
Selected Drug-Likeness Properties of 2-Arylidene-indan-1,3-dione Derivatives—Chemical Compounds with Potential Anti-Cancer Activity

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
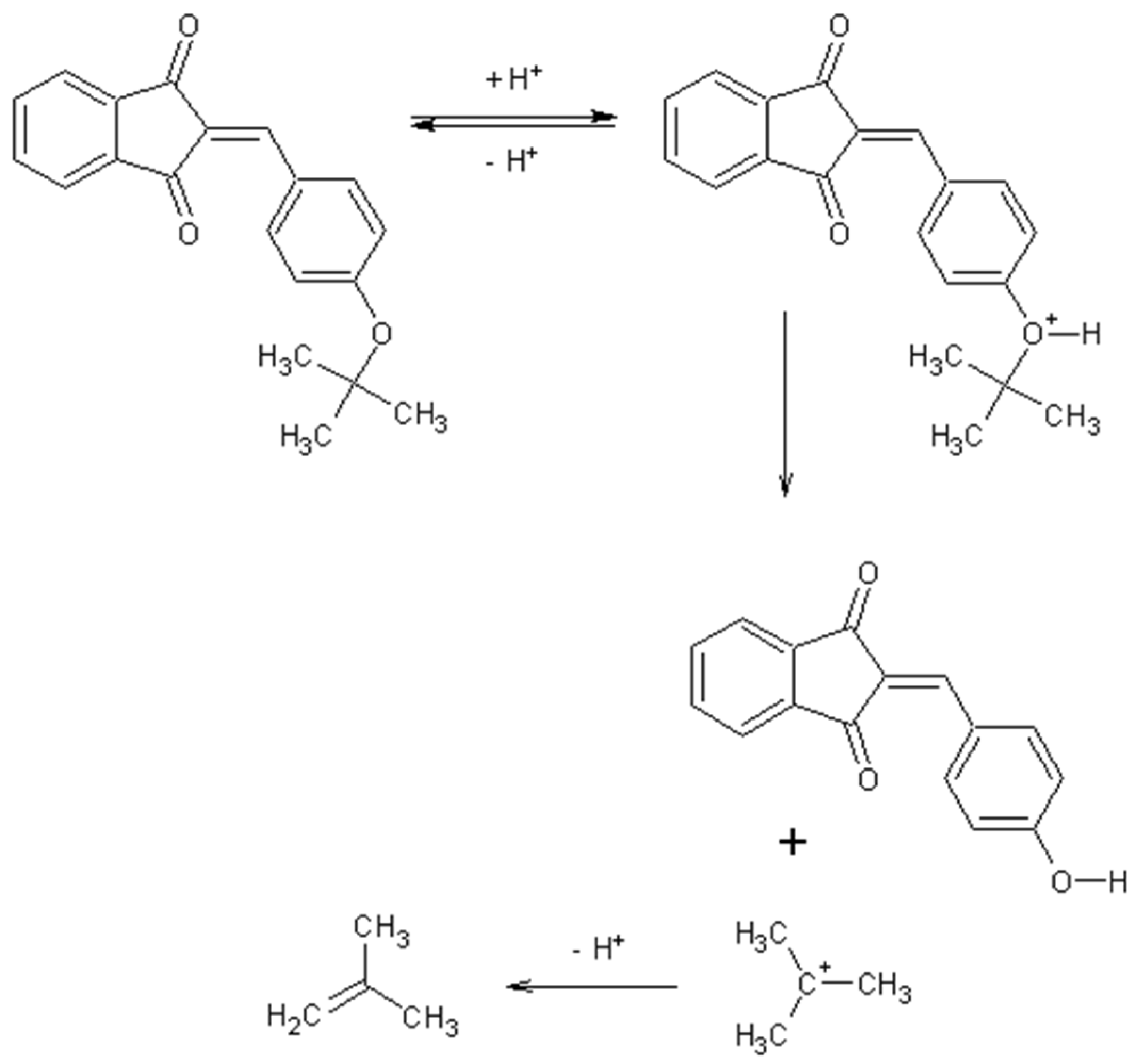
2.1. Synthesis Mechanism
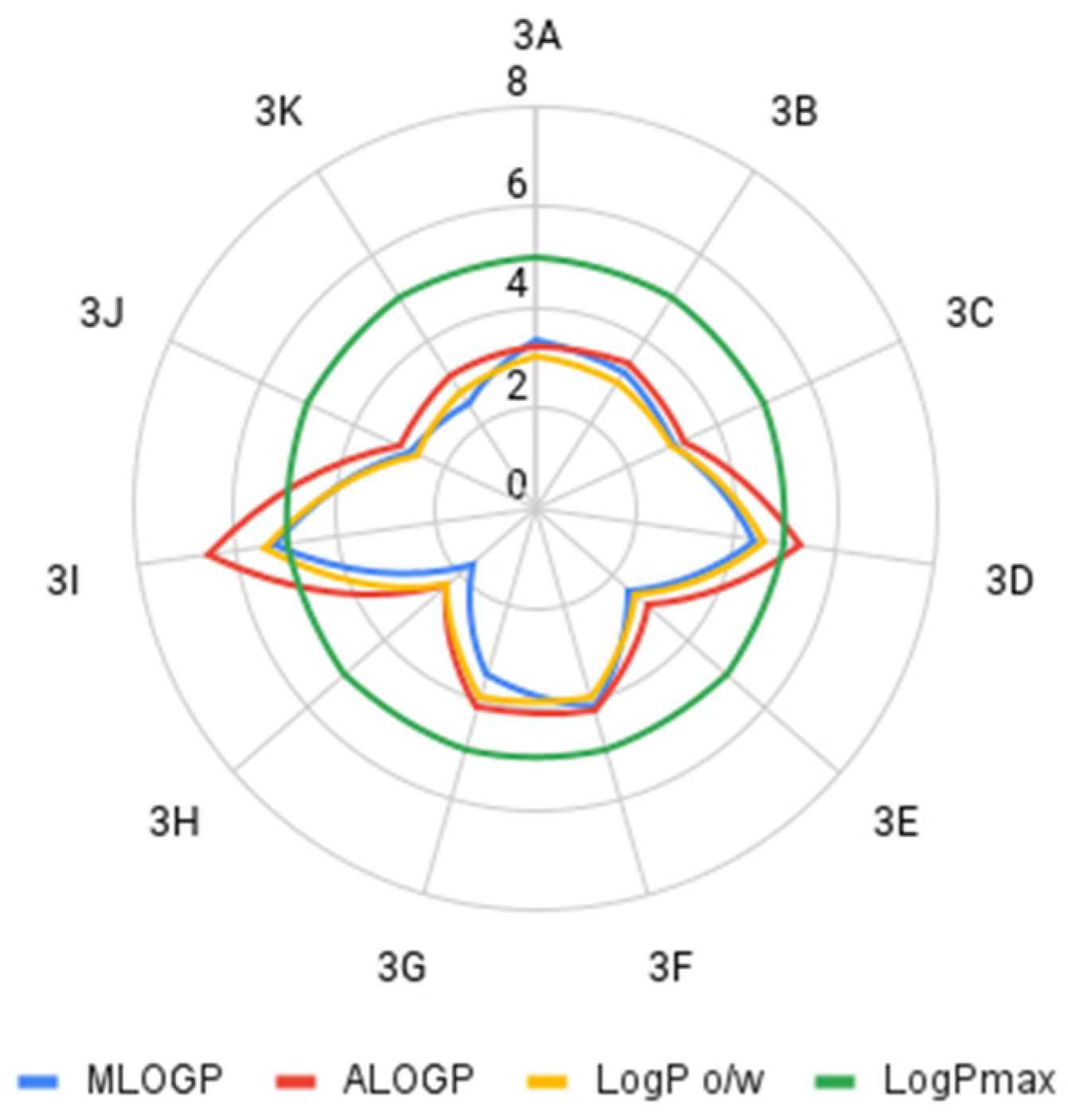
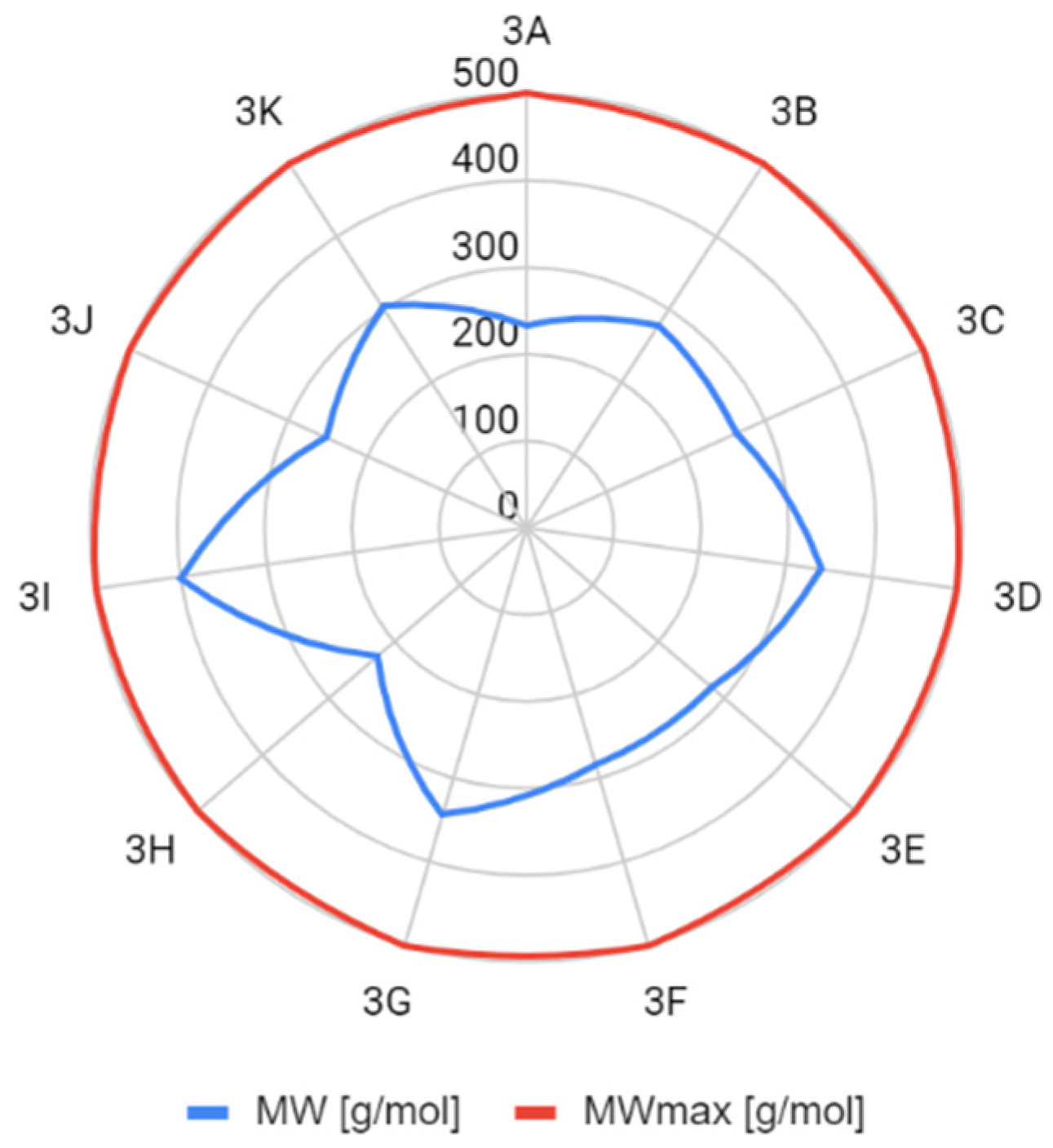
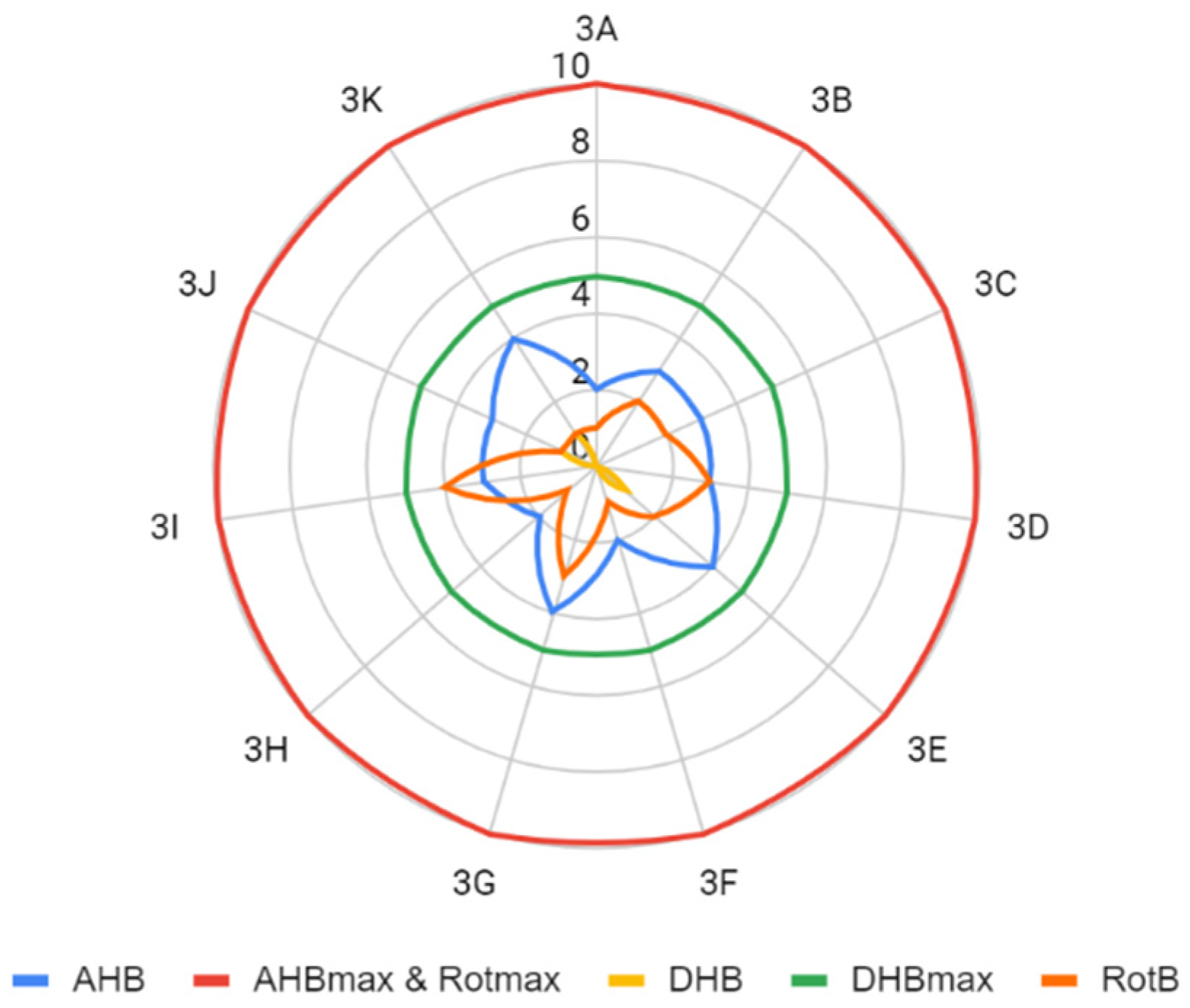
2.2. Assumptions of the Lipiński’s and Veber’s Rules
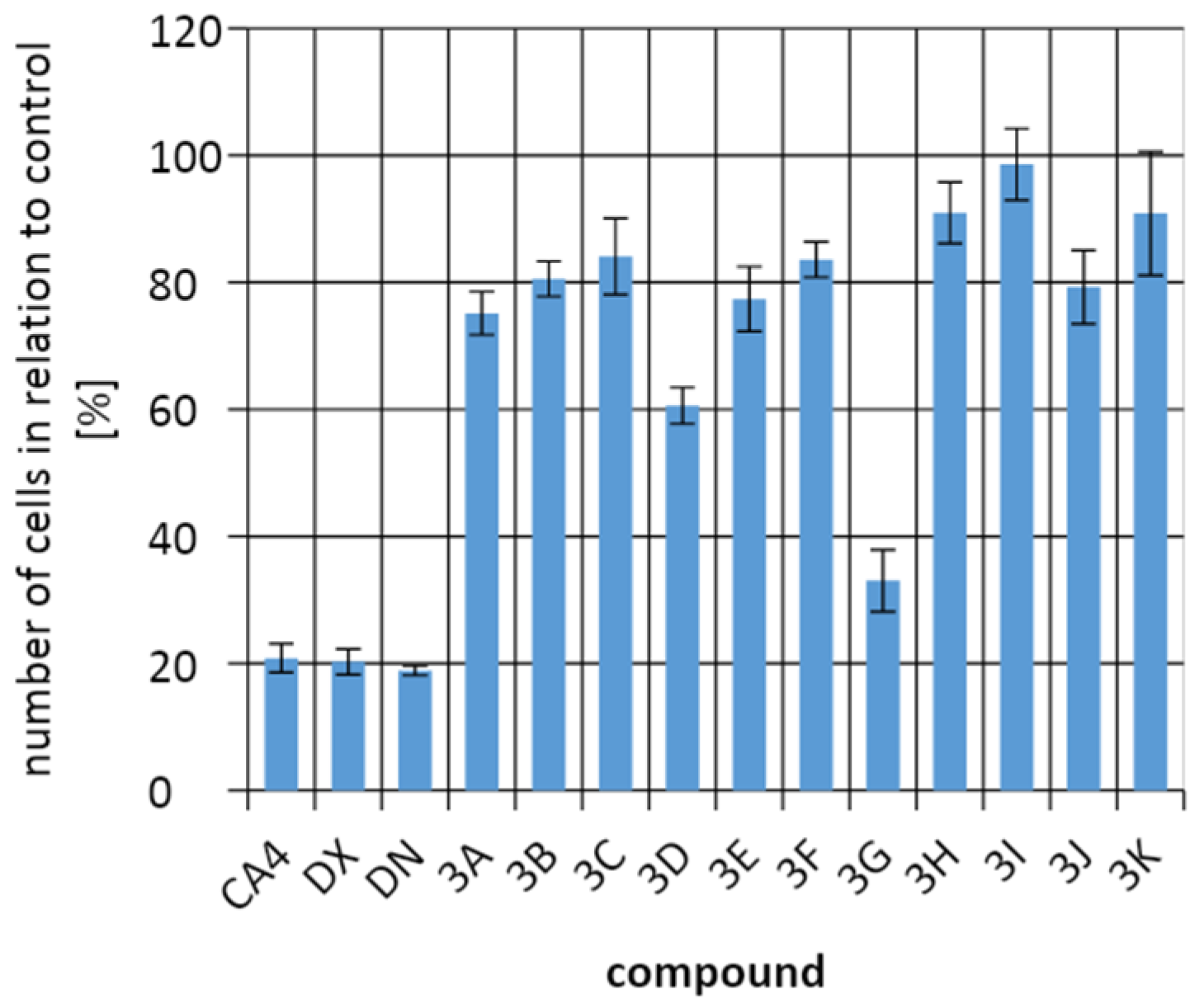
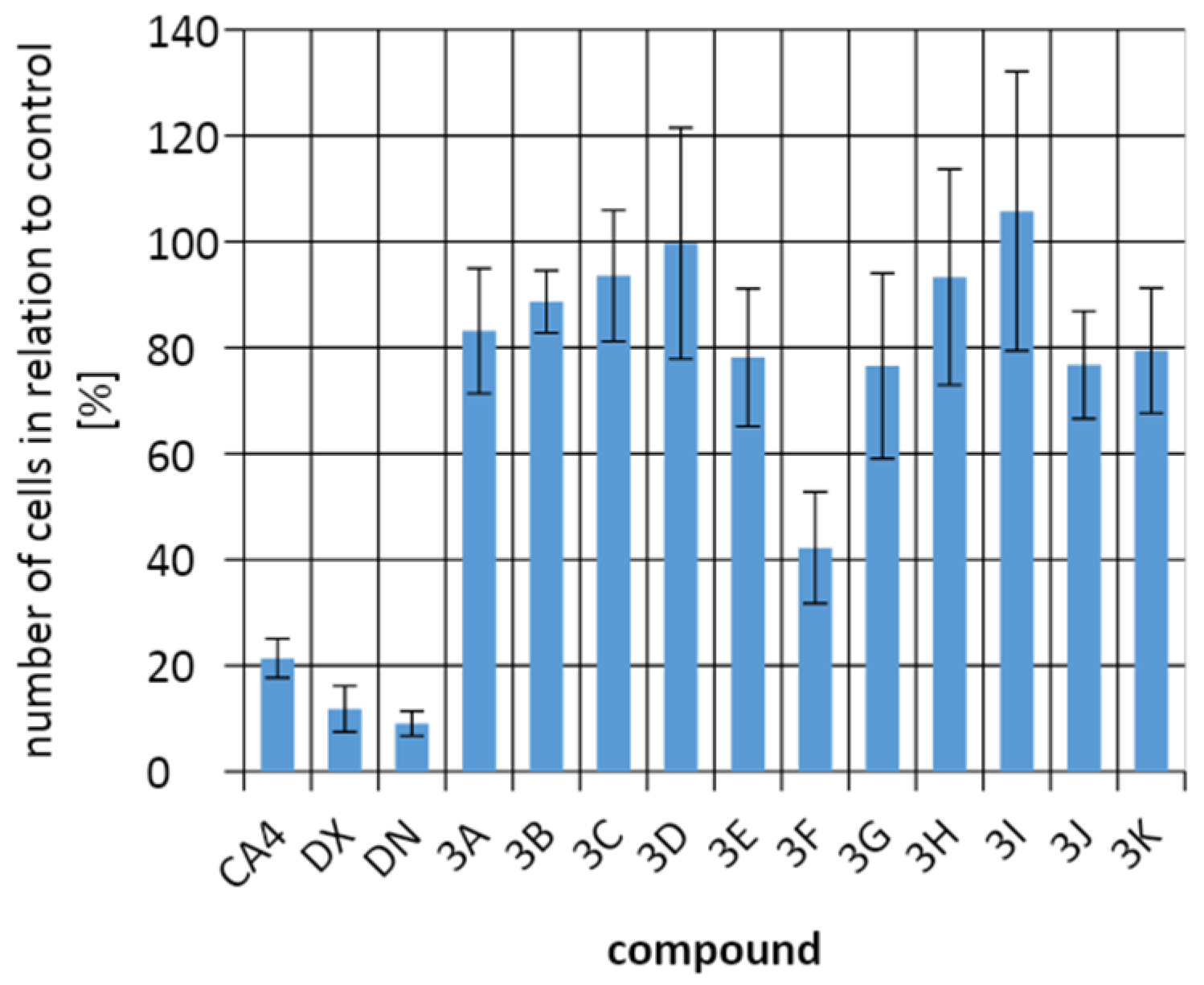
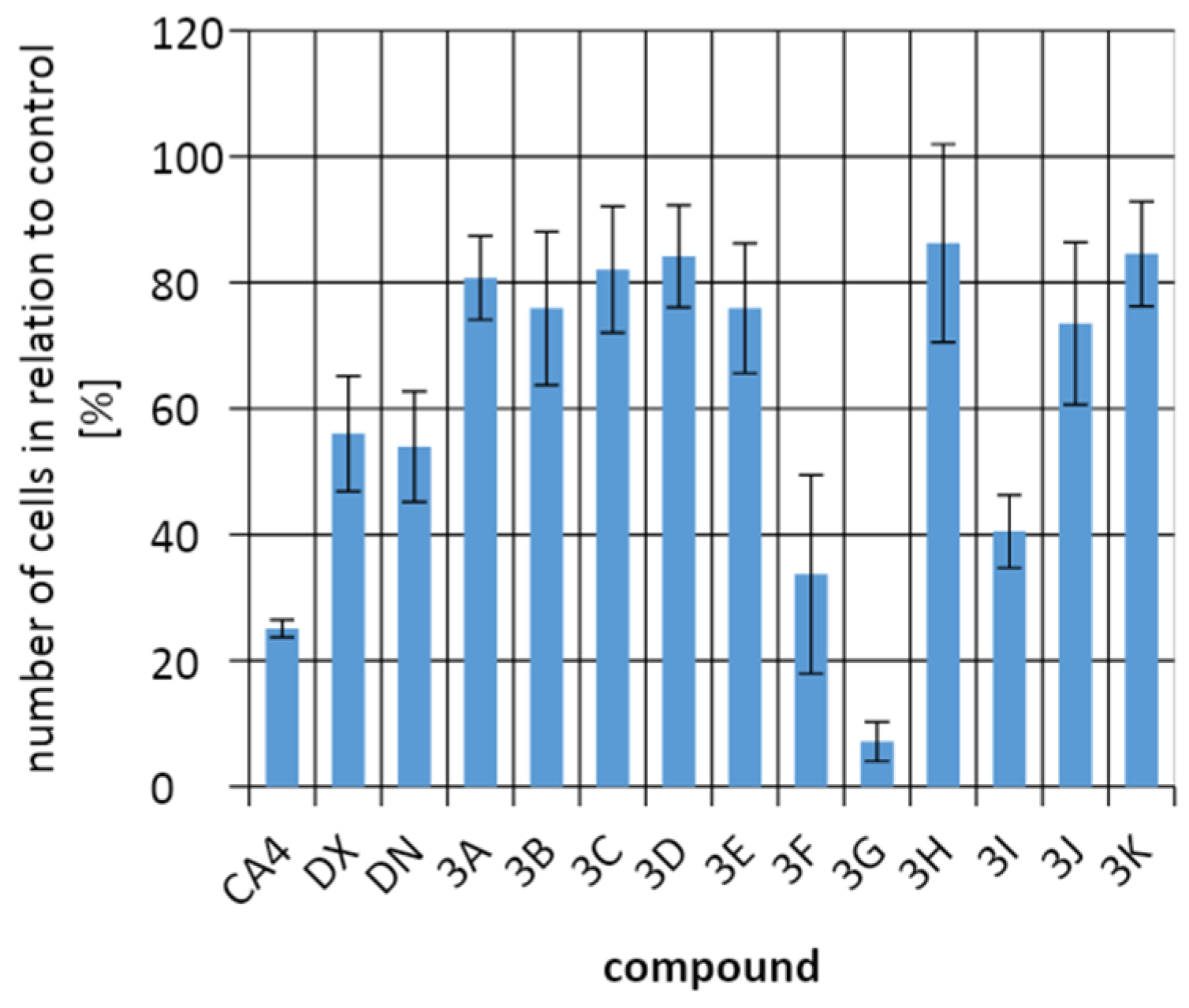
2.3. Biological Activity
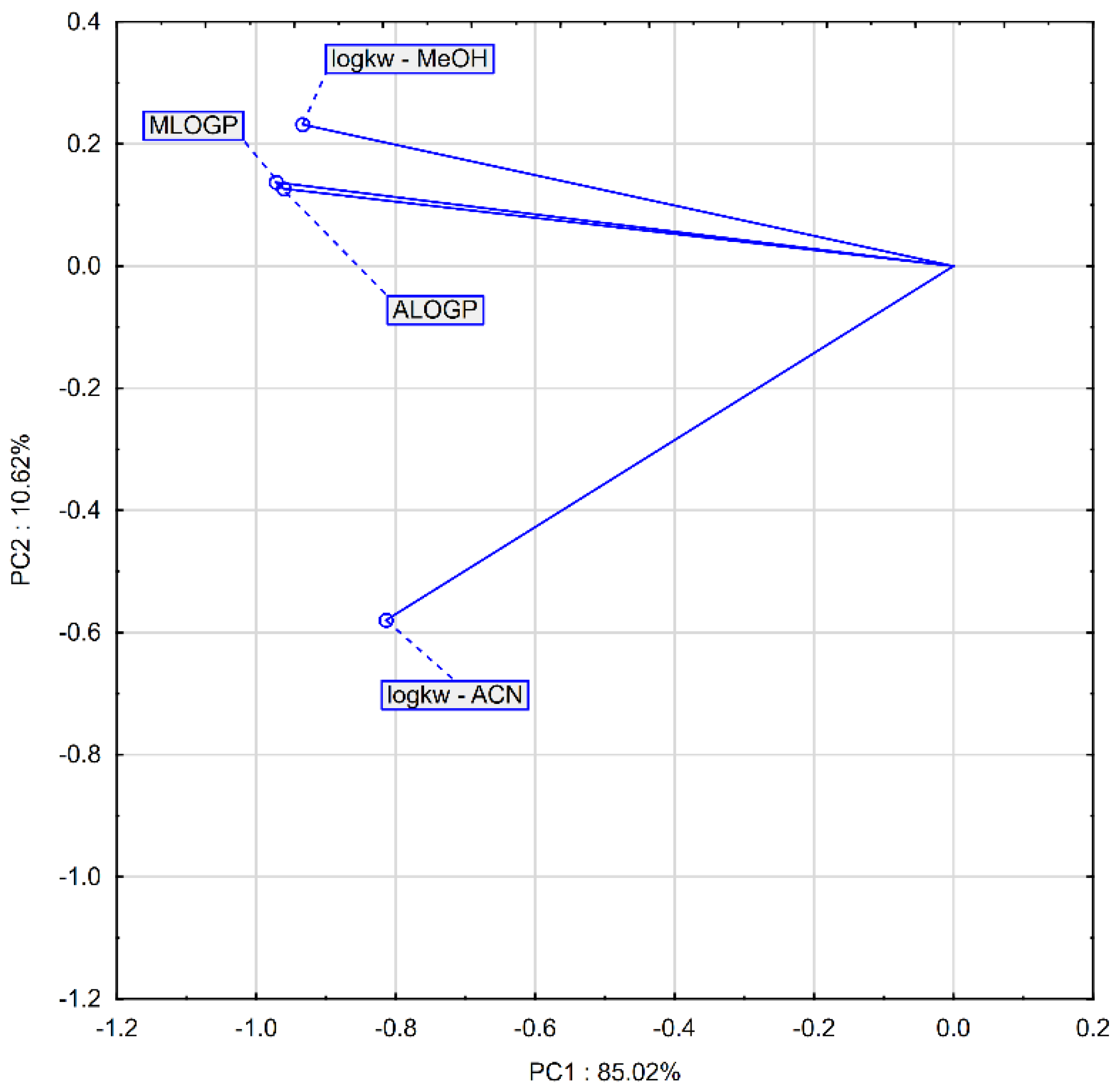
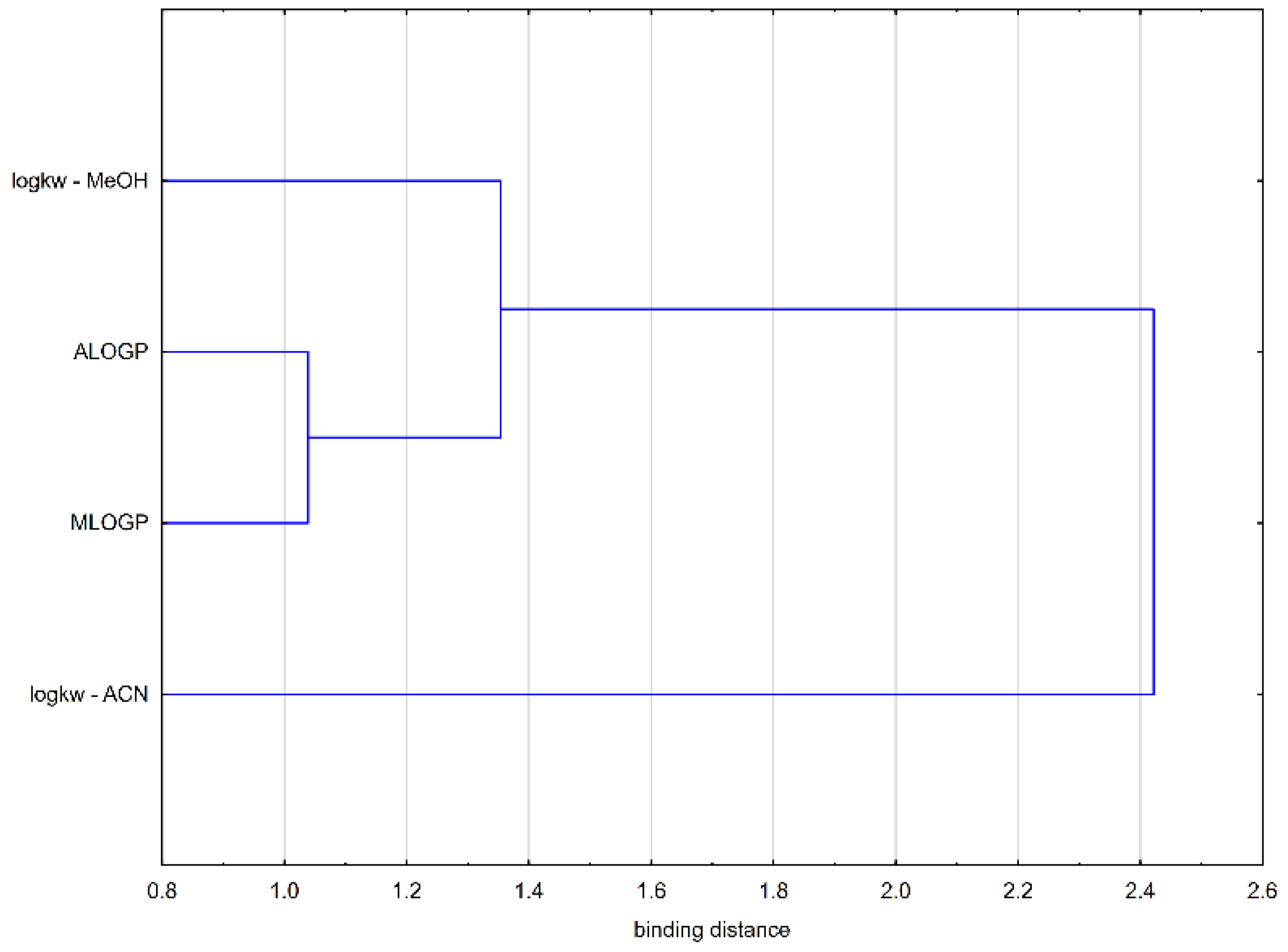
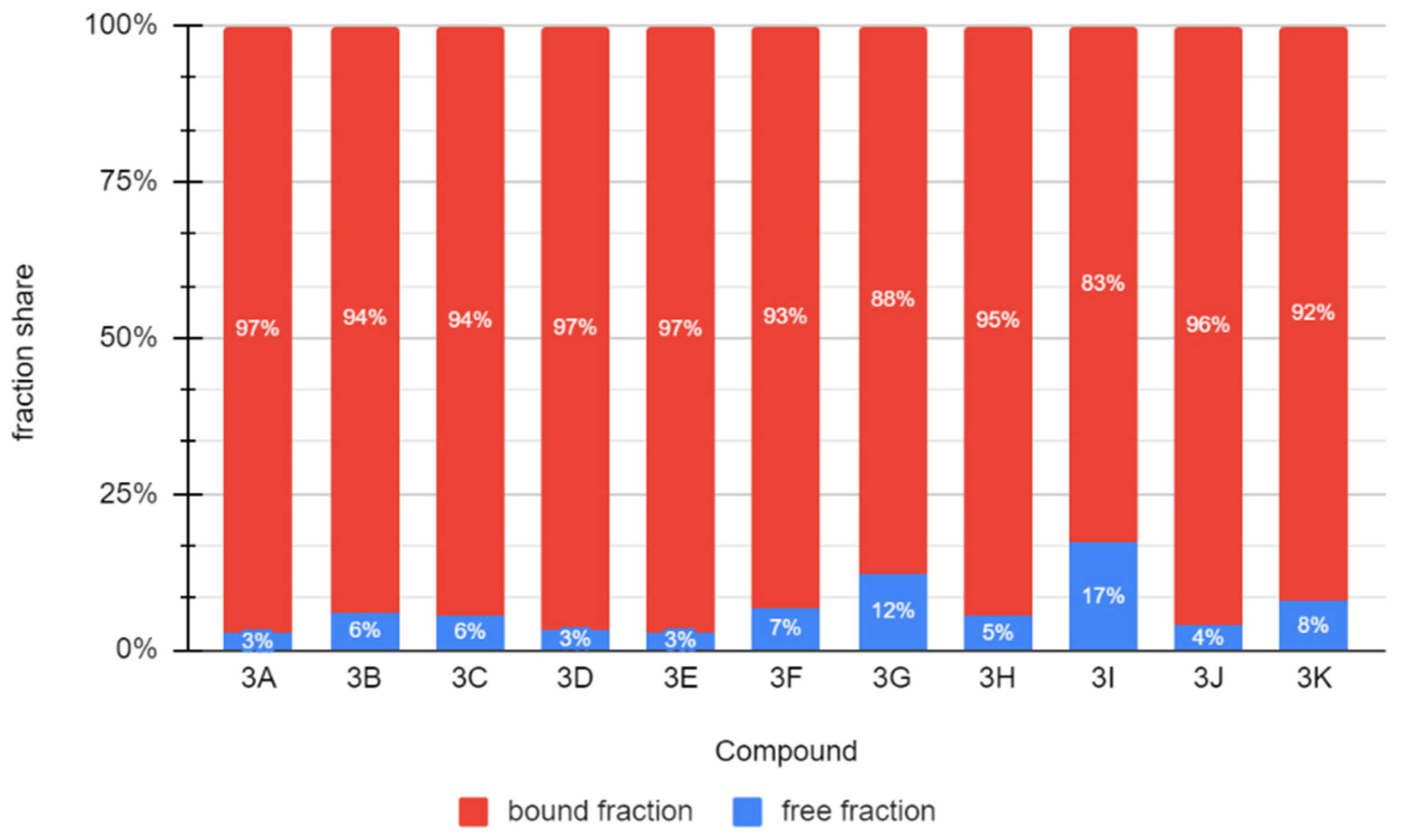
2.4. Binding to Albumin
2.5. Toxicity
3. Discussion
4. Materials and Methods
4.1. Synthesis, Purification, and Structure Conformation—General Procedure
4.2. Chromatographic Examination of Lipophilicity
4.3. Geometry Optimization of Structures
4.4. Testing of Antiproliferative Activity
4.5. Measurement of the Ability to Bind to Albumin
4.6. Toxicity Simulation
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Appendix A
- 2-benzylidene-1H-indene-1,3(2H)-dione (3A)The product was obtained as a yellow powder; 0.645 g (27.53%). m.p. 152–153 °C. C16H10O2 (calcd 234.26). ESI-MS: (M + H)+ = 235.10. 1H-NMR (CDCl3), δ [ppm]: δ 8.45 (m, 2H -Ar), 8.00 (m, 2H -Ar), 7.89 (s, 1H =CH-), 7.80 (m, 2H -Ar), 7.53 (m, 3H -Ar). 13C-NMR (CDCl3), δ [ppm]: 190.43 (aliphatic C in ring), 189.18 (aliphatic C in ring), 147.14 (=CH-), 142.71, 140.24, 135.57, 134.30, 133.34, 129.35, 129.00, 123.50.
- 2-{[4-(dimethylamino)phenyl]methylidene}-1H-indene-1,3(2H)-dione (3B)The product was obtained as a red powder; 1.938 g (69.96%). m.p. 203–205 °C. C18H15NO2 (calcd 277.34). ESI-MS: (M + H)+ = 278.20. 1H-NMR (CDCl3), δ [ppm]: δ 8.51 (d, J = 10 Hz, 2H -Ar), 7.91(m, 2H -Ar), 7.77 (s, 1H =CH-), 7.70 (m, 2H -Ar), 6.73 (d, J = 10 Hz, 2H -Ar), 3.13 (s, 6H -CH3). 13C-NMR (CDCl3), δ [ppm]: 191.27 (aliphatic C in ring), 190.14 (aliphatic C in ring), 154.01, 147.61 (=CH-), 142.40, 138.09, 134.54, 123.31, 122.64, 40.31 (N-CH2).
- 2-(4-methoxybenzylidene)-1H-indene-1,3(2H)-dione (3C)The product was obtained as a red/orange powder; 1.663 g (62.92%); m.p. 156–157 °C. C17H12O3 (calcd 264.29). ESI-MS: (M + H)+ = 265.15. 1H-NMR (CDCl3), δ [ppm]: δ 8.52 (d, J = 10 Hz, 2H -Ar), 7.96 (m, 2H -Ar), 7.82 (s, =CH-), 7.76 (m, 2H -Ar), 6.99 (m, 2H -Ar), 3.89 (s, -CH3). 13C-NMR (CDCl3), δ [ppm]: 190.95 (aliphatic C in ring), 189.65 (aliphatic C in ring), 164.20, 146.97 (=CH-), 142.53, 140.12, 137.34, 135.09, 126.67, 123.21, 114.52, 55.73 (-OCH3).
- 2-{[4-(4-methylphenoxy)phenyl]methylidene}-1H-indene-1,3(2H)-dione (3D)The product was obtained as a yellow powder; 0.518 g (60.99%); m.p. 146–147 °C. C23H16O3 (calcd 340.39). ESI-MS: (M + H)+ = 341.20. 1H-NMR (CDCl3), δ [ppm]: δ 8.54 (d, J = 10 Hz, 2H -Ar), 8.01 (m, 2H -Ar), 7.88 (s, =CH-), 7.83 (m, 2H -Ar), 7.25 (m, 2H -Ar), 7.05 (m, 4H -Ar), 2.4 (s, -CH3). 13C-NMR (CDCl3), δ [ppm]: 190.78 (aliphatic C in ring), 189.48 (aliphatic C in ring), 163.09, 152.77, 146.53 (=CH-), 142.55, 140.14, 135.29, 135.09, 134.81, 130.73, 127.74, 120.58, 117.25, 20.96 (CH3).
- 2-[(3-hydroxy-4-methoxyphenyl)methylidene]-1H-indene-1,3(2H)-dione (3E)The product was obtained as a lemon powder; 2.084 g (74.35%); m.p. 227–228 °C. C17H12O4 (calcd 280.29). ESI-MS: (M + H)+ = 281.15. 1H-NMR (CDCl3), δ [ppm]: δ 8.27 (s, 1H = CH-), 8.06 (m, 1H -Ar), 7.98 (m, 2H -Ar), 7.77 (m, 2H -Ar), 6.96 (d, J = 5 Hz, 1H -Ar), 5.66 (s, 1H -Ar), 3.99 (s, 3H -Ar), 3.85 (s, 1H -OH). 13C-NMR (CDCl3), δ [ppm]: 190.90 (aliphatic C in ring), 189.46 (aliphatic C in ring), 151.40, 147.28, 145.61 (=CH-), 142.63, 140.14, 135.28, 135.04, 123.31, 123.26, 110.53, 56.30 (-CH3).
- 2-[(naphthalen-1-yl)methylidene]-1H-indene-1,3(2H)-dione (3F)The product was obtained as an orange powder; 0.883 g (62.10%); m.p. 175–176 °C. C20H12O2 (calcd 284.32). ESI-MS: (M + H)+ = 285.10. 1H-NMR (CDCl3), δ [ppm]: δ 8.78 (s, =CH-), 8.74 (d, J = 10 Hz, 1H -Ar), 8.24 (d, J = 10 Hz, 1H -Ar), 8.02 (m, 3H -Ar), 7.91 (d, J = 5 Hz, 1H -Ar), 7.81 (m, 2H -Ar), 7.62 (m, 2H -Ar), 7.55 (t, J = 5 Hz, 1H -Ar). 13C-NMR (CDCl3), δ [ppm]: 190.32 (aliphatic C in ring), 188.81 (aliphatic C in ring), 143.22, 142.69, 140.23 (=CH-), 135.60, 135.43, 133.73, 133.66, 132.89, 132.22, 130.24, 129.29, 128.76, 127.80, 126.48, 125.30, 123.76, 123.53.
- 2-[(4,7-dimethoxynaphthalen-1-yl)methylidene]-1H-indene-1,3(2H)-dione (3G)The product was obtained as a brown-beige powder; 1.305 g (37.89%); m.p. 223–225 °C. C22H16O4 (calcd 344.38). ESI-MS: (M + H)+ = 345.15. 1H-NMR (CDCl3), δ [ppm]: δ 9.20 (d, J = 5 Hz, 1H -Ar), 8.68 (s, 1H =CH-), 8.24 (d, J = 10 Hz, 1H -Ar), 7.99 (m, 2H -Ar), 7.77 (m, 2H -Ar), 7.54 (s, 1H -Ar), 7.15 (d, J = 5 Hz, 1H -Ar), 6.87 (d, J = 5 Hz, 1H -Ar), 4.09 (s, 3H -CH3), 4.00 (s, 3H -CH3). 13C-NMR (CDCl3), δ [ppm]: 191.31 (aliphatic C in ring), 189.49 (aliphatic C in ring), 161.23, 160.06, 143.04, 142.53 (=CH-), 140.00, 136.83, 135.12, 134.90, 126.69, 125.08, 123.15, 123.07, 120.86, 120.56, 117.71, 102.69, 56.12 (-OCH3), 55.67 (-OCH3).
- 2-[(furan-3-yl)methylidene]-1H-indene-1,3(2H)-dione (3H)The product was obtained as a brown powder; 0.612 g (27.30%); m.p. 152–153 °C. C14H8O3 (calcd 224.22). ESI-MS: (M + H)+ = 225.10. 1H-NMR (CDCl3), δ [ppm]: δ 8.64 (s, 1H =CH-), 7.97 (m, 2H -Ar), 7.78 (m, 2H -Ar), 7.72 (s, 1H furan ring), 7.52 (m, 1H furan ring), 7.38 (m, 1H furan ring). 13C-NMR (CDCl3), δ [ppm]: 190.34 (aliphatic C in ring), 189.53 (aliphatic C in ring), 152.45, 144.53, 142.48, 140.40, 135.40, 135.18 (=CH-), 135.00, 127.76, 123.39, 123.24, 121.99, 113.08.
- 2-{[4-(diphenylamino)phenyl]methylidene}-1H-indene-1,3(2H)-dione (3I)The product was obtained as a brick-red powder; 1.617 g (80.57%); m.p. 211–212 °C. C28H19NO2 (calcd 401.48). ESI-MS: (M + H)+ = 402.20. 1H-NMR (CDCl3), δ [ppm]: δ 8.40 (d, J = 10 Hz, 2H -Ar), 7.96 (m, 2H -Ar), 7.80 (s, 1H =CH-), 7.77 (m, 2H -Ar), 7.38 (m, 2 × 2H -Ar), 7.22 (m, 2 × 3H -Ar), 7.03 (d, J = 10 Hz, 2H -Ar). 13C-NMR (CDCl3), δ [ppm]: 191.41 (aliphatic C in ring), 189.79 (aliphatic C in ring), 152.87, 146.73, 145.91, 142.52, 140.13, 136.95, 134.92, 134.68, 129.90, 126.71, 125.65, 122.99, 122.96, 119.02.
- 2-[(4-hydroxyphenyl)methylidene]-1H-indene-1,3(2H)-dione (3J)The product was obtained as a red powder; 0.980 g (78.40%); m.p. 238–240 °C. C16H10O3 (calcd 250.26). ESI-MS: (M + H)+ = 251.10. 1H-NMR (DMSO-d6), δ [ppm]: δ 6.94 (d, J = 14 Hz, 2H -Ar); 7.74 (s, 1H =CH); 7.92 (m, 4H -Ar); 8.53 (d, J = 14 Hz, 2H -Ar); 10.91 (s, 1H -OH). 13C-NMR (DMSO-d6), δ [ppm]: 189.98 (aliphatic C in ring), 189.02 (aliphatic C in ring), 163.31, 146.30 (=CH-), 141.66, 137.60, 135.55, 135.38, 122.75, 122,70, 116.00.
- 2-[(6-hydroxy-2H-1-benzopyran-3-yl)methylidene]-1H-indene-1,3(2H)-dione (3K)The product was obtained as a brown-black powder; 0.116 g (15.24%); m.p. > 260 °C. C19H12O4 (calcd 304.31). ESI-MS: (M + H)+ = 305.1. 1H-NMR (DMSO-d6), δ [ppm]: δ 5.33 (s, 2H); 6.70 (s, 1H -Ar); 6.76 (d, J = 1.4 Hz, 2H -Ar); 7.40 (s, 1H -Ar); 7.71 (s, 1H =CH); 7.94 (m, 4H -Ar), 9.31 (s, 1H -OH). 13C-NMR (DMSO-d6), δ [ppm]: 189.35 (aliphatic C in ring), 188.55 (aliphatic C in ring), 152.05, 148.25, 142.62 (=CH-), 140.86, 135.79, 130.08, 122.93, 122.85, 116.58, 114.34, 66.55.
References
- Pluskota, R.; Koba, M. Indandione and Its Derivatives—Chemical Compounds with High Biological Potential. Mini-Rev. Med. Chem. 2018, 18, 1321–1330. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H.; Lipinski, C.A. The rule of five should not impede anti-parasitic drug development. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 248–249. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, E.; Pajak, K.; Jóźwiak, K. Lipophilicity-methods of determination and its role in medicinal chemistry. Acta Pol. Pharm. 2013, 70, 3–18. [Google Scholar] [PubMed]
- Bakht, M.A.; Yar, M.S.; Abdel-Hamid, S.G.; Alqasoumi, S.I.; Samand, A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5862–5869. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional Structure—Directed Quantitative Structure—Activity Relationships I. Partition Coefficients as a Measure of Hydrophobicity. J. Comput. Chem. 1986, 7, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tugrak, M.; Gul, H.I.; Sakagami, H.; Gulcin, I.; Supuran, C.T. New Azafluorenones with Cytotoxic and Carbonic Anhydrase Inhibitory Properties: 2-Aryl-4-(4-hydroxyphenyl)-5H-indeno[1,2-b]pyridin-5-ones. Bioorg. Chem. 2018, 81, 433–439. [Google Scholar] [CrossRef]
- Mitka, K.; Kowalski, P.; Pawelec, D. Synthesis of Novel Indane-1,3-dione Derivatives and Their Biological Evaluation as Anticoagulant Agents. Croat. Chem. Acta 2009, 82, 613–618. [Google Scholar]
- Oliveira, A.F.C.D.S.; de Souza, A.P.M.; de Oliveira, A.S.; da Silva, M.L.; de Oliveira, F.M.; Santos, E.G.; da Silva, Í.E.P.; Ferreira, R.S.; Villela, F.S.; Martins, F.T.; et al. Zirconium catalyzed synthesis of 2-arylidene Indan-1,3-diones and evaluation of their inhibitory activity against NS2B-NS3 WNV protease. Eur. J. Med. Chem. 2018, 149, 98–109. [Google Scholar] [CrossRef]
- Kumar, P.S.; Kumar, K.B.; Obadiah, A.; Kumar, S.J.; Mohanapriya, R.; Durairaj, A.; Ramanathan, S.; Vasanthkumar, S. Synthesis, Molecular Docking, Cytotoxicity and Antioxidant Activity Evaluation of Isoindoline-1,3-dione Derivatives. Asian J. Chem. 2019, 31, 2548–2556. [Google Scholar] [CrossRef]
- Yang, M.; Fazio, S.; Munch, D.; Drumm, P. Impact of methanol and acetonitrile on separations based on π–π interactions with a reversed-phase phenyl column. J. Chromatogr. A 2005, 1097, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Tchaikovskaya, O.N.; Basyl, O.K.; Sultimova, N.B. Proton-acceptor and proton-donor properties of phenol and its substitutes. Russ. Phys. J. 2005, 48, 1245–1250. [Google Scholar] [CrossRef]
- Aldeghi, M.; Malhotra, S.; Selwood, D.L.; Chan, A.W.E. Two- and three-dimensional rings in drugs. Chem. Biol. Drug Des. 2014, 83, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Kosmalski, T.; Studzińska, R.; Redka, M.; Pluskota, R.; Modzelewska-Banachiewicz, B. Lipophilicity Study of 1-(Benzofuran-2-yl)ethan-1-one Oxime and its Substituted O-Benzyl Ethers. J. Braz. Chem. Soc. 2017, 28, 2100–2105. [Google Scholar] [CrossRef]
- Ryckmans, T.; Edwards, M.P.; Horne, V.A.; Correia, A.M.; Owen, D.R.; Thompson, L.R.; Tran, I.; Tutt, M.F.; Young, T. Rapid assessment of a novel series of selective CB(2) agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- van de Weert, M.; Stella, L. Fluorescence quenching and ligand binding: A critical discussion of a popular methodology. J. Mol. Struct. 2011, 998, 144–150. [Google Scholar] [CrossRef]
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: A new approach. Biochem. Pharmacol. 2002, 64, 355–374. [Google Scholar] [CrossRef]
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR modelling of rat acute toxicity on the basis of PASS prediction. Mol. Inform. 2011, 30, 241–250. [Google Scholar] [CrossRef]
- Maunz, A.; Gütlein, M.; Rautenberg, M.; Vorgrimmler, D.; Gebele, D.; Helma, C. Lazar: A modular predictive toxicology framework. Front. Pharmacol. 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipiński’s Rule | Veber’s Rule | ||
|---|---|---|---|
| Feature | Value | Feature | Value |
| LogP | <5 | number of rotating bonds (RotB) | ≤10 |
| molecular weight (MW) | <500 Da | ||
| number of hydrogen bond acceptors (AHB) | <10 | polar surface area (PSA) | <140 Å2 |
| number of hydrogen bond donors (DHB) | <5 | ||
| Compound | MeOH | ACN | Δ * | ||||
|---|---|---|---|---|---|---|---|
| logkw | R2 | −S | logkw | R2 | −S | ||
| 3A | 3.3210 | 0.9907 | 3.3278 | 2.2433 | 0.9974 | 2.4449 | 1.0777 |
| 3B | 3.2790 | 0.9955 | 3.1597 | 2.2394 | 0.9982 | 2.3613 | 1.0396 |
| 3C | 3.3554 | 0.9968 | 3.2782 | 2.3042 | 0.9976 | 2.4879 | 1.0512 |
| 3D | 5.6763 | 0.9966 | 5.4798 | 3.5726 | 0.9993 | 3.5570 | 2.1037 |
| 3E | 2.2321 | 0.9917 | 2.4114 | 1.5356 | 0.9919 | 1.9201 | 0.6965 |
| 3F | 3.9640 | 0.9967 | 3.8549 | 2.6107 | 0.9987 | 2.7449 | 1.3533 |
| 3G | 4.6064 | 0.9972 | 4.6064 | 2.8533 | 0.9991 | 2.8762 | 1.7531 |
| 3H | 2.5521 | 0.9959 | 2.6243 | 1.9162 | 0.9950 | 2.2011 | 0.6359 |
| 3I | 5.3524 | 0.9994 | 5.0483 | 3.8489 | 0.9995 | 3.7589 | 1.5035 |
| 3J | 2.3814 | 0.9952 | 2.5802 | 3.7694 | 0.9997 | 3.7694 | 1.3880 |
| 3K | 2.9717 | 0.9946 | 3.0667 | 1.7720 | 0.9967 | 2.1213 | 1.1997 |
| Compound | MLOGP * | ALOGP ** | LogP o/w *** |
|---|---|---|---|
| 3A | 3.322 | 3.236 | 3 |
| 3B | 3.235 | 3.399 | 3 |
| 3C | 2.998 | 3.22 | 2.99 |
| 3D | 4.402 | 5.283 | 4.6 |
| 3E | 2.458 | 2.953 | 2.61 |
| 3F | 4.116 | 4.145 | 3.91 |
| 3G | 3.451 | 4.112 | 3.89 |
| 3H | 1.664 | 2.34 | 2.31 |
| 3I | 5.218 | 6.569 | 5.45 |
| 3J | 2.754 | 2.969 | 2.58 |
| 3K | 2.462 | 3.134 | 2.76 |
| Compound | LD50 * | Toxicity Class ** |
|---|---|---|
| 3A | 1113.0 | harmful |
| 3B | 1707.0 | harmful |
| 3C | 3167.0 | unclassified |
| 3D | 2937.0 | unclassified |
| 3E | 2608.0 | unclassified |
| 3F | 742.5 | harmful |
| 3G | 1707.0 | harmful |
| 3H | 1264.0 | harmful |
| 3I | 2176.0 | unclassified |
| 3J | 2373.0 | unclassified |
| 3K | 577.4 | harmful |
| indan-1,3-dione | 297.8 | harmful |
| doxorubicin | 110.8 | toxic |
| Compound | Acute Toxicity [mg/L] | Overcoming the Blood–Brain Barrier * | Carcinogenicity ** | Mutagenicity *** | |
|---|---|---|---|---|---|
| Fathead minnow | Daphnia magna | ||||
| 3A | 0.236 | 6.58 | penetrating | non-carcinogenic | mutagenic |
| 3B | 21.4 | 9.65 | non-penetrating | carcinogenic | non-mutagenic |
| 3C | 32.1 | 3.59 | non-penetrating | non-carcinogenic | mutagenic |
| 3D | 69.8 | 42.2 | penetrating | non-carcinogenic | mutagenic |
| 3E | 31.7 | 9.4 | non-penetrating | non-carcinogenic | mutagenic |
| 3F | 4.51 | 2.82 | non-penetrating | non-carcinogenic | mutagenic |
| 3G | 64.5 | 9.3 | penetrating | carcinogenic | mutagenic |
| 3H | - **** | - **** | penetrating | non-carcinogenic | non-mutagenic |
| 3I | - **** | 10.6 | non-penetrating | non-carcinogenic | mutagenic |
| 3J | 13.7 | 12.3 | penetrating | non-carcinogenic | mutagenic |
| 3K | 24.4 | 23.4 | non-penetrating | non-carcinogenic | mutagenic |
| indan-1,3-dione | 52.9 | 25.7 | penetrating | non-carcinogenic | non-mutagenic |
| Doxorubicin | - ***** | - ***** | non-penetrating | carcinogenic | mutagenic |
 | ||
| 1 2A–2K 3A–3K 4 | ||
 |  |  |
| A | B | C |
 |  |  |
| D | E | F |
 |  |  |
| G | H | I |
 |  | |
| substrate | ||
 | ||
| product | ||
| J * | K | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pluskota, R.; Jaroch, K.; Kośliński, P.; Ziomkowska, B.; Lewińska, A.; Kruszewski, S.; Bojko, B.; Koba, M. Selected Drug-Likeness Properties of 2-Arylidene-indan-1,3-dione Derivatives—Chemical Compounds with Potential Anti-Cancer Activity. Molecules 2021, 26, 5256. https://doi.org/10.3390/molecules26175256
Pluskota R, Jaroch K, Kośliński P, Ziomkowska B, Lewińska A, Kruszewski S, Bojko B, Koba M. Selected Drug-Likeness Properties of 2-Arylidene-indan-1,3-dione Derivatives—Chemical Compounds with Potential Anti-Cancer Activity. Molecules. 2021; 26(17):5256. https://doi.org/10.3390/molecules26175256
Chicago/Turabian StylePluskota, Robert, Karol Jaroch, Piotr Kośliński, Blanka Ziomkowska, Agnieszka Lewińska, Stefan Kruszewski, Barbara Bojko, and Marcin Koba. 2021. "Selected Drug-Likeness Properties of 2-Arylidene-indan-1,3-dione Derivatives—Chemical Compounds with Potential Anti-Cancer Activity" Molecules 26, no. 17: 5256. https://doi.org/10.3390/molecules26175256