
Novel Oxidovanadium Complexes with Redox-Active R-Mian and R-Bian Ligands: Synthesis, Structure, Redox and Catalytic Properties

 , , , ,
, , , ,  and
and
Abstract
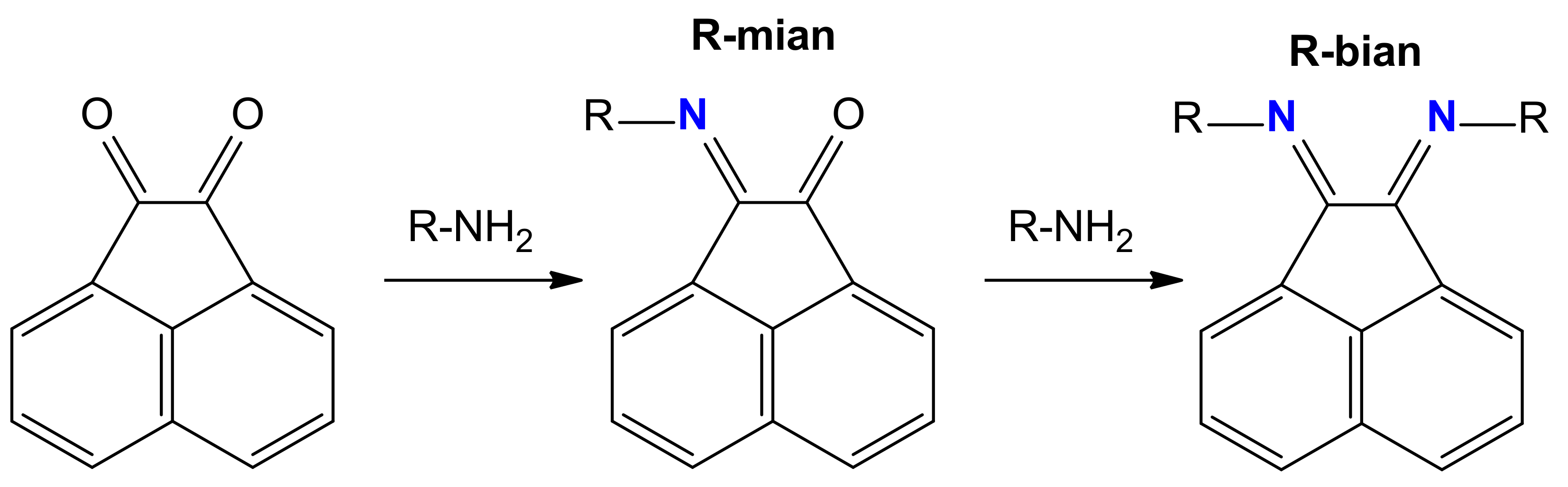
:1. Introduction
2. Results and Discussion
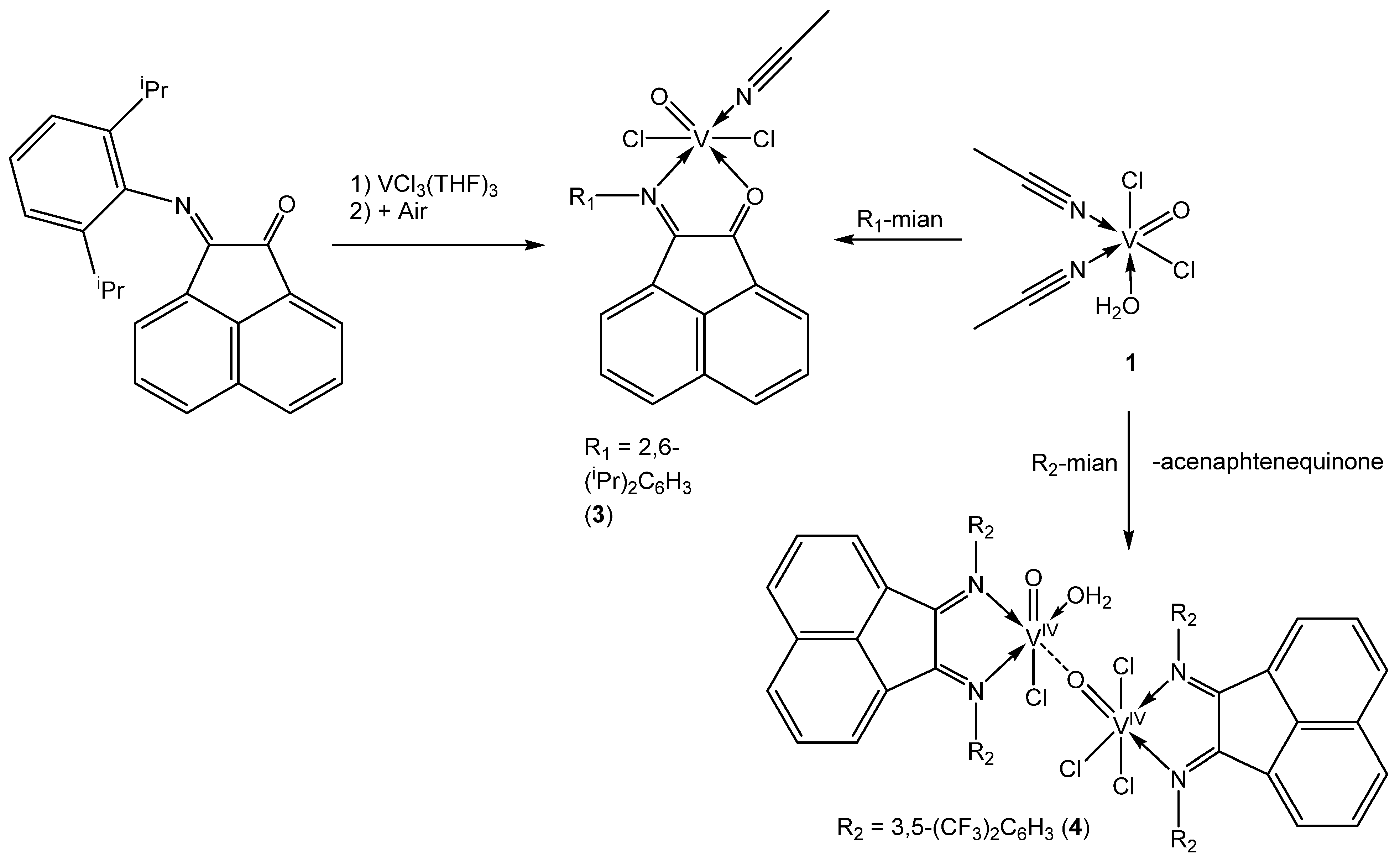
2.1. Synthesis of 1–4
2.2. IR Spectra of 1–4
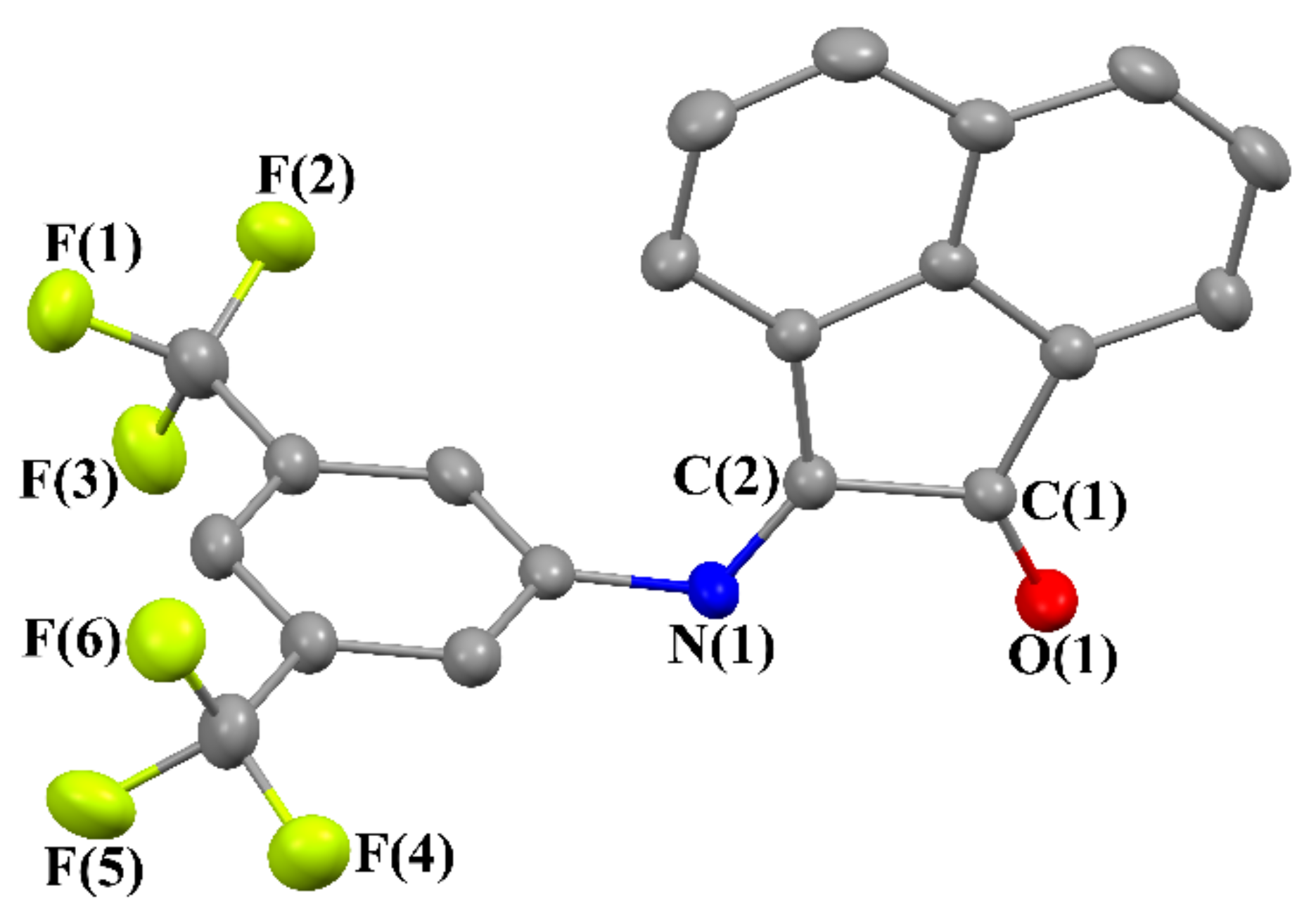
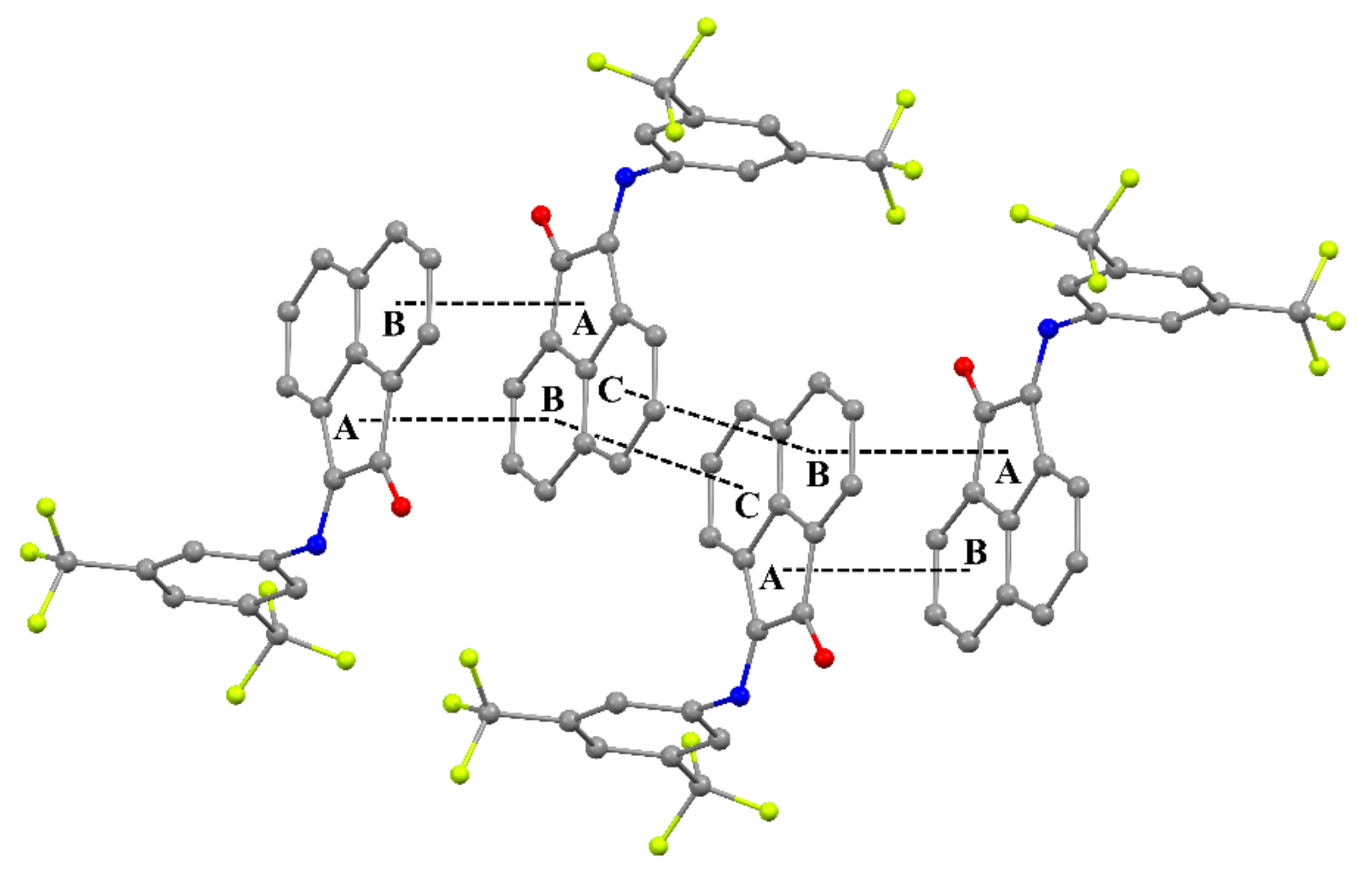
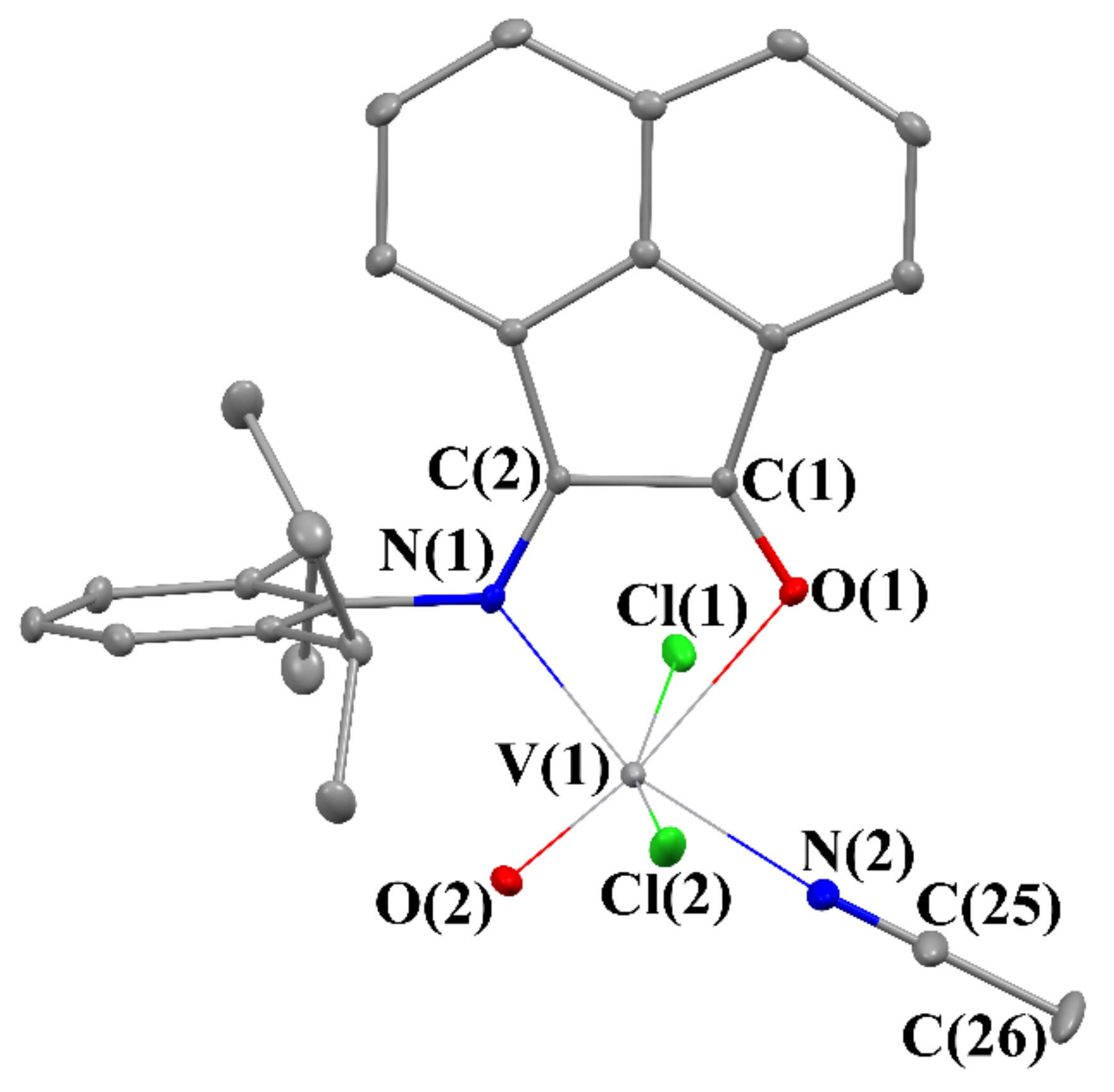
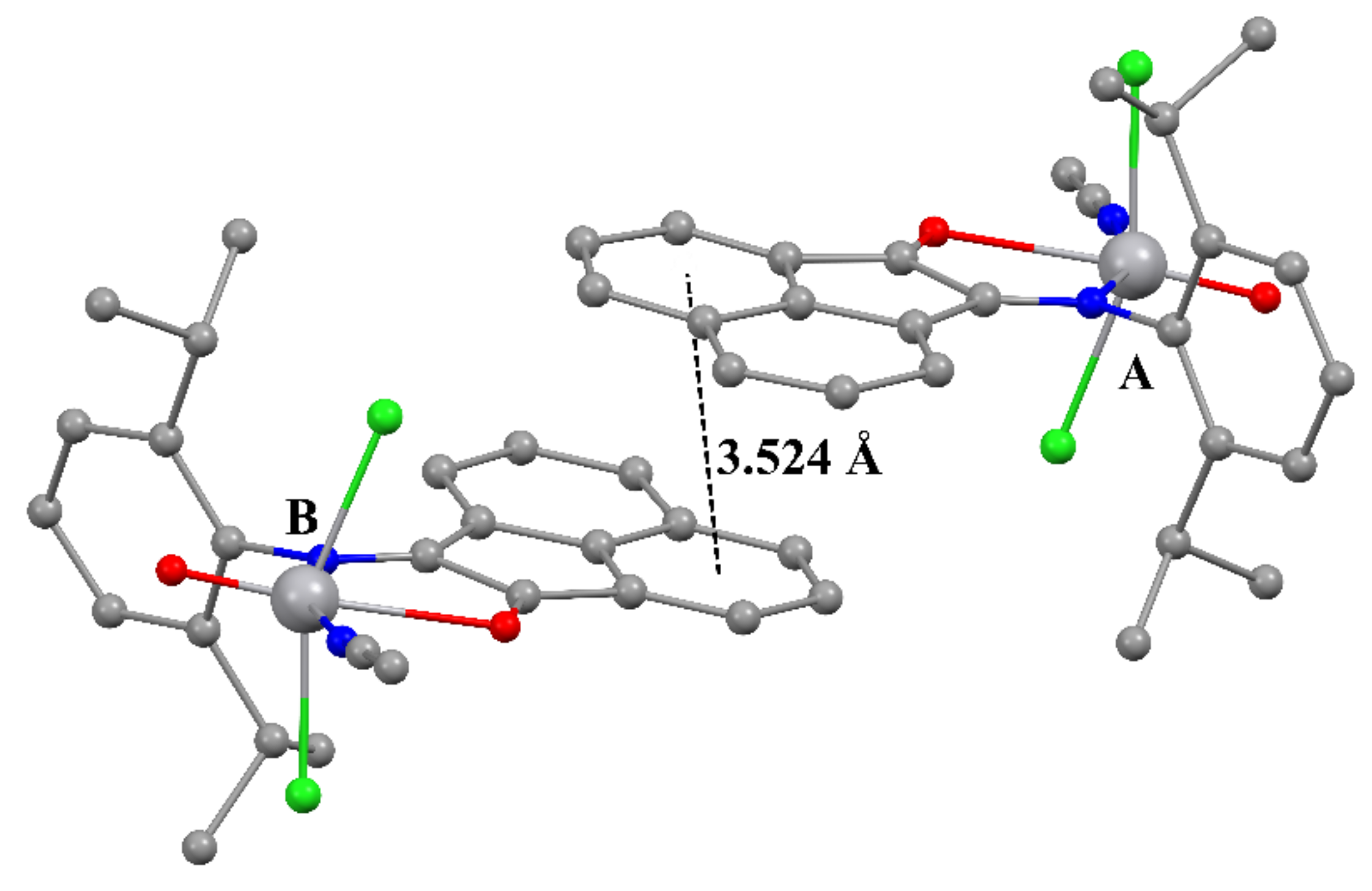
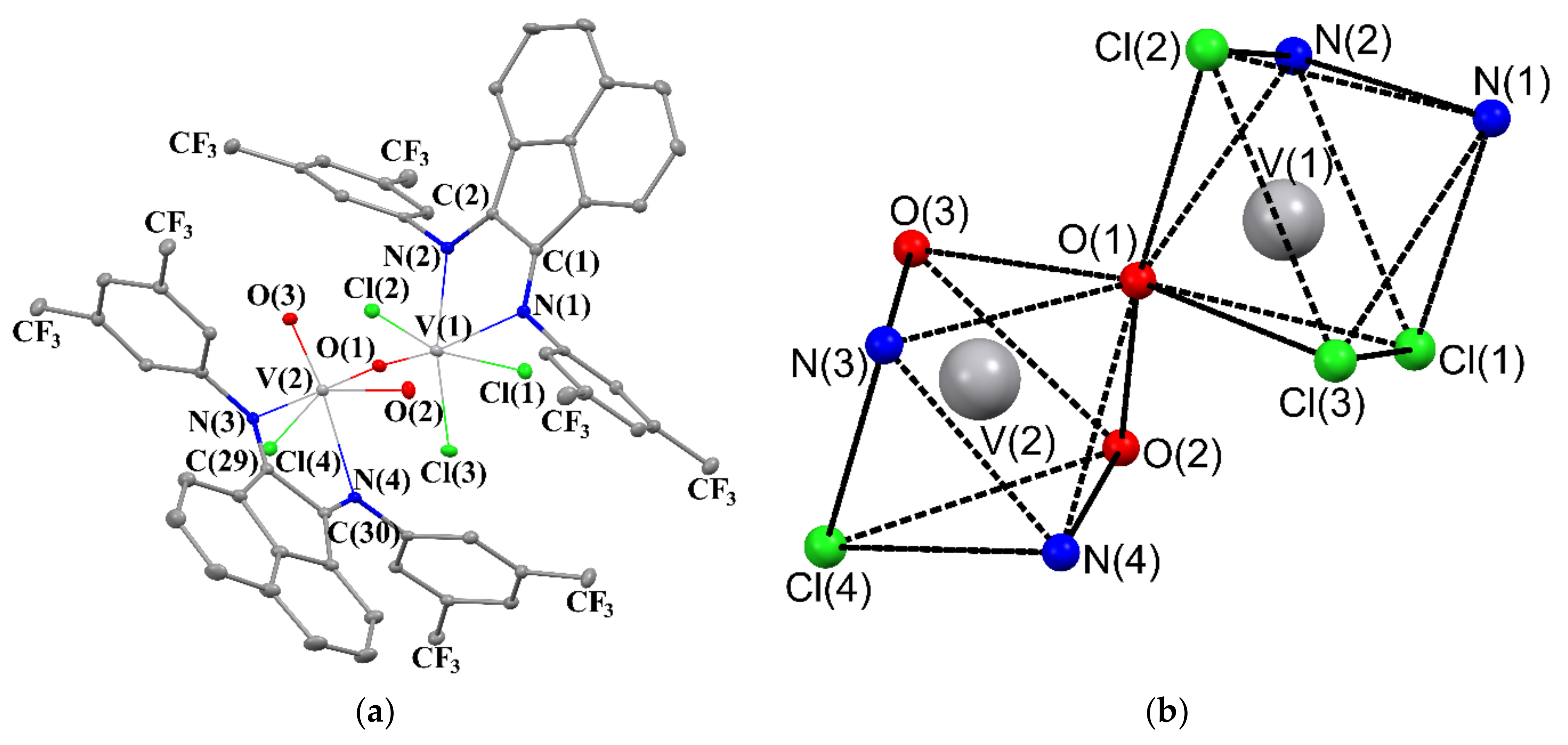
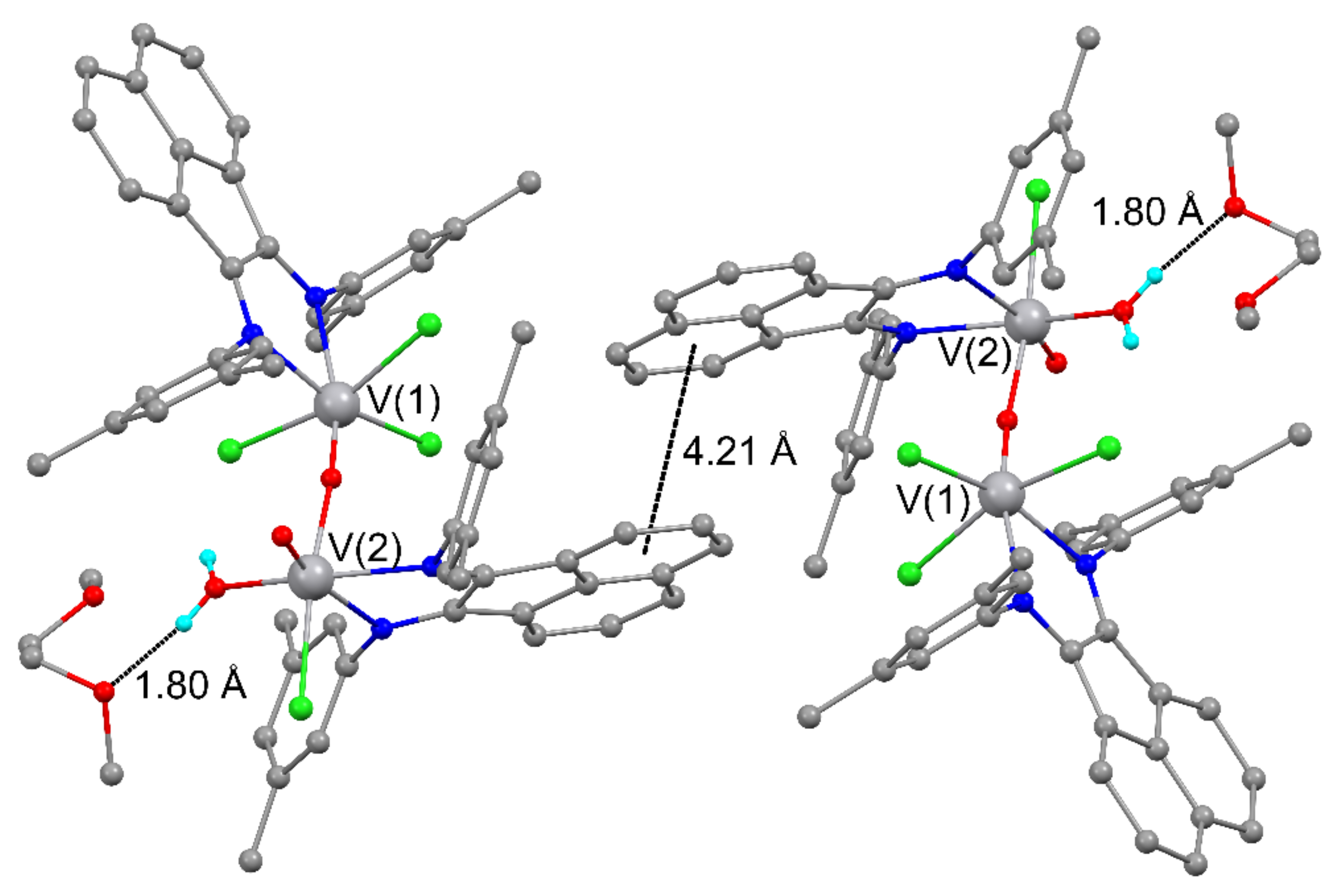
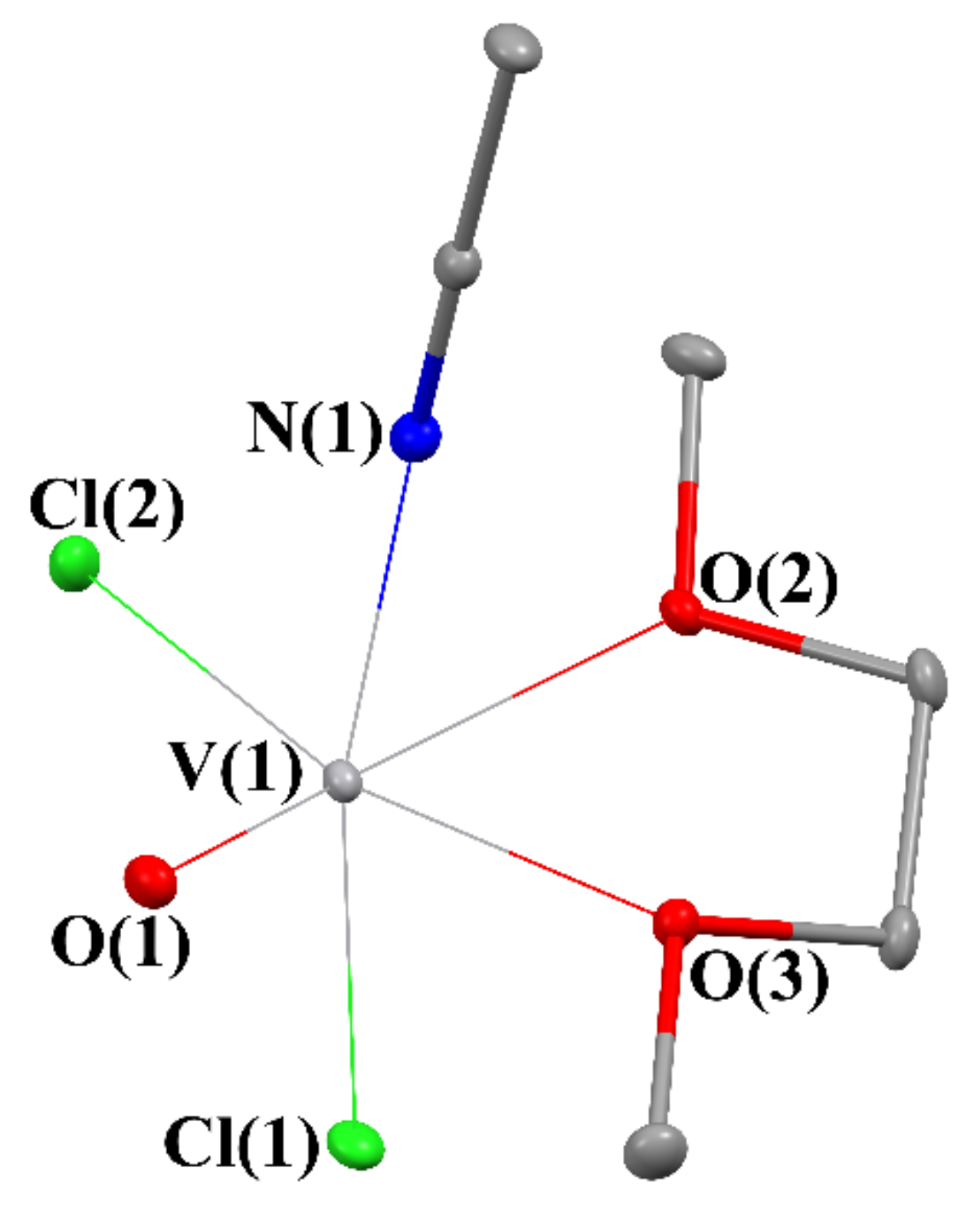
2.3. Crystal Structures of 2–5
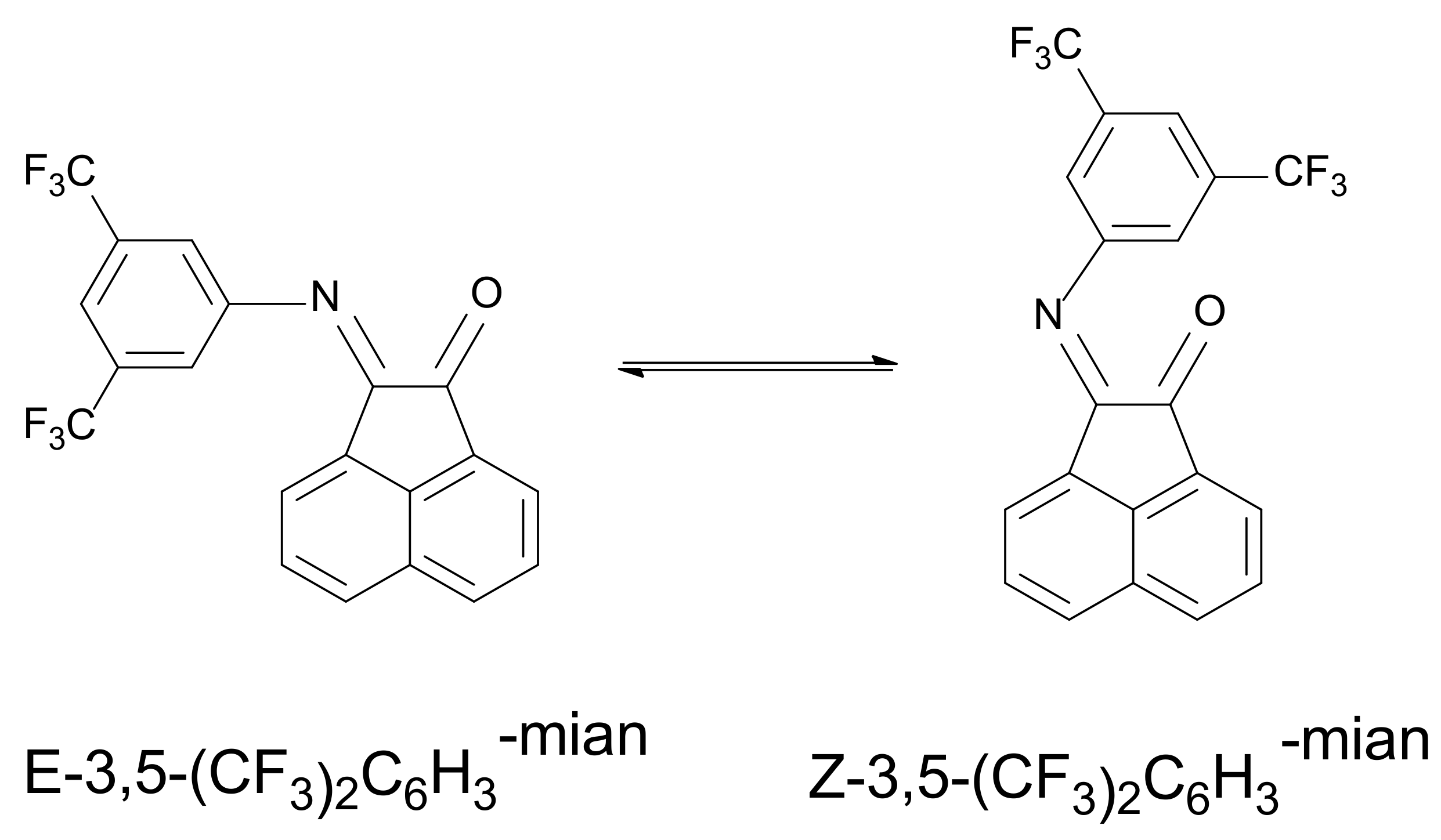
2.4. NMR Spectra of 3,5-(CF3)2C6H3-mian (2)
2.5. Redox Properties of dpp-mian and 2–4
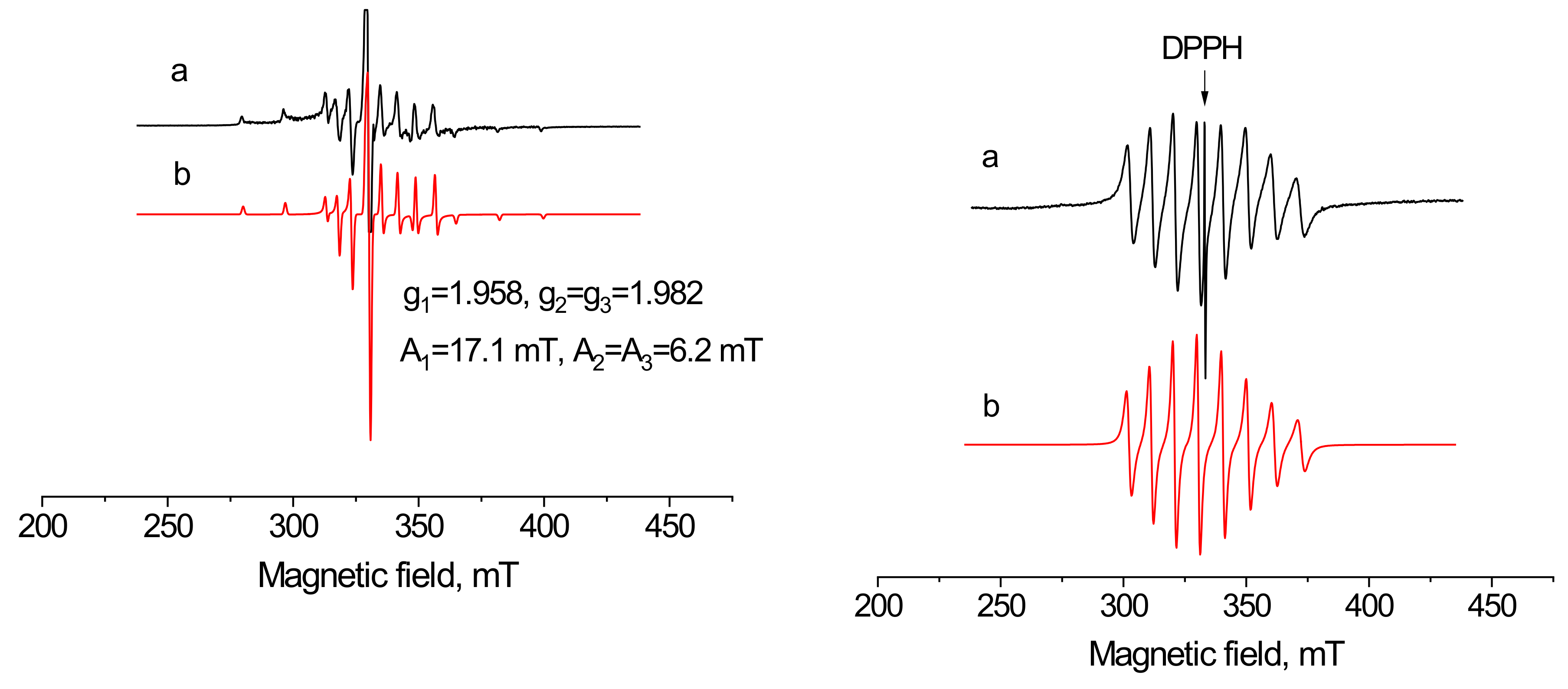
2.6. The EPR Spectra of 3 and 4
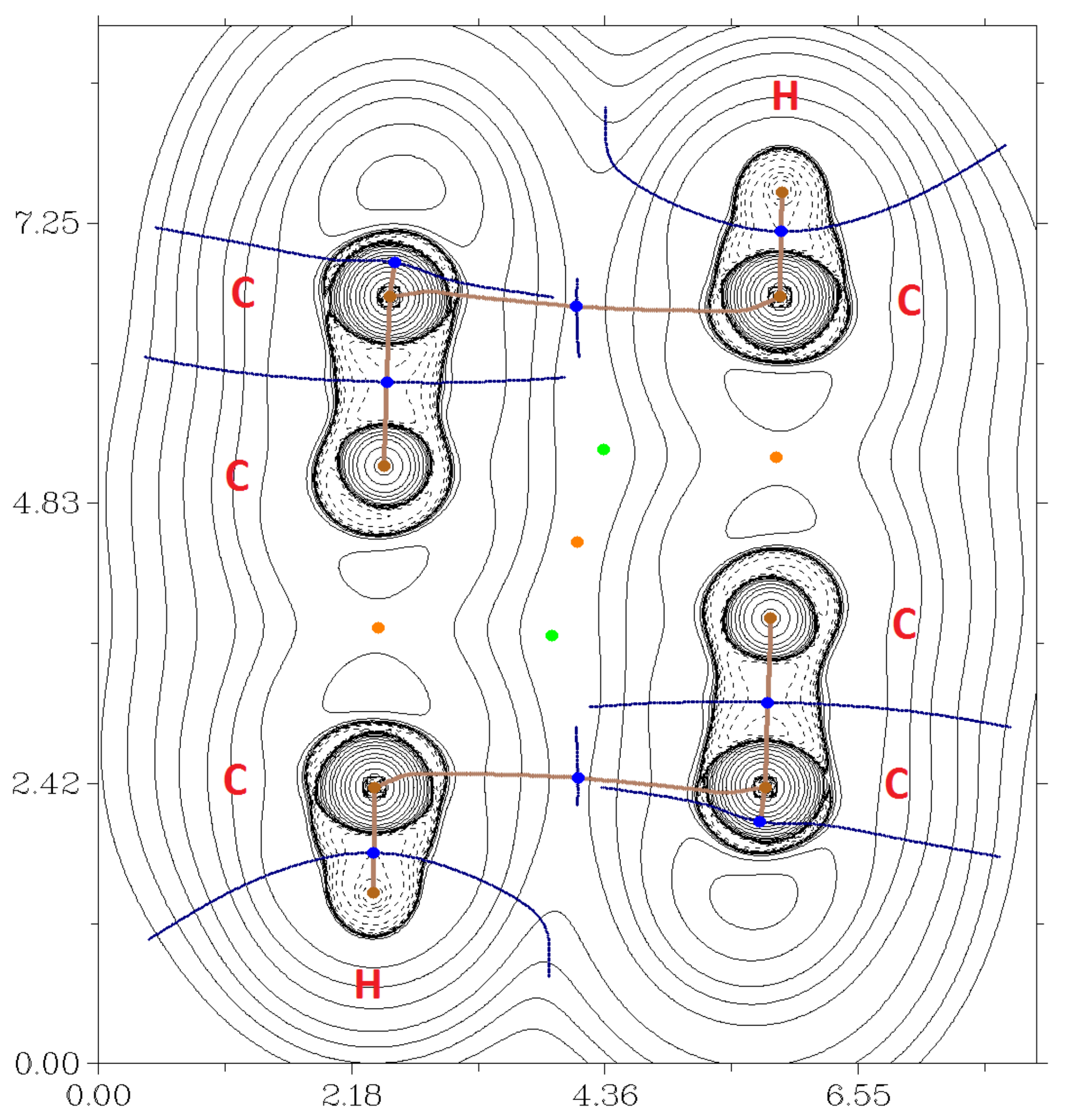
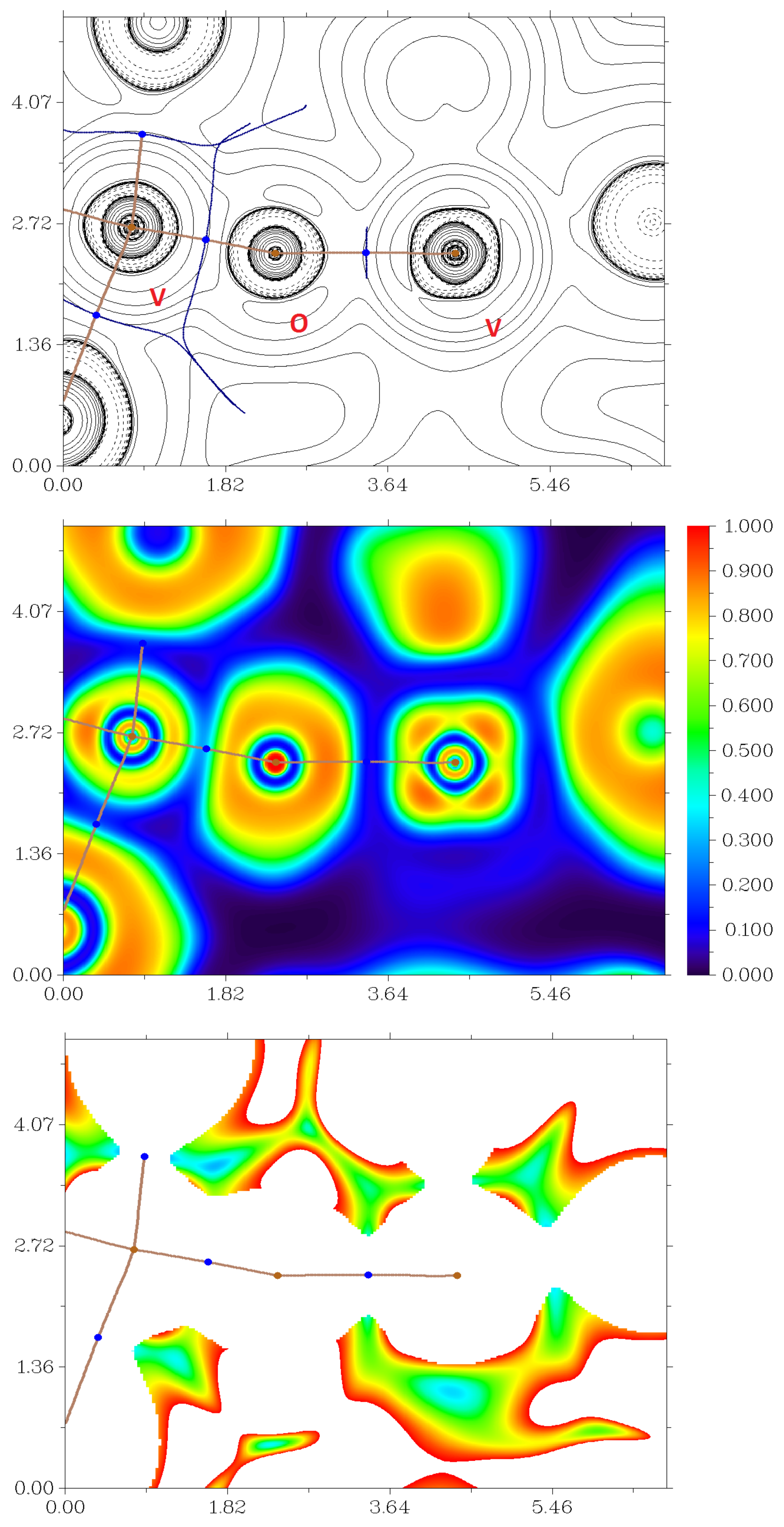
2.7. DFT Calculations
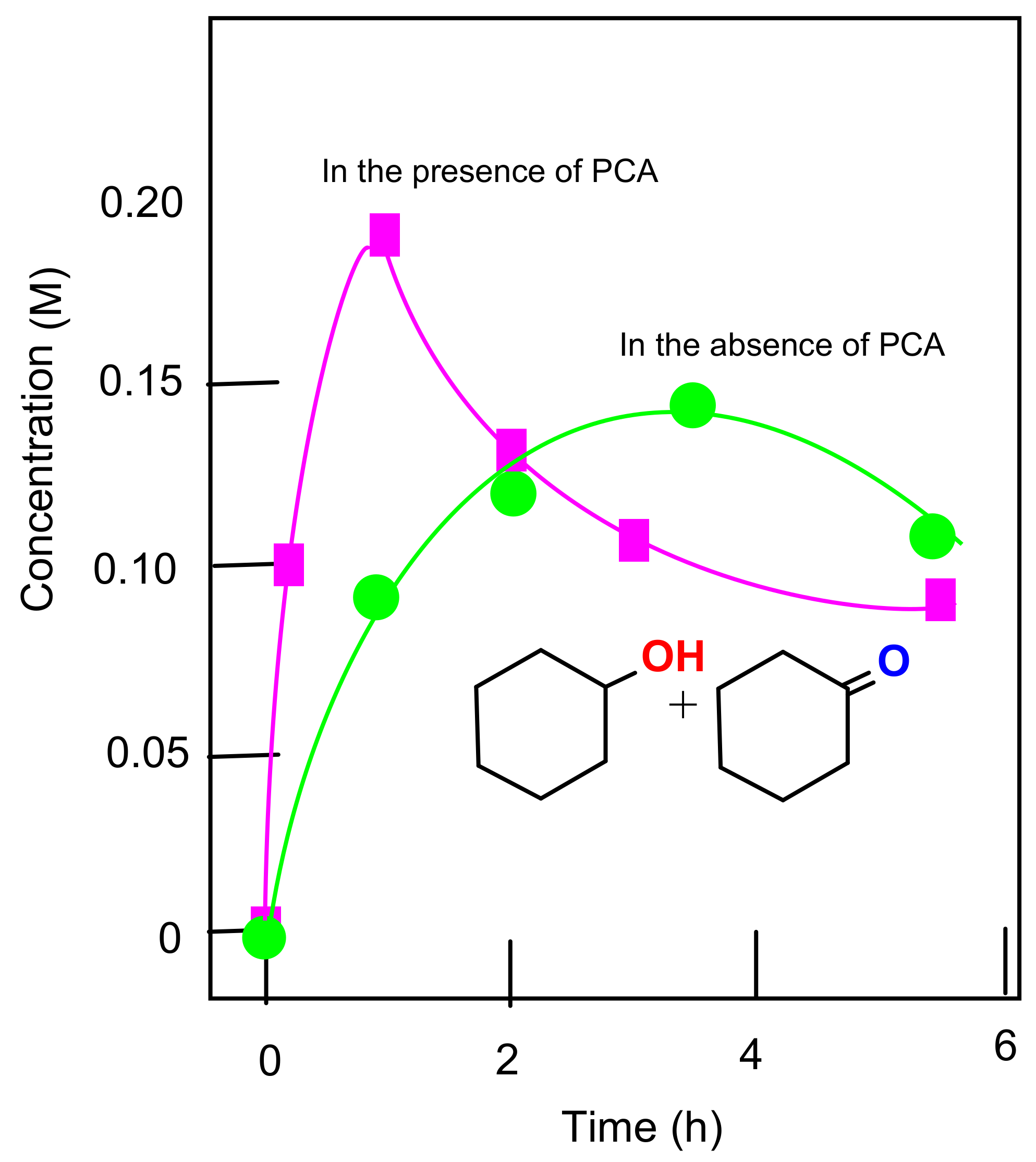
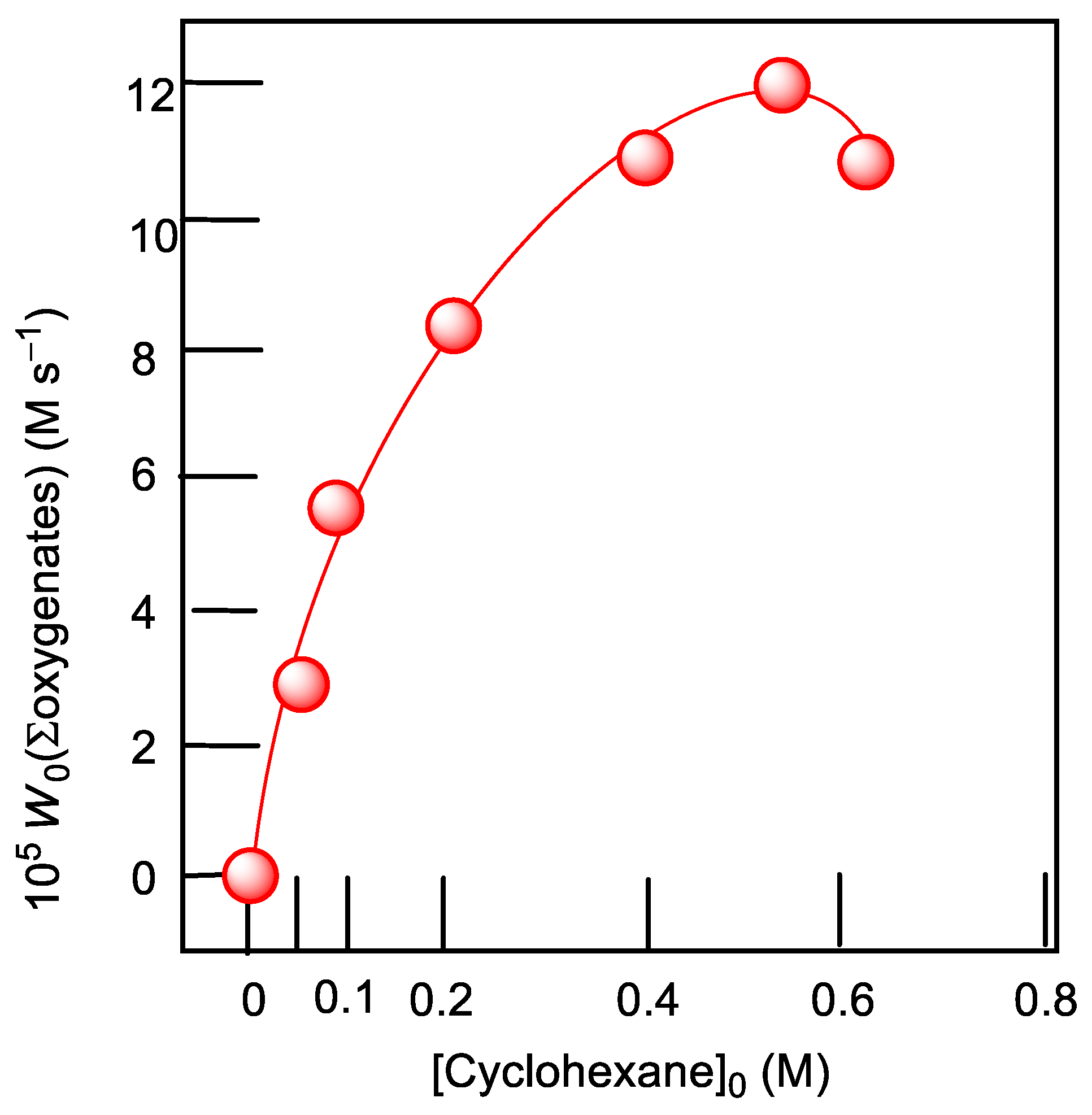
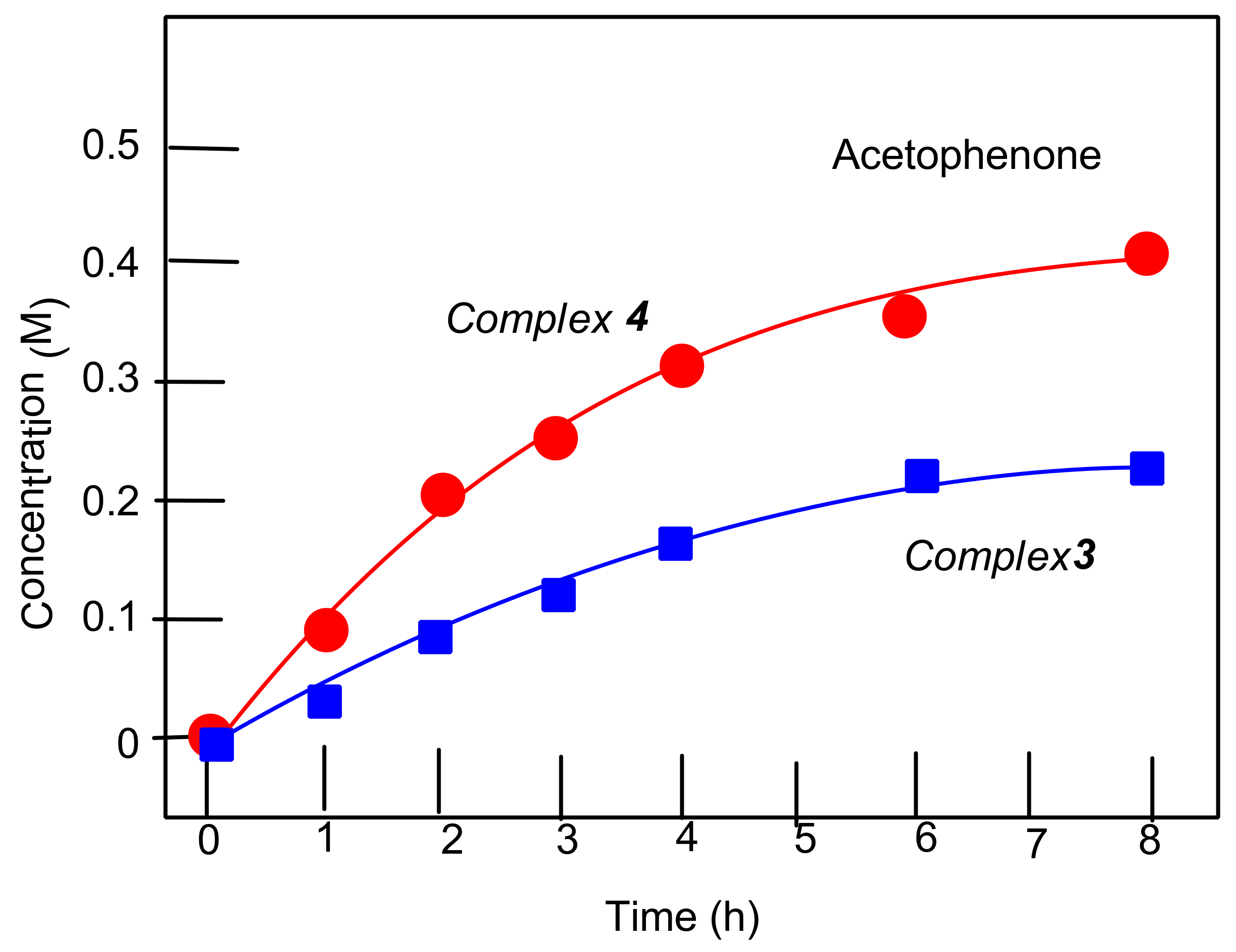
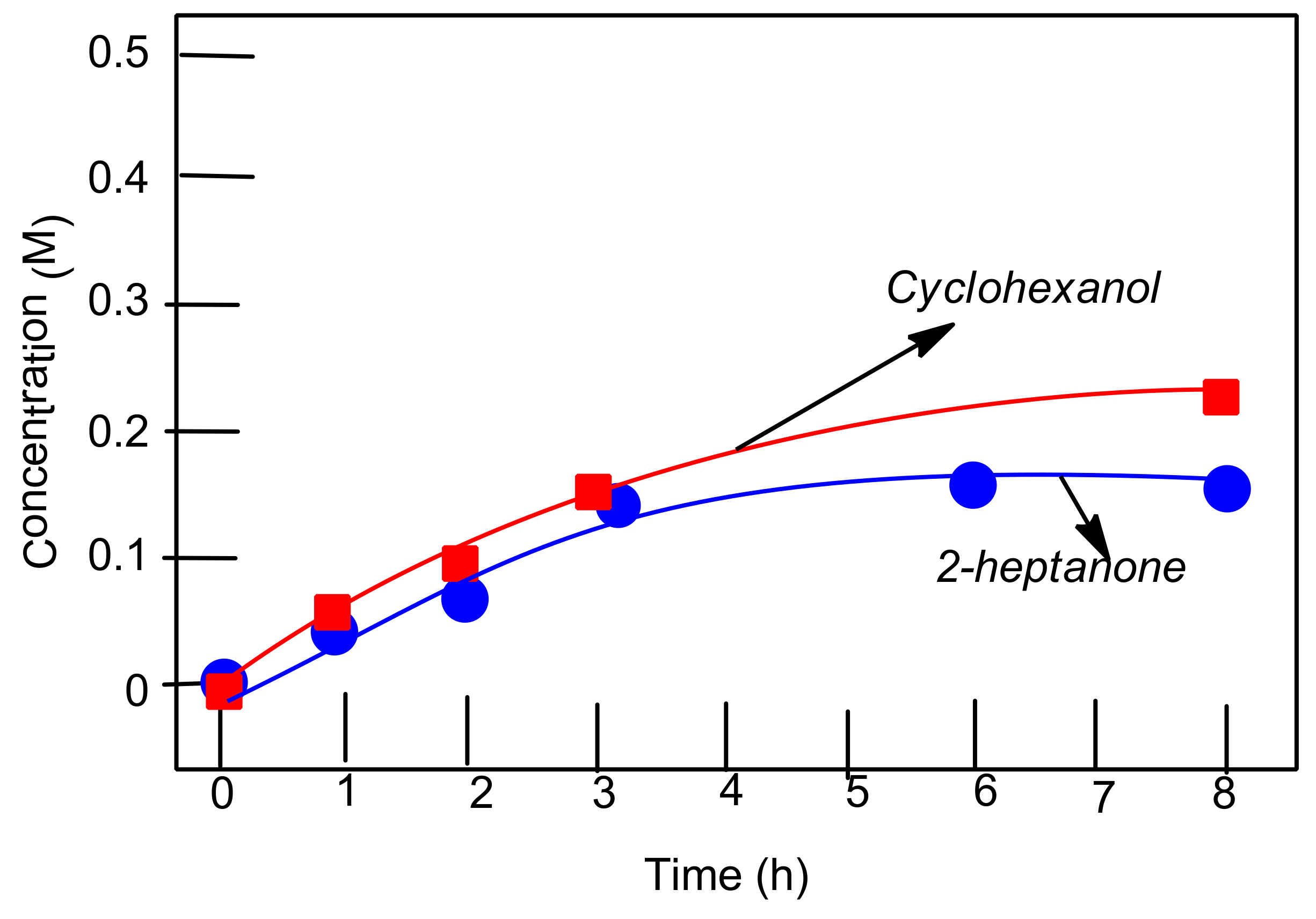
2.8. Oxygenation of Alkanes and Alcohols
3. Conclusions
4. Experimental Section
4.1. General Procedures
4.2. Physical Measurements
4.3. X-ray Data Collection and Structure Refinement
4.4. Synthesis of [VOCl2(CH3CN)2(H2O)] (1)
4.5. Synthesis of 3,5-(CF3)2C6H3-mian (2)
4.6. Synthesis of [VOCl2(dpp-mian)(CH3CN)] (3)
4.6.1. Method A
4.6.2. Method B
4.7. Synthesis of [{VOCl(3,5-(CF3)2C6H3-bian)(H2O)}{VOCl3(3,5-(CF3)2C6H3-bian)}]·2.85DME (4)
Method B
4.8. Synthesis of [VOCl2(CH3CN)(DME)] (5)
4.9. Computational Details
4.10. Alkane Oxygenation Reactions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lyaskovskyy, V.; de Bruin, B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Praneeth, V.K.K.; Ringenberg, M.R.; Ward, T.R. Redox-Active Ligands in Catalysis. Angew. Chem. Int. Ed. 2012, 51, 10228–10234. [Google Scholar] [CrossRef] [PubMed]
- Deibel, N.; Schweinfurth, D.; Hohloch, S.; Fiedler, J.; Sarkar, B. Donor–acceptor systems of Pt(ii) and redox-induced reactivity towards small molecules. Chem. Commun. 2012, 48, 2388–2390. [Google Scholar] [CrossRef] [PubMed]
- Sanz, C.; Ferguson, M.; McDonald, R.; Patrick, B.O.; Hicks, R.G. Classical and non-classical redox reactions of Pd(ii) complexes containing redox-active ligands. Chem. Commun. 2014, 50, 11676–11678. [Google Scholar] [CrossRef] [PubMed]
- Heyduk, A.F.; Zarkesh, R.A.; Nguyen, A. Designing Catalysts for Nitrene Transfer Using Early Transition Metals and Redox-Active Ligands. Inorg. Chem. 2011, 50, 9849–9863. [Google Scholar] [CrossRef]
- Kaim, W. Manifestations of Noninnocent Ligand Behavior. Inorg. Chem. 2011, 50, 9752–9765. [Google Scholar] [CrossRef]
- McKinnon, M.; Ngo, K.T.; Sobottka, S.; Sarkar, B.; Ertem, M.Z.; Grills, D.C.; Rochford, J. Synergistic Metal–Ligand Redox Cooperativity for Electrocatalytic CO2 Reduction Promoted by a Ligand-Based Redox Couple in Mn and Re Tricarbonyl Complexes. Organometallics 2018, 38, 1317–1329. [Google Scholar] [CrossRef]
- Rajabimoghadam, K.; Darwish, Y.; Bashir, U.; Pitman, D.; Eichelberger, S.; Siegler, M.A.; Swart, M.; Garcia-Bosch, I. Catalytic Aerobic Oxidation of Alcohols by Copper Complexes Bearing Redox-Active Ligands with Tunable H-Bonding Groups. J. Am. Chem. Soc. 2018, 140, 16625–16634. [Google Scholar] [CrossRef]
- Dinda, S.; Genest, A.; Rösch, N. O2 Activation and Catalytic Alcohol Oxidation by Re Complexes with Redox-Active Ligands: A DFT Study of Mechanism. ACS Catal. 2015, 5, 4869–4880. [Google Scholar] [CrossRef]
- Villa, M.; Miesel, D.; Hildebrandt, A.; Ragaini, F.; Schaarschmidt, D.; Von Wangelin, A.J. Synthesis and Catalysis of Redox-Active Bis(imino)acenaphthene (BIAN) Iron Complexes. ChemCatChem 2017, 9, 3203–3209. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Yambulatov, D.S.; Skatova, A.A.; Baranov, E.V.; Demeshko, S.; Bogomyakov, A.S.; Ovcharenko, V.I.; Zueva, E.M. Ytterbium and Europium Complexes of Redox-Active Ligands: Searching for Redox Isomerism. Inorg. Chem. 2017, 56, 9825–9833. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Maslova, O.V.; Morozov, A.G.; Dechert, S.; Demeshko, S.; Meyer, F. Genuine Redox Isomerism in a Rare-Earth-Metal Complex. Angew. Chem. Int. Ed. 2012, 51, 10584–10587. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.H.; Ziller, J.W.; Guan, Z. Axial Donating Ligands: A New Strategy for Late Transition Metal Olefin Polymerization Catalysis. J. Am. Chem. Soc. 2008, 130, 7538–7539. [Google Scholar] [CrossRef] [PubMed]
- Fedushkin, I.L.; Maslova, O.V.; Baranov, E.; Shavyrin, A. Redox Isomerism in the Lanthanide Complex [(dpp-Bian)Yb(DME)(μ-Br)]2 (dpp-Bian = 1,2-Bis[(2,6-diisopropylphenyl)imino]acenaphthene). Inorg. Chem. 2009, 48, 2355–2357. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Huang, Y.-T.; Liu, H.; Liu, R.-Z.; Shen, D.-S.; Liu, N.; Liu, F.-S. α-Hydroxyimine palladium complexes: Synthesis, molecular structure, and their activities towards the Suzuki–Miyaura cross-coupling reaction. J. Organomet. Chem. 2013, 729, 95–102. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Lukoyanov, A.N.; Baranov, E. Lanthanum Complexes with a Diimine Ligand in Three Different Redox States. Inorg. Chem. 2018, 57, 4301–4309. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Skatova, A.A.; Chudakova, V.A.; Fukin, G.K. Four-Step Reduction of dpp-bian with Sodium Metal: Crystal Structures of the Sodium Salts of the Mono-, Di-, Tri- and Tetraanions of dpp-bian. Angew. Chem. Int. Ed. 2003, 42, 3294–3298. [Google Scholar] [CrossRef]
- Bendix, J.; Clark, K.M. Delocalization and Valence Tautomerism in Vanadium Tris(iminosemiquinone) Complexes. Angew. Chem. Int. Ed. 2016, 55, 2748–2752. [Google Scholar] [CrossRef]
- Clark, K.M.; Bendix, J.; Heyduk, A.F.; Ziller, J.W. Synthesis and Characterization of a Neutral Titanium Tris(iminosemiquinone) Complex Featuring Redox-Active Ligands. Inorg. Chem. 2012, 51, 7457–7459. [Google Scholar] [CrossRef] [PubMed]
- Fomenko, I.S.; Gushchin, A.L.; Shul’Pina, L.S.; Ikonnikov, N.S.; Abramov, P.A.; Romashev, N.F.; Poryvaev, A.S.; Sheveleva, A.M.; Bogomyakov, A.S.; Shmelev, N.Y.; et al. New oxidovanadium(iv) complex with a BIAN ligand: Synthesis, structure, redox properties and catalytic activity. New J. Chem. 2018, 42, 16200–16210. [Google Scholar] [CrossRef]
- Abramov, P.A.; Dmitriev, A.A.; Kholin, K.; Gritsan, N.; Kadirov, M.K.; Gushchin, A.; Sokolov, M.N. Mechanistic study of the [(dpp-bian)Re(CO)3Br] electrochemical reduction using in situ EPR spectroscopy and computational chemistry. Electrochim. Acta 2018, 270, 526–534. [Google Scholar] [CrossRef]
- Williams, B.S.; Leatherman, M.D.; White, P.S.; Brookhart, M. Reactions of Vinyl Acetate and Vinyl Trifluoroacetate with Cationic Diimine Pd(II) and Ni(II) Alkyl Complexes: Identification of Problems Connected with Copolymerizations of These Monomers with Ethylene. J. Am. Chem. Soc. 2005, 127, 5132–5146. [Google Scholar] [CrossRef]
- Romain, C.; Rosa, V.; Fliedel, C.; Bier, F.; Hild, F.; Welter, R.; Dagorne, S.; Avilés, T. Highly active zinc alkyl cations for the controlled and immortal ring-opening polymerization of ε-caprolactone. Dalton Trans. 2012, 41, 3377–3379. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, F.; Zangrando, E.; Carfagna, C.; Müller, C.; Vogt, D.; Hagar, M.; Ragaini, F.; Milani, B. Catalyst activity or stability: The dilemma in Pd-catalyzed polyketone synthesis. Dalton Trans. 2013, 42, 14583–14602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherian, A.E.; Rose, J.M.; Lobkovsky, A.E.B.; Coates, G.W. AC2-Symmetric, Living α-Diimine Ni(II) Catalyst: Regioblock Copolymers from Propylene. J. Am. Chem. Soc. 2005, 127, 13770–13771. [Google Scholar] [CrossRef] [PubMed]
- Flapper, J.; Reek, J.N.H. Templated Encapsulation of Pyridyl-Bian Palladium Complexes: Tunable Catalysts for CO/4-tert-Butylstyrene Copolymerization. Angew. Chem. Int. Ed. 2007, 46, 8590–8592. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gomes, C.; Gomes, P.; Duarte, M.T.; Fan, Z. New tetradentate N,N,N,N-chelating α-diimine ligands and their corresponding zinc and nickel complexes: Synthesis, characterisation and testing as olefin polymerisation catalysts. Dalton Trans. 2011, 40, 3365–3380. [Google Scholar] [CrossRef] [PubMed]
- Tempel, D.J.; Johnson, L.K.; Huff, R.L.; White, P.S.; Brookhart, M. Mechanistic Studies of Pd(II)−α-Diimine-Catalyzed Olefin Polymerizations1. J. Am. Chem. Soc. 2000, 122, 6686–6700. [Google Scholar] [CrossRef]
- Rose, J.M.; Cherian, A.E.; Coates, G.W. Living Polymerization of α-Olefins with an α-Diimine Ni(II) Catalyst: Formation of Well-Defined Ethylene−Propylene Copolymers through Controlled Chain-Walking. J. Am. Chem. Soc. 2006, 128, 4186–4187. [Google Scholar] [CrossRef]
- Lukoyanov, A.N.; Ulivanova, E.A.; Razborov, D.A.; Khrizanforova, V.V.; Budnikova, Y.H.; Makarov, S.; Rumyantcev, R.; Ketkov, S.Y.; Fedushkin, I.L. One-Electron Reduction of 2-Mono(2,6-diisopropylphenylimino)acenaphthene-1-one (dpp-mian). Chem. Eur. J. 2019, 25, 3858–3866. [Google Scholar] [CrossRef]
- Razborov, D.A.; Lukoyanov, A.N.; Baranov, E.; Fedushkin, I.L. Addition of phenylacetylene to a magnesium complex of monoiminoacenaphtheneone (dpp-mian). Dalton Trans. 2015, 44, 20532–20541. [Google Scholar] [CrossRef] [PubMed]
- Razborov, D.A.; Lukoyanov, A.N.; Moskalev, M.V.; Baranov, E.; Fedyushkin, I.L. Gallium Complexes with Acenaphthene-1-Imino-2-one: Synthesis and Reactivity. Russ. J. Coord. Chem. 2018, 44, 380–387. [Google Scholar] [CrossRef]
- Bhattacharjee, J.; Sachdeva, M.; Banerjee, I.; Panda, T.K. Zinc catalyzed Guanylation reaction of Amines with Carbodiimides/ Isocyanate leading to Guanidines/Urea derivatives formation. J. Chem. Sci. 2016, 128, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Anga, S.; Rej, S.; Naktode, K.; Pal, T.; Panda, T.K. Syntheses and solid state structures of zinc (II) complexes with Bi-dentate N-(Aryl)imino-acenapthenone (Ar-BIAO) ligands. J. Chem. Sci. 2015, 127, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Anga, S.; Paul, M.; Naktode, K.; Kottalanka, R.K.; Panda, T.K. Cobalt (II) and Copper (I) Complexes of Rigid Bidentate [N-(2, 6-Diisopropyl-phenyl)imino]acenapthenone Ligand: Synthesis and Structural Studies. Z. Für Anorg. Und Allg. Chem. 2012, 638, 1311–1315. [Google Scholar] [CrossRef]
- Anga, S.; Pal, T.; Kottalanka, R.K.; Paul, M.; Panda, T.K. Synthesis and Structures of Dimeric Zinc Complexes Supported by Unsymmetrical Rigid Bidentate Imino- acenapthenone Ligand. Can. Chem. Trans. 2013, 1, 105–115. [Google Scholar] [CrossRef]
- Carrington, S.J.; Chakraborty, I.; Mascharak, P.K. Exceptionally rapid CO release from a manganese(i) tricarbonyl complex derived from bis(4-chloro-phenylimino)acenaphthene upon exposure to visible light. Dalton Trans. 2015, 44, 13828–13834. [Google Scholar] [CrossRef]
- Hazari, A.S.; Ray, R.; Hoque, A.; Lahiri, G.K. Electronic Structure and Multicatalytic Features of Redox-Active Bis(arylimino)acenaphthene (BIAN)-Derived Ruthenium Complexes. Inorg. Chem. 2016, 55, 8160–8173. [Google Scholar] [CrossRef] [PubMed]
- Hazari, A.S.; Das, A.; Ray, R.; Agarwala, H.; Maji, S.; Mobin, S.M.; Lahiri, G.K. Tunable Electrochemical and Catalytic Features of BIAN- and BIAO-Derived Ruthenium Complexes. Inorg. Chem. 2015, 54, 4998–5012. [Google Scholar] [CrossRef]
- Bristow, S.; McAvilley, S.C.; Clegg, W.; Collison, D. The preparation and characterization of thiourea complexes of vanadium(IV): The crystal structure of dichlorobis-(1,3-dimethylimidazolidine-2-thione)-oxovanadium(IV). Polyhedron 1989, 8, 87–90. [Google Scholar] [CrossRef]
- Gray, B.M.; Hector, A.L.; Levason, W.; Reid, G.; Webster, M.; Zhang, W.; Jura, M. Synthesis, spectroscopic and structural characterisation of vanadium(IV) and oxovanadium(IV) complexes with arsenic donor ligands. Polyhedron 2010, 29, 1630–1638. [Google Scholar] [CrossRef]
- Fomenko, I.S.; Mikhailov, A.A.; Vorobyev, V.; Kuratieva, N.V.; Kostin, G.A.; Schaniel, D.; Nadolinny, V.A.; Gushchin, A.L. Solution and solid-state light-induced transformations in heterometallic vanadium-ruthenium nitrosyl complex. J. Photochem. Photobiol. A Chem. 2021, 407, 113044. [Google Scholar] [CrossRef]
- Fomenko, I.S.; Vincendeau, S.; Manoury, E.; Poli, R.; Abramov, P.A.; Nadolinny, V.A.; Sokolov, M.N.; Gushchin, A.L. An oxidovanadium(IV) complex with 4,4′-di-tert-butyl-2,2′-bipyridine ligand: Synthesis, structure and catalyzed cyclooctene epoxidation. Polyhedron 2020, 177, 114305. [Google Scholar] [CrossRef]
- Fomenko, I.S.; Gushchin, A.L. Mono- and binuclear complexes of group 5 metals with diimine ligands: Synthesis, reactivity and prospects for application. Russ. Chem. Rev. 2020, 89, 966–998. [Google Scholar] [CrossRef]
- Fomenko, I.S.; Gushchin, A.L.; Abramov, P.A.; Sokolov, M.N.; Shul’Pina, L.S.; Ikonnikov, N.S.; Kuznetsov, M.L.; Pombeiro, A.J.L.; Kozlov, Y.N.; Shul’Pin, G.B. New Oxidovanadium(IV) Complexes with 2,2′-bipyridine and 1,10-phenathroline Ligands: Synthesis, Structure and High Catalytic Activity in Oxidations of Alkanes and Alcohols with Peroxides. Catalysts 2019, 9, 217. [Google Scholar] [CrossRef] [Green Version]
- Fomenko, I.S.; Nadolinnyi, V.A.; Efimov, N.N.; Kokovkin, V.V.; Gushchin, A.L. Binuclear Oxidovanadium(IV) Complex with the Bridging Chloranilate Ligand: Synthesis and Magnetic Properties. Russ. J. Coord. Chem. 2019, 45, 776–781. [Google Scholar] [CrossRef]
- Fomenko, Y.S.; Gushchin, A.L.; Tkachev, A.V.; Vasilyev, E.S.; Abramov, P.A.; Nadolinny, V.A.; Syrokvashin, M.M.; Sokolov, M.N. Fist oxidovanadium complexes containing chiral derivatives of dihydrophenanthroline and diazafluorene. Polyhedron 2017, 135, 96–100. [Google Scholar] [CrossRef]
- Sutton, D.; Einstein, F.W.B.; Enwall, E.; Morris, D.M. Crystal and molecular structure and vibrational spectrum of the vanadium(V) oxide trinitrate-acetonitrile complex, VO(NO3)3.CH3CN. Inorg. Chem. 1971, 10, 678–686. [Google Scholar] [CrossRef]
- Tsuchida, E. Oxovanadium(III–V) mononuclear complexes and their linear assemblies bearing tetradentate Schiff base ligands: Structure and reactivity as multielectron redox catalysts. Coord. Chem. Rev. 2003, 237, 213–228. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Y.; Yuan, D.; Yao, Y.; Shen, Q. Zirconium complexes stabilized by amine-bridged bis(phenolato) ligands as precatalysts for intermolecular hydroamination reactions. Dalton Trans. 2015, 44, 20352–20360. [Google Scholar] [CrossRef] [PubMed]
- Schmiege, B.M.; Carney, M.J.; Small, B.L.; Gerlach, D.L.; Halfen, J.A. Alternatives to pyridinediimine ligands: Syntheses and structures of metal complexes supported by donor-modified?—Diimine ligands. Dalton Trans. 2007, 2547–2562. [Google Scholar] [CrossRef]
- Kovach, J.; Peralta, M.; Brennessel, W.; Jones, W.D. Synthesis and X-ray crystallographic characterization of substituted aryl imines. J. Mol. Struct. 2011, 992, 33–38. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2013, 114, 2170–2191. [Google Scholar] [CrossRef]
- Xu, S.-Y.; Chen, X.-M.; Huang, L.-C.; Li, F.; Gao, W. Vanadium chlorides supported by BIAN (BIAN = bis(arylimo)-acenaphthene) ligands: Synthesis, characterization, and catalysis on ethylene polymerization. Polyhedron 2019, 164, 146–151. [Google Scholar] [CrossRef]
- Razborov, D.A.; Lukoyanov, A.; Makarov, V.M.; Samsonov, M.; Fedushkin, I.L. Complexes of gallium(III), antimony(III), titanium(IV), and cobalt(II) with acenaphthenequinonimine. Russ. Chem. Bull. 2015, 64, 2377–2385. [Google Scholar] [CrossRef]
- Kabanos, T.A.; Keramidas, A.; Papaioannou, A.; Terzis, A. Model Investigations for Vanadium-Protein Interactions: Synthesis and X-ray Structures of mer-[VOCl3(Hpycan)] and [VOCl2(CH3CN)(Hpycan)] {Hpycan = N-(2-Nitrophenyl)pyridine-2-carboxamide}. Inorg. Chem. 1994, 33, 845–846. [Google Scholar] [CrossRef]
- Zefirov, Y.V.; Zorky, P.M. New applications of van der Waals radii in chemistry. Russ. Chem. Rev. 1995, 64, 415–428. [Google Scholar] [CrossRef]
- Preuss, F.; Hornung, G.; Frank, W. Komplexbildung des tert-Butyliminovanadium(V)-trichlorids mit O-Donor-Liganden. Z. Für Anorg. Und Allg. Chem. 1995, 621, 1663–1671. [Google Scholar] [CrossRef]
- Romashev, N.F.; Gushchin, A.L.; Fomenko, I.S.; Abramov, P.A.; Mirzaeva, I.V.; Kompan’Kov, N.B.; Kal’Nyi, D.B.; Sokolov, M.N. A new organometallic rhodium(I) complex with dpp-bian ligand: Synthesis, structure and redox behaviour. Polyhedron 2019, 173, 114110. [Google Scholar] [CrossRef]
- Gushchin, A.L.; Romashev, N.F.; Shmakova, A.A.; Abramov, P.A.; Ryzhikov, M.R.; Fomenko, I.S.; Sokolov, M.N. Novel redox active rhodium(iii) complex with bis(arylimino)acenaphthene ligand: Synthesis, structure and electrochemical studies. Mendeleev Commun. 2020, 30, 81–83. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii of Metals in Covalent Compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Baykov, S.V.; Filimonov, S.I.; Rozhkov, A.V.; Novikov, A.S.; Ananyev, I.V.; Ivanov, D.M.; Kukushkin, V.Y. Reverse Sandwich Structures from Interplay between Lone Pair−π-Hole Atom-Directed C···dz2[M] and Halogen Bond Interactions. Cryst. Growth Des. 2019, 20, 995–1008. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Zolotarev, A.A.; Boyarskiy, V.P.; Kukushkin, V.Y. (Isocyano Group π-Hole)⋅⋅⋅[d-M II ] Interactions of (Isocyanide)[M II ] Complexes, in which Positively Charged Metal Centers (d8-M=Pt, Pd) Act as Nucleophiles. Chem. A Eur. J. 2019, 25, 8590–8598. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Krykova, M.A.; Ivanov, D.M.; Novikov, A.S.; Sinelshchikova, A.A.; Volostnykh, M.V.; Konovalov, M.A.; Grigoriev, M.S.; Gorbunova, Y.G.; Kukushkin, V.Y. Reverse Arene Sandwich Structures Based upon π-Hole⋅⋅⋅[MII] (d8 M=Pt, Pd) Interactions, where Positively Charged Metal Centers Play the Role of a Nucleophile. Angew. Chem. Int. Ed. 2019, 58, 4164–4168. [Google Scholar] [CrossRef]
- Rozhkov, A.; Novikov, A.S.; Ivanov, D.M.; Bolotin, D.S.; Bokach, N.A.; Kukushkin, V.Y. Structure-Directing Weak Interactions with 1,4-Diiodotetrafluorobenzene Convert One-Dimensional Arrays of [MII(acac)2] Species into Three-Dimensional Networks. Cryst. Growth Des. 2018, 18, 3626–3636. [Google Scholar] [CrossRef]
- Ivanov, D.; Kirina, Y.V.; Novikov, A.S.; Starova, G.; Kukushkin, V.Y. Efficient π-stacking with benzene provides 2D assembly of trans-[PtCl2(p-CF3C6H4CN)2]. J. Mol. Struct. 2016, 1104, 19–23. [Google Scholar] [CrossRef]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-Garcia, J.; Cohen, A.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Shul’Pin, G.B.; Attanasio, D.; Suber, L. Efficient H2O2 Oxidation of Alkanes and Arenes to Alkyl Peroxides and Phenols Catalyzed by the System Vanadate-Pyrazine-2-Carboxylic Acid. J. Catal. 1993, 142, 147–152. [Google Scholar] [CrossRef]
- Levitsky, M.M.; Bilyachenko, A.N.; Shul’Pin, G.B. Oxidation of C-H compounds with peroxides catalyzed by polynuclear transition metal complexes in Si- or Ge-sesquioxane frameworks: A review. J. Organomet. Chem. 2017, 849–850, 201–218. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Shul’Pin, G.B. Pyrazinecarboxylic acid and analogs: Highly efficient co-catalysts in the metal-complex-catalyzed oxidation of organic compounds. Coord. Chem. Rev. 2013, 257, 732–754. [Google Scholar] [CrossRef]
- Shul’Pin, G.B.; Shul’Pina, L.S. The Vanadate–Pyrazinecarboxylic Acid–Hydrogen Peroxide Reagent and Similar Systems for Efficient Oxidations with Peroxides. In Vanadium Catalysis; Royal Society of Chemistry: London, UK, 2020; Chapter 4; pp. 72–76. [Google Scholar] [CrossRef]
- Shul’Pin, G.B.; Kozlov, Y.N.; Shul’Pina, L.S.; Petrovskiy, P.V. Oxidation of alkanes and alcohols with hydrogen peroxide catalyzed by complex Os3(CO)10(µ-H)2. Appl. Organomet. Chem. 2010, 24, 464–472. [Google Scholar] [CrossRef]
- Shul’Pin, G.B. Metal-catalyzed hydrocarbon oxygenations in solutions: The dramatic role of additives: A review. J. Mol. Catal. A Chem. 2002, 189, 39–66. [Google Scholar] [CrossRef]
- Shul’Pin, G.B.; Kozlov, Y.N.; Shul’Pina, L.S. Metal Complexes Containing Redox-Active Ligands in Oxidation of Hydrocarbons and Alcohols: A Review. Catalysts 2019, 9, 1046. [Google Scholar] [CrossRef] [Green Version]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Data Collection, Reduction and Correction Program; CrysAlisPro 1.171.38.46; Rigaku Oxford Diffraction: Oxford, UK, 2015.
- SAINT, Data Reduction and Correction Program; Bruker AXS: Madison, WI, USA, 2014.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr. Sect. A Found. Adv. 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Reporting and evaluating absolute-structure and absolute-configuration determinations. J. Appl. Crystallogr. 2000, 33, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Kassandrova, O.N.; Lebedev, V.V. Obrabotka Rezul’tatov Nabljudenij; Nauka: Moscow, Russia, 1970. [Google Scholar]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Reduction, E1/2 (ΔE [b], mV) | Oxidation, Epa |
|---|---|---|
| dpp-mian | −1.16 (160), −1.94 (irrev.) | 1.64 |
| 2 | −0.85 (155), −1.67 (irrev.) | - |
| 3 | −1.23 (irrev.), −1.62 (130) | 1.12, 1.62 |
| 4 | −0.98 (irrev.), −1.56 (110) | 1.08, 1.52 |
| Contact * | ρ(r) | ∇2ρ(r) | λ2 | Hb | V(r) | G(r) | Eint ** |
|---|---|---|---|---|---|---|---|
| 2 | |||||||
| C18···C46 3.360 Å | 0.005 | 0.016 | −0.005 | 0.001 | −0.002 | 0.003 | 0.6 |
| C9···C55 3.360 Å | 0.005 | 0.016 | −0.005 | 0.001 | −0.002 | 0.003 | 0.6 |
| C12···C51 3.398 Å | 0.006 | 0.016 | −0.006 | 0.001 | −0.003 | 0.003 | 0.9 |
| C14···C49 3.398 Å | 0.006 | 0.016 | −0.006 | 0.001 | −0.003 | 0.003 | 0.9 |
| 3 | |||||||
| C12···C78 3.394 Å | 0.006 | 0.017 | −0.006 | 0.001 | −0.003 | 0.004 | 0.9 |
| C11···C76 3.467 Å | 0.006 | 0.015 | −0.006 | 0.001 | −0.003 | 0.003 | 0.9 |
| C19···C72 3.491 Å | 0.006 | 0.015 | −0.006 | 0.001 | −0.003 | 0.003 | 0.9 |
| C15···C70 3.549 Å | 0.005 | 0.013 | −0.005 | 0.001 | −0.002 | 0.003 | 0.6 |
| Contact * | ρ(r) | ∇2ρ(r) | λ2 | Hb | V(r) | G(r) |
|---|---|---|---|---|---|---|
| V1–O7 1.640 Å | 0.233 | 0.932 | −0.233 | −0.141 | −0.516 | 0.375 |
| V2···O7 2.013 Å | 0.076 | 0.446 | −0.076 | 0.005 | −0.102 | 0.107 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukoyanov, A.N.; Fomenko, I.S.; Gongola, M.I.; Shul’pina, L.S.; Ikonnikov, N.S.; Shul’pin, G.B.; Ketkov, S.Y.; Fukin, G.K.; Rumyantcev, R.V.; Novikov, A.S.; et al. Novel Oxidovanadium Complexes with Redox-Active R-Mian and R-Bian Ligands: Synthesis, Structure, Redox and Catalytic Properties. Molecules 2021, 26, 5706. https://doi.org/10.3390/molecules26185706
Lukoyanov AN, Fomenko IS, Gongola MI, Shul’pina LS, Ikonnikov NS, Shul’pin GB, Ketkov SY, Fukin GK, Rumyantcev RV, Novikov AS, et al. Novel Oxidovanadium Complexes with Redox-Active R-Mian and R-Bian Ligands: Synthesis, Structure, Redox and Catalytic Properties. Molecules. 2021; 26(18):5706. https://doi.org/10.3390/molecules26185706
Chicago/Turabian StyleLukoyanov, Anton N., Iakov S. Fomenko, Marko I. Gongola, Lidia S. Shul’pina, Nikolay S. Ikonnikov, Georgiy B. Shul’pin, Sergey Y. Ketkov, Georgy K. Fukin, Roman V. Rumyantcev, Alexander S. Novikov, and et al. 2021. "Novel Oxidovanadium Complexes with Redox-Active R-Mian and R-Bian Ligands: Synthesis, Structure, Redox and Catalytic Properties" Molecules 26, no. 18: 5706. https://doi.org/10.3390/molecules26185706