, P.J.; Prudêncio, M.; Moreira, R.; Santos, M.M. Isolation, spectroscopic characterization, X-ray, theoretical studies as well as in vitro cytotoxicity of Samarcandin. Bioorg. Chem. 2016, 66, 27–32. [Google Scholar] [CrossRef]
- Khan, J.; Sakib, S.A.; Mahmud, S.; Khan, Z.; Islam, M.N.; Sakib, M.A.; Emran, T.B.; Simal-Gandara, J. Identification of potential phytochemicals from Citrus limon against main protease of SARS-CoV-2: Molecular docking, molecular dynamic simulations and quantum computations. J. Biomol. Struct. Dyn. 2021, 2021, 1–12. [Google Scholar] [CrossRef]
- Saleem, S.; Muhammad, G.; Hussain, M.A.; Altaf, M.; Bukhari, S.N.A. Withania somnifera L.: Insights into the phytochemical profile, therapeutic potential, clinical trials, and future prospective. Iran. J. Basic Med. Sci. 2020, 23, 1501–1526. [Google Scholar] [CrossRef] [PubMed]
- White, P.T.; Subramanian, C.; Motiwala, H.F.; Cohen, M.S. Natural Withanolides in the Treatment of Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 329–373. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P.; Chikhale, R.; Dey, Y.N.; Pasha, I.; Chand, S.; Gurav, N.; Ayyanar, M.; Patil, B.M.; Gurav, S. Withanolides from Withania somnifera as an immunity booster and their therapeutic options against COVID-19. J. Biomol. Struct. Dyn. 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Liu, J.; Du, R.; Yu, Q.; Gong, L.; Jiang, H.; Rong, R. Qualitative and Quantitative Analysis for the Chemical Constituents of Tetrastigma hemsleyanum Diels et Gilg Using Ultra-High Performance Liquid Chromatography/Hybrid Quadrupole-Orbitrap Mass Spectrometry and Preliminary Screening for Anti-Influenza Virus. Evid. Based. Complement. Alternat. Med. 2019, 2019, 9414926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Świderek, K.; Moliner, V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 M(pro) by QM/MM computational methods. Chem. Sci. 2020, 11, 10626–10630. [Google Scholar] [CrossRef]
- Dutta, M.; Nezam, M.; Chowdhury, S.; Rakib, A.; Paul, A.; Sami, S.A.; Uddin, M.Z.; Rana, M.S.; Hossain, S.; Effendi, Y.; et al. Appraisals of the Bangladeshi Medicinal Plant Calotropis gigantea Used by Folk Medicine Practitioners in the Management of COVID-19: A Biochemical and Computational Approach. Front Mol. Biosci. 2021, 8, 625391. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, S.; Paul, G.K.; Afroze, M.; Islam, S.; Gupt, S.B.R.; Razu, M.H.; Biswas, S.; Zaman, S.; Uddin, M.S.; Khan, M.; et al. Efficacy of Phytochemicals Derived from Avicennia officinalis for the Management of COVID-19: A Combined In Silico and Biochemical Study. Molecules 2021, 28, 2210. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; So, R.; Lv, H.; Mok, C.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Huo, J.; Zhao, Y.; Ren, J.; Zhou, D.; Duyvesteyn, H.; Ginn, H.M.; Carrique, L.; Malinauskas, T.; Ruza, R.R.; Shah, P.; et al. Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike. Cell Host Microbe 2020, 28, 445–454.e6. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Nicholls, R.A.; Emsley, P.; Gražulis, S.; Merkys, A.; Vaitkus, A.; Murshudov, G.N. AceDRG: A stereochemical description generator for ligands. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73 Pt 2, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Serillon, D.; Bo, C.; Barril, X. Testing automatic methods to predict free binding energy of host-guest complexes in SAMPL7 challenge. J. Comput. Aided. Mol. Des. 2021, 35, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Ugone, V.; Sanna, D.; Ruggiu, S.; Sciortino, G.; Garribba, E. Covalent and non-covalent binding in vanadium–protein adducts. Inorg. Chem. Front. 2021, 8, 1189–1196. [Google Scholar] [CrossRef]
- Rakib, A.; Nain, Z.; Islam, M.A.; Sami, S.A.; Mahmud, S.; Islam, A.; Ahmed, S.; Siddiqui, A.B.F.; Babu, S.M.O.F.; Hossain, P.; et al. A molecular modelling approach for identifying antiviral selenium-containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: An in silico investigation. Brief. Bioinform. 2021, 22, 1476–1498. [Google Scholar] [CrossRef]
- De Andrade, F.d.C.P.; Mendes, A.N. Computational analysis of eugenol inhibitory activity in lipoxygenase and cyclooxygenase pathways. Sci. Rep. 2020, 10, 16204. [Google Scholar] [CrossRef]
- Mhatre, S.; Naik, S.; Patravale, V. A molecular docking study of EGCG and theaflavin digallate with the druggable targets of SARS-CoV-2. Comput. Biol. Med. 2021, 129, 104137. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Das, P.; Purohit, R. Structural based study to identify new potential inhibitors for dual specificity tyrosine-phosphorylation- regulated kinase. Comput. Methods Programs Biomed. 2020, 194, 105494. [Google Scholar] [CrossRef]
- Arthur, D.E.; Uzairu, A. Molecular docking studies on the interaction of NCI anticancer analogues with human Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit. J. King Saud Univ. Sci. 2019, 31, 1151–1166. [Google Scholar] [CrossRef]
- Olasupo, S.B.; Uzairu, A.; Shallangwa, G.; Uba, S. QSAR modeling, molecular docking and ADMET/pharmacokinetic studies: A chemometrics approach to search for novel inhibitors of norepinephrine transporter as potent antipsychotic drugs. J. Iran. Chem. Soc. 2020, 17, 1953–1966. [Google Scholar] [CrossRef] [Green Version]
- De Freitas, R.F.; Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef] [PubMed]
- Niemann, T.; Stange, P.; Strate, A.; Ludwig, R. When hydrogen bonding overcomes Coulomb repulsion: From kinetic to thermodynamic stability of cationic dimers. Phys. Chem. Chem. Phys. 2019, 21, 8215–8220. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided. Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Geada, I.L.; Ramezani-Dakhel, H.; Jamil, T.; Sulpizi, M.; Heinz, H. Insight into induced charges at metal surfaces and biointerfaces using a polarizable Lennard-Jones potential. Nat. Commun. 2018, 9, 716. [Google Scholar] [CrossRef]
- Stevens, K.; Thamwattana, N.; Tran-Duc, T. New functional Lennard-Jones parameters for heterogeneous molecules. J. Appl. Phys. 2020, 128, 204301. [Google Scholar] [CrossRef]
- Chowdhury, K.H.; Chowdhury, M.R.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Reza, A.S.M.A.; Emran, T.B.; Simal-Gandara, J. Drug Repurposing Approach against Novel Coronavirus Disease (COVID-19) through Virtual Screening Targeting SARS-CoV-2 Main Protease. Biology 2020, 10, 2. [Google Scholar] [CrossRef]
- Nocentini, A.; Bonardi, A.; Gratteri, P.; Cerra, B.; Gioiello, A.; Supuran, C.T. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J. Enzym. Inhib. Med. Chem. 2018, 33, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Diana, R.; Caruso, U.; di Costanzo, L.; Bakayoko, G.; Panunzi, B. A Novel DR/NIR T-Shaped AIEgen: Synthesis and X-Ray Crystal Structure Study. Crystals 2020, 10, 269. [Google Scholar] [CrossRef] [Green Version]
- Tallei, T.E.; Fatimawali; Yelnetty, A.; Idroes, R.; Kusumawaty, D.; Emran, T.B.; Yesiloglu, T.Z.; Sippl, W.; Mahmud, S.; Alqahtani, T.; et al. An Analysis Based on Molecular Docking and Molecular Dynamics Simulation Study of Bromelain as Anti-SARS-CoV-2 Variants. Front. Pharmacol. 2021, 12, 2192. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kan, R.; Ji, H.; Wu, S.; Zhao, W.; Shuian, D.; Liu, J.; Li, J. Identification of tuna protein-derived peptides as potent SARS-CoV-2 inhibitors via molecular docking and molecular dynamic simulation. Food Chem. 2021, 342, 128366. [Google Scholar] [CrossRef]
- Herschlag, D.; Pinney, M.M. Hydrogen Bonds: Simple after All? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Q. Effect of conjugated (EK)(10) peptide on structural and dynamic properties of ubiquitin protein: A molecular dynamics simulation study. J. Mater. Chem. B 2020, 8, 6934–6943. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Mahmud, S.; Sultana, R.; Dong, W. Identification and in silico molecular modelling study of newly isolated Bacillus subtilis SI-18 strain against S9 protein of Rhizoctonia solani. Arab. J. Chem. 2020, 13, 8600–8612. [Google Scholar] [CrossRef]
- Ali, S.A.; Hassan, M.I.; Islam, A.; Ahmad, F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 456–476. [Google Scholar] [CrossRef]
- Mahmud, S.; Uddin, M.; Paul, G.K.; Shimu, M.; Islam, S.; Rahman, E.; Islam, A.; Islam, M.S.; Promi, M.M.; Emran, T.B.; et al. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Brief. Bioinform. 2021, 22, 1402–1414. [Google Scholar] [CrossRef]
- Mondal, I.; Basak, T.; Banerjee, S.; Chattopadhyay, S. A theoretical insight on the rigid hydrogen-bonded network in the solid state structure of two zinc(ii) complexes and their strong fluorescence behaviors. CrystEngComm 2020, 22, 3005–3019. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel. J. Cheminform. 2011, 3, 1–14. Available online: https://jcheminf.biomedcentral.com/track/pdf/10.1186/1758-2946-3-33 (accessed on 26 July 2021).
- Morris, G.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Tumilaar, S.G.; Siampa, J.P.; Fatimawali; Kepel, B.J.; Niode, N.J.; Idroes, R.; Rakib, A.; Emran, T.B.; Tallei, T.E. The potential of leaf extract of Pangium edule Reinw as HIV-1 protease inhibitor: A computational biology approach. J. Appl. Pharm. Sci. 2021. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Mackerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013. [CrossRef] [PubMed] [Green Version]
- Price, D.J.; Brooks, C.L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qiu, Y.; Baron, R.; Molinero, V. Coarse-Graining of TIP4P/2005, TIP4P-Ew, SPC/E, and TIP3P to Monatomic Anisotropic Water Models Using Relative Entropy Minimization. J. Chem. Theory Comput. 2014, 10, 4104–4120. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, C.; Dinesh, D.C.; Panwar, U.; Abhirami, R.; Boura, E.; Singh, S.K. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 Guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J. Biomol. Struct. Dyn. 2020, 39, 4582–4593. [Google Scholar] [CrossRef]
- Hadda, T.B.; Berredjem, M.; Almalki, F.A.; Rastija, V.; Bader, A.; Jamalis, J.; Emran, T.B.; Abu-Iznei, T.; Esharkawy, E.; Rodriguez, L.C.; et al. How to face COVID-19: Proposed treatments based on Remdesivir and Hydroxychloroquin in presence of zinc-sulfate and POM theory. J. Biomol. Struct. Dyn. 2021, 2021, 1–14. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands’ Chemical Name | PubChem ID | Binding Affinity to the Receptors (kcal/mol) | |
|---|---|---|---|
| 6LU7 | 6YLA | ||
| Lopinavir | 92727 | −5.2 | - |
| Chelidimerine | 190990 | −10.2 | −8.2 |
| Withanolide G | 21679023 | −8.6 | −8.4 |
| Badrakemin acetate | 1771505 | −8.6 | −8.0 |
| Samarcandin | 71587098 | −8.5 | −7.4 |
| Catechin gallate | 6419835 | −8.6 | −6.1 |
| Somniferine | 14106343 | −8.3 | −6.7 |
| Withanone | 21679027 | −8.2 | −7.8 |
| Adlumidine | 120734 | −8.2 | −6.8 |
| Pelargonidin 3-glucoside | 443648 | −8.1 | −6.2 |
| Norsanguinarine | 97679 | −7.5 | −7.0 |
| Sanguinarine | 5154 | −7.7 | −6.8 |
| Fumariline | 159888 | −7.8 | −6.4 |
| Astragalin | 5282102 | −7.9 | −4.4 |
| Rutin | 5280805 | −7.4 | −4.1 |
| Cyanidin 3,5-di-O-glucoside | 441688 | −6.9 | −2.8 |
| Cyanidin 3-O-rutinoside | 441674 | −6.9 | −4.0 |
| Kaempferitrin | 5486199 | −6.1 | −4.6 |
| Harpagoside | 5281542 | −6.1 | −3.9 |
| Pinoresinol 4-O-b-d-glucopyranoside | 486614 | −4.9 | −7.1 |
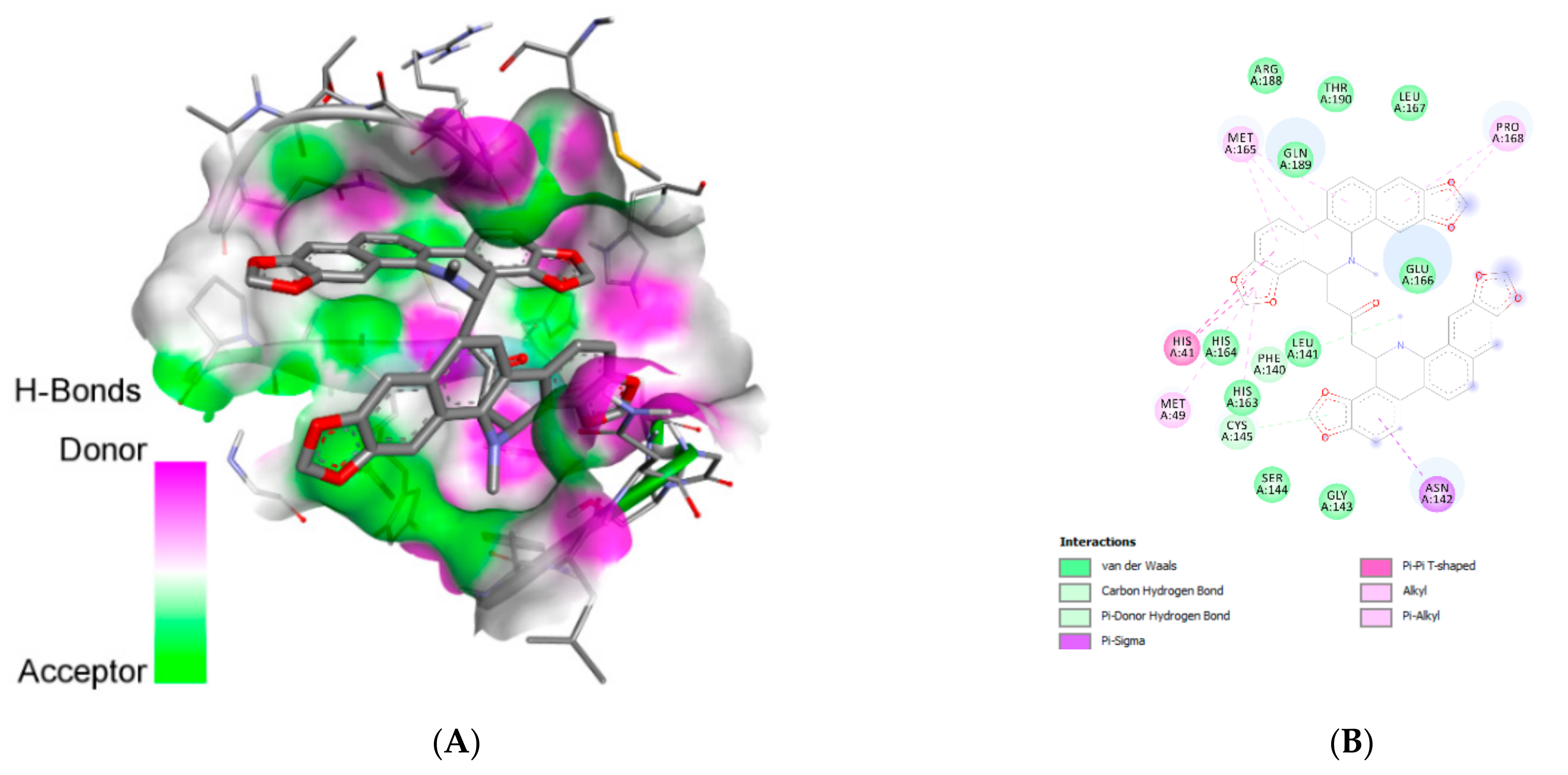
| Ligand | Interacting Residues | Category | Type of Interaction |
|---|---|---|---|
| Lopinavir | Glu:A166 | H-bond | Conventional |
| Asn:A142 | H-bond | Conventional | |
| Gln:A189 | H-bond | Conventional | |
| Gly:A143 | H-bond | Conventional | |
| Asn:A142 | H-bond | Carbon | |
| Met:A49 | Hydrophobic | Pi-sigma | |
| Pro:A168 | Hydrophobic | Alkyl/pi-alkyl | |
| His:A163 | Hydrophobic | Alkyl/pi-alkyl | |
| Cys:A145 | Hydrophobic | Alkyl/pi-alkyl | |
| His:A163 | Hydrophobic | Pi-sulfur | |
| Cys:A145 | Hydrophobic | Pi-sulfur | |
| Chelidimerine | Cys:A145 | H-bond | Pi-donor/carbon |
| Phe:A140 | H-bond | Pi-donor/carbon | |
| His:A41 (2) | Hydrophobic | Pi-pi T-shaped | |
| Met:A49 | Hydrophobic | Alkyl/pi-alkyl | |
| Cys:A145 | Hydrophobic | Alkyl/pi-alkyl | |
| Met:A165 (3) | Hydrophobic | Alkyl/pi-alkyl | |
| Pro:A168 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Asn:A142 | Hydrophobic | Pi-sigma | |
| Catechin gallate | Glu:A166 | H-bond | Conventional |
| Asp:A187 | H-bond | Conventional | |
| Gln:A192 | H-bond | Conventional | |
| Cys:A145 | H-bond | Conventional | |
| Leu:A141 | H-bond | Conventional | |
| His:A163 | H-bond | Conventional | |
| Phe:A140 | H-bond | Conventional | |
| Met:A165 (3) | Hydrophobic | Alkyl/pi-alkyl | |
| Pro:A168 | Hydrophobic | Alkyl/pi-alkyl | |
| His:A41 | Hydrophobic | Pi-pi T-shaped | |
| Cys:A145 | Hydrophobic | Pi-sulfur | |
| Badrakemin acetate | His:A41 | H-bond | Conventional |
| Gln:A192 | H-bond | Conventional | |
| Cys:A145 (3) | Hydrophobic | Alkyl/pi-alkyl | |
| His:A163 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| His:A41 | Hydrophobic | Alkyl/pi-alkyl | |
| Met:A165 (2) | Hydrophobic | Pi-sulfur | |
| His:A41 | Hydrophobic | Pi-sigma | |
| Withanolide G | Asn:A142 | H-bond | Conventional |
| Gly:A143 | H-bond | Conventional | |
| Glu:A166 | H-bond | Conventional | |
| Asn:A142 | H-bond | Carbon | |
| Cys:A145 (3) | Hydrophobic | Alkyl/pi-alkyl | |
| His:A163 | Hydrophobic | Alkyl/pi-alkyl | |
| Pro:A168 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Met:A165 | Hydrophobic | Alkyl/pi-alkyl | |
| Samarcandin | Cys:A145 | H-bond | Conventional |
| Gly:A143 | H-bond | Conventional | |
| Glu:A166 (2) | H-bond | Conventional | |
| Cys:A145 | H-bond | Pi-donor | |
| Leu:A167 | Hydrophobic | Alkyl | |
| Pro:A168 (2) | Hydrophobic | Alkyl | |
| Somniferine | Gln:A189 | H-bond | Conventional |
| Glu:A166 (2) | H-bond | Conventional | |
| Met:A165 | H-bond | Carbon | |
| His:A41 | Hydrophobic | Alkyl/pi-alkyl | |
| Cys:A145 | Hydrophobic | Alkyl/pi-alkyl | |
| Pro:A168 | Hydrophobic | Alkyl/pi-alkyl | |
| Adlumidine | Glu:A166 | H-bond | Conventional |
| Cys:A145 | H-bond | Conventional | |
| Glu:A166 | H-bond | Carbon | |
| Leu:A167 | H-bond | Carbon | |
| Phe:A140 | H-bond | Carbon | |
| Glu:A166 (2) | Hydrophobic | Pi-anion | |
| Cys:A145) | Hydrophobic | Pi-alkyl | |
| Withanone | Glu:A166 | H-bond | Conventional |
| Cys:A145 | H-bond | Conventional | |
| Gly:A143 | H-bond | Conventional | |
| Ser:A144 | H-bond | Conventional | |
| His:A41 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Met:A49 | Hydrophobic | Alkyl/pi-alkyl | |
| Cys:A145 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Pelargonidin-3 glucoside | His:A163 | H-bond | Conventional |
| His:A164 | H-bond | Conventional | |
| Glu:A166 (2) | H-bond | Conventional | |
| Thr:A190 | H-bond | Conventional | |
| Gln:A189 | Hydrophobic | Pi-sigma | |
| His:A41 | Hydrophobic | Pi-pi T-shaped | |
| Pro:A168 | Hydrophobic | Pi-alkyl | |
| Met:A165 | Hydrophobic | Pi-alkyl | |
| Met:A165 (2) | Hydrophobic | Pi-sulfur | |
| Astragalin | Gln:A189 (2) | H-bond | Conventional |
| Tyr:A54 | H-bond | Conventional | |
| Glu:A166 (2) | H-bond | Conventional | |
| Thr:A190 | H-bond | Conventional | |
| Met:A165 (3) | Hydrophobic | Pi-alkyl | |
| Met:A49 | Hydrophobic | Pi-alkyl | |
| His:A41 | Hydrophobic | Pi-pi T-shaped | |
| Fumariline | Glu:A166 | H-bond | Conventional |
| Glu:A166 | H-bond | Carbon | |
| Gln:A192 | H-bond | Carbon | |
| Leu:A167 | H-bond | Carbon | |
| Gln:A189 | Hydrophobic | Amide-pi stacked | |
| Pro:A168 | Hydrophobic | Pi-sigma | |
| Leu:A167 | Hydrophobic | Pi-alkyl | |
| Pro:A168 | Hydrophobic | Pi-alkyl | |
| Met:A165 | Hydrophobic | Pi-alkyl | |
| Sanguinarine | Tyr:A54 | H-bond | Conventional |
| Met:A49 | H-bond | Carbon | |
| Asp:A187 | H-bond | Carbon | |
| Cys:A145 | H-bond | Carbon | |
| Glu:A166 | H-bond | Carbon | |
| Met:A49 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Cys:A145 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| His:A163 | Hydrophobic | Pi-cation | |
| Norsanguinarine | His:A163 | H-bond | Conventional |
| Leu:A141 | H-bond | Carbon | |
| Ser:A144 | H-bond | Carbon | |
| Met:A49 (2) | Hydrophobic | Pi-alkyl | |
| Cys:A145 | Hydrophobic | Pi-alkyl | |
| Met:A165 | Hydrophobic | Pi-alkyl | |
| His:A41 (2) | Hydrophobic | Pi-pi T-shaped | |
| Met:A165 | Hydrophobic | Pi-sigma | |
| Rutin | Asp:A187 | H-bond | Conventional |
| His:A41 | H-bond | Conventional | |
| Arg:A188 | H-bond | Conventional | |
| Thr(A190) | H-bond | Conventional | |
| Glu:A166 (2) | H-bond | Conventional | |
| Glu:A166 | H-bond | Carbon | |
| Pro:A168 | H-bond | Carbon | |
| Cys:A145 (2) | Hydrophobic | Pi-cation/pi-sulfur | |
| Met:A49 | Hydrophobic | Pi-cation/pi-sulfur | |
| His:A49 | Hydrophobic | Pi-cation/pi-sulfur | |
| Met:A165 | Hydrophobic | Pi-alkyl | |
| Cyanidin 3,5-di-O-glucoside | Gln:A192 | H-bond | Conventional |
| Glu:A166 (2) | H-bond | Conventional | |
| Gly:A143 | H-bond | Conventional | |
| Leu:A141 | H-bond | Conventional | |
| Glu:A166 | H-bond | Carbon | |
| His:A41 | Hydrophobic | Pi-pi T-shaped | |
| Gln:A189 | Hydrophobic | Pi-sigma | |
| Met:A165 (2) | Hydrophobic | Pi-alkyl | |
| Pro:A168 | Hydrophobic | Pi-alkyl | |
| Met:A165 (2) | Hydrophobic | Pi-sulfur | |
| His:A164 | Unfavorable | Donor-donor | |
| Cyanidin 3-O-rutinoside | Asn:A142 (2) | H-bond | Conventional |
| Cys:A145 | H-bond | Conventional | |
| His:A164 | H-bond | Conventional | |
| Asp:A187 | H-bond | Conventional | |
| Met:A49 | H-bond | Conventional | |
| Thr:A190 | H-bond | Conventional | |
| Gln:A189 | H-bond | Carbon | |
| Met:A165 (2) | Hydrophobic | Pi-alkyl | |
| His:A41 | Hydrophobic | Pi-pi T-shaped | |
| His:A163 | Unfavorable | Donor-donor | |
| Gln:A192 | Unfavorable | Donor-donor | |
| Kaempferitrin | Ser:A144 | H-bond | Conventional |
| Cys:A145 | H-bond | Conventional | |
| Leu:A141 | H-bond | Conventional | |
| Glu:A166 | H-bond | Conventional | |
| Asn:A142 | H-bond | Conventional | |
| Glu:A166 | H-bond | Pi-donor | |
| Met:A49 | Hydrophobic | Alkyl/pi-alkyl | |
| Ala:A191 | Hydrophobic | Alkyl/pi-alkyl | |
| Met:A165 | Hydrophobic | Pi-sulfur | |
| Glu:A166 | Hydrophobic | Pi-lone pair | |
| Harpagoside | Glu:A166 (3) | H-bond | Conventional |
| Asn:A142 | H-bond | Conventional | |
| Leu:A141 | H-bond | Carbon | |
| Met:A49 | Hydrophobic | Pi-sulfur | |
| Met:A165 | Hydrophobic | Pi-sulfur | |
| His:A41 | Hydrophobic | Pi-pi T-shaped |
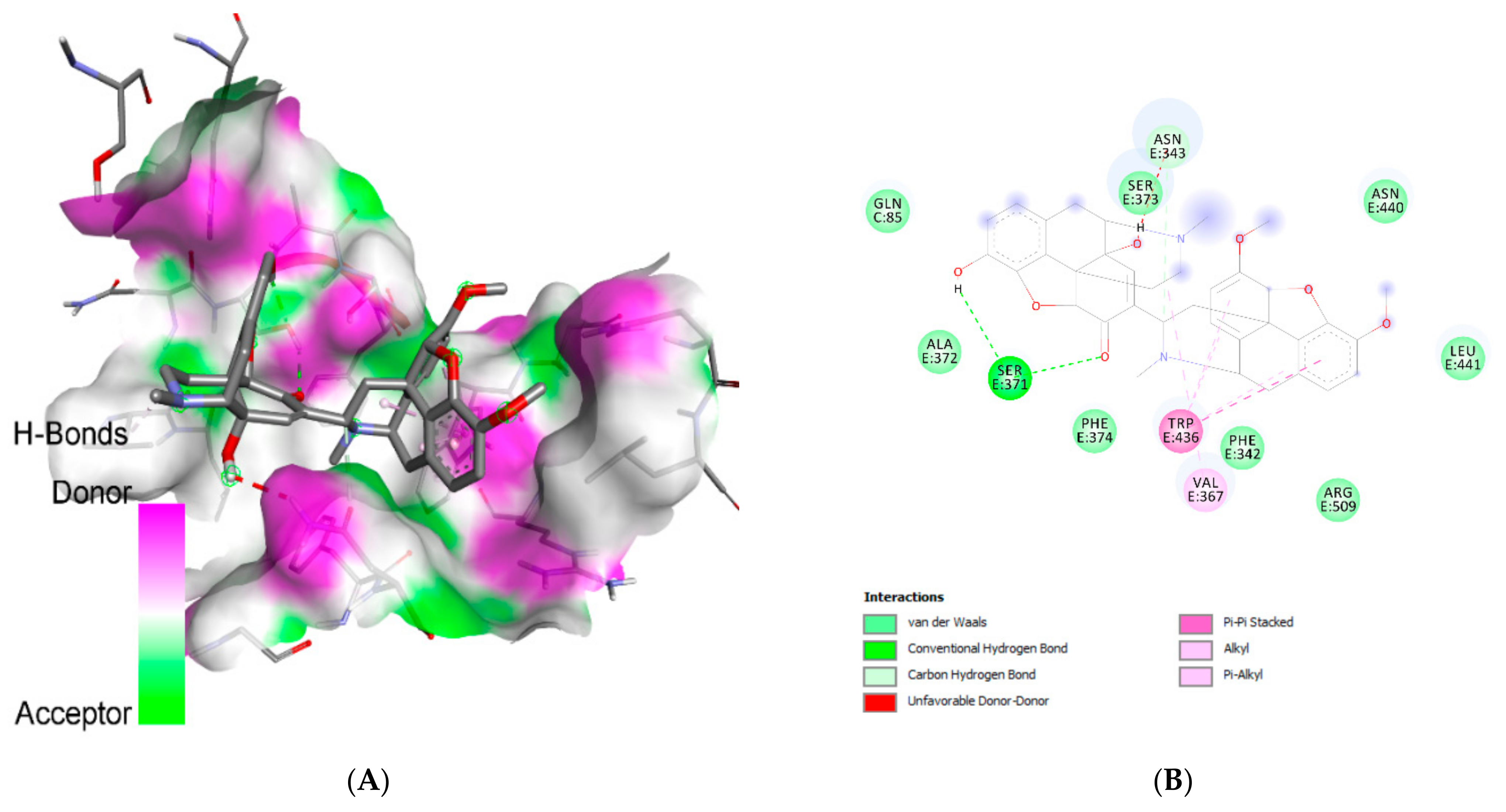
| Ligand | Interacting Residues | Category | Type of Interaction |
|---|---|---|---|
| Withanolide G | Asn:E370 | H-bond | Conventional |
| Asp:C66 | H-bond | Conventional | |
| Arg:C67 | H-bond | Conventional | |
| Gln:C85 | H-bond | Conventional | |
| Val:E367 (4) | Hydrophobic | Alkyl/pi-alkyl | |
| Phe:E374 | Hydrophobic | Alkyl/pi-alkyl | |
| Chelidimerine | Ser:E371 | H-bond | Carbon |
| Asp:E364 | H-bond | Carbon | |
| Val:E367 (4) | Hydrophobic | Alkyl/pi-alkyl | |
| Phe:E374 | Hydrophobic | Pi-sigma | |
| Phe:E338 | Hydrophobic | Pi-sigma | |
| Badrakemin acetate | Glu:E340 | H-bond | Conventional |
| Gly:E339 | H-bond | Conventional | |
| Asn:E343 | H-bond | Pi-donor | |
| Val:E367 (3) | Hydrophobic | Alkyl/pi-alkyl | |
| Leu:E335 (3) | Hydrophobic | Alkyl/pi-alkyl | |
| Leu:E368 | Hydrophobic | Alkyl/pi-alkyl | |
| Phe:E338 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Asn:E343 | Hydrophobic | Pi-lone pair | |
| Withanone | Gly:E339 | H-bond | Pi-donor |
| Trp:E436 (3) | Hydrophobic | Alkyl/pi-alkyl | |
| Leu:E368 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Phe:E342 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Phe:E374 | Hydrophobic | Alkyl/pi-alkyl | |
| Val:E367 | Hydrophobic | Alkyl/pi-alkyl | |
| Ser:E371 | Unfavorable | Donor-donor | |
| Samarcandin | Asn:E343 | H-bond | Conventional |
| Asp:E364 | H-bond | Conventional | |
| Leu:E335 | Hydrophobic | Pi-alkyl | |
| Val:E367 | Hydrophobic | Pi-alkyl | |
| Val:E367 | Hydrophobic | Pi-sigma | |
| Phe:E338 | Hydrophobic | Pi-sigma | |
| Pinoresinol 4-O-b-d-glucopyranoside | Asp:E364 | H-bond | Conventional |
| Cys:E336 | H-bond | Conventional | |
| Asn:E343 (2) | H-bond | Conventional | |
| Ser:E371 | H-bond | Conventional | |
| Val:E367 (2) | Hydrophobic | Pi-alkyl | |
| Leu:E368 | Hydrophobic | Pi-alkyl | |
| Val:E367 | Hydrophobic | Pi-sigma | |
| Phe:E338 | Hydrophobic | Pi-sigma | |
| Norsanguinarine | Asp:C66 | H-bond | Conventional |
| Val:E367 | H-bond | Conventional | |
| Asp:C66 | H-bond | Carbon | |
| Arg:C67 | Hydrophobic | Pi-alkyl | |
| Val:E367 | Hydrophobic | Pi-alkyl | |
| 2Asn:E370 | Hydrophobic | Amide-pi stacked | |
| Sanguinarine | Asn:E343 | H-bond | Conventional |
| Leu:E335 | Hydrophobic | Alkyl/pi-alkyl | |
| Val:E367 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Val:E367 | Hydrophobic | Pi-sigma | |
| Phe:E342 | Hydrophobic | Pi-pi stacked | |
| Adlumidine | Asn:E343 | H-bond | Conventional |
| Val:E367 | H-bond | Carbon | |
| Leu:E368 | H-bond | Carbon | |
| Val:E367 | Hydrophobic | Pi-sigma | |
| Phe:E374 | Hydrophobic | Pi-alkyl | |
| Phe:E374 | Hydrophobic | Pi-pi T-shaped | |
| Trp:E436 | Hydrophobic | Pi-pi T-shaped | |
| Phe:E338 | Hydrophobic | Pi-pi T-shaped | |
| Somniferine | Ser:E371 (2) | H-bond | Conventional |
| Asn:E343 | H-bond | Carbon | |
| Trp:E436 (3) | Hydrophobic | Alkyl/pi-alkyl | |
| Val:E367 | Hydrophobic | Alkyl/pi-alkyl | |
| TrpE436 | Hydrophobic | Pi-pi stacked | |
| Asn:E343 | Unfavorable | Donor-donor | |
| Fumariline | Gly:E339 | H-bond | Conventional |
| Leu:E335 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Val:E367 (2) | Hydrophobic | Alkyl/pi-alkyl | |
| Phe:E338 | Hydrophobic | Pi-sigma | |
| Pelargonidin-3 glucoside | Asn:E343 (2) | H-bond | Conventional |
| Ser:E371 | H-bond | Conventional | |
| Asn:E364: | H-bond | Conventional | |
| Cys:E336 | H-bond | Conventional | |
| Phe:E338 | Hydrophobic | Pi-sigma | |
| Val:E367 | Hydrophobic | Pi-sigma | |
| Val:E367 (2) | Hydrophobic | Pi-alkyl | |
| Leu:E368 | Hydrophobic | Pi-alkyl | |
| Catechin gallate | Ser:E375 | H-bond | Conventional |
| Asn:E440 (2) | H-bond | Conventional | |
| Asn:E437 | H-bond | Conventional | |
| Arg:E509 | H-bond | Conventional | |
| Phe:E342 | H-bond | Conventional | |
| Trp:E436 | Hydrophobic | Pi-alkyl | |
| Leu:E441 | Hydrophobic | Pi-sigma | |
| Trp:E436 | Hydrophobic | Pi-pi T-shaped |
| Parameters | Chelidimerine | Withanolide G | Badrakemin Acetate | Samarcandin | Catechin Gallate | Somniferine | Withanone | Cyanidin 3-O-Rutinoside |
|---|---|---|---|---|---|---|---|---|
| Molecular weight | 720.7 g/mol | 454.6 g/mol | 424.5 g/mol | 400.5 g/mol | 442.4 g/mol | 608.7 g/mol | 470.6 g/mol | 595.5 g/mol |
| H-bond acceptor | 11 | 5 | 5 | 5 | 10 | 9 | 6 | 14 |
| H-bond donor | 0 | 2 | 0 | 2 | 7 | 2 | 2 | 10 |
| CNS | −2.718 | −2.894 | −1.638 | −2.044 | −3.743 | −3.073 | −2.719 | −4.943 |
| CYP2D6 substrate | No | No | No | No | No | No | No | No |
| CYP3A4 substrate | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| CYP1A2 inhibitor | No | No | No | No | No | No | No | No |
| CYP2C19 inhibitor | No | No | Yes | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | Yes | No | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | Yes | No | No | No | No | No |
| Carcinogenicity | Carcinogenic | Carcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic |
| Hepatotoxicity | No | No | No | Yes | No | No | No | No |
| p-glycoprotein substrate | Yes | Yes | No | Yes | No | Yes | Yes | No |
| Acute oral toxicity | Class IV, LD50 1408 mg/kg | Class IV, LD50 400 mg/kg | Class V, LD50 3200 mg/kg | Class V, LD50 3200 mg/kg | Class IV, LD50 1000 mg/kg | Class IV, LD50 1100 mg/kg | Class II, LD50 7 mg/kg | Class V, LD50 5000 mg/kg |
| Lipinski rule of five | No | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Ligands’ Chemical Name | Predicted IC50 | |
|---|---|---|
| 6LU7 | 6YLA | |
| Lopinavir | 136.22 µM | - |
| Chelidimerine | 25.90 nM | 843.38 nM |
| Withanolide G | 455.10 nM | 614.03 nM |
| Badrakemin acetate | 478. 95 nM | 1.23 µM |
| Samarcandin | 578.21 nM | 3.57 µM |
| Catechin gallate | 433.52 nM | 29.17 µM |
| Somniferine | 776.89 nM | 12.30 µM |
| Withanone | 895.84 nM | 1.68 µM |
| Adlumidine | 953.86 nM | 9.48 µM |
| Pelargonidin 3-glucoside | 1.06 µM | 25.10 µM |
| Norsanguinarine | 2.91 µM | 6.60 µM |
| Sanguinarine | 2.05 µM | 10.24 µM |
| Fumariline | 1.77 µM | 19.31 µM |
| Astragalin | 1.49 µM | 508.13 µM |
| Rutin | 3.59 µM | 894.70 µM |
| Cyanidin 3,5-di-O-glucoside | 8.43 µM | 1.09 mM |
| Cyanidin 3-O-rutinoside | 8.12 µM | 8.59 mM |
| Kaempferitrin | 31.75 µM | 381.16 mM |
| Harpagoside | 31.75 µM | 1.30 mM |
| Pinoresinol 4-O-b-d-glucopyranoside | 245.63 µM | 5.53 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mousavi, S.S.; Karami, A.; Haghighi, T.M.; Tumilaar, S.G.; Fatimawali; Idroes, R.; Mahmud, S.; Celik, I.; Ağagündüz, D.; Tallei, T.E.; et al. In Silico Evaluation of Iranian Medicinal Plant Phytoconstituents as Inhibitors against Main Protease and the Receptor-Binding Domain of SARS-CoV-2. Molecules 2021, 26, 5724. https://doi.org/10.3390/molecules26185724
Mousavi SS, Karami A, Haghighi TM, Tumilaar SG, Fatimawali, Idroes R, Mahmud S, Celik I, Ağagündüz D, Tallei TE, et al. In Silico Evaluation of Iranian Medicinal Plant Phytoconstituents as Inhibitors against Main Protease and the Receptor-Binding Domain of SARS-CoV-2. Molecules. 2021; 26(18):5724. https://doi.org/10.3390/molecules26185724
Chicago/Turabian StyleMousavi, Seyyed Sasan, Akbar Karami, Tahereh Movahhed Haghighi, Sefren Geiner Tumilaar, Fatimawali, Rinaldi Idroes, Shafi Mahmud, Ismail Celik, Duygu Ağagündüz, Trina Ekawati Tallei, and et al. 2021. "In Silico Evaluation of Iranian Medicinal Plant Phytoconstituents as Inhibitors against Main Protease and the Receptor-Binding Domain of SARS-CoV-2" Molecules 26, no. 18: 5724. https://doi.org/10.3390/molecules26185724